Back to Journals » Drug Design, Development and Therapy » Volume 20
Tumor Assembloids as Three-Dimensional Platforms for Modeling Drug Delivery Barriers: Construction Strategies, Applications, and Translational Challenges
Authors Zhang J ![]() , Luo X
, Luo X ![]() , Sun Z, Jin B
, Sun Z, Jin B ![]() , Zhao Y
, Zhao Y ![]()
Received 21 April 2026
Accepted for publication 3 July 2026
Published 14 July 2026 Volume 2026:20 618870
DOI https://doi.org/10.2147/DDDT.S618870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Jiankang Zhang,1 Xinying Luo,1 Zhongyi Sun,1 Bohao Jin,1 Yunqi Zhao1– 4
1Department of Natural Sciences, Wenzhou-Kean University, Wenzhou, Zhejiang, People’s Republic of China; 2International Frontier Interdisciplinary Research Institute, Wenzhou-Kean University, Wenzhou, Zhejiang, People’s Republic of China; 3Zhejiang Bioinformatics International Science and Technology Cooperation Center, Wenzhou-Kean University, Wenzhou, Zhejiang, People’s Republic of China; 4Department of Biological Sciences, Kean University, Union, NJ, USA
Correspondence: Yunqi Zhao, Department of Natural Sciences, Wenzhou-Kean University, Wenzhou, Zhejiang, People’s Republic of China, Email [email protected]
Abstract: A major limitation of conventional two and three-dimensional preclinical in vitro cancer models is their inability to reproduce the drug-delivery barriers. Tumor assembloids, which integrate patient-derived cancer cells with stromal, endothelial, and immune components in three-dimensional architectures, provide a manipulable framework for simulating these multicellular impediments. This review specifically examines the application of tumor assembloids to investigate drug delivery constraints, including stromal exclusion, vascular transport, immune-mediated resistance, penetration gradients, and spatially heterogeneous drug exposure. We compared three major construction strategies, including self-assembly, 3D bioprinting, and microfluidic compartmentalization, and evaluated their respective strengths for drug assessment. We also discussed assembloids’ current limitations, including reproducibility, incomplete physiological dynamics, insufficient spatial analytics, and the need for standardized benchmarking. Overall, tumor assembloids represent promising mechanistic platforms for studying tumor drug delivery barriers. However, broader clinical application will necessitate rigorous validation and harmonized assay standards.
Keywords: tumor assembloids, organoid, drug delivery barriers, tumor microenvironment, precision oncology
Introduction
The successful development of anticancer therapeutics depends on the reliability of preclinical models. For anticancer therapeutics, that standard is especially demanding. A useful model must not only estimate in vivo efficacy, but also capture the delivery bottlenecks imposed by the native tumor milieu.1 Most commonly used systems do this only in part. Conventional two-dimensional (2D) cultures are easy to scale and convenient for screening. They are particularly susceptible to culture-induced genetic and transcriptional evolution during repeated passaging, which can alter drug sensitivity and contribute to inconsistent pharmacological readouts.2 In comparison, patient-derived organoids (PDOs) more closely resemble the clinical setting by retaining clonal heterogeneity and enabling medium-scale drug testing.3 They generally retain a higher degree of genomic and phenotypic similarity to the parental tumor, although prolonged culture, selective media conditions, and clonal expansion may still introduce model-specific drift.4,5 They usually still lack the stromal, immune, and vascular elements that shape drug access within tumors. That absence matters because without those compartments, it becomes difficult to reproduce the physical obstruction to penetration and the context-dependent resistance imposed by the microenvironment.6 Animal models preserve vascular and immune integrity, but they introduce a different problem. Species-specific differences in extracellular matrix (ECM) structure and immune infiltration can alter local drug transport in ways that are not trivially comparable to human tumors. They are also expensive and inherently low-throughput, making them not suited for personalized treatment selection or large-scale compound screening.7,8
These limitations stem largely from the absence of an authentic human tumor microenvironment (TME). The TME is more than just a passive setting. It actively contributes to the modulation of how drugs are transported and the responses to treatment. Cancer-associated fibroblasts (CAFs), for instance, synthesize and reorganize a dense ECM, which can obstruct the passage of therapeutic agents prior to their arrival at malignant cells.9 Through its control over drug extravasation, the vascular endothelium introduces a new dimension of regulation for distributing medication into the tumor interstitium.10 Immune cell populations add further complexity by establishing biochemical barriers to drug efficacy while also serving as mediators or targets of therapeutic interventions. These major physical and cellular barriers in solid tumors are demonstrated in Figure 1. Numerous traditional frameworks usually manage to capture just scattered components of this elaborate multicellular system. Hence, they often do not manage to replicate the synchronized transportation hurdles and cellular interactions that dictate treatment results in human tumors, which could reveal the ongoing differences between preclinical data and clinical results.11,12
 |
Figure 1 Major physical and cellular barriers in the tumor microenvironment. Drug-delivery performance can be assessed by drug penetration depth, vascular permeability, drug extravasation, spatial drug distribution, immune-cell infiltration, and therapeutic response. |
The assembloid concept emerged from the broader organoid field as a strategy to overcome a central limitation of single-lineage organoids. This limitation lies in their incomplete representation of interactions among distinct tissue compartments. Early assembloid studies were especially influential in developmental neuroscience. Researchers fused region-specific human brain spheroids or organoids within this field to model processes such as interneuron migration, axonal projection, and circuit formation. For example, human forebrain spheroids were assembled to generate functionally integrated forebrain models that recapitulate aspects of interneuron migration and neural connectivity.13 This principle was later extended beyond the nervous system to epithelial-stromal tissue reconstruction. Typical examples include bladder assembloids, which combine epithelial stem-cell-derived structures with stromal components to reproduce multilayered tissue architecture.14 In cancer research, tumor assembloids represent a further evolution of this concept. Rather than modeling tumor cells alone, these constructs integrate patient-derived malignant cells alongside stromal, endothelial, and immune components to recapitulate core features of the tumor microenvironment.15 In this review, tumor assembloids are defined as spatially organized 3D tumor constructs.16 They comprise malignant cells and at least one supplementary tumor microenvironmental component, like stromal, endothelial, or immune cells. This operational definition is consistent with the broader assembloid concept, in which distinct organoids or cell types are integrated to model inter-compartmental interactions. This definition sets tumor assembloids apart from several alternative in vitro models. Spheroids are essentially pure aggregates of tumor cells. Conventional organoids maintain epithelial and tumor cell heterogeneity but commonly omit non-malignant microenvironmental compartments. Unlike the above two models, basic co-cultures cannot reliably develop ordered tissue-like structures. As for organ-on-chip systems, these are microengineered perfused devices capable of accommodating assembloids, yet they are not assembloids per se.17
Compared with 2D cultures, spheroids and conventional patient-derived organoids, tumor assembloids are better suited for modeling multicellular drug-delivery barriers.18 Two-dimensional cultures mainly reflect cell-intrinsic drug sensitivity. Spheroids feature improved 3D architecture yet insufficient microenvironmental complexity, whereas conventional patient-derived organoids retain tumor heterogeneity but commonly lack stromal, vascular, and immune compartments. Animal and patient-derived xenograft models retain systemic physiology but are low-throughput, costly, and affected by species-specific stromal and immune differences.19 Thus, tumor assembloids occupy an intermediate position between reductionist in vitro systems and complex in vivo models. They provide experimentally controllable platforms to investigate stromal exclusion, vascular transport, immune-mediated resistance, and spatially heterogeneous drug exposure.
