")
Back to Journals » OncoTargets and Therapy » Volume 14
Urolithin A Inhibits Epithelial–Mesenchymal Transition in Lung Cancer Cells via P53-Mdm2-Snail Pathway
Authors Cheng F, Dou J, Zhang Y, Wang X, Wei H, Zhang Z, Cao Y, Wu Z
Received 16 February 2021
Accepted for publication 22 April 2021
Published 17 May 2021 Volume 2021:14 Pages 3199—3208
DOI https://doi.org/10.2147/OTT.S305595
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Feng Cheng,1,* Jintao Dou,1,2,* Yong Zhang,1,3,* Xiang Wang,1,4,* Huijun Wei,5 Zhijian Zhang,1,5 Yuxiang Cao,4,6 Zhihao Wu1,5,7,8
1Research Laboratory of Tumor Microenvironment, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 2School of Anesthesiology, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 3School of Clinical Medicine, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 4School of Laboratory Medicine, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 5School of Preclinical Medicine, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 6Provincial Engineering Laboratory for Screening and Re-Evaluation of Active Compounds of Herbal Medicines in Southern Anhui, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 7Anhui Province Key Laboratory of Active Biological Macro-Molecules Research, Wannan Medical College, Wuhu, 241001, People’s Republic of China; 8Key Laboratory of Non-Coding RNA Transformation Research of Anhui Higher Education Institution, Wannan Medical College, Wuhu, 241001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhihao Wu
Research Laboratory of Tumor Microenvironment, Wannan Medical College, Wuhu, 241001, People’s Republic of China
Email [email protected]
Purpose: The epithelial-to-mesenchymal transition (EMT) is a fundamental process in tumor progression that endows cancer cells with migratory and invasive potential. Snail, a zinc finger transcriptional repressor, plays an important role in the induction of EMT by directly repressing the key epithelial marker E-cadherin. Here, we assessed the effect of urolithin A, a major metabolite from pomegranate ellagitannins, on Snail expression and EMT process.
Methods: The role of Snail in urolithin A-induced EMT inhibition in lung cancer cells was explored by wound healing assay and cell invasion assay. The qRT-PCR and CHX assay were performed to investigate how urolithin A regulates Snail expression. Immunoprecipitation assays were established to determine the effects of urolithin A in mdm2-Snail interaction. In addition, the expression of p53 was manipulated to explore its effect on the expression of mdm2 and Snail.
Results: The urolithin A dose-dependently upregulated epithelial marker and decreased mesenchymal markers in lung cancer cells. In addition, exposure to urolithin A decreased cell migratory and invasive capacity. We have further demonstrated that urolithin A inhibits lung cancer cell EMT by decreasing Snail protein expression and activity. Mechanistically, urolithin A disrupts the interaction of p53 and mdm2 which leads Snail ubiquitination and degradation.
Conclusion: We conclude that urolithin A could inhibit EMT process by controlling mainly Snail expression. These results highlighted the role of pomegranate in regulation of EMT program in lung cancer.
Keywords: urolithin A, ellagitannin metabolite, epithelial–mesenchymal transition, Snail, mdm2, lung cancer
Introduction
Lung cancer is the leading cause of cancer-related death worldwide with non-small cell lung cancer (NSCLC) accounting for more than 80% of cases.1,2 More often, patients with NSCLC are diagnosed as advanced or metastatic disease, and despite improvements in targeted therapies and immunotherapeutics, the five-year survival rate for lung cancer is still as low as 15%.1 Therefore, understanding the molecular mechanisms that contribute to tumor metastasis, and seeking the drugs targeting tumor metastasis may have important clinical and social significance.
Epithelial-mesenchymal transition (EMT) plays a significant role in the metastatic process of cancer by facilitating cell migration and invasion.3 The hallmark of EMT is to acquire mesenchymal traits through the regulation of core EMT related proteins, such as decreasing the expression levels of epithelial markers E-cadherin and increasing mesenchymal markers N-cadherin and Vimentin expression levels.4,5 A paramount premise of these events is the upregulation of transcriptional repressors, such as the zinc-finger proteins Snail, Zeb1 and Slug, as well as the bHLH factors Twist.6,7 These proteins bind to the E-box element in the promoter of the E-cadherin gene, where they recruit histone deacetylases (HDACs) and other co-repressors to facilitate chromatin condensation and subsequent transcriptional repression of E-cadherin expression.8,9 Previous reports demonstrated that constitutive activation of Snail in numerous cancers, including lung cancer, is one of the major mechanisms of tumor progression and metastasis. 10−12 Several key signaling pathways, including Wnt, TGFβ, and Akt, are known to be involved in inducing Snail expression in transcription level, whereas phosphorylation and ubiquitination are associated with Snail degradation in post-translation level.13–17 Recently, it has been reported that mdm2, murine double minute 2, serves as a mediator which promotes Snail degradation via ubiquitination.18 Mdm2 has long been regarded as E3 ubiquitin ligase, which regulates the stability of p53 protein.19 Tumor suppressor p53 is related to cell cycle arrest, DNA repair and apoptosis.20 Considering the above, mdm2 may serve as a novel molecular target to inhibit cancer cell proliferation and EMT process by combining p53 and Snail.
