Back to Journals » ImmunoTargets and Therapy » Volume 14
A High-Throughput, Three-Dimensional Multiple Myeloma Model Recapitulating Tumor-Stroma Interactions for CAR-Immune Cell-Mediated Cytotoxicity Assay
Authors Luanpitpong S ![]() , Janan M, Poohadsuan J, Rodboon N, Samart P, Rungarunlert S, Issaragrisil S
, Janan M, Poohadsuan J, Rodboon N, Samart P, Rungarunlert S, Issaragrisil S
Received 19 November 2024
Accepted for publication 18 March 2025
Published 30 March 2025 Volume 2025:14 Pages 321—338
DOI https://doi.org/10.2147/ITT.S503984
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Sudjit Luanpitpong,1– 3 Montira Janan,1– 3 Jirarat Poohadsuan,1 Napachai Rodboon,1 Parinya Samart,1 Sasitorn Rungarunlert,4 Surapol Issaragrisil1,5,6
1Siriraj Center of Excellence for Stem Cell Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 2Siriraj Cell Factory for Cancer Immunotherapy, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 3Blood Products and Cellular Immunotherapy Research Group, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 4Department of Preclinic and Applied Animal Science, Faculty of Veterinary Science, Mahidol University, Nakhon Pathom, Thailand; 5Division of Hematology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 6BDMS Center of Excellence for Hematology, Wattanosoth Cancer Hospital, Bangkok, Thailand
Correspondence: Sudjit Luanpitpong; Surapol Issaragrisil, Siriraj Center of Excellence for Stem Cell Research, Faculty of Medicine Siriraj Hospital, Mahidol University, 2 Siriraj Hospital, Bangkok, 10700, Thailand, Tel +6624192907 ; +6624194446, Email [email protected]; [email protected]
Background: Multiple myeloma (MM) is characterized by an excessive proliferation of clonal plasma cells in the bone marrow (BM). Components in BM niche contribute to the immunosuppressive tumor microenvironment (TME), but three-dimensional (3D) MM models that recreate the complex TME and enable high-throughput cytotoxicity assay of chimeric antigen receptor (CAR)-engineered immune cells are still lacking.
Methods: Stable, luciferase (Luc)-labeled target MM cells were generated using Luc/RFP dual reporter system to track MM growth. 3D spheroids were formed in a 96-well plate in the presence or absence of cancer-associated fibroblast (CAF)-like stromal cells activated by MM-derived conditioned medium and the cytotoxicity of CAR-immune cells, which were represented by third-generation anti-CD138 CAR-NK-92 cells, was evaluated by luciferase assay using a multimode microplate reader. Immune cell infiltration was visualized under a fluorescence microscope by using multiple fluorescent dyes.
Results: We first showed that luciferase assay provides a relatively simple and robust means to specifically monitor Luc-labeled tumor cell growth in a coculture system, allowing the high-throughput assessment of CAR-immune cytotoxicity. Through this assay, we demonstrated that CAF-like stromal cells impaired NK cell effector function in 2D culture and 3D spheroids, likely via paracrine signaling and physical barrier function. Importantly, we showed that 3D spheroids consisting of MM cells and CAF-like stromal cells provide a more comprehensive, physiologically relevant immuno-oncology model. Our established model could also be used to investigate the trafficking and infiltration of immune cells into the core of spheroids. Herein, we showed that CAR incorporation did improve the ability of NK cells to infiltrate 3D spheroids.
Conclusion: Our established 3D spheroid model, which partially recapitulates the complex TME with immunosuppressive environment, is suitable for high-throughput screening of CAR-immune cytotoxicity and could be important in accelerating immuno-oncology drug discovery for MM since there is a pressing need to establish innovative CAR-immune cells.
Keywords: 3D culture, multiple myeloma, tumor microenvironment, stromal cells, cancer-associated fibroblasts, CAR
Introduction
Immunotherapy harnessing the power of immune system to combat cancer is heralded as the fifth pillar of cancer therapy, alongside surgery, chemotherapy, radiotherapy, and targeted therapy. The cellular and molecular mechanisms of cancer immune escape have led to the development of immune checkpoint inhibitors (ICIs) against CTLA-4, PD-1, and PD-L1, which have been widely used for the treatment of a broad spectrum of advanced cancers, such as metastatic melanoma, non-small cell lung cancer, renal carcinoma, and Hodgkin lymphoma, in the past decade.1,2 Adoptive cell therapy (ACT) is a more complex approach to immunotherapy involving the genetic engineering of patient’s own or donor immune cells to express tumor antigen-specific chimeric antigen receptor (CAR) or T-cell receptor (TCR) to enhance their capability to recognize and target tumor cells in vivo.3
CAR-T cells have emerged as a standard of care for relapsed/refractory B-cell malignancies, ie, acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL), since the approval of the first two anti-CD19 CAR-T cell products by the US Food and Drug Administration (FDA) in 2017.4,5 Most recently, two anti-B cell maturation antigen (BCMA) CAR-T cells have been approved for the treatment of relapsed/refractory multiple myeloma (MM), and a total of six products are available thus far.6 Despite the remarkable clinical success of CAR-T cells in certain hematologic malignancies, there are still various major limitations and challenges that restrict their efficacy and/or widespread use, eg, laborious manufacturing process, severe life-threatening toxicities, tumor antigen escape, restricted trafficking, and limited tumor infiltration.7 Several types of CAR-immune cells have been rapidly developed and are currently undergoing clinical studies as alternatives to CAR-T cells, including CAR-natural killer (NK) cells, CAR-NKT cells, and CAR-macrophages (CAR-M), all of which have a greater off-the-shelf potential, a likely lower production cost, and safer clinical profiles.8,9 Likewise, multiple engineering strategies have been explored to design better CARs to improve therapeutic efficacy and tumor specificity, and overcome antigen escape and immunosuppression in the tumor microenvironment (TME), for example, screening and verification of new target antigens, introducing the dependency of functional activation on multiple tumor antigens, targeting tumor stroma, etc.10,11 Hence, there is a pressing need for high-throughput screening models for evaluating the cytotoxicity of CAR-immune cells to identify the potential therapeutic candidates for further preclinical and clinical studies.
MM is the second most common hematologic malignancy worldwide and is characterized by excessive proliferation of clonal plasma cells in the bone marrow (BM) and overproduction of monoclonal immunoglobulins.12,13 As mentioned, CAR-T cells present promising advances in the treatment of MM; however, the durability of responses remains a therapeutic challenge.14,15 A significant proportion of MM patients eventually experience relapse although the majority of patients (> 70%) initially respond to anti-BCMA CAR-T cells. In conventional two-dimensional (2D) cell culture system, individual MM cell lines are propagated in suspension, and thus interactions between different cell populations and mechanical cues are not present, unlike in the BM.16,17 It is widely accepted that three-dimensional (3D) cell culture models better mimic the tumor architecture and microenvironment, providing more physiological relevance. Although the importance of BM environment in supporting MM growth has been previously reported,13,18 3D MM models that recreate the complex TME for immuno-oncology research, particularly for the high-throughput assessment of the cytotoxicity of immune cells, are still lacking. The evaluation of immune cytotoxicity was initially based on live/dead staining and subsequent evaluation/visualization by flow cytometry or imaging, which are laborious. Previously, we designed a third-generation CAR targeting CD138 (syndecan-1), which is highly expressed in MM, and established anti-CD138 CAR-NK-92 (CAR138 NK-92) cells with high and selective cytotoxicity against CD138-positive MM cells as evaluated in the 2D culture.19 In the present study, CAR138 NK-92 cells were therefore used as a representative CAR-immune cells for testing against the established high-throughput 3D MM model. Our preclinical in vitro model could be important in accelerating immuno-oncology drug discovery for MM since there is a pressing need for identification and optimization of novel CAR-immune cells.
Materials and Methods
Cell Culture
Human NK-92 cells, and hematologic cancer cell lines, including human mantle cell lymphoma (MCL) Jeko-1 cells and human MM NCI-H929 and MM.1R cells were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Normal, human diploid lung fibroblasts, WI-38-40 (JCRB0518), and immortalized, human BM-derived mesenchymal stem cell (MSC) line, UE6E7T-3 (JCRB1136), were obtained from Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan), and human embryonic kidney (HEK) 293FT cells were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Mycoplasma contamination was checked every eight weeks using MycoAlertTM PLUS mycoplasma detection kit (Lonza, Cologne, Germany), and any cell lines found positive were discarded.
NK-92 cells were cultured in Minimum Essential Medium Alpha (MEM α; Gibco, Thermo Fisher Scientific) containing 100 U/mL recombinant human (rh) IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany), 12.5% horse serum (Invitrogen, Carlsbad, CA, USA), and 12.5% fetal bovine serum (FBS; Merck Millipore, Darmstadt, Germany). HEK293FT cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), while WI-38-40 and UE6E7T-3 cells were cultured in MEM and DMEM with low glucose, respectively. All other cells were cultured in RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
CAR Constructs and Anti-CD19 and Anti-CD138 CAR-NK-92 Cells
Third-generation CARs targeting CD19 (anti-CD19 CAR) and CD138 (anti-CD138 CAR) with a CD3ζ signaling domain and costimulatory domains CD28 and 4–1BB were designed, constructed and cloned into lentiviral vector Lenti-EF1a-AT-Free by Creative Biolabs (Shirley, NY, USA). The inserts were confirmed by Sanger sequencing. Stable, anti-CD19 CAR and anti-CD138 CAR-expressing NK-92 cells, designated as CAR19 NK-92 and CAR138 NK-92 cells, respectively, were subsequently established as previously described.19 The CAR expression on CAR19 NK-92 and CAR138 NK-92 cells were routinely checked by flow cytometry based on F(ab’)2 fragment and target antigen-specific detection to ensure their expression and functionality as previously described.19
Luciferase Reporter and Lentiviral Production
Lentiviral dual reporter UbC-RFP-T2A-luciferase (Luc) plasmid was obtained from System Biosciences (Mountain View, CA, USA). To produce lentiviral particles, the Luc/RFP reporter plasmid was incubated with HEK293FT packaging cells in conjunction with pCMV.dR8.2 dvpr lentiviral packaging and pCMV-VSV-G envelope plasmids (Addgene #8454 and #8455)20 at a ratio of 12:5:1 using Lipofectamine 3000 (Thermo Fisher Scientific). HEK293FT cells were checked for the RFP positivity by flow cytometry at 24 and 48 h posttransfection, and the particles were harvested, pooled, and concentrated using Amicon Ultra-15 centrifugal filters (Merck Millipore, Tullagreen, Ireland) at 4500 × g for 1 h.