Tumor assembloids offer one promising route toward closing this translational divide. These 3D constructs combine patient-derived cancer cells with stromal and immune components, such as cancer-associated fibroblasts, endothelial cells, and autologous immune cells. When properly established, they can reproduce many key features of solid tumors in their native state. These include extracellular matrix architecture, immune microenvironmental features, and restricted perfusion typical of in vivo tumors.14 A drug introduced into such a system must contend with physical and cellular barriers not unlike those encountered in vivo. Tumor assembloids can generate several distinct categories of therapeutic readouts that should not be interpreted interchangeably. Tumor cells possess inherent drug sensitivity.20 This trait is not affected by the surrounding microenvironment. Tumor resistance can also be induced by stromal components. Fibroblasts, immune cells and extracellular matrix interact with tumor cells in this process. Such interactions weaken the effects of treatment. Vascular transport involves drug extravasation and passage through endothelial barriers.21 Drug delivery ultimately evaluates three key aspects. These are physical penetration, spatial distribution and intratumoral concentration gradients. This review, we emphasize that the principal advantage of tumor assembloids lies in reconstructing the latter three microenvironment-dependent processes rather than merely measuring intrinsic drug sensitivity. Whether this translates to genuinely more predictive efficacy readouts remains an open question, though early evidence is encouraging.22 Worth noting is a subtler consequence, the very barriers that blunt drug penetration also appear to shelter genetically heterogeneous subclones whose resistance profiles diverge widely. Monolayer cultures strip away this spatial complexity almost entirely, but assembloids do not.23
The field has not lacked for reviews, but their focal points differ in ways that leave an important gap. Mei et al surveyed organoid and assembloid platforms largely as immunotherapy testbeds and briefly touched on drug delivery assessment as one application area.24 Onesto et al turned to cell-cell interaction modeling, with neurodevelopment as the principal application.25 Brain assembloids in neuroscience were studied by Wu and Nowakowski.26 Liu et al used a taxonomic approach, sorting assembloids into four categories based on assembly strategy across organ systems.27 While these reviews collectively acknowledge that assembloids can be applied to drug delivery studies, none have systematically organized tumor assembloid platforms according to the specific types of delivery barriers they reconstruct. In particular, a framework that categorizes platforms by their capacity to model penetration resistance, vascular transport, stromal exclusion, and spatially heterogeneous drug exposure is currently lacking. This review discusses three fabrication paradigms: self-assembly, 3D bioprinting, and microfluidic compartmentalization. In each case, we assess how consistently the resulting construct reproduces the spatial and cellular barriers that influence drug penetration, local distribution, and therapeutic outcome. We close by confronting the technical limitations that continue to constrain the field, sketching a path toward measurement standardization, and weighing the realistic prospects for moving assembloid-derived insights into the clinic.
The Tumor Microenvironment and Drug Delivery Barriers
Immune Cells in the TME
The TME is not a passive participant in cancer progression. It operates as a dynamic ecosystem whose cellular composition shifts with disease stage, treatment pressure, and host genetics.28 Within this ecosystem, immunosuppressive populations, such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), coexist with cytotoxic T lymphocytes and natural killer cells. The balance between these compartments dictates whether the local milieu suppresses or licenses anti-tumor immunity.29 TAMs illustrate the point well. Through TGF-β and IL-10 secretion, they can blunt T cell cytotoxicity directly, an effect reinforced by checkpoint engagement via the PD-1/PD-L1 axis.30
How does this immune landscape shape drug delivery? Two interrelated mechanisms stand out. TAMs and CAFs jointly deposit and crosslink ECM constituents, collagen and fibronectin, which raise stromal density and impede penetration of both small-molecule chemotherapeutics and large biologics into the tumor core.31 Layered on top of this physical barrier is a biochemical one. Immunosuppressive myeloid- and fibroblast-rich TMEs can impair therapeutic efficacy through ECM remodeling, immune exclusion, and altered local signaling.32,33 A less appreciated contributor is the ATP-binding cassette (ABC) transporter family expressed on select immune subsets. These efflux pumps may further erode local drug bioavailability by extruding therapeutics from the microenvironment.34
The relationship, however, runs in both directions. Checkpoint inhibitors that re-activate cytotoxic T cells can trigger transient vascular normalization, widening the perfusion window and, paradoxically, improving co-delivery of cytotoxic agents.35 This synergy is conspicuously absent in most simplified preclinical models, underscoring a basic mismatch between in vivo complexity and conventional in vitro platforms. Assembloid architectures that embed patient-derived immune cells within a structured stromal scaffold offer one route toward closing that gap,36 though whether they faithfully recapitulate the temporal dynamics of immune-drug crosstalk remains an open question.
The foregoing discussion underscores that immune populations are not merely bystanders in the TME. They actively shape the physical and biochemical landscape through which drugs must travel. This dual role of simultaneously modulating ECM density, vascular permeability, and local drug metabolism means that any preclinical model lacking these immune components will systematically underestimate the delivery barriers a therapeutic agent faces in vivo. This limitation is precisely what conventional organoid platforms encounter, as discussed below.