Natural compounds called ellagitannins (ETs) are common in some fruits.21 In particular, for humans, the pomegranate is a main source of ellagitannins.22 ETs are hydrolyzed in the gut to release ellagic acid (EA), which is further processed by the microflora into urolithins through the loss of one of its two lactones and by successive removal of hydroxyl groups.23 In the species investigated to date (including humans), urolithin A (UA), urolithin B (UB), urolithin C (UC) and urolithin D (UD) are the redundant measurable metabolites that are considered to be the end-products of both ETs and EA.24 Many studies suggest that urolithin A (UA) is the main functional substance in antioxidant and anti-inflammatory activities after the body consumes foods rich in ellagitannin or ellagic acid.25,26 Recently, growing studies have found that urolithin A downregulates multiple tumor pathways in colon, prostate, and bladder cancer through downregulating several oncogenes such as mTOR and K-Ras, upregulating tumor suppressor genes such as p53 and EGFR.27–30 Regardless of significant progress in tumor proliferation, the relationship between urolithin A and tumor metastasis is not clear. Our present study has successfully shown that urolithin A upregulates E-cadherin and decreased N-cadherin as well as Vimentin via mdm2-Snail pathway. We also demonstrated that urolithin A upregulates mdm2 by inhibiting the interaction of p53 and mdm2. Furthermore, p53 is crucial for Snail degradation by urolithin A. Taken together, our results indicate that urolithin A inhibits the process of EMT via p53-mdm2-Snail pathway.
Materials and Methods
Cell Culture and Reagents
The human lung cancer A549 (p53+/+), H460 (p53+/+) and H1299 (p53-/-) cells were purchased from National Collection of Authenticated Cell Cultures (Shanghai, China) and cultured with DMEM (GibcoThermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (Gibco) at 37 °C in a humidified atmosphere of 5% CO2. Urolithin A (no. SML1791) and Cycloheximide (no.239763) were purchased from Sigma (Sigma-Aldrich Co., St Louis, MO, USA). Proteasome Inhibitor I (no. 539161) was purchased from Calbiochem (Sigma-Aldrich). Hydroxychloroquine Sulfate (no. S4430) was purchased from Selleck Chemicals (USA)
MTT Assay
Cell viability was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Beyotime Institute of Biotechnology, Shanghai, China) assay. Cells were pretreated with urolithin A (0, 10, 20μM) for 5 h, and then the medium was aspirated. MTT assay was used to detect cell viability after 24h or 48h of continuous culture. After treatment, cells were washed with PBS and incubated with fresh medium with MTT solution (0.5mg/mL) for 4h at 37°C, the resulting formazan crystals were dissolved in DMSO and absorbance at wavelength of 570nm was taken on a plate reader using BioTek citation 5 (BioTek, Winooski, VT, USA).
Quantitative Real-Time RT–PCR Analysis
Cells were treated with different concentrations of urolithin A in six-well plates. Trizol was used to isolate the total mRNA according to the manufacturer’s protocols. The RNA was re-transcribed using Reverse Transcription Kit (TaKaRaBio, DaLian, China). The cDNA was mixed with TB Green Master Mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), and the mixture was subjected to amplification using Roche LightCycler 96 (Roche). The thermocycling conditions for the qPCR were as follows: 95°C for 30 sec, followed by 42 cycles of 95°C for 10 sec and 60°C for 30 sec, and finally 1 min at 50°C. The primers used were: Snail forward: 5′-AGGCAGCTATTTCAGCCTCC-3′; Snail reverse:5ʹ-CACATCGGTCAGACCAG AGC-3; β-actin forward: 5′- CCTTCCTGGGCATGGAGTCCT-3′; β-actin reverse: 5ʹ-GGAGCAATGATCTTGATCTTC-3′. p53 forward: 5′- GTCAGATCCTAGCGTCGAG −3′;p53 reverse: 5′- GGGACAGCATCAAATCATC −3′; mdm2 forward: 5′- AATCATCGGACTCAGGTA −3′; mdm2 reverse: 5′- CTTCACTAAGGCTATAATCTTC −3′.
Dual Luciferase Reporter Assays
After the plasmid was co-transfected 36–48 h, the medium was discarded and the cells were washed with 1mL 1x PBS. Dilute 5x PLB (lysis solution) into 1x PLB (ready for use) with deionized water, and put it at room temperature before use. Add 100μL 1x PLB diluted to each well, and shake with a shaker for 20–30min to ensure that the lysate buffer lyse cells completely. Add respectively 20 μL supernatant and 20 μL Luciferase Assay Reagent to each well of the 96-well white opaque enzyme-labeled plate. Microplate reader was used to detect Firefly luciferase reaction intensity. Then add 20 μL Stop Assay Reagent to test Renilla luciferase activity.
Plasmid and Short Interfering RNA (siRNA) Transfection
Cells seeded in six-well plates were grown to 90% confluence before plasmids transfection and transfection of plasmids was done with PolyJet DNA Transfection Reagent (SignaGen Laboratories, Gaithersburg, MD, USA) according to the manufacturer’s instructions. The plasmids of Snail promoter (no.31694), Snail cDNA (no.16218), mdm2 cDNA (no.16233),p53 cDNA (no.69003),p53 shRNA (no.28222) were purchased from Addgene. The transfection with siRNA using GenMute siRNA Transfection Reagent (SignaGen Laboratories) when cells seeded in a six-well plate were grown to 30%–50% confluence. All the siRNAs were purchased from GENERAL BIOSYSTEMS (Chuzhou, China). After transfection for 48 h, cells were deprived of serum and growth factors for 10 h and then treated with urolithin A for indicated time. The sequences of the siRNAs are as follow: Snail, 5ʹ- CAAATACTGCAACAAGGAA-3ʹ; mdm2, 5ʹ-GCCUGGCUCUGUGUGUAAUdTdT-3ʹ.