Generation of Stable Luc-Labeled Target Cells
Target tumor cells were transduced with lentiviral particles containing the Luc/RFP reporter using spinoculation technique. Briefly, cells at 5×105 cells/mL were transduced with concentrated viral particles in the presence of polybrene (8 μg/mL) (Miltenyi Biotec), followed by a spinoculation at 800 × g for 2 h at room temperature. At 24 h posttransduction, the cells were replenished with fresh medium and cultured for an additional 48 h. After which, the cells were enriched by fluorescence-activated cell sorting (FACS) for RFP-positive cells using FACSAria cell sorter (BD Biosciences, San Jose, CA, USA). Cell transduction and sorting were repeated thrice to ensure that more than 80% of the cells in culture were positive for RFP. The enriched cells were then checked for luciferase activity by a multimode microplate reader or bioluminescence imaging as described below. To verify the stability of luciferase expression, the cells were maintained in cultured for two months plus three freeze-thaw cycles.
Luciferase Activity by Multimode Microplate Reader and Bioluminescence Imaging
Luc-labeled tumor cells were seeded into a 96-well plate at the indicated cell number and treated with 150 μg/mL IVISbrite D-luciferin, potassium salt (Revvity, Waltham, MA, USA) for at least 5 min (2D culture) or 1 h (3D culture) prior to the measurement of luciferase activity by luminescence detection using VICTOR Nivo S multimode microplate reader with a luminescence filter (700 nm IR Blocker; Revvity) using VICTOR Nivo 5.0.1 software. The luminescence results were presented as counts. For bioluminescence imaging, the cells were similarly treated with IVISbrite D-luciferin and monitored on an IVIS Spectrum imaging system (PerkinElmer, Waltham, MA, USA) using Living Image Software 4.3.0. The luminescence results were presented as total flux (photons (p)/sec).
Luciferase-Based NK Cytotoxicity Assay
2D or 3D culture of Luc-labeled target cells were cocultured with wild type (WT) or CAR-NK-92 cells at various effector-to-target (E:T) ratios for the indicated times (0−24 h). The luminescence signals were subsequently detected by a multimode microplate reader. The percentage of killing was calculated using the following formula: % specific killing = [(average signal from well with target cells alone) − (signal from well with effector and target cells)]/(average signal from well with target cells alone) × 100.
Flow Cytometry-Based NK Cytotoxicity Assay
Target tumor cells were labeled with PKH67 green fluorescent lipophilic dye for 5 min at room temperature and were mixed with various E:T ratios of CAR-NK-92 cells for the indicated times (0−24 h). After which, target cell death was evaluated by Annexin V/7-AAD assay. The mixture of effector and target tumor cells was harvested, washed, and stained with PE-conjugated Annexin V and 7-AAD in binding buffer supplemented with 5 mmol/L calcium chloride for 15 min at room temperature. Samples were immediately analyzed by the FACScalibur flow cytometer (BD Biosciences) with a specific gating strategy to determine the percentage of cell death of the target tumor cells, which was defined as Annexin V- and/or 7-AAD-positive cells in the PKH67-positive subpopulation.
Collection of MM-Derived Conditioned Medium and Activation of the Stromal Cells
Human MM NCI-H929-Luc cells at a density of 0.5−1×106 cells/mL were cultured in RPMI 1640 medium containing 10% FBS in a T75 flask with a total volume of 20 mL for 48 h to generate the conditioned medium (MM-CM). The collected MM-CM was centrifuged at 2500 rpm at 25 °C for 10 min to remove cell pellets and stored at − 80 °C until use. For stromal cell activation, 2×105 WI-38-40 or UE6E7T-3 cells were plated into a 6-well plate and left overnight. The normal culture medium was removed and replaced with 4 mL MM-CM for an additional 48 h.
qPCR Analysis
Total RNA was isolated using TRI Reagent® (Molecular Research Center, Cincinnati, Ohio, USA) and 2 μg of total RNA was converted to complementary DNA (cDNA) using the RevertAid First Strand cDNA synthesis kit (Thermo Fisher Scientific). The qPCR reactions consisted of 1X Select Master Mix (Thermo Fisher Scientific), 200 nM of forward and reverse primers (see also Supplementary Table S1 for primer sequences), and 1 μL of diluted template cDNA (~40 ng) were run on the CFX384 Touch Real-Time PCR detection system (Bio-Rad, Hercules, CA, USA). The cycle parameters started with an activation step at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 15 sec and annealing/extension at 60 °C for 1 min. Relative expression of each gene was normalized to the housekeeping GAPDH gene product. Heatmap and hierarchical cluster analysis were performed using Bio-Rad CFX Manager 3.0 software.
Formation of 3D Tumor Spheroids
3D tumor spheroids were generated in the presence or absence of activated stromal cells, designated as 3D MM/CAF and bare 3D MM spheroids, respectively. Briefly, 5×103 Luc-labeled MM cells with or without activated WI-38-40 cells at 5 or 10×103 cells per well (1:1 or 1:2 ratio) were resuspended in 1.2% methylcellulose (MC) (H4100; Stem Cell Technologies, Vancouver, Canada) supplemented with 30% FBS, 1% bovine serum albumin (BSA), 100 μM 2-mercaptoethanol, and 2 mm GlutaMax (Gibco). The mixture was then plated in ultralow adherent, round bottom 96-well plates (Thermo Fisher Scientific) and cultured for 5−6 days. The growth of MM cells was measured by luciferase activity as described above.
Immune Cell Infiltration
To visualize different subpopulations in the 3D spheroids, target tumor cells were prelabeled with PKH26 red-fluorescent, lipophilic membrane dye (Sigma-Aldrich, St. Louis, MO), whereas activated WI-38-40 cells were prelabeled with PKH67 green-fluorescent dye (Sigma-Aldrich) before being resuspended in the MC-based matrix, as described above. On day 6 of culture, effector WT or CAR138 NK-92 cells were prelabeled with Vybrant DyeCycle Violet stain (Thermo Fisher Scientific, Waltham, MA) and added to the spheroids for a 24-h incubation. Visualization was conducted using a Leica DMi8 inverted fluorescence microscope equipped with a Leica DFC6200 camera and Leica Application Suite X (LAS X) imaging software (version 3.7.6) (Leica Microsystems, Wetzlar, Germany). For each sample, a minimum of 150 z-stacks were acquired at an interval of 8 μm. Measurement of region of interest (ROI) was performed on the merged z-stack micrographs using LAS X.
Statistical Analysis
The data represent means ± S.D. from three or more independent experiments or as indicated. Statistical analysis was performed by Student’s t-test or one-way ANOVA followed by Tukey’s multiple comparison test at a significance level of P < 0.05 using GraphPad Prism software 8.0. Descriptive statistics, ie, 95% confidence interval (CI; upper and lower bounds) for cytotoxicity data are summarized in Supplementary Tables S2−S5.
Results
Characterization of Luc-Labeled Target Cells
One of the most common assays for CAR-immune cytotoxicity is the flow cytometry-based assay using Annexin V and/or propidium iodide (PI) or 7-AAD dyes. However, to distinguish between tumor and effector immune cells, the additional staining of either tumor or effector cells with a fluorescent cell staining dye, eg, PKH67 or carboxyfluorescein succinimidyl ester (CSFE), is needed.19,21,22 For high-throughput applications, we generated various Luc-labeled tumor cells, including Jeko-1-Luc, MM.1R-Luc, and NCI-H929-Luc cells, through multiple rounds of lentiviral transduction of Luc/RFP reporter transgene and sequential FACS sorting, which allowed the direct measurement of luciferase activity upon exposure to D-luciferin substrate (Figure 1A). The enriched Luc-labeled cells were checked after being maintained in culture for more than two months or at > 20 passages after FACS sorting. Figure 1B shows a linear correlation between luminescence signal from the microplate reader and cell number (R2 > 0.99). The linear correlation was also found between IVIS bioluminescence intensity and cell number (R2 > 0.99) (Figure 1C; see also Supplementary Figure S1 for results of MM.1R-Luc cells), and hence these Luc-labeled tumor cells can be used for in vivo xenograft mouse models as well.