Limitations of Organoids in Modeling Drug Delivery Barriers
PDOs maintain the mutational profile and extensive phenotypic characteristics of the donor, which helps explain the rapid adoption of this platform.37 However, most conventional PDO systems fail to fully preserve the immunological microenvironment. In many standard culture formats, key immune populations such as macrophages, dendritic cells, and T lymphocytes are absent or rapidly lost during propagation. Within an in vivo tumor context, these cell populations are not mere bystanders. They actively engage in the phagocytosis of therapeutic carriers, modulate local cytokine networks, and significantly impact the tissue’s resistance to molecular diffusion.38 The absence of these cells results in the elimination of a critical layer of pharmacokinetic regulation that cannot be compensated for by any degree of medium optimization.39
There exists a more nuanced issue. In actual tumors, the interstitial fluid undergoes shifts, cells collectively migrate, and the extracellular matrix is remodeled over timescales of hours, all of which create drug-concentration gradients that are both spatially steep and temporally variable.40 An organoid situated in a well does not replicate this dynamic. The process of diffusion occurs passively, the matrix experiences minimal remodeling, and the resultant concentration profile is considerably more uniform than what a solid tumor would generate. For systemic agents, this disparity might be acceptable However, for approaches reliant on localized flow, such as intraperitoneal perfusion or localized infusion, this is not the case. The vascular system introduces yet another disparity, arguably the most significant one. A drug entering a perfused tumor must cross the endothelium, contend with interstitial pressure that can exceed 30 mmHg in some carcinomas, and distribute under flow conditions that are heterogeneous over short distances across the vascular bed.41 Several microphysiological systems have begun to address this pressure-related barrier more directly.42 Tumor-microenvironment-on-chip platforms contain capillary, interstitial, and lymphatic compartments. The fluid pressure inside each compartment can be adjusted separately. This feature enables researchers to explore the regulatory effects of pressure gradients. The regulated processes include nanoparticle extravasation, interstitial diffusion and lymphatic drainage. In such systems, elevated interstitial pressure or an unfavorable capillary-to-interstitial pressure gradient can markedly reduce nanoparticle penetration into the tumor matrix.43 Other spheroid-on-chip and organ-on-chip platforms adopt continuous perfusion or hydrostatic pressure differences. These methods produce environments similar to interstitial flow. Researchers can carry out quantitative analysis with these systems. The analyzed indexes include hydraulic permeability, matrix-dependent transport and flow-modulated tumor cell behavior. These models provide useful comparators for assembloid research because most current tumor organoid and assembloid cultures remain static or only weakly perfused.44 Therefore, although assembloids better preserve multicellular tumor architecture, the direct measurement and dynamic control of interstitial pressure remain underdeveloped. Integrating pressure-controlled microfluidic modules with patient-derived tumor assembloids may represent an important next step for modeling drug delivery barriers under more physiologically relevant transport conditions.
Then there is the matrix itself. Most groups still embed organoids in Matrigel, a basement-membrane extract that is soft, biochemically narrow, and nowhere near the collagen-dense, crosslinked stroma of a desmoplastic tumor.45 The practical consequences, including diffusion resistance, are underestimated, sometimes by a wide margin. The cellular picture is equally incomplete. A native tumor is not just cancer cells. It is fibroblasts laying down matrix, pericytes stabilizing vessels, and immune cells surveilling and sometimes sabotaging therapy.32 Standard organoid protocols strip virtually all of that away. What remains is a useful but narrow window. Tumor-cell-autonomous drug response, divorced from the stromal context that, frustratingly often, determines whether a drug works or fails in the clinic.
Tumor Assembloid Construction Strategies
Tumor assembloids were developed to address the limitations described above. By integrating patient-derived tumor cells with stromal partners, CAFs, endothelial cells, and immune populations within a structured 3D matrix, these platforms functionally reconstruct an in vitro representation of the native TME. This reconstruction facilitates investigation of two interconnected factors implicated in therapeutic failure, including multicellular drug delivery barriers and intrinsic tumor heterogeneity.
Regarding the first factor, the coordinated actions of CAFs and endothelial cells replicate the physical and biological barriers that restrict drug penetration in vivo, a process that remains difficult to simulate in 2D monolayers.31,46 Regarding the second, assembloids maintain the cellular diversity of a patient’s tumor within a 3D microenvironmentally stratified context, supporting the emergence of distinct subpopulations and clonal behaviors that contribute to treatment resistance.24 Three complementary engineering strategies have been developed to realize these models: co-culture-based self-assembly, precision 3D bioprinting, and microfluidic organ-on-a-chip systems (Table 1).
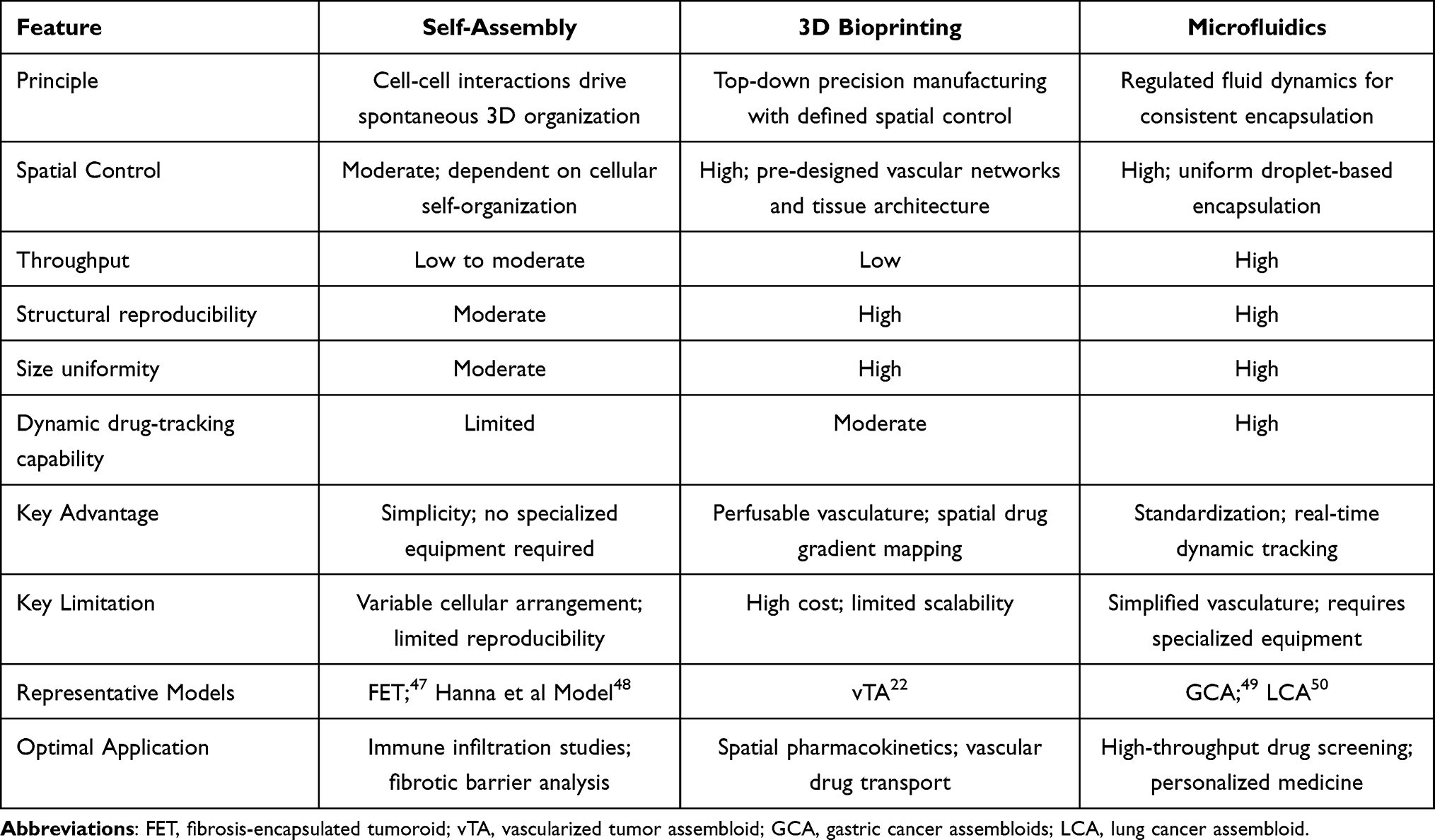 |
Table 1 Comparison of Three Major Tumor Assembloid Construction Strategies |
Self-Assembly Through Co-Culture for Multicellular Crosstalk
Self-assembly exploits native cell-cell adhesion to generate 3D architectures without specialized equipment, which is a practical advantage for routine adoption. The Fibrosis-Encapsulated Tumoroid (FET) model reported by Jang et al47 offers an instructive case. A tumor-cell spheroid is first allowed to compact, then deliberately coated with CAFs in a second seeding step, yielding a bilayered construct, a proliferative core wrapped in a fibrotic shell. Because layering is sequential rather than stochastic, the FET circumvents the spatial randomness inherent in conventional mixed co-cultures35 and delivers batch-to-batch uniformity sufficient for longitudinal drug screens.51