Western Blot
Cells were scraped and homogenized with Sample Buffer, Laemmli 2x Concentrate (S3401; SIGMA). Protein per sample was separated by polyacrylamide gel electrophoresis and then transferred to nitrocellulose (NC) membrane (GE Healthcare, Piscataway, NJ, USA) and detected with the antibodies. The signals were scanned by AI 680 (GE). Anti-p53 (no.2527S), Anti-Snail (no.3895S), Anti-N-cadherin (no.13116S), Anti-Ubiquitin (no.3933S), Anti-LC3 A/B (no.12741S) and Anti-p62 (no.88588S) were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-mdm2 (no. A0345) and Anti-E-cadherin (no.A3044) were purchased from Abclonal (Wuhan, China). Anti-β-Actin (no. A1978) and anti-Vimentin (no.V6630) were purchased from Sigma (Sigma, Victoria, BC, Canada).
Wound Healing Assay
About 5×105 cells were added to each hole of the six-well plate, and the specific number varied according to different cells, so that it could be filled overnight. On the second day, the corresponding concentrations of urolithin A were added and cultured in a 5% CO2 incubator at 37 °C for 5 hours, and the 200 μL pipette tips were used to tilt the line. The cells were washed 5 times with PBS to remove the floating cells, and then the serum-free medium was added for the rest time of culture. The field of view was watched with a microscope and pictures were taken. Then photographs were taken every 12 hours.
Cell Invasion Assays
The cells were first treated with indicated concentrations of urolithin A for 5 h and then digested, centrifuged, resuspended serum-free, and diluted into 1×105 cell suspensions/mL. Add 500 μL complete medium containing 20% FBS to Transwell plate and place the compartment into plate. Cell suspension of 200 μL was added into Transwell chamber (ECM550, Millipore, USA) and Transwell plate was cultured in CO2 (5%) incubator at 37°C for 24 h. The compartment was taken out, the medium was washed with PBS and stained with crystal violet for 10 min. The crystal violet on the surface was washed off with tap water, and the cells on the inoculated side of the upper chamber were wiped clean with a cotton swab. The non-cellular inoculated side was photographed under a microscope. Five fields were randomly selected for each treatment, and the cell number was used to reflect the invasion ability of cells.
Immunoprecipitation (IP) Assays
After indicated treatment, an appropriate amount of cell IP lysis buffer (containing protease inhibitors) was added, and cells were lysed on ice or 4°C for 30 min. The lysate was centrifuged at 12,000 g for 30 min, and then take supernatant. A small amount of lysate was taken for Western blot analysis. Add 1 ug of corresponding antibody and 10–50 μL Protein A/G beads into the remaining lysate, and shake slowly at 4oC and incubate overnight. After the immunoprecipitation reaction, the samples were centrifuged at 4°C at a speed of 3,000 g for 5 min. The Protein G beads (SC-2002, Santa Cruz, CA, USA) was centrifuged into the bottom of the tube. Carefully blot out the supernatant and use 1 mL of splitting buffer for Protein G beads. Wash with liquid 3–4 times, then add in 15 μL 2x SDS sample buffer, and boil for 10 minutes. Western Blot analysis was performed by SDS-PAGE.
Cell Morphology Assays
Indicated cells seeded in six-well plate were grown to 60%–70% confluence before serum-free starvation. Cells were deprived of serum and growth factors for 10 h and then treated with urolithin A for 24 h. Cells were photographed by a phase-contrast microscope at 100 magnification (Olympus, Shinjuku-ku, Japan).
Statistical Analysis
Statistical analyses were performed with analysis of variance (ANOVA) using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA, USA) and are presented as mean ± s.d. from triplicated independent experiments. A significant difference was considered when the P-value from a two-tailed test was <0.05.
Results
Urolithin A Inhibits EMT in Lung Cancer Cells
We sought to determine the effects of urolithin A on cell migratory and invasive potential. To this end, we have chosen the lung carcinoma cell lines A549 and H460, both bearing K-Ras mutation, showing a more mesenchymal phenotype in the culture even in the absence of serum. Urolithin A treatment caused a different phenotype, as cells adopt their cuboidal epithelial morphology, characterized by the much fewer dynamic protrusions of filopodia and lamellipodia (Figure 1A). We next performed a wounding assay, in which both A549 and H460 cells migrate into wound created by scratching confluent cultures. Urolithin A treatment led cells to invade the wound poorly compare with untreated cells (Figure 1B). Urolithin A has been previously reported to induce cell death in human prostate cancer cell lines,32 we found Urolithin A also reduced cell viability in A549 and H460 cells in dose-dependent manner (Figure S1A). However, under the experimental condition of wounding assay, the cell viability was not affected by Urolithin A (Figure S1B), indicating that inhibition of cell migration by Urolithin A in wound assays was not caused by cell death. Furthermore, the cell-invasive potential of both cell lines was similarly blocked by urolithin A treatment judged by the numbers of cell that penetrated the Matrigel-coated chamber (Figure 1C). To confirm these results implicating that urolithin A might regulate EMT-like program, we analyzed some proteins of EMT markers by Western blot. Cells were treated with various concentrations of urolithin A for 10 h (Figure 1D; Figure S1C). Consistent with the possible inhibition of EMT program, urolithin A upregulated epithelial marker E-cadherin and downregulated mesenchymal markers Vimentin and N-cadherin in a dose-dependent manner.