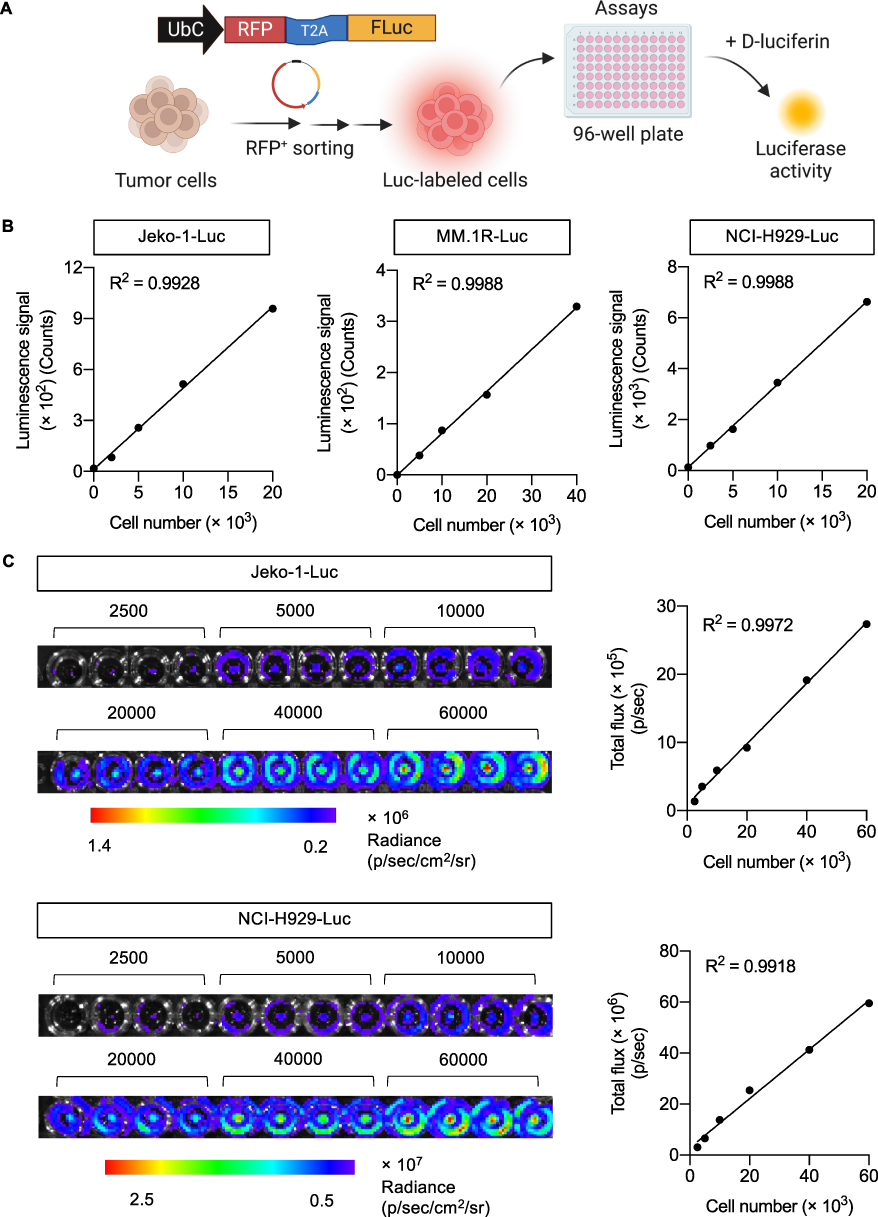 |
Figure 1 Luciferase activity in Luc-labeled tumor cells linearly correlates with cell number. (A) Schematic diagram of the generation of Luc-labeled tumor cells and measurement of luciferase activity in 96-well plate following the addition of luciferase substrate D-luciferin. (B and C) Luciferase activity in human MCL Jeko-1-Luc, MM NCI-H929-Luc, and MM MM.1R-Luc cells was assessed by luminescence microplate reader (B) and IVIS bioluminescence imaging (C). Data showed a linear correlation between luminescence signal (count or total flux) and cell number in both detection systems (see also Supplementary Figure S1). Figure 1A created in BioRender. Luanpitpong, S. (2025) https://BioRender.com/ f56a368. |
CAR-NK Cells Show Dose-Dependent Cytotoxicity Against Luc-Labeled Target Cells
Next, CAR-immune cytotoxicity against the established Luc-expressing tumor cells in 2D culture was measured. The cytotoxic functions of CAR138 NK-92 cells (effector cells, E) against CD138-positive NCI-H929-Luc MM cells (target cells, T) were determined by luciferase assay using a multimode microplate reader in comparison to the conventional flow cytometry-based Annexin V/7-AAD assay at various E:T ratios, as schematically illustrated in Figure 2A. The cytotoxicity of CAR19 NK-92 cells (E) against CD19+ Jeko-1-Luc cells (T) was also included as CD19 is considered a standard target antigen. We assessed the expression of surface CD19 and CD138 in Jeko-1-Luc and NCI-H929-Luc cells, respectively, to confirm that expression of target antigen in Luc-labeled cells was not significantly different from that in parental cells (Figure 2B). Figure 2C shows that both CAR19 and CAR138 NK-92 cells were highly cytotoxic toward their corresponding target tumor cells in a dose-dependent manner. Percentages of specific killing in Jeko-1-Luc cells were comparable between luciferase assay and Annexin V/7-AAD assay, although the luciferase assay deemed to be more sensitive in NCI-H929-Luc cells. A significant increase in surface CD107a expression, a functional marker of NK cell activity, in CAR138 NK-92 cells upon NCI-H929 exposure at various E:T ratios further supported the higher cytotoxicity of CAR138 NK-92 cells toward NCI-H929-Luc cells (Supplementary Figure S2).
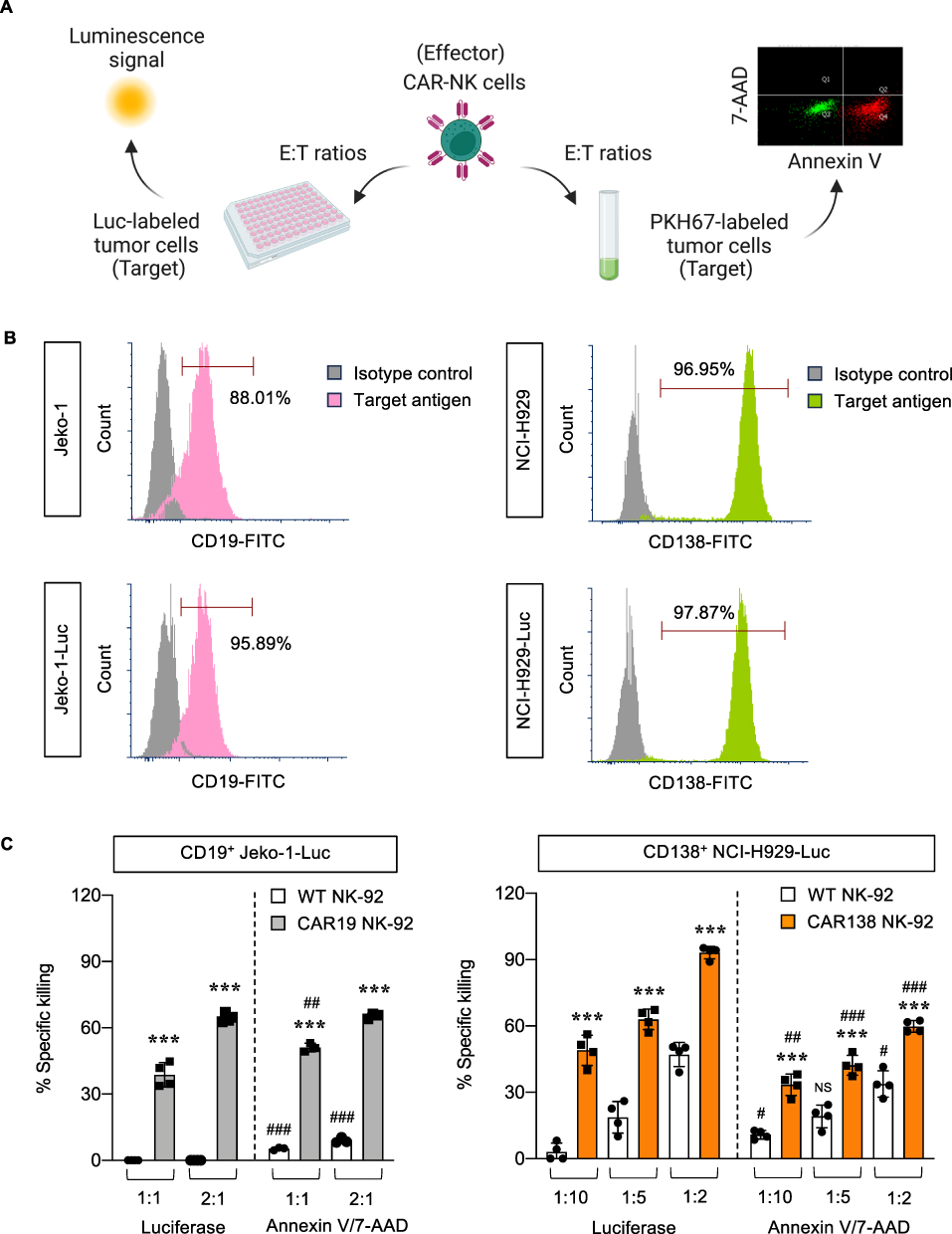 |
Figure 2 CAR-NK cytotoxicity against Luc-labeled tumor cells in 2D culture. (A) Schematic diagram of the parallel evaluation of CAR-NK cytotoxicity by luciferase assay using microplate reader and Annexin V/7-AAD assay using flow cytometry. (B) Flow cytometric analysis of target antigen expression in Luc-labeled tumor cells, ie, CD19 in Jeko-1-Luc cells and CD138 in NCI-H929-Luc cells. (C) Percentages of specific killing of Jeko-1-Luc (left) and NCI-H929-Luc (right) cells in response to CAR19 and CAR138 NK-92 cells, respectively, at indicated E:T ratios at 4 h in comparison to their WT counterparts, as measured by luciferase and Annexin V/7-AAD assays. ***P < 0.001 versus WT NK-92 cells at the same concentration; ##P < 0.01, ###P < 0.001 versus data from luciferase assay under the same conditions; two-sided Student’s t-test. Figure 2A created in BioRender. Luanpitpong, S. (2025) https://BioRender.com/ r44v780. |
MM-CM Activates a Cancer-Associated Fibroblast-Like Phenotype in Stromal Cells
It is well-accepted that stromal cells in the TME plays a fundamental role in tumor growth and progression of numerous cancers.23,24 Cancer-associated fibroblasts (CAFs), the most prominent cell type in the TME, can be activated or differentiated from stromal cells, including normal fibroblasts and MSCs under the influence of tumor cells via paracrine signaling and/or direct cell-cell interaction.23,25 CAFs have long been considered the protumorigenic component of the TME.26,27 Here, we collected the MM-CM from NCI-H929-Luc cells at 0.5 or 1×106 cells/mL to activate UE6E7T-3 MSCs and WI-38-40 normal fibroblasts, as schematically illustrated in Figure 3A. The inflammatory and myofibroblast markers, which are generally expressed on inflammatory CAFs (iCAFs) and myofibroblastic CAFs (myCAFs), respectively,23,27 were determined by qPCR at 48 h. Figure 3B shows that UE6E7T-3 cells expressed higher mRNA levels of iCAF-like markers than WI-38-40 cells at the basal state and upon activation by MM-CM. WI-38-40 cells, by contrast, expressed relatively higher expression of myCAF-like markers. When analyzed separately, we found that MM-CM induced the expression of both iCAF- and myCAF-like markers in WI-38-40 cells and, to a lesser extent, in UE6E7T-3 cells (Figure 3C and D). The dose-dependent activation of CAF markers in response to MM-CM in WI-38-40 cells included PDPN, IL1A, IL6, ACTA2, and MYL9, and in UE6E7T-3 cells included IL1A, IL6, and TAGLN. Additionally, Western blot analysis revealed an activation of common CAF markers, ie, FAP and S100A4, in both MM-CM-exposed UE6E7T-3 and WI-38-40 cells (Supplementary Figure S3). Meanwhile, the level of IL-6, but not TGF-β, was found to increase in UE6E7T-3 cells by MM-CM, supporting its iCAF-like phenotype. Although the CAF profiles are not fully characterized herein, these results confirmed the acquisition of activated fibroblasts and MSCs with a CAF-like phenotype by MM cells via paracrine signaling. Coculture of NCI-H929-Luc cells and activated stromal cells in 2D culture were found to promote the growth of NCI-H929-Luc cells at 24−72 h, thereby validating the tumor-promoting role of activated stromal cells used herein (Supplementary Figure S4).