Where the FET model interrogates stromal barriers, the multi-compartment platform of Hanna et al48 addresses a distinct yet related bottleneck, such as immune cell infiltration. Their design reconstructs the stepwise passage, stromal invasion, matrix migration, basement-membrane crossing, and eventual cytotoxicity that 2D transwell assays collapse into a single readout.48 Critically, ECM compositions in both the tumor and stromal compartments were tuned to approximate physiological stiffness ranges, enabling quantitative dissection of how matrix rigidity gates immune penetration.52 Although the authors focused on immune-cell dynamics, the same physical barriers that retard lymphocyte trafficking would, in principle, attenuate small-molecule diffusion as well, an inference worth testing explicitly.
Taken together, these two self-assembly platforms address complementary facets of tumor impermeability. The FET model isolates fibrotic drug exclusion, while the Hanna system captures immune-infiltration kinetics. Their convergence suggests a modular experimental framework for dissecting and ultimately disrupting the physical and immunological barriers that limit therapeutic efficacy in solid tumors.
3D Bioprinting for Spatial Control of Drug Distribution
Unlike self-assembly co-cultures, 3D bioprinting imposes tissue architecture by design. The vascularized tumor assembloid (vTA) reported by Lv et al22 illustrates what this top-down control affords. Through a mold-replacement and assembly route, endothelialized microchannels of defined diameter and spacing are first fabricated. These channels are then integrated with tumor spheroids in a 3D matrix, thereby producing an interconnected tumor-vasculature-stroma construct. This sidesteps two persistent limitations of conventional models. One is the difficulty of generating perfusable, spatially ordered microvasculature and the other is the structural randomness that confounds quantitative drug-penetration readouts.53,54 Neufeld et al demonstrated a perfusable 3D-bioprinted glioblastoma platform capable of recapitulating tumor microenvironmental architecture and transport phenomena.53
What makes the vTA particularly informative is its ability to recapitulate the sequential pharmacokinetic barriers a drug encounters in vivo, including intravascular transit, transendothelial extravasation, interstitial diffusion, and finally access to distal tumor cells.22,55 A measurable concentration gradient emerges across the engineered tissue. The cells adjacent to the vasculature experience higher drug exposure and correspondingly greater cytotoxicity, whereas those in peripheral regions receive sub-therapeutic doses and survive at elevated rates.22 This gradient, captured spatially rather than inferred from bulk viability assays, offers direct in vitro evidence for the treatment-resistant niches that limited penetration creates in solid tumors.
The model’s utility extends beyond structural recapitulation. By resolving drug distribution profiles within a controlled geometry, the vTA enables early-stage identification of candidates likely to fail owing to poor tissue penetration, a failure mode that conventional screens rarely capture. It also provides a testbed for next-generation delivery strategies such as nanoparticle carriers and vascular-disrupting agents. Lv et al further validated the platform for hyperthermic intraperitoneal chemotherapy (HIPEC), a locoregional modality for which the gap between standard in vitro assays and clinical drug-distribution behavior has been especially wide.22
It should be noted that the vTA is, to date, one of the very few published examples of 3D-bioprinted tumor assembloids specifically designed for drug delivery barrier modeling. While the broader bioprinting field has produced vascularized tissue constructs for other applications, the specific application to tumor drug-penetration studies remains at an early stage. This limited evidence base means that the advantages described above include spatial gradient mapping and HIPEC validation, await independent replication across additional tumor types and bioprinting configurations before the strategy can be considered broadly validated.
Microfluidic Organ-on-a-Chip for Dynamic Drug Delivery Analysis
Microfluidic platforms offer a route to replicate, at least in part, the physiological dynamics governing drug delivery in vivo. Xu et al exploited high-throughput droplet microfluidics to co-encapsulate gastric cancer cells and patient-derived stromal populations within a GelMA-Gelatin-Matrigel (GGM) composite hydrogel, producing gastric cancer assembloids (GCAs) with narrow size distributions and reproducible cellular architecture.49 Such standardization is non-trivial. The inter-model variability has long plagued conventional 3D cultures and eroded confidence in their drug-screening readouts.
The GCA system specifically targets a blind spot in preclinical oncology. Endpoint assays capture only a single temporal slice, obscuring the kinetics of absorption, distribution, and target engagement that ultimately dictate therapeutic outcome.56 Because the GGM microspheres are optically transparent and dimensionally uniform, live-cell imaging and fluorescent drug tracking become feasible. Researchers can watch a compound diffuse through patient-derived ECM in real time and map the resulting intratumoral concentration gradients. This spatiotemporal resolution, still rare among organoid platforms, opens the door to disentangling two failure modes that conventional assays routinely conflate: inadequate penetration through stromal barriers versus intrinsic cellular resistance.57
A conceptually parallel strategy was adopted by Zhang’s group for lung cancer.50 Their lung cancer assembloid (LCA) relies on a microinjection module, silicone tubing coupled to a T-junction chip, to shear cell-laden GelMA-Matrigel bioink into monodisperse droplets (400–500 μm), which are then UV-crosslinked into stable microgels. The throughput is striking. Under optimized conditions, roughly 200 assembloids can be generated from as little as 10 μL of clinical microsample within one minute, although whether this throughput is reproducible across heterogeneous clinical specimens remains to be systematically evaluated. Histological architecture, genomic and transcriptomic signatures of parental tumors are retained, and CAF heterogeneity is reconstructed rather than averaged out. Patient-specific responses to targeted agents, chemotherapy, and combination immunotherapy could therefore be interrogated within a single experiment.