 |
Figure 1 Urolithin A inhibits EMT in lung cancer cells. (A) A549 and H460 cells were exposed to 10 μM urolithin A for 24 h. Cell morphology was assessed by phase-contrast microscopy (magnification 100×), scale bars 200 μm. (B) Wound Healing Assay reveals a dose-dependent (urolithin A 0, 10, 20 μM) (upper panel) and time-dependent (0, 12, 24 h) (lower panel) decrease in cell migration, scale bars represent 200 μm. The quantification was present in right panel. (*P < 0.05, **P < 0.01, ***P < 0.001 for difference from untreated control by ANOVA with Dunnett’s correction for multiple comparisons). (C) A549 and H460 were exposed to different concentrations of urolithin A (0, 10, 20 μM) treatment for 24 h. The cell invasiveness was assessed by Transwell chamber, scale bars indicate 200 μm. (**P < 0.01, ***P < 0.001 for difference from untreated control by ANOVA with Dunnett’s correction for multiple comparisons). (D) A549 and H460 cells were treated with different concentrations of urolithin A (0, 5, 10, 15, 20, 25 μM). Western blot examined epithelial marker E-cadherin, mesenchymal markers N-cadherin, Vimentin. |
Snail is Required for Inhibition of EMT by Urolithin A in Lung Cancer Cells
In epithelial cells, E-cadherin plays an important role in maintaining the epithelial integrity. Several EMT-inducing transcriptional factors such as Snail, Slug, Twist1 and Zeb1 repress E-Cadherin expression to initiate cell migratory and invasive program. Our previous study has demonstrated that Snail plays a key role in EMT process in lung cancer cells.31 Interestingly, exposure to urolithin A markedly decreased the level of Snail protein in a dose-dependent manner, but not that of Slug, Twist and Zeb1 (Figure 2A; Figure S2A). To investigate whether Snail is related to urolithin A-induced EMT inhibition, we next measured the effect of manipulation of Snail levels on the urolithin A-dependent E-cadherin upregulation. As shown in Figure 2B, urolithin A induced E-cadherin levels in a dose-dependent manner in both A549 and H460 cells transfected with control siRNA; however, this induction was markedly enhanced in cells when Snail levels was diminished by siRNA-mediated knockdown (Figure 2B; Figure S2B and S2C). Significantly, overexpressing of Snail attenuated the basal and urolithin A-induced expression of E-cadherin (Figure 2C; Figure S2D), indicating that Snail is an essential mediator of E-cadherin downregulation induced by urolithin A in A549 and H460 cells. The dominant role of Snail in mediating inhibition of EMT program by urolithin A is established by the fact that urolithin A decreased cell migratory and invasive potential even in the cells overexpressing the Snail protein (Figure 2D, E).
 |
Figure 2 Snail is required for inhibition of EMT by urolithin A in lung cancer cells. (A) Western blot demonstrates decreased Snail expression following 5 h of urolithin A (0, 5, 10, 15, 20 and 25 μM) stimulation in H1299 and A549 cell lines compared with Slug, Twist and Zeb1. (B) The cells transfected with a control or Snail-specific siRNA. At 48 h post-transfection, cells were stimulated with urolithin A for additional 10 h. Western blotting shows that the expression of E-cadherin was increased in cells transfected with a Snail siRNA. (C) A549 and H460 cells were transfected with a Snail cDNA. After 48 h, cells were untreated or treated with the indicated amounts of urolithin A for 10 h. Western blotting shows that the urolithin A-induced levels of E-Cadherin decreased further in the cells transfected with a Snail cDNA. (D) The cell migration of A549 and H460 after transfection of Snail cDNAs and urolithin A treatment (urolithin A 0, 10 μM) was assessed by the Wound healing assay. The quantification was present in right panels. (*P<0.01, **P<0.01, ***P<0.001 for the difference from the control cells). (E) The cell invasion and motility of A549 and H460 after transfection of Snail cDNAs and urolithin A treatment were assessed by the Cell Invasion Assay. (**P<0.01, ***P<0.001 for the difference from the control cells). |
Urolithin A Induces Snail Degradation via mdm2-Mediated Ubiquitination
Since inhibition of EMT-like program by urolithin A is dependent on Snail, we first investigated whether urolithin A influences mRNA levels of Snail. Interestingly, RT-PCR data revealed no change in Snail mRNA level in the presence of urolithin A (Figure 3A). Furthermore, urolithin A did not cause discernible change in the Snail promoter activity (Figure 3B), suggesting the Snail is regulated post-transcriptionally by urolithin A. To test this possibility, we examined Snail stability by CHX chase experiments. As shown in Figure 3C, compared with the untreated cells, cells treated with urolithin A exhibited markedly decreased half-life of Snail proteins (Figure S3A). Previous research has suggested that urolithin A exhibits a pro-autophagy effect in certain cell types.33 Therefore, we next addressed the role of autophagy in urolithin A-induced Snail degradation. Indeed, urolithin A is capable of inducing autophagy in both A549 cells and H460 cells (Figure 3D; Figure S3B), as indicated by increased levels of microtubule-associated protein 1 light chain-3B (LC3B, a lipid form of LC3), which is a general marker for autophagy flux, and reduced levels of p62, which is autophagy adaptor residing in the late lysosome (compare lanes 1, 2 and 3). Consistent with previous study, p62 levels was accumulated when autophagy was inhibited by hydroxychloroquine (HCQ) treatment (compare lanes 4 and 5), which inhibits autophagosome fuses with lysosome. Remarkably, reduction of Snail by urolithin A was not rescued by HCQ, indicating urolithin A-induced autophagy is irrelevant for Snail regulation by urolithin A. Next, we test the hypothesis that ubiquitination degradation was involved in deduction of Snail induced by urolithin A. Remarkably, an effective proteasome inhibitor (PII) rescues the levels of Snail protein even in the presence of urolithin A (Figure 3E; Figure S3C). Furthermore, in immunoprecipitation assays, we observed that urolithin A significantly enhanced Snail ubiquitination compared with control (Figure 3).