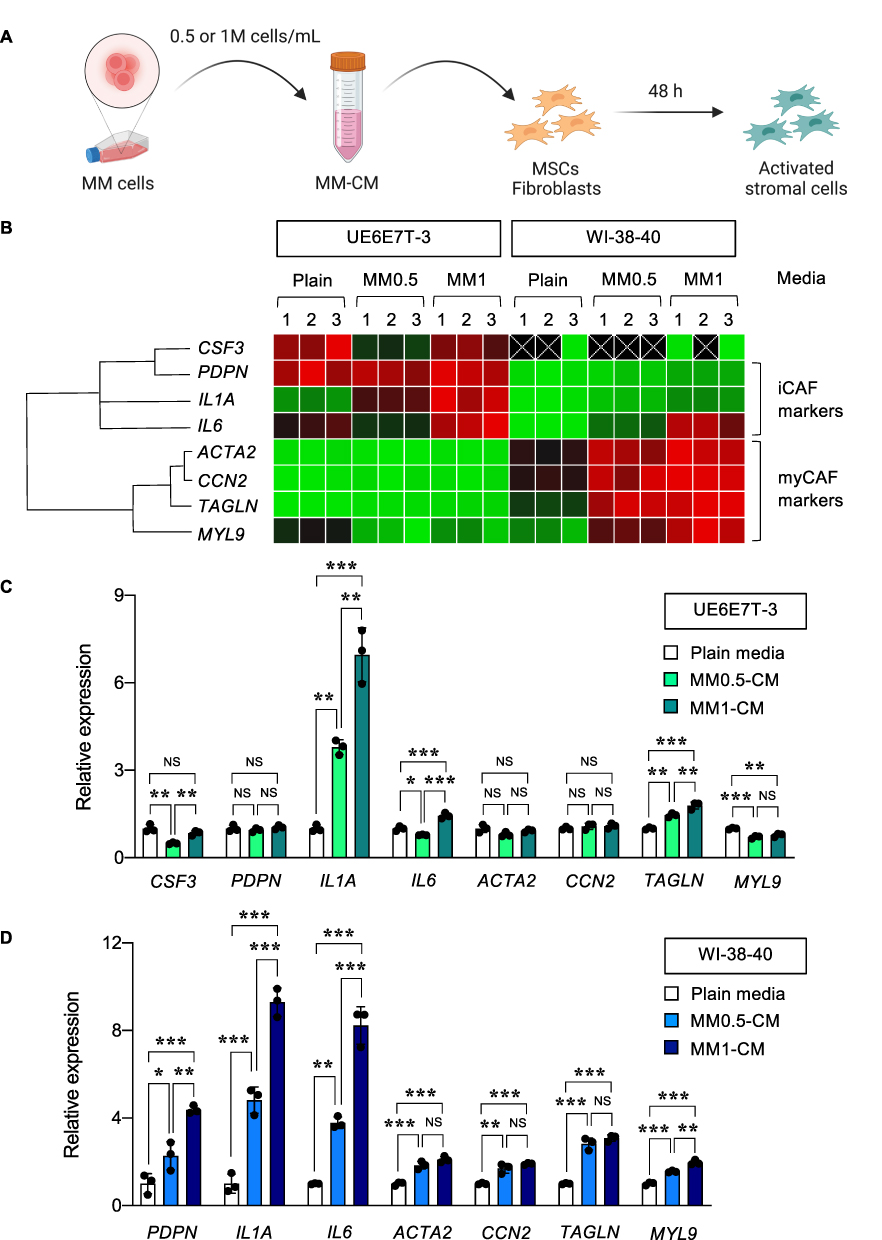 |
Figure 3 Activation of stromal cells with a CAF-like phenotype by MM-CM. (A) Schematic diagram showing the collection of MM-CM from NCI-H929 cells and activation of stromal cells, ie, UE6E7T-3 MSCs and WI-38-40 fibroblasts. (B−D) qPCR analysis of UE6E7T-3 and WI-38-40 cells upon exposure to various concentrations of MM-CM, ie, MM0.5-CM and MM1-CM. (B) Data from three biological replicates were normalized to the house-keeping gene GAPDH, averaged and represented in a heatmap on a three-color scale of red (upper), black (midpoint) and green (lower). With different gene expression patterns, the qPCR results of activated UE6E7T-3 (C) and WI-38-40 cells (D) were separately plotted and analyzed. **P < 0.01, ***P < 0.001 versus UE6E7T-3 or WI-38-40 cells in plain media; one-way ANOVA with Tukey’s post-test. Figure 3A created in BioRender. Luanpitpong, S. (2025) https://BioRender.com/ t14d651. |
Activated Stromal Cells Protected MM Cells Against CAR138 NK Cells
CAFs have previously been shown to promote an immunosuppressive TME in various cancers.23,25 In MM, CAFs have previously been shown to inhibit the antitumor activity of anti-BCMA CAR-T cells in vivo.28 We next determined the impact of activated stromal cells with a CAF-like phenotype on the cytotoxicity of CAR138 NK-92 cells toward MM cells in 2D coculture. NCI-H929-Luc MM target (T) cells were cultured in direct contact with MM-CM-activated UE6E7T-3 or WI-38-40 stromal cells at 1:1 ratio, after which they were exposed to WT or CAR138 NK-92 (E) cells at various E:T ratios, ie, 1:10, 1:5, and 1:2, and NK cytotoxicity was measured by luciferase assay at 4−24 h, as schematically illustrated in Figure 4A. Figure 4B shows that both activated UE6E7T-3 or WI-38-40 cells protected NCI-H929-Luc cells from the antitumor activity of WT NK-92 cells at various E:T ratios and time points; however, only activated WI-38-40 cells had strong protective effects against CAR138 NK-92 cells. Collectively, our data confirmed that stromal cells activated by MM-CM impaired NK cell function and that activated WI-38-40 cells exhibited more profound effect than UE6E7T-3 cells, consistent with the qPCR data showing the striking upregulation of CAF markers in activated WI-38-40 cells (Figure 3D).