These two models occupy complementary niches. The GCA platform leans toward mechanistic dissection of drug transport dynamics. The LCA model is oriented more toward clinically actionable predictions of treatment response. Shared strengths, including minimal tissue requirements, rapid fabrication, and compatibility with real-time imaging, address several persistent shortcomings of static 3D culture systems, though broader clinical validation across diverse tumor subtypes remains an open task.
Applications and Case Studies
Tumor assembloids offer several advantages over conventional in vitro models for preclinical drug development. At present, the evidence supports their value primarily as mechanistically informative platforms rather than as broadly validated clinical predictors, though emerging data are encouraging.
Beyond the specific models discussed in the preceding construction section, the broader literature provides additional evidence for the utility of assembloid-based drug evaluation. A central strength is the ability to model the multicellular drug barrier. When patient-derived tumor cells are co-cultivated alongside CAFs and endothelial cells in a 3D matrix, the resulting assembloids reproduce the dual in vivo barrier: the dense CAF-secreted ECM restricts drug diffusion, while the endothelial lining functions as a selective biological barrier governing extravasation.31,58 For instance, esophageal adenocarcinoma assembloids incorporating patient-derived CAFs have demonstrated that fibroblast co-culture alters both the histological phenotype and drug sensitivity of the tumor compartment compared to organoid monocultures, highlighting the indispensable role of stromal context in preclinical drug evaluation.59 The FET model, for instance, demonstrated that CAF-derived fibrotic encapsulation increased drug resistance compared to non-encapsulated tumoroids.47 The vTA model further showed that drug concentration gradients across the vascular–interstitial interface resulted in spatially heterogeneous cytotoxicity, with distal tumor cells receiving sub-therapeutic doses.22 These observations suggest that assembloids may help identify specific barriers responsible for drug delivery failure in individual tumors.
Assembloids also preserve the intrinsic spatial and functional heterogeneity of the native TME. By maintaining a patient’s cellular repertoire within a 3D context, these models support the emergence of distinct cellular subpopulations and clonal dynamics that mirror the evolutionary pressures driving acquired resistance in vivo.23 When challenged with drugs, assembloids reveal which subpopulations are eliminated and which persist, offering mechanistic insight into partial responses and relapse. The LCA model, for example, demonstrated concordance between assembloid-predicted drug responses and actual clinical outcomes in patients receiving targeted therapy and chemotherapy combinations.50 More recently, gastric cancer assembloid models incorporating matched stromal subtypes have shown that different CAF subpopulations can differentially modulate drug resistance, with some agents losing efficacy in assembloids compared to organoid monocultures, underscoring the importance of stromal heterogeneity in treatment prediction.36 Although encouraging, such concordance data remain limited in scale and should be validated in larger, prospectively designed patient cohorts before assembloids can be considered robust surrogate platforms for treatment selection.
Furthermore, assembloids may provide a practical ex vivo framework for evaluating patient-specific drug delivery barriers.42,60 By using patient-derived tumor and stromal components, these platforms allow investigators to compare how different therapeutic agents penetrate, distribute, and act within a microenvironment that more closely reflects the donor tumor.61 In this context, patient-specific testing should be viewed not as a separate personalized medicine application, but as a translational extension of barrier-focused drug delivery modeling. The GCA and LCA models illustrate how microfluidic-based assembloids can support ex vivo assessment of penetration efficiency, stromal restriction, and spatially heterogeneous drug response while preserving key features of the patient-derived tumor microenvironment.49,50
Tumor assembloids serve as promising research platforms. They are applicable to advanced drug delivery systems, not just traditional small-molecule drugs. Recent advances in nanomedicine have shed new light on this topic. One representative example is lysyl oxidase-modified lipid nanovesicles, which are developed to enhance tumor penetration and therapeutic outcomes. These findings further demonstrate that physiologically realistic 3D models are critical for analyzing the transport and distribution of nanoparticles.62 The assessment focuses on three key capacities. These include penetration into dense stromal matrices, extravasation through engineered vascular barriers, and uniform distribution inside tumor tissues. Antibody-drug conjugates (ADCs) bring new factors to consider. These factors include target accessibility and receptor heterogeneity. They also involve payload release in complex multicellular tumor microenvironments. Similarly, cell-based and biomaterial-assisted delivery platforms may benefit from assembloid models that simultaneously capture stromal, immune, and vascular constraints.63,64 Systematic validation of these applications is still insufficient. Tumor assembloids can recapitulate drug delivery barriers seen in clinical settings.65 These applications match well with this strength of the models. They also point to a key direction for future translational research.
Persistent Challenges and Translational Hurdles
Methodological progress notwithstanding, tumor assembloids remain constrained by technical and translational barriers that limit their near-term adoption in drug discovery and the clinic.14
Chief among these is the absence of standardized protocols. Assembloid fabrication sits at the intersection of cell sourcing, printing or fluidic architecture, and culture optimization. These are variables that laboratories currently tune in isolation. The consequences are not trivial. Recent patient-specific and co-culture studies show that heterogeneity in CAF states remodels tumor microenvironment mechanics and can alter stromal protection, thereby changing ex vivo drug-response readouts.66,67 Across the three dominant fabrication routes, self-assembly, 3D bioprinting, and microfluidics, no consensus parameter sets exist. Drug-response measurements can vary substantially with changes in media composition, passage number, chip design, organoid size, extracellular matrix, and assay endpoint, and only a minority of studies currently report strong concordance with clinical outcomes.68 These findings, it should be noted, originate from a narrow evidence base. Multi-center benchmarking has yet to be undertaken.
Equally limiting is the fundamentally static character of most assembloid cultures. Engineered microvasculature sustains rudimentary perfusion yet omits the hemodynamic forces, shear gradients, and adaptive remodeling intrinsic to living tissue.69 Systemic inputs such as neural signaling and lymphatic clearance, both of which shape drug distribution and metabolism in vivo, are rarely recapitulated.25 The native TME, by contrast, is in constant flux. ECM turnover, immune cell trafficking, and phenotypic state transitions proceed concurrently.32,33 This mismatch raises a pointed concern. If assembloids cannot mirror treatment-induced adaptive resistance, a process driven by precisely such dynamic crosstalk, their predictive ceiling for longitudinal drug response may be lower than assumed.70 Specific technical approaches to address these dynamic limitations are discussed in the Future Perspectives section.