 |
Figure 3 Urolithin A induces Snail degradation via mdm2-mediated ubiquitination. (A) A549 and H460 cells were treated with urolithin A (0, 10 and 20 μM) for 5 h. The expression of Snail gene was detected by RT-PCR. (ns means no statistical difference). (B) A549 and H460 cells were co-transfected with a plasmid of the Snail promoter luciferase reporter gene with a plasmid of control Renilla luciferase reporter gene. At 36 h after transfection, cells were treated with urolithin A (0, 5, 10 and 20 μM) for 5 h, and luciferase activity was detected using the dual luciferase reporter system. (ns means no statistical difference). (C) Cells were treated with CHX (Cycloheximide, 50 μg/mL) for the indicated time in the presence or absence of urolithin A. Western blot was used to determine Snail protein levels. (D) Western blotting analysis of Snail, p62 and LC3A/B after cells were pre-treated with 20 μM HCQ for 1 h and then treated with urolithin A (0, 10 and 20 μM) for 5 h in A549 and H460 cells. (E) Western blotting analysis of Snail, after cells were pre-treated with 20 μM PII for 1 h and then treated with urolithin A (0, 10 and 20 μM) for 5 h in A549 and H460 cells. (F) Cells were treated with urolithin A after which cell lysates were immunoprecipitated with anti-Snail antibody and then Western blotted with anti-Ubiquitin. (G) Western blot examined mdm2 expression flowing 5 h of urolithin A (0, 5, 10, 15, 20 and 25 μM) stimulation in A549 and H460 cells. (H and I) After transfection with mdm2 cDNA (H) or mdm2 siRNA (I) for 48 h, A549 and H460 cells were treated with urolithin A (0, 10 and 20 μM) for 5 h. Western blot was carried out for analysis of Snail levels. (J) Cells were treated with urolithin A for 5 h after which cell lysates were immunoprecipitated with anti-mdm2 antibody and then Western blotted with anti-Snail. |
Previous study determined that mdm2, a p53 E3-ubiquitin ligase, plays crucial roles in the regulation of Snail degradation via ubiquitination (Lim et al 2010). To ascertain whether urolithin A acts through mdm2 to downregulate Snail expression, we first measured the protein levels of mdm2. Interestingly, urolithin A increased mdm2 levels in a dose-dependent manner (Figure 3G; Figure S3D). Consistent with the demonstrated ability of mdm2 to regulate Snail levels, Snail expression is suppressed significantly in the cells transfected with mdm2 cDNA (Figure 3H; Figure S3E). Conversely, when cells were transfected with mdm2 siRNA, urolithin A-induced Snail degradation were predictably rescued (Figure 3I; Figure S3F; S3G), suggesting that urolithin A regulates the levels of Snail protein via a mdm2-dependent process. In this regard, co-IP assay showed that the association of Snail with mdm2 is remarkably enhanced in the presence of urolithin A (Figure 3J). Taken together, these data indicate that urolithin A increases the levels of E3-ubiquitin ligase mdm2 to control Snail stability.
Urolithin A Upregulates mdm2 by Inhibiting the Interaction of p53 and mdm2
Next, we further explore the mechanism of mdm2 regulation by urolithin A. Mdm2 is well-documented transcriptional target of p53, which also ubiquitinates p53 and commits it to degradation in a negative feedback loop. Recent study has established urolithin A inhibits the p53-mdm2 interaction, which is sufficient to induce mdm2 accumulation by preventing p53 degradation (Mohammed Saleem et al 2020). Consistently, the expression of mdm2 in p53-null H1299 cells showed no discernible changes by urolithin A (Figure 4A; Figure S4A). Significantly, urolithin A increased endogenous p53 levels in a dose-dependent manner in p53-wild-type A549 and H460 cells (Figure 4B; Figure S4B). Furthermore, co-IP experiment showed the p53 polyubiquitination and interaction between p53 and mdm2 were markedly decreased by urolithin A treatment (Figure 4C).