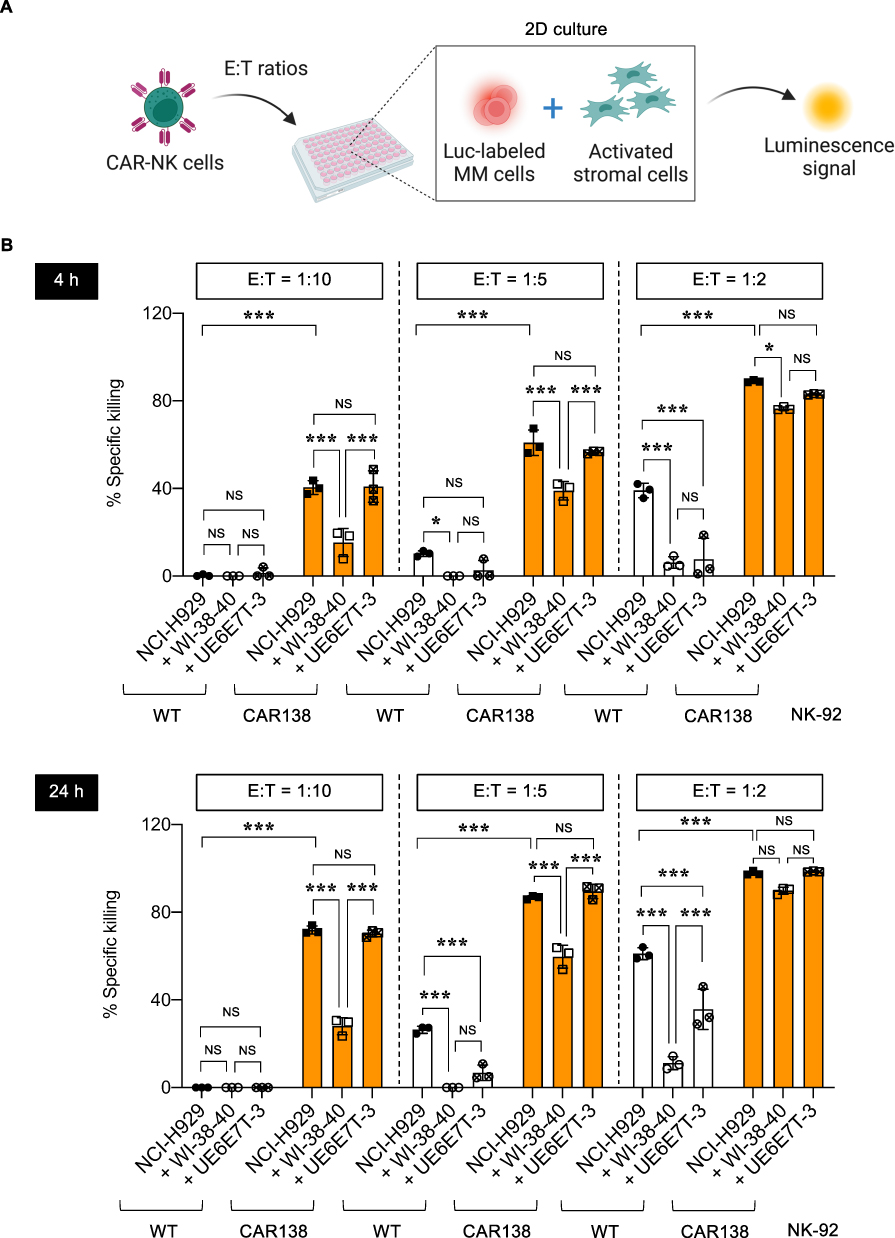 |
Figure 4 Activated stromal cells impaired CAR-NK cytotoxicity in 2D culture. (A) Schematic diagram of NK cytotoxicity by luciferase assay in 2D coculture of Luc-labeled MM cells and activated stromal cells. (B) Percentages of specific killing of NCI-H929-Luc cells in response to WT and CAR138 NK-92 cells in the presence or absence of MM1-CM-activated WI-38-40 and UE6E7T-3 cells at various E:T ratios at 4−24 h. *P < 0.05, ***P < 0.001 versus indicated group under the same E:T ratio; one-way ANOVA with Tukey’s post-test. FIgure 4A created in BioRender. Luanpitpong, S. (2025) https://BioRender.com/ i56f301. |
Growth Kinetics of 3D Tumor Spheroids by Luciferase Assay
We initially established 3D tumor spheroids of human MM cells, in the absence of stromal cells, using MC-based matrix in a 96-well plate, which is applicable for further high-throughput applications, and cell growth kinetics were quantified by luciferase assay, as schematically illustrated in Figure 5A. NCI-H929-Luc cells were seeded at different densities, ranging from 3000 to 7500 cells, and visualized at various times under an inverted microscope to verify the optimal culture conditions. Figure 5B shows that round tumor spheroids were formed and that spheroid sizes correlated well with the initial cell seeding and time in culture, indicating the allowance of MM cell proliferation in 3D culture for up to 5 days. To generate the growth curve, the number of NCI-H929-Luc cells in 3D spheroids was calculated from the standard curve of luminescence signal versus cell number and plotted over time. As the spheroids grew over time, the luminescence signal and calculated cell number increased, with larger spheroids correlating to higher signals (Figure 5C). These results validated the ability to track the size and growth of 3D spheroids by luciferase assay.
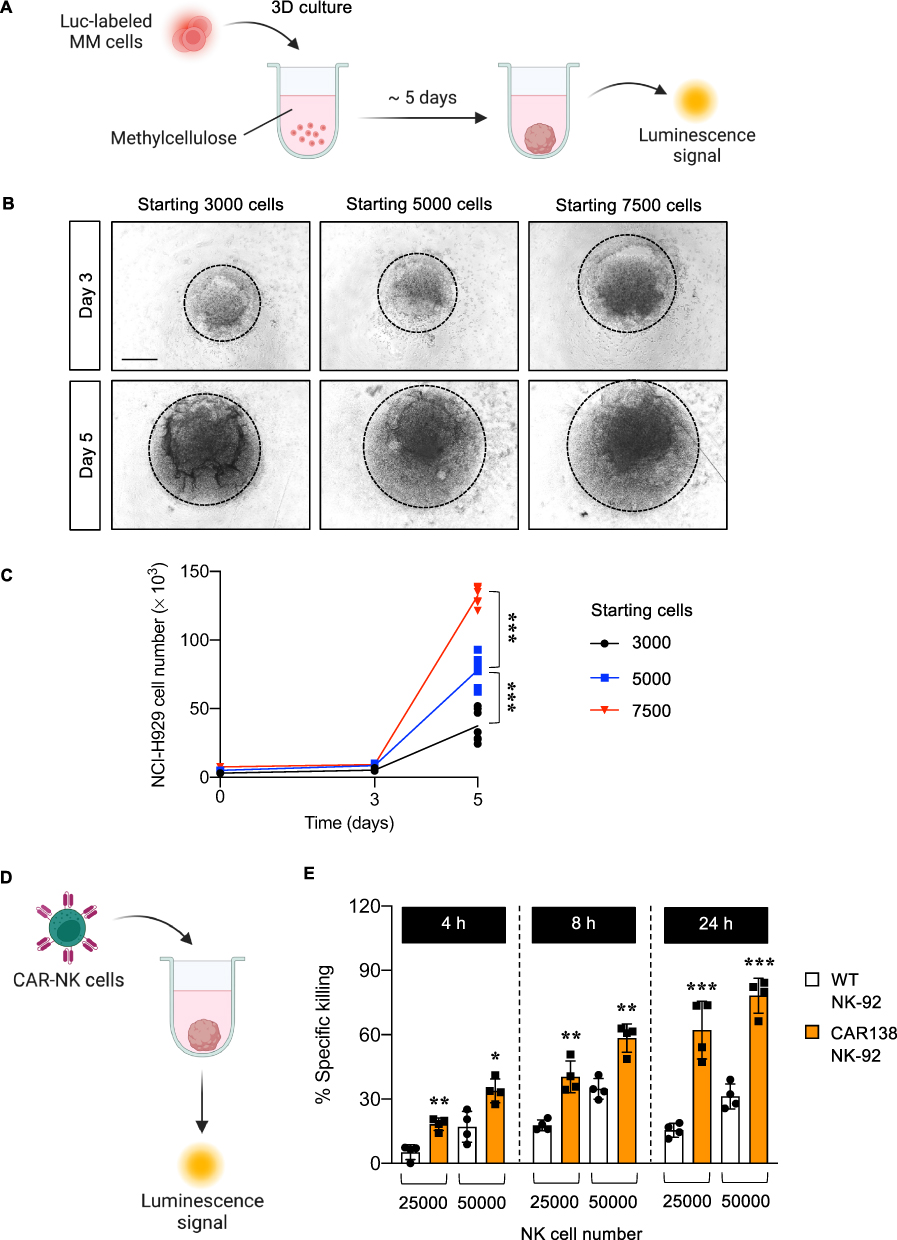 |
Figure 5 Growth kinetics of 3D MM spheroids and its response to CAR-NK cells. (A) Schematic diagram of 3D MM spheroid formation. (B) Morphology of generated 3D MM spheroids from NCI-H929-Luc cells with different seeding numbers, ranging from 3000−7500 cells, on days 3 and 5 of culture. Scale bar = 500 μm. (C) Numbers of NCI-H929-Luc cells on days 3 and 5 of culture, as calculated from the standard curve of luminescence signal versus cell number. ***P < 0.001 versus 3D MM spheroids from the indicated starting number; two-sided Student’s t-test. (D) Schematic diagram of NK cytotoxicity by luciferase assay in 3D MM spheroids. (E) Percentages of specific killing of NCI-H929-Luc cells in 3D MM spheroids in response to WT and CAR138 NK-92 cells at 25000−50000 cells at 4−24 h. *P < 0.05, **P < 0.01, ***P < 0.001 versus WT NK-92 cells under the same conditions; two-sided Student’s t-test. Figure 5A and D created in BioRender. Luanpitpong, S. (2025) https://BioRender.com/ e35q840. |
Cytotoxicity of CAR138 NK Cells in 3D Tumor Spheroids
Having demonstrated the formation of 3D spheroids of MM cells and its growth detection by luciferase assay, we next investigated the cytotoxicity of CAR-immune cells toward these spheroids. 3D MM spheroids at the initial seeding of 5000 NCI-H929-Luc cells were formed for 5 days, after which they were exposed to CAR138 NK-92 cells at 25000 and 50000 cells, as schematically illustrated in Figure 5D, equivalent to the E:T ratios of approximately 1:3 and 2:3, respectively, for 4−24 h. Similar to the findings in 2D culture model, CAR138 NK-92 cytotoxicity was significantly greater than that of WT NK-92 cells at various times and concentrations and its effect was in a dose- and time-dependent manner (Figure 5E). Additionally, CAR138 NK-92 cells were evident to be more cytotoxic in 2D culture than in the 3D spheroids. For example, CAR138 NK-92 cells at the E:T ratio of 1:2 would kill most (~ 90%), if not all, NCI-H929-Luc cells in 2D culture at 4 h, while 24 h was required for CAR138 NK-92 cells at the higher E:T ratio of 2:3 for equivalent cell killing toward 3D spheroids.