A related gap lies in analytical resolution. Drug delivery unfolds as a kinetic cascade: penetration, distribution, pharmacological action, and metabolism. Their current measurement strategies capture only in fragments. Most studies default to endpoint transcriptomic or genomic snapshots; integrated multi-omic coverage spanning proteomics, metabolomics, and lipidomics is largely absent. Complex paracrine circuits among heterogeneous cell populations further compound the problem, introducing analytical noise that obscures cell-type-specific drug responses and complicates target deconvolution.
Practical considerations deserve frank acknowledgment as well. Generating patient-derived assembloids demands time and resources that may collide with clinical decision windows, particularly in fast-progressing malignancies. A rigorous cost-benefit framework, one that weighs predictive gain against logistical overhead, will be essential to delineate where assembloids add the most value. Their relationship with patient-derived xenograft models is complementary rather than competitive. PDX platforms preserve systemic complexity, whereas assembloids offer faster iteration and tighter experimental control. Clarifying the boundary between the two will sharpen the translational case for each.
The relationship between assembloids and patient-derived xenograft (PDX) models warrants careful delineation, as the two platforms are complementary rather than competitive. PDX models preserve systemic complexity, including intact vasculature, immune surveillance (in humanized variants), and multi-organ pharmacokinetics, making them well-suited for in vivo efficacy and toxicity evaluation. However, PDX models are inherently low-throughput, require months for establishment, introduce species-specific stromal replacement that can alter drug transport, and are poorly suited for rapid, individualized treatment selection. Assembloids, by contrast, offer faster turnaround (days to weeks), direct use of patient-derived cells without species confounders, and tighter experimental control over microenvironmental variables. Their principal weakness, the absence of systemic pharmacokinetics and authentic perfusion, is precisely where PDX excels. An optimal preclinical workflow might therefore employ assembloids for early-stage mechanistic screening and barrier-specific drug evaluation, reserving PDX for later-stage in vivo validation. Clarifying this division of labor will sharpen the translational case for each platform.
Establishing patient-derived assembloids and completing the full experimental process normally takes 2 to 3 weeks, starting from tumor sample collection until drug response results are obtained (Figure 2). In contrast, PDX models typically demand several months of experimental cycle, making the assembloid platform far more time-efficient. Novel platforms built on microfluidic chips and microspheres have further shortened the experimental cycle to 10–14 days. Thanks to this notable time compression, drug screening guided by assembloids can be carried out within the time frame available for clinical treatment decisions. Nevertheless, whether such turnaround times can be consistently achieved across diverse tumor types and routine clinical settings remains to be established through larger prospective studies.
 |
Figure 2 The 21-day workflow of patient-derived tumor assembloids. |
Toward a Standardization Framework
Translating assembloid platforms into clinical practice hinges on protocols that are not merely reproducible within a single group but comparable across sites. Drawing on the challenges discussed above, we outline a four-pillar framework and flag where consensus remains thin (Figure 3).
 |
Figure 3 Proposed standardization roadmap for tumor assembloid technology. |
The first pillar concerns cellular input quality. As a practical starting point, future benchmarking efforts may benefit from reporting predefined viability thresholds upon receipt (eg, approximately 90% for tumor fractions and 85% for stromal compartments), together with passage number, recovery rate, and post-thaw performance. These thresholds are extrapolated from established good-practice standards for primary cell isolation in organoid culture and tissue engineering, where viability below 85–90% upon receipt is commonly associated with compromised engraftment and phenotypic drift; however, they have not been formally validated in the assembloid context and should be viewed as provisional reporting benchmarks rather than established field-wide standards. Passage number is seldom reported, despite evidence that secretome profiles may shift within as few as three passages. Cell identity warrants confirmation through standardized immunophenotyping, such as α-SMA and FAP for cancer-associated fibroblasts, CD31 for endothelial populations, and lineage-resolved panels for immune subsets.
Matrix formulation constitutes the second pillar, and arguably the most under-specified. Variables such as collagen type, fiber density, and Matrigel lot number, as well as scaffold-specific parameters such as the degree of GelMA substitution, can each modulate drug diffusion barriers in ways that confound cross-study comparison when left unreported. Mechanical readouts, including storage modulus and mesh size, should accompany every formulation to anchor these variables quantitatively.
Third, fabrication parameters demand definition tailored to the construction strategy employed. Self-assembly routes require documentation of seeding ratios, sequential layering intervals, and culture vessel geometry. Bioprinting requires explicit control over bioink rheology, print speed, and z-resolution. Microfluidic encapsulation, in turn, depends on flow rate, droplet diameter, and crosslinking kinetics. None of these parameters is yet governed by a community-accepted reporting template.
Fourth, validation metrics need harmonization if results are to travel between laboratories. We propose that future studies routinely report the structural integrity and dimensional consistency, histological verification of spatial organization, and quantitative drug penetration depth. When patient-matched data are available, analyze their correlation with clinical response. Establishing reference assembloid lines derived from well-characterized cell banks would offer an inter-laboratory calibration point, conceptually analogous to what the NCI-60 panel once provided for monolayer screening. However, the analogy has clear limits: NCI-60 standardized a single culture modality with a single cell type per line, whereas assembloids involve multiple cell types, diverse matrix formulations, and distinct fabrication strategies. A reference assembloid panel would therefore need to specify not only the cell sources but also the matrix composition, construction protocol, and recommended readout endpoints for each entry. This added complexity means that assembloid standardization will likely require a staged approach, beginning with a few well-characterized tumor types, rather than a single comprehensive panel.
Realizing this framework will demand a coordinated, multi-center effort. Tumor-type-specific adaptation is inevitable What a shared reporting architecture provides is not rigidity but a common language. That would materially strengthen both reproducibility and the clinical credibility of assembloid-based drug evaluation.
Discussion and Future Perspective
Having identified the key challenges constraining current tumor assembloid platforms in the preceding sections, we now turn to specific technical and methodological advances that may address these limitations.
Vascular modeling illustrates one of the most pressing opportunities for improvement. The current perfusable channels are geometrically reasonable yet lacks physiologically relevant hemodynamic regulation. Endothelial cells sit in a near-stagnant medium and never encounter the shear gradients or pulsatile waveforms that govern transendothelial transport in vivo. Imposing physiological flow regimes would significantly sharpen extravasation readouts. Such refinements may partly explain why assembloid-derived IC50 values do not always align with clinical pharmacokinetic behavior. The stromal compartment poses a subtler difficulty. CAF populations are functionally heterogeneous. Their activation state drifts with mechanical load, TGF-β flux, and matrix stiffness, yet many current protocols rely on a limited fibroblast phenotype and do not capture dynamic state transitions over time. The incorporation of regulatory feedback mechanisms, stiffness-sensitive promoter networks, or adjustable cytokine gradients has the potential to facilitate the co-evolution of the stroma with the tumor, ultimately reflecting the dynamics observed in patients. Layer in lymphatic drainage, innervation, and controlled immune trafficking, and one begins to approximate the paracrine geometry that makes immunotherapy response so notoriously context-dependent.