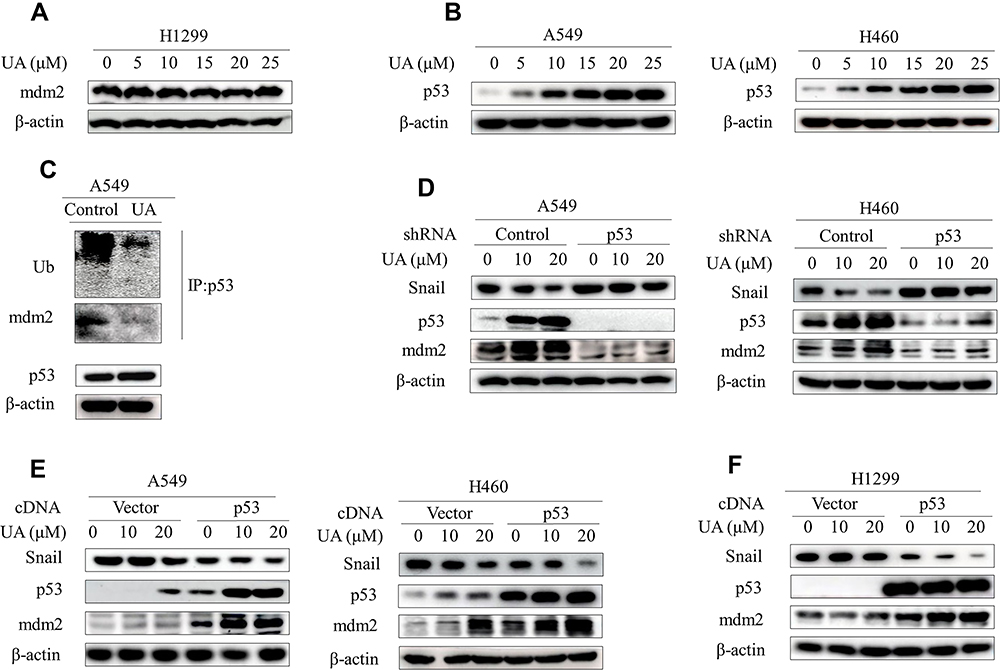 |
Figure 4 Urolithin A upregulates mdm2 by inhibiting the interaction of p53 and mdm2. (A) H1299 cells were treated with different concentrations of urolithin A (0, 5, 10, 15, 20 and 25 μM) for 5 h. Western blot examined the expression of mdm2. (B) Western blot demonstrates expression of p53 following 5 h of urolithin A (0, 5, 10, 15, 20 and 25 μM) stimulation in indicated lung cancer cell lines. (C) Cells were treated with urolithin A for 5 h after which cell lysates were immunoprecipitated with anti-p53 antibody and then Western blotted with anti-Ubiquitin and anti-mdm2 antibodies. (D) Transfection of A549 and H460 cells with p53 shRNA for 48h, and then treated with different concentrations of urolithin A for 5 h, the expression levels of mdm2 and Snail were analyzed by immunoblotting. (E and F) Indicated cells were transfected with p53 cDNA. After 48 h, cells were treated with urolithin A (0, 10 and 20 μM) for 5 h. The levels of mdm2 and Snail were detected by Western blotting. |
Our results thus suggested a model whereby urolithin A stabilizes p53 to increase mdm2 levels, which in turn ubiquitinates Snail protein. In this scenario, p53 should be required for urolithin A-induced deduction of Snail protein. Indeed, knockdown of endogenous p53 dramatically rescued the urolithin A-induced Snail degradation (Figure 4D; Figure S4C and D). In contrast, urolithin A-induced Snail degradation were significantly enhanced when cells were transfected with p53 expression vector (Figure 4E; Figure S4E). Significantly, urolithin A increased mdm2 levels and decreased Snail expression in p53-null H1299 cells when transfected with wild-type p53 cDNA (Figure 4F; Figure S4F). Taken together, our results indicated that the p53 plays a key role in the regulation of urolithin A-induced Snail degradation through upregulation of mdm2.
Discussion
EMT is a fundamental process in development.34 However, cancer cells take advantage of this process to elicit a complex phenotypic switch that endows tumor cells to survive during invasion and metastasis.35 Therefore, it is important to seek the regents which has both the effects of anti-proliferation and inhibiting EMT. Interestingly, the present study has shown urolithin A, a metabolite of intestinal flora, serves as a natural drug in regulation of EMT. Previous studies have found that urolithin A has a significant effect on antioxidation and inflammation response.25,26 Recently, urolithin A is an extensively studied natural reagent with anti-cancer ability in inhibiting cell proliferation.28 However, none of the studies has examined the role in regulation of metastasis. In this study, we used several NSCLC cell lines to address the role of urolithin A in EMT process. We demonstrated that urolithin A dose-dependently increased epithelial marker E-cadherin and decreased mesenchymal markers N-cadherin and Vimentin. The results of wound healing assay and cell invasion assay indicated urolithin A suppresses cell migration and invasion in A549 and H460 cells. We also found that Snail is critical for mediating urolithin A-induced E-cadherin upregulation. Mechanistically, urolithin A suppresses the interaction of p53 and mdm2 leading to p53 upregulation. p53 in turn promotes the transcriptional activity of mdm2 gene and enhances expression of mdm2 expression. By binding to Snail, mdm2 catalyzes its ubiquitylation and subsequent degradation, which cause E-cadherin upregulation.
p53 is generally thought to trigger DNA repair and apoptosis. Since over 50% of human cancers harbor p53 mutations, mutational inactivation is a major molecular mechanism behind p53 dysfunction.36 Although wild-type p53 exists in a few cancer cell lines, its function is inhibited by some oncogenes, such as Kras.37 In our study, two non-small-cell lung cancer (NSCLC) cell lines, A549 and H460 harboring p53 wild-type, are used to investigate the effect of anti-tumor. Previous studies demonstrated urolithin A induces cell senescence via p53-p21 pathway in the LNCaP cell line.32,38 Other study has shown the effects of urolithin A on mdm2 in different p53 genetic type CaP cells.32 Although the results of our study on urolithin A regulation of mdm2 levels are similar with the previous results in wt-p53 cell lines, the results vary in p53 null cell lines. The protein expression of mdm2 was below detection limit in another NSCLC cell line H1299 (p53−/−) as compared to A549 (p53+/+) and H460 (p53+/+). Interestingly, overexpression of p53 with urolithin A treatment increases mdm2 expression in a dose-dependent manner in H1299 cells, suggesting the requirement of p53 in mdm2 expression. Therefore, we speculate that urolithin A may act as a molecular target antagonist to directly inhibit the interaction between p53 and mdm2.