Activated Stromal Cells Promoted MM Growth and Impaired CAR138 NK Cell Function in 3D Spheroids
We further determined the impact of activated stromal cells on tumor growth in 3D culture. 3D MM/CAF spheroids were formed in the presence of CAF-like, activated WI-38-40 fibroblasts at the seeding tumor-to-stroma ratios of 1:1 and 1:2, as schematically illustrated in Figure 6A, and the number of NCI-H929-Luc cells was evaluated by luciferase assay on day 5 of culture in comparison to those in the bare 3D MM spheroids without stromal cells. Figure 6B shows that activated WI-38-40 cells promoted the 3D growth of NCI-H929-Luc cells in a dose-dependent manner, supporting the tumor-promoting role of CAFs in the TME and validating the utility of using activated WI-38-40 cells in 3D culture. Next, the cytotoxicity of CAR138 NK-92 cells against 3D MM/CAF spheroids, which were generated from the seeding tumor-to-stroma ratio of 1:1, was determined to evaluate the functions of CAR-immune cells under the more complex TME, as schematically illustrated in Figure 6C. CAR138 NK-92 cells at 25000 and 50000 cells were added to the 3D MM/CAF spheroids on day 6 of culture and the specific tumor killing was evaluated by luciferase assay at 4−24 h. Figure 6D shows that CAR138 NK-92 cells induced higher NCI-H929 cell death rate than WT NK-92 cells in 3D MM/CAF spheroids at various times and doses, similar to the results obtained from the bare 3D MM spheroids and 2D culture. Importantly, activated WI-38-40 cells were found to protect NCI-H929-Luc cells against WT and CAR138 NK-92 cells, particularly at the lower dose and shorter time. A similar pattern of MM protection by activated WI-38-40 cells against WT NK-92 cells and CAR138 NK-92 cells were observed in 3D MM/CAF spheroids composed of MM.1R cells (Supplementary Figure S5). Altogether, our findings suggest that 3D MM spheroids with activated stromal cells may offer a more physiological representation of the complex TME in vivo.
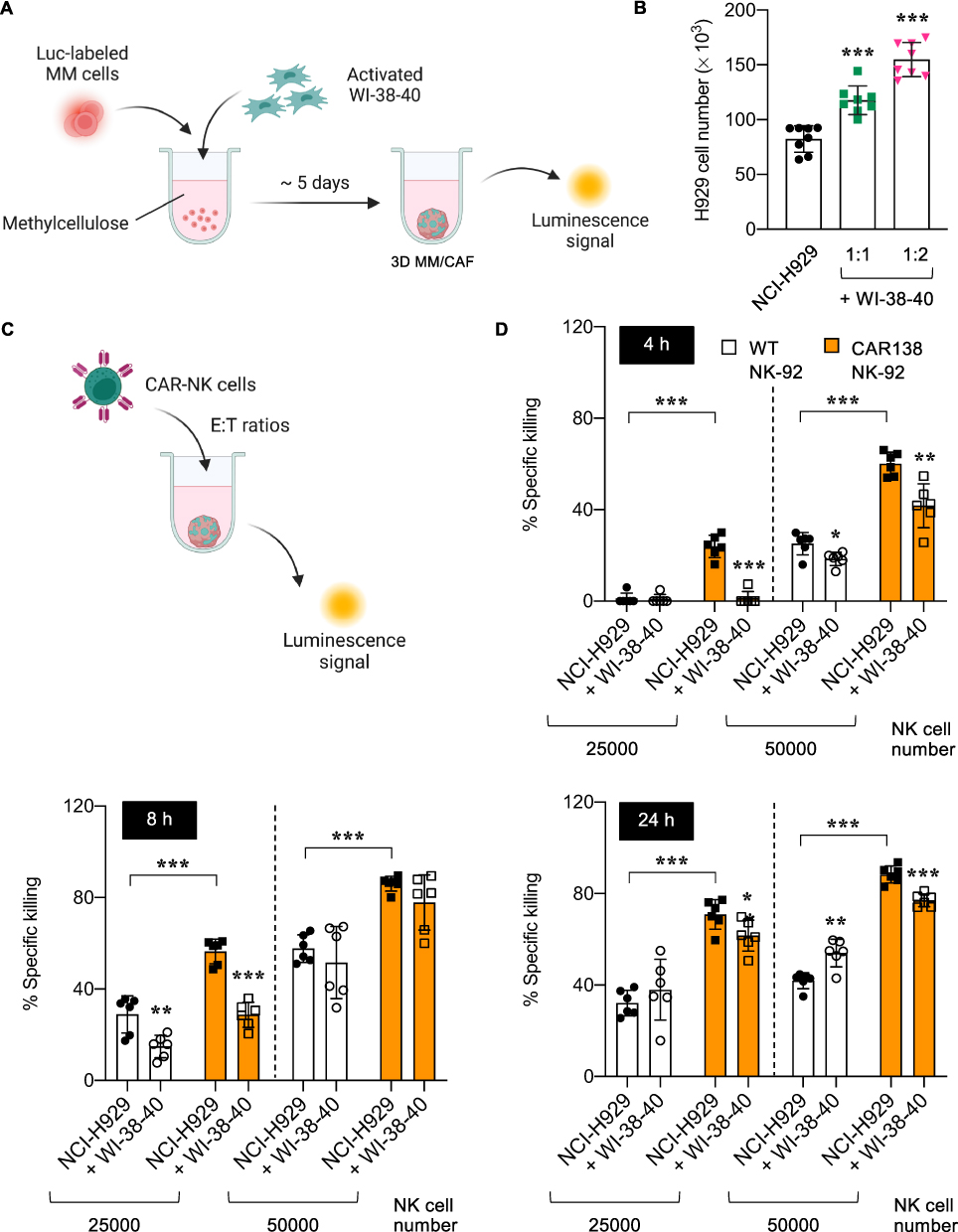 |
Figure 6 Formation of 3D MM/CAF spheroids and its response to CAR-NK cells. (A) Schematic diagram showing the formation of 3D spheroids composed of MM cells and MM-CM-activated WI-38-40 fibroblasts, designated as 3D MM/CAF spheroids. (B) Numbers of NCI-H929-Luc cells in bare 3D MM spheroids and 3D MM/CAF spheroids generated from different ratios of activated WI-38-40 cells on day 5 of culture, as calculated from the standard curve of luminescence signal versus cell number. (C) Schematic diagram of NK cytotoxicity by luciferase assay in 3D MM/CAF spheroids. (D) Percentages of specific killing of NCI-H929-Luc cells in 3D MM/CAF spheroids in response to WT and CAR138 NK-92 cells at 25000−50000 cells at 4−24 h. *P < 0.05, **P < 0.01, ***P < 0.001 versus bare 3D MM spheroids without activated stromal cells under the same conditions or as indicated; two-sided Student’s t-test. Figure 6A and C created in BioRender. Luanpitpong, S. (2025) https://BioRender.com/ b89n777. |
Infiltration of CAR138 NK-92 Cells Into 3D Spheroids
One of the challenges of CAR-T cell therapy is the physical barriers that limit the infiltration of immune cells into tumor masses.10,29,30 Hence, immune infiltration, which can be monitored in 3D spheroids but not in the more common 2D culture, is important for designing better CAR-immune cells. We herein tracked the infiltration of WT and CAR138 NK-92 (blue) cells into the 3D MM spheroids of NCI-H929-Luc (red) cells formed with or without activated WI-38-40 (green) cells at 24 h after incubation using an advanced inverted fluorescence microscope. ROIs were categorized as follows: ROI1, the peripheral zone of tumor cells in a spheroid; ROI2, the core of tumor cells in a spheroid. Figure 7 shows that the bare 3D MM and 3D MM/CAF spheroids without NK-92 cells exhibited an intact, round morphology, whereas those with either type of NK-92 cells exhibited a reduced spheroid volume and damaged structure, indicating a cytolytic activity in the peripheral zone of the spheroids. The excessive infiltration of CAR138 NK-92 cells, but not WT NK-92 cells, into the core of bare 3D MM spheroids was noticeable from the presence of blue cells in the ROI2. We also observed that the presence of CAF-like, activated WI-38-40 fibroblasts inhibited the infiltration of CAR138 NK-92 cells into the core zone of spheroids, yet CAR138 NK-92 cells infiltrated more efficiently into the inner core of spheroids than WT NK-92 cells.