Computational approaches are likely to play an increasingly important role in addressing these biological and analytical challenges. It is indeed possible. However, the field must openly recognize its limitations. Real-time image-based monitoring of culture viability is already feasible at a modest scale. Parsing heterogeneous death phenotypes across thousands of replicates, however, remains far more challenging, demanding segmentation models trained on data that, for 3D tumor cultures, barely exist. Multi-omic integration faces a similar bootstrapping problem. Aligning spatial transcriptomics with proteomic and metabolomic layers from one assembloid is computationally tractable in principle. Nevertheless, no validated framework currently handles the registration artifacts and batch effects unique to organoid-scale specimens. Spatial pharmacodynamic modeling, predicting local drug exposure as a function of tissue architecture, remains largely aspirational. The governing parameters (tortuosity, interstitial pressure heterogeneity, binding-site density gradients) are themselves poorly constrained experimentally. Until predictive outputs from such pipelines survive prospective clinical benchmarking with interpretable confidence intervals, their utility will stay confined to hypothesis generation rather than therapeutic guidance.
Finally, the regulatory landscape is evolving in ways that may accelerate the translational path for assembloid platforms. The FDA Modernization Act 2.0 (2022) removed the longstanding requirement for animal testing prior to human clinical trials, explicitly permitting alternative methods, including cell-based assays and organ-on-a-chip systems.71 This regulatory shift creates a formal opening for assembloid-derived efficacy and safety data to contribute to Investigational New Drug (IND) applications. However, realizing this potential will require that the field demonstrate reproducibility and predictive validity at a level that regulatory agencies find actionable, an endeavor that is inseparable from the standardization efforts outlined in the last section.
From a regulatory perspective, tumor assembloids should currently be viewed as emerging New Approach Methodologies (NAMs), rather than as fully qualified substitutes for animal studies or clinical evidence.72 Recent regulatory moves by the FDA have opened up greater opportunities for human-relevant in vitro test systems.73 This progress ties closely to the agency’s Alternative Methods Program. The program is designed to broaden formal qualification routes for alternative testing approaches and boost the predictive performance of nonclinical studies. It also facilitates the regulatory acceptance of testing technologies built around the 3R principles (replacement, reduction and refinement of animal experiments). In March 2026, FDA also issued draft guidance on the use of NAMs in drug development (Docket Number: FDA-2025-D-6131), emphasizing validation frameworks, study design, reporting principles, and human-relevant predictivity. However, this guidance remains non-binding and does not qualify any specific model type, including assembloids, for a defined regulatory use. Therefore, tumor assembloids are most practically positioned for regulatory applications in the near term. They serve as fit-for-purpose supplementary evidence to back multiple research goals.74 These applications include mechanism-of-action exploration, drug penetration evaluation, candidate drug prioritization and the mitigation of translational risks.75,76 Such supporting data gains higher regulatory credibility only after strict validation against cross-verified assays, in vivo animal findings, pharmacokinetic/pharmacodynamic indicators and paired clinical trial results.
In pharmaceutical R&D, assembloid assays can also be aligned with the logic of The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) quality guidelines. ICH Q2(R2) puts forward requirements for the validation of analytical procedures. Validation is meant to confirm each method’s fitness for its intended use. Meanwhile, such validation can be implemented in line with corresponding clinical development stages for analytical methods deployed in clinical trials. The above requirement applies to test systems built with tumor assembloids. It requires a predefined set of validation performance parameters for these assays. These parameters cover endpoint specificity/selectivity, intra-assay and inter-assay precision, cross-operator and cross-laboratory reproducibility, accuracy or consistency versus reference assays, quantifiable reporting range, plus robustness against intentional changes of experimental conditions, including cell proportion, matrix formulation, organoid dimension, perfusion speed, imaging cutoff value and drug incubation duration.77,78
ICH Q14 establishes a supplementary framework targeting analytical procedure development. The formulation of this framework follows a specific design philosophy. It attaches importance to prior knowledge, analytical procedure control strategy as well as lifecycle management. When working with assembloids, the above idea can be converted into an assembloid assay target profile. This document specifies the intended context of use together with critical assay attributes and acceptable performance criteria. It also sets out system suitability criteria and a relevant change-control strategy prior to routine application. ICH Q8(R2) reinforces the implementation of quality-by-design principles. It establishes a correlation between critical material attributes, key process parameters and corresponding assay performance. ICH Q9(R1) lays out a systematic framework for risk identification and risk reduction. This framework addresses common risks, including patient-sample variation, batch fluctuation of ECM, passage-induced performance shift, heterogeneity of stromal cells, and variation arising from microfluidic manipulation or bioprinting processes. ICH Q10 facilitates full lifecycle governance of analytical workflows. Relevant measures consist of standardized document management, deviation disposition, technology transfer implementation, corrective action deployment and ongoing continuous improvement.
Accordingly, future regulatory translation of tumor assembloids will depend on moving from descriptive model construction toward standardized, validated, and lifecycle-managed assay systems. Researchers can follow a feasible implementation pathway for assay development. The first step is to confirm the intended context of use for the established model. Next, developers define core critical quality attributes. These indicators cover cell identity, cell viability, spatial structure, ECM stiffness, vascular permeability and immune cell retention capacity. Subsequent work contains endpoint verification of drug distribution or drug response. The whole validation complies with performance specifications aligned with ICH Q2(R2) requirements. Teams carry out risk assessment for manufacturing and detection variables referencing ICH Q9(R1) guidelines, and sustain long-term method management within a quality system consistent with ICH Q10. Adoption of this framework cannot guarantee final regulatory approval from authorities. It can nevertheless improve the overall quality of data obtained from assembloid models. Optimized data gains better interpretability and cross-laboratory transferability, and becomes eligible for formal communication with the FDA. Relevant communication can proceed via established pathways for drug development tools or novel alternative methods. Suitable options include context-of-use-driven qualification initiatives like the Innovative Science and Technology Approaches for New Drugs (ISTAND) program whenever needed.
Conclusion
This review has surveyed tumor assembloids as experimental platforms for dissecting drug delivery barriers within the tumor microenvironment. Three fabrication strategies address complementary facets of TME reconstruction. Self-assembly, the most accessible route, captures fibrotic stroma and immune-cell infiltration with minimal infrastructure. 3D bioprinting introduces spatial control sufficient to resolve vascular transport gradients. Microfluidics, in turn, couples throughput with standardization, a prerequisite for clinically meaningful screening campaigns. What distinguishes assembloids from earlier organoid formats is their capacity to co-embed multicellular transport barriers and patient-derived tumor heterogeneity in one construct, opening a window onto drug penetration profiles and resistance trajectories that conventional monolayer or spheroid assays largely obscure.