Snail, a key transcriptional repressor, is related with cancer progress. It is reported that K-ras activated Snail could suppress the function of p53 by binding it.37 Another study has shown that p53- mdm2 pathway could regulate Snail degradation.18 In the model, mdm2-mediated Snail degradation is required for p53-mdm2-Snail complex. However, our study established a new model for degradation of Snail by mdm2, independent of p53-mdm2-Snail complex. First, IP experiments shown urolithin A suppresses the interaction of p53-mdm2 and enhances the interaction of mdm2-Snail. Furthermore, overexpression of mdm2 obviously decreased Snail protein level in p53 null H1299 cells.
Conclusion
Our data show Snail plays a significant role in regulation of EMT. Moreover, mdm2 is a latent target of urolithin A for cancer therapy by inhibiting proliferation and metastasis in wild type p53 cell lines. We clearly explain how urolithin A, a natural anticancer drug, overcomes the side effects of EMT to escape tumor metastasis in lung cancer cells. Moreover, this theory also supports that urolithin A can be used as a first-line chemotherapeutic drug combination candidate to prevent drug resistance.
Acknowledgments
This work was Supported by National Natural Science Foundation of China (81872371 to Zhihao Wu), Open Project Program of State Key Laboratory of Molecular Oncology (SKL-KF-2019-11 to Zhihao Wu) and Natural Science Foundation of the Higher Education Institutions of Anhui Province (KJ2018A0248 Huijun Wei). Jintao Dou, Yong Zhang and Xiang Wang are team members of Precision Medicine Beidou Research Group.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Molina JR, Yang P, Cassivi SD, et al. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic Proc. 2008;83(5):584–594. doi:10.12677/ACRPO.2016.53004
3. Chaffer CL, San Juan BP, Lim E, et al. Emt, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35(4):645–654. doi:10.1007/s10555-016-9648-7
4. Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer. 2016;15(18). doi:10.1186/s12943-016-0502-x
5. Luo T, Wang L, Wu P, et al. Downregulated vimentin and upregulated e-cadherin in t1 stage non-small-cell lung cancer: does it suggest a mesenchymal-epithelial transition? Neoplasma. 2017;64(5):693–699. doi:10.4149/neo_2017_506
6. Nieszporek A, Skrzypek K, Adamek G, et al. Molecular mechanisms of epithelial to mesenchymal transition in tumor metastasis. Acta Biochimica Polonica. 2019;66(4):509–520. doi:10.18388/abp.2019_2899
7. Konrad L, Dietze R, Riaz MA, et al. Epithelial-mesenchymal transition in endometriosis-when does it happen? J Clin Med. 2020;9(6). doi:10.3390/jcm9061915
8. Sundararajan V, Tan M, Tan TZ. Snai1 recruits hdac1 to suppress snai2 transcription during epithelial to mesenchymal transition. Sci Rep. 2019;9(1):8295. doi:10.1038/s41598-019-44826-8
9. von Burstin J, Eser S, Paul MC, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a snail/hdac1/hdac2 repressor complex. Gastroenterology. 2009;137(1):
10. Zhang Y, Zhang X, Ye M, et al. Fbw7 loss promotes epithelial-to-mesenchymal transition in non-small cell lung cancer through the stabilization of snail protein. Cancer Lett. 2018;419:75–83. doi:10.1016/j.canlet.2018.01.047
11. Smith BN, Odero-Marah VA. The role of snail in prostate cancer. Cell Adh Migr. 2012;6(5):433–441. doi:10.4161/cam.21687
12. Hojo N, Huisken AL, Wang H, et al. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci Rep. 2018;8(1):8704. doi:10.1038/s41598-018-27021-z
13. Yang S, Liu Y, Li MY, et al. Foxp3 promotes tumor growth and metastasis by activating wnt/β-catenin signaling pathway and emt in non-small cell lung cancer. Mol Cancer. 2017;16(1):124. doi:10.1186/s12943-017-0700-1
14. Leng Z, Li Y, Zhou G, et al. Krüppel-like factor 4 regulates stemness and mesenchymal properties of colorectal cancer stem cells through the tgf-β1/smad/snail pathway. J Cell Mol Med. 2020;24(2):1866–1877. doi:10.1111/jcmm.14882
15. Meng Q, Shi S. Abrogation of glutathione peroxidase-1 drives emt and chemoresistance in pancreatic cancer by activating ros-mediated akt/gsk3β/snail signaling. Oncogene. 2018;37(44):5843–5857. doi:10.1038/s41388-018-0392-z
16. Ryu KJ, Park SM, Park SH, et al. P38 stabilizes snail by suppressing dyrk2-mediated phosphorylation that is required for gsk3β-βtrcp-induced snail degradation. Cancer Res. 2019;79(16):4135–4148. doi:10.1158/0008-5472.CAN-19-0049