 |
Figure 7 Infiltration of CAR-NK cells into 3D spheroids. 3D spheroids of MM NCI-H929-Luc cells (red), prelabeled with PKH26 dye, were formed in the presence or absence of MM-CM-activated WI-38-40 fibroblasts (green), prelabeled with PKH67 dye. On day 6 of culture, bare 3D MM spheroids and 3D MM/CAF spheroids were exposed to WT and CAR138 NK-92 cells (blue), prelabeled with Vybrant DyeCycle Violet dye, at 25000 cells. Infiltration of NK cells into the core of spheroids was visualized by an inverted fluorescence microscope at 24 h. ROIs were categorized as follows: ROI1 (yellow), the peripheral zone of tumor cells in a spheroid; and ROI2 (Orange), the core of tumor cells in a spheroid. The dashed white line delineates the area in the core zone with minimal infiltrated NK cells, and its area measurement (mm2) is indicated. Scale bar = 500 μm. |
Discussion
To date, six second-generation CAR-T cell products targeting one of the two antigens, either CD19 in certain B-cell malignancies or BCMA in MM, have been granted FDA approval, namely Kymriah (tisagenlecleucel), Yescarta (axicabtagene ciloleucel), Tecartus (brexucabtagene), Breyanzi (lisocabtagene maraleucel), Abecma (idecabtagene vicleucel), and Carvykti (ciltacabtagene autoleucel).31,32 Despite this progress over a relatively short time span, various challenges and limitations to effective CAR-T cell therapy in hematologic malignancies and most solid tumors remain. Investigators are attempting to improve the safety associated with the currently approved products through innovations in CAR designs, eg, implementing on/off switches and suicide gene system, introducing the requirement of multiple tumor antigens, etc.10,31,33 To overcome tumor antigen escape—that is, a partial or complete loss of the target antigen in tumor cells after initial response—multitarget strategies created by mixing different CAR-T cell products targeting single tumor antigens or by transducing T cells with multiple CAR constructs or bispecific CAR constructs may be used. The critical barriers that diminish CAR-T cell efficacy in solid tumors include the antigen heterogeneity, the physical barrier of the TME and its immunosuppressive network, and the suboptimal trafficking into tumor masses.7,10,29 Investigators have developed new CARs not only for CAR-T cells to address these challenges but also for several non-T, CAR-immune cells with diverse trafficking and killing mechanisms.9,31,34 That has raised the prospect that novel CAR-immune cells with reduced toxicity and enhanced efficacy toward solid tumors and incurable hematologic cancers could be provided. There has been a considerable increase in targeting CAFs and the TME using immunotherapy.28,35 Accordingly, there is a pressing need for high-throughput screening models with enhanced physiological relevance and TME complexity for evaluating the cytotoxicity of CAR-immune cells to identify potential therapeutic candidates for further preclinical and clinical studies.
MM, although it is a hematologic malignancy, is among those tumors whose survival and expansion of malignant cell clones heavily depend on various direct and indirect signaling pathways provided by the surrounding TME.13,25 Numerous studies have demonstrated that the dynamic interactions between the TME and MM cells affect its progression and dissemination.36,37 Like in solid tumors, barriers to CAR-T cell activity in MM could be possibly linked to the immunosuppressive TME and T cell exhaustion/dysfunction.15,25,38 Elevated expression of CAF markers was detected in the BM of patients with MM progression.39 However, 3D MM models for immuno-oncology research that mimic the TME, which consists of CAFs or other stromal cells, and allow high-throughput screening have not been well established. In this study, we demonstrated the formation of 3D spheroids of Luc-labeled MM cells with CAF-like stromal cells, which was activated by exposing WI-38-40 fibroblasts to MM-CM through paracrine signaling, in a 96-well plate format. Cytotoxicity of representative CAR-immune cells, which in this case were third-generation CAR138 NK-92 cells, was then evaluated by a high-throughput luciferase assay instead of a conventional flow cytometry-based Annexin V/7-AAD assay. Notably, monitoring of NK cytotoxicity in 3D spheroids by Annexin V/7-AAD assay can be time-consuming because it requires additional steps of fluorescent dye prelabeling and destruction of 3D spheroids postincubation. We validated herein the ability of luciferase assay to track the growth of MM cells and detect the cytotoxicity of CAR-immune cells in 2D culture and in 3D spheroids. As the luciferase assay could be performed without lysing the cells and D-luciferin did not affect cell viability, repeated measurements at multiple time points could be taken in the same experimental well. Unsurprisingly, CAR138 NK-92 cells displayed greater antitumor activity than WT NK-92 cells in both 2D and 3D cultures at various times and concentrations. Importantly, activated stromal cells with a CAF-like phenotype impaired NK cell effector function in both 2D and 3D cultures, indicating that the activated stromal cells did provide the immunosuppressive TME, which resembled real scenarios in vivo.28,39 We postulated that the protective effect of activated stromal cells against WT and CAR138 NK-92 cells was likely through physical barrier function and the secretion of cytokines and other soluble factors based on the observation that activated tumor cells impaired NK cell function in 2D coculture, where the cells were lightly in contact.
In the past decade, 3D tumor spheroids have been widely used to assess tumor responses to chemotherapy and radiotherapy, considering that they are a better predictor of in vivo responses.40,41 Tumor cells in 3D spheroids are generally more resistant to the treatment than those in 2D culture, attributable to the physical barriers and numerous signals mediated by cell-cell interaction, the surrounding extracellular matrix (ECM), and hypoxic core.42 We observed that CAR138 NK-92 cells were less effective at combating MM cells in the 3D spheroids, especially 3D MM/CAF spheroids, when compared to 2D culture, suggesting that 3D spheroids with activated stromal cells could capture tumor-stromal cell interactions and are more closely resemble the complexity of immunosuppressive TME observed in MM. Not only did activated stromal cells impair CAR-NK cell effector function, but also promoted MM growth, consistent with previous studies reporting the tumor-promoting role of BM-derived CAFs isolated from MM patients.28,39 The limitation of our 3D spheroid study is the relatively high variations of tumor response, resulting in an insufficient statistical power (~ 70%) in a certain condition, ie, when comparing the specific tumor killing of 25000 WT NK-92 cells in bare 3D MM spheroids versus 3D MM/CAF spheroids at 24 h, though we were able to achieve statistical significance (Supplementary Table S5).
Accumulating evidence indicates that tumor infiltration is one of the parameters that dictates the success of cellular therapy.29,43,44 We for the first time visually demonstrated that CAR138 NK-92 cells, but not WT NK-92 cells, were able to infiltrate into the core of 3D MM spheroids and, to a lesser extent, 3D MM/CAF spheroids—the degree of immune cell infiltration correlated well with their cytotoxic potential. To exert antitumor activity, CAR-immune cells must traffic to BM niche, where the majority of MM cells reside, to promote effective antigen recognition.13,25 We postulated that the activated stromal cells created a physical barrier that impeded the infiltration and mobility of effector immune cells, even with CAR incorporation, as activated stromal cells localized mostly in the core zone where the infiltration of CD138 NK-92 cells was limited. Still, the established 3D spheroid model has some limitations. Our data revealed that although activated WI-38-40 cells promoted MM cell growth in 3D spheroids, they themselves did not proliferate well in the MC-based matrix, thereby increasing the ratio of tumor-to-stroma in 3D MM/CAF spheroids by approximately 10 times the ratio in those 2D culture experiments. Hence, the observed tumor-protective effect of activated stromal cells was much weaker in 3D MM/CAF spheroids than in 2D coculture. Further studies are required to identify the matrix for spheroid formation that supports both tumor and stromal cell growth. Additionally, it is worthwhile to incorporate endothelial cells into the system to recreate even more complex interactions in the TME, which is lacking in the current model, and further explore potential novel therapeutic strategies, ie, combination therapy targeting MM cells, tumor stroma, and angiogenesis.45,46
Conclusion
Taken together, our findings indicate that the established 3D spheroid model composed of MM cells and activated stromal cells recapitulates some of the complexity of TME with immunosuppressive environment. We further revealed that this model is suitable for high-throughput screening of CAR-immune cytotoxicity using luciferase assay and offers the possibility to evaluate immune cell infiltration, thus overcoming the current drawbacks of conventional cytotoxicity assays and of 2D culture. Targeting CAFs has been investigated as a promising approach to enhance the efficacy of CAR-T cells in MM and to overcome the CAR-T exhaustion/dysfunction involving relapse.15,28 Our established 3D MM/CAF spheroids, though require further in vivo validation for its predictive efficiency, might prove useful for better understanding of mechanisms underlying CAR-immune cytotoxicity and interactions/crosstalk among MM cells, stromal cells, and immune cells as well as for screening of other strategies targeting MM cells and/or CAFs, eg, by therapeutic monoclonal antibodies. The protocols described here may be applicable to other types of tumor cells as well.
Data Sharing Statement
Data are provided within the manuscript or supplementary information file.
Ethics Approval and Consent to Participate
Cell lines used in this study were purchased from ATCC, JCRB, and Thermo Fisher Scientific.
Acknowledgments
We extend our sincere appreciation for the instrumentation support provided for the Leica DMi8 inverted fluorescence microscope equipped with a Leica DFC6200 camera, funded by Mahidol University (Basic Research Fund: fiscal year 2022). We thank scientists Phakhin Jantahiran and Nattarun Chaisilp from Faculty of Veterinary Science, Mahidol University for their kind assistance with imaging, and Sirinart Buasamrit for her administrative assistance. We would also like to acknowledge Dr. Orawan Supapueng, Division of Clinical Epidemiology, Department of Research and Development, Faculty of Medicine Siriraj Hospital, Mahidol University for her advice with statistical analysis. Schematic illustrations in Figures 1-6 were created with BioRender.com under an academic license (to S.L).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas. S.L. and S.I. supervised the project. The original draft of the article was written by S.L., and all authors revised or critically reviewed the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by Specific League Funds from Mahidol University (to S.L.) and in part by a grant from The National Research Council of Thailand and Mahidol University (N42A650372 to S.L.).
Disclosure
The authors declare no competing interests in this work.