Significant obstacles remain. Protocol harmonization across laboratories is still lacking. Dynamic physiological cues, such as cyclic perfusion, immune-cell trafficking, and matrix remodeling, are only partially captured, and multi-omic readouts have yet to be integrated at the single-assembloid level. Closing the gap between preclinical fidelity and clinical predictivity will hinge less on any single technical advance than on sustained, multi-center commitment to rigorous benchmarking, transparent reporting of failure modes, and head-to-head validation of assembloid-derived predictions against matched patient outcomes. At present, evidence supports the value of tumor assembloids as mechanistically informative models of stromal, vascular, and immune-associated drug delivery barriers. Their use for individualized treatment prediction remains a promising but secondary translational application that will require larger-scale prospective validation before clinical implementation.
Data Sharing Statement
No datasets were generated or analyzed in this review. All data discussed herein are available in the cited original publications.
Funding
This research was supported by the Zhejiang Provincial Natural Science Foundation of China under Grant No. ZCLTGY24C1001 and Wenzhou-Kean University International Frontier Interdisciplinary Research Institute Talent Program (No. TP2026004).
Disclosure
The authors report there are no competing interests to declare.
References
1. Shi Y, van der Meel R, Chen X, et al. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10(17):7921–16. doi:10.7150/thno.49577
2. Ben-David U, Siranosian B, Ha G, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560(7718):325–330. doi:10.1038/s41586-018-0409-3
3. Li X, Fu G, Zhang L, et al. Assay establishment and validation of a high-throughput organoid-based drug screening platform. Stem Cell Res Ther. 2022;13(1):219. doi:10.1186/s13287-022-02902-3
4. Choi SY, Shim J, Gu DE, et al. Clonal evolution of long-term expanding head and neck cancer organoid: impact on treatment response for personalized therapeutic screening. Oral Oncol. 2023;146:106571. doi:10.1016/j.oraloncology.2023.106571
5. Thalheim T, Siebert S, Quaas M, et al. Epigenetic drifts during long-term intestinal organoid culture. Cells. 2021;10(7):1718. doi:10.3390/cells10071718
6. Jiang X, Oyang L, Peng Q, et al. Organoids: opportunities and challenges of cancer therapy. Front Cell Dev Biol. 2023;11:1232528. doi:10.3389/fcell.2023.1232528
7. Chuprin J, Buettner H, Seedhom MO, et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol. 2023;20(3):192–206.
8. Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol. 2014;87(1):150–161. doi:10.1016/j.bcp.2013.06.020
9. Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. doi:10.1016/j.ccr.2014.04.005
10. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31(17):2205–2218. doi:10.1200/JCO.2012.46.3653
11. De Vita A, Recine F, Mercatali L, et al. Myxofibrosarcoma primary cultures: molecular and pharmacological profile. Ther Adv Med Oncol. 2017;9(12):755–767. doi:10.1177/1758834017737472
12. De Vita A, Recine F, Miserocchi G, et al. The potential role of the extracellular matrix in the activity of trabectedin in UPS and L-sarcoma: evidences from a patient-derived primary culture case series in tridimensional and zebrafish models. J Exp Clin Cancer Res. 2021;40(1):165. doi:10.1186/s13046-021-01963-1
13. Birey F, Andersen J, Makinson CD, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545(7652):54–59. doi:10.1038/nature22330
14. Kim E, Choi S, Kang B, et al. Creation of bladder assembloids mimicking tissue regeneration and cancer. Nature. 2020;588(7839):664–669. doi:10.1038/s41586-020-3034-x
15. Mun S, Lee HJ, Kim P. Rebuilding the microenvironment of primary tumors in humans: a focus on stroma. Exp Mol Med. 2024;56(3):527–548. doi:10.1038/s12276-024-01191-5
16. Li Z, Rao Q, Ma N, et al. Assembloids: the next dimension in organoid research. Cell Biomaterials;2026:100488. doi:10.1016/j.celbio.2026.100488
17. Liu K, Chen X, Fan Z, et al. From organoids to organoids-on-a-chip: current applications and challenges in biomedical research. Chin Med J. 2025;138(7):792–807. doi:10.1097/CM9.0000000000003535
18. Zhang Y, Payab N, Weigelin B, et al. From 2D cultures to 3D systems: evolving cancer models at the interface of functional precision medicine and theranostics. Theranostics. 2026;16(8):4042–4057. doi:10.7150/thno.127053
19. Liu M, Yang X. Patient-derived xenograft models: current status, challenges, and innovations in cancer research. Genes Dis. 2025;12(5):101520. doi:10.1016/j.gendis.2025.101520
20. Landry BD, Leete T, Richards R, et al. Tumor-stroma interactions differentially alter drug sensitivity based on the origin of stromal cells. Mol Syst Biol. 2018;14(8):e8322. doi:10.15252/msb.20188322
21. Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211. doi:10.3389/fonc.2013.00211
22. Lv Q, Wang Y, Xiong Z, et al. Microvascularized tumor assembloids model for drug delivery evaluation in colorectal cancer-derived peritoneal metastasis. Acta Biomater. 2023;168:346–360. doi:10.1016/j.actbio.2023.06.034
23. Pauli C, Hopkins BD, Prandi D, et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017;7(5):462–477. doi:10.1158/2159-8290.CD-16-1154
24. Mei J, Liu X, Tian HX, et al. Tumour organoids and assembloids: patient-derived cancer avatars for immunotherapy. Clin Transl Med. 2024;14(4):e1656. doi:10.1002/ctm2.1656
25. Onesto MM, Kim JI, Pasca SP. Assembloid models of cell-cell interaction to study tissue and disease biology. Cell Stem Cell. 2024;31(11):1563–1573. doi:10.1016/j.stem.2024.09.017
26. Wu SR, Nowakowski TJ. Exploring human brain development and disease using assembloids. Neuron. 2025;113(8):1133–1150. doi:10.1016/j.neuron.2025.02.010
27. Xing Y, Jian Y, Zhao X, et al. Evolving from organoid to assembloid with enhanced cellular interactions edit. Test Journal. 2025;1001.
28. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
29. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–354. doi:10.1038/nature12626
30. Li X, Wang Z, Mao S, et al. Integrating CAR-T therapy with PD-1/PD-L1 blockade: mechanisms, synergy, and optimized strategies in NSCLC. iScience. 2026;29(2):114607. doi:10.1016/j.isci.2025.114607
31. Neal JT, Li X, Zhu J, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175(7):1972–1988.e16. doi:10.1016/j.cell.2018.11.021
32. de Visser KE, Joyce JA. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41(3):374–403. doi:10.1016/j.ccell.2023.02.016
33. Du W, Xia X, Hu F, et al. Extracellular matrix remodeling in the tumor immunity. Front Immunol. 2023;14:1340634. doi:10.3389/fimmu.2023.1340634
34. Fan