17. Zhou BP, Deng J, Xia W, et al. Dual regulation of snail by gsk-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi:10.1038/ncb1173
18. Lim SO, Kim H, Jung G. P53 inhibits tumor cell invasion via the degradation of snail protein in hepatocellular carcinoma. FEBS Lett. 2010;584(11):2231–2236. doi:10.1016/j.febslet.2010.04.006
19. Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi:10.1038/387296a0
20. Hafner A, Bulyk ML, Jambhekar A, et al. The multiple mechanisms that regulate p53 activity and cell fate. 2019;20(4):199–210. doi:10.1038/s41580-019-0110-x
21. Sánchez-González C, Ciudad CJ, Noé V, et al. Health benefits of walnut polyphenols: an exploration beyond their lipid profile. Crit Rev Food Sci Nutr. 2017;57(16):3373–3383. doi:10.1080/10408398.2015.1126218
22. Altieri F, Cairone F, Giamogante F, et al. Influence of ellagitannins extracted by pomegranate fruit on disulfide isomerase pdia3 activity. Nutrients. 2019;11:1. doi:10.3390/nu11010186
23. Evtyugin DD, Magina S. Recent advances in the production and applications of ellagic acid and its derivatives. A review. Molecules. 2020;25(12). doi:10.3390/molecules25122745
24. Olennikov DN, Kashchenko NI, Chirikova NK. In vitro bioaccessibility, human gut microbiota metabolites and hepatoprotective potential of chebulic ellagitannins: a case of padma hepaten® formulation. Nutrients. 2015;7(10):8456–8477. doi:10.3390/nu7105406
25. Jing T, Liao J, Shen K, et al. Protective effect of urolithin a on cisplatin-induced nephrotoxicity in mice via modulation of inflammation and oxidative stress. Food Chem Toxicol. 2019;129(108–114). doi:10.1016/j.fct.2019.04.031
26. Fu X, Gong LF, Wu YF, et al. Urolithin a targets the pi3k/akt/nf-κb pathways and prevents il-1β-induced inflammatory response in human osteoarthritis: in vitro and in vivo studies. Food Funct. 2019;10(9):6135–6146. doi:10.1039/C9FO01332F
27. Totiger TM, Srinivasan S, Jala VR, et al. Urolithin a, a novel natural compound to target pi3k/akt/mtor pathway in pancreatic cancer. Mol Cancer Ther. 2019;18(2):301–311. doi:10.1158/1535-7163.MCT-18-0464
28. Qiu Z, Zhou J, Zhang C, et al. Antiproliferative effect of urolithin a, the ellagic acid-derived colonic metabolite, on hepatocellular carcinoma hepg2.2.15 cells by targeting lin28a/let-7a axis. Braz J Med Biol Res. 2018;, 51(7):e7220. doi:10.1590/1414-431x20187220
29. Norden E, Heiss EH. Urolithin a gains in antiproliferative capacity by reducing the glycolytic potential via the p53/tigar axis in colon cancer cells. Carcinogenesis. 2019;40(1):93–101. doi:10.1093/carcin/bgy158
30. González-Sarrías A, Espín JC, Tomás-Barberán FA, et al. Gene expression, cell cycle arrest and mapk signalling regulation in caco-2 cells exposed to ellagic acid and its metabolites, urolithins. Mol Nutr Food Res. 2009;53(6):686–698. doi:10.1002/mnfr.200800150
31. Li X, Zhang Z, Zhang Y, et al. Upregulation of lactate-inducible snail protein suppresses oncogene-mediated senescence through p16(ink4a) inactivation. J Exp Clin Cancer Res. 2018;37(1):39. doi:10.1186/s13046-018-0701-y
32. Mohammed Saleem YI, Albassam H, Selim M. Urolithin a induces prostate cancer cell death in p53-dependent and in p53-independent manner. Eur J Nutr. 2020;59(4):1607–1618. doi:10.1007/s00394-019-02016-2
33. Ahsan A, RongZheng Y, LiWu X, et al. Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci Ther. 2019;25(9):976–986. doi:10.1111/cns.13136
34. Xing J, Tian XJ. Investigating epithelial-to-mesenchymal transition with integrated computational and experimental approaches. Phys Biol. 2019;16(3):031001. doi:10.1088/1478-3975/ab0032
35. Antony J, Thiery JP, Huang RY. Epithelial-to-mesenchymal transition: lessons from development, insights into cancer and the potential of emt-subtype based therapeutic intervention. Phys Biol. 2019;16(4):041004. doi:10.1088/1478-3975/ab157a
36. Pitolli C, Wang Y, Mancini M, et al. Do mutations turn p53 into an oncogene? Int J Mol Sci. 2019;20:24. doi:10.3390/ijms20246241
37. Lee SH, Lee SJ, Chung JY, et al. P53, secreted by k-ras-snail pathway, is endocytosed by k-ras-mutated cells; implication of target-specific drug delivery and early diagnostic marker. Oncogene. 2009;28(19):2005–2014. doi:10.1038/onc.2009.67
38. Sánchez-González C, Ciudad CJ, Izquierdo-Pulido M, et al. Urolithin a causes p21 up-regulation in prostate cancer cells. Eur J Nutr. 2016;55(3):1099–1112. doi:10.1007/s00394-015-0924-z
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.