References
1. Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China cancer immunotherapy workshop in Beijing. J Hematol Oncol. 2018;11(1):142. doi:10.1186/s13045-018-0684-3
2. Pan C, Liu H, Robins E, et al. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol. 2020;13(1):29. doi:10.1186/s13045-020-00862-w
3. Hiltensperger M, Krackhardt AM. Current and future concepts for the generation and application of genetically engineered CAR-T and TCR-T cells. Front Immunol. 2023;14:1121030. doi:10.3389/fimmu.2023.1121030
4. Dreger P, Dietrich S, Schubert ML, et al. CAR T cells or allogeneic transplantation as standard of care for advanced large B-cell lymphoma: an intent-to-treat comparison. Blood Adv. 2020;4(24):6157–6168. doi:10.1182/bloodadvances.2020003036
5. Wang JY, Wang L. CAR-T cell therapy: where are we now, and where are we heading? Blood Sci. 2023;5(4):237–248. doi:10.1097/BS9.0000000000000173
6. Mishra AK, Gupta A, Dagar G, et al. CAR-T-cell therapy in multiple myeloma: b-cell maturation antigen (BCMA) and beyond. Vaccines. 2023;11(11):1721. doi:10.3390/vaccines11111721
7. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi:10.1038/s41408-021-00459-7
8. Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):168. doi:10.1186/s13045-020-00998-9
9. Hadiloo K, Taremi S, Heidari M, Esmaeilzadeh A. The CAR macrophage cells, a novel generation of chimeric antigen-based approach against solid tumors. Biomark Res. 2023;11(1):103. doi:10.1186/s40364-023-00537-x
10. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167.
11. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T cell therapy and the tumor microenvironment: current challenges and opportunities. mol Ther Oncolytics. 2022;25:69–77. doi:10.1016/j.omto.2022.03.009
12. Geng J, Zhao J, Fan R, et al. Global, regional, and national burden and quality of care of multiple myeloma, 1990-2019. J Glob Health. 2024;14:04033. doi:10.7189/jogh.14.04033
13. Suzuki K, Nishiwaki K, Yano S. Treatment strategies considering micro-environment and clonal evolution in multiple myeloma. Cancers. 2021;13(2):215. doi:10.3390/cancers13020215
14. Zhang X, Zhang H, Lan H, Wu J, Xiao Y. CAR-T cell therapy in multiple myeloma: current limitations and potential strategies. Front Immunol. 2023;14:1101495. doi:10.3389/fimmu.2023.1101495
15. Ledergor G, Fan Z, Wu K, et al. CD4+ CAR T-cell exhaustion associated with early relapse of multiple myeloma after BCMA CAR T-cell therapy. Blood Adv. 2024;8(13):3562–3575. doi:10.1182/bloodadvances.2023012416
16. Shimizu S, Yoshioka R, Hirose Y, Sugai S, Tachibana J, Konda S. Establishment of two interleukin 6 (B cell stimulatory factor 2/interferon beta 2)-dependent human bone marrow-derived myeloma cell lines. J Exp Med. 1989;169(1):339–344. doi:10.1084/jem.169.1.339
17. Helfrich MH, Livingston E, Franklin IM, Soutar RL. Expression of adhesion molecules in malignant plasma cells in multiple myeloma: comparison with normal plasma cells and functional significance. Blood Rev. 1997;11(1):28–38. doi:10.1016/S0268-960X(97)90004-7
18. Kawano Y, Moschetta M, Manier S, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263(1):160–172. doi:10.1111/imr.12233
19. Luanpitpong S, Poohadsuan J, Klaihmon P, Issaragrisil S. Selective cytotoxicity of single and dual anti-CD19 and anti-CD138 chimeric antigen receptor-natural killer cells against hematologic malignancies. J Immunol Res. 2021;2021:5562630. doi:10.1155/2021/5562630
20. Stewart SA, Dykxhoorn DM, Palliser D, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9(4):493–501. doi:10.1261/rna.2192803
21. Kiesgen S, Messinger JC, Chintala NK, Tano Z, Adusumilli PS. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat Protoc. 2021;16(3):1331–1342. doi:10.1038/s41596-020-00467-0
22. Yang Y, Badeti S, Tseng HC, et al. Superior expansion and cytotoxicity of human primary NK and CAR-NK cells from various sources via enriched metabolic pathways. Mol Ther Methods Clin Dev. 2020;18:428–445. doi:10.1016/j.omtm.2020.06.014
23. Mao X, Xu J, Wang W, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. mol Cancer. 2021;20(1):131. doi:10.1186/s12943-021-01428-1
24. Samart P, Heenatigala Palliyage G, Issaragrisil S, Luanpitpong S, Rojanasakul Y. Musashi-2 in cancer-associated fibroblasts promotes non-small cell lung cancer metastasis through paracrine IL-6-driven epithelial-mesenchymal transition. Cell Biosci. 2023;13(1):205. doi:10.1186/s13578-023-01158-5
25. García-Ortiz A, Rodríguez-García Y, Encinas J, et al. The role of tumor microenvironment in multiple myeloma development and progression. Cancers. 2021;13(2):217. doi:10.3390/cancers13020217
26. Luanpitpong S, Wang L, Castranova V, et al. Induction of cancer-associated fibroblast-like cells by carbon nanotubes dictates its tumorigenicity. Sci Rep. 2016;6(1):39558. doi:10.1038/srep39558
27. Biffi G, Oni TE, Spielman B, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9(2):282–301. doi:10.1158/2159-8290.CD-18-0710
28. Sakemura R, Hefazi M, Siegler EL, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood. 2022;139(26):3708–3721. doi:10.1182/blood.2021012811
29. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. 2017;8:1850. doi:10.3389/fimmu.2017.01850
30. Castelletti L, Yeo D, van Zandwijk N, Rasko JEJ. Anti-Mesothelin CAR T cell therapy for malignant mesothelioma. Biomark Res. 2021;9(1):11. doi:10.1186/s40364-021-00264-1
31. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. 2023;614(7949):635–648.
32. Mazinani M, Rahbarizadeh F. CAR-T cell potency: from structural elements to vector backbone components. Biomark Res. 2022;10(1):70. doi:10.1186/s40364-022-00417-w
33. Celichowski P, Turi M, Charvátová S, et al. Tuning CARs: recent advances in modulating chimeric antigen receptor (CAR) T cell activity for improved safety, efficacy, and flexibility. J Transl Med. 2023;21(1):197. doi:10.1186/s12967-023-04041-6
34. Hadiloo K, Tahmasebi S, Esmaeilzadeh A. CAR-NKT cell therapy: a new promising paradigm of cancer immunotherapy. Cancer Cell Int. 2023;23(1):86. doi:10.1186/s12935-023-02923-9
35. Liu T, Han C, Wang S, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):86. doi:10.1186/s13045-019-0770-1
36. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3(1):17046. doi:10.1038/nrdp.2017.46
37. Forster S, Radpour R. Molecular impact of the tumor microenvironment on multiple myeloma dissemination and extramedullary disease. Front Oncol. 2022;12:941437. doi:10.3389/fonc.2022.941437
38. Sheykhhasan M, Ahmadieh-Yazdi A, Vicidomini R, et al. CAR T therapies in multiple myeloma: unleashing the future. Cancer Gene Ther. 2024;31(5):667–686. doi:10.1038/s41417-024-00750-2
39. Frassanito MA, Rao L, Moschetta M, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia. 2014;28(4):904–916. doi:10.1038/leu.2013.254
40. Nayak P, Bentivoglio V, Varani M, Signore A. Three-dimensional in vitro tumor spheroid models for evaluation of anticancer therapy: recent updates. Cancers. 2023;15(19):4846. doi:10.3390/cancers15194846
41. Manduca N, Maccafeo E, De Maria R, Sistigu A, Musella M. 3D cancer models: one step closer to in vitro human studies. Front Immunol. 2023;14:1175503. doi:10.3389/fimmu.2023.1175503
42. Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol. 2014;12(4):207–218. doi:10.1089/adt.2014.573
43. Giannattasio A, Weil S, Kloess S, et al. Cytotoxicity and infiltration of human NK cells in in vivo-like tumor spheroids. BMC Cancer. 2015;15(1):351. doi:10.1186/s12885-015-1321-y
44. Zboralski D, Hoehlig K, Eulberg D, Frömming A, Vater A. Increasing tumor-infiltrating T cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol Res. 2017;5(11):950–956. doi:10.1158/2326-6066.CIR-16-0303
45. Neumeister P, Schulz E, Pansy K, Szmyra M, Deutsch AJ. Targeting the microenvironment for treating multiple myeloma. Int J mol Sci. 2022;23(14):7627. doi:10.3390/ijms23147627
46. Rao L, De Veirman K, Giannico D, et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: a preclinical study. Oncotarget. 2018;9(17):13366–13381. doi:10.18632/oncotarget.24351
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
CD73 Dysregulates Monocyte Anti-Tumor Activity in Multiple Myeloma

Zhou L, Liu X, Guan T, Xu H, Wei F
Cancer Management and Research 2023, 15:729-738
Published Date: 19 July 2023
Nanomedicine Targeting Cancer-Associated Fibroblasts in Prostate Cancer: From Biological Mechanisms to Integrated Theranostic Strategies
Qian F, Zhou J, Tang Y, Chen J, Zuo J, Gou K, Tan C, Fang J, Zhao Z, Zhao J, Wen L, Fu S, Yang J, Wang C, Zhang Z
International Journal of Nanomedicine 2026, 21:588669
Published Date: 17 March 2026
Cancer-Associated Fibroblasts in the Treatment of Intrahepatic Cholangiocarcinoma: Present and Prospects
Yuan Y, Zhang R, Qu C, Huang N, Wang L, Liu M
Journal of Hepatocellular Carcinoma 2026, 13:581943
Published Date: 23 March 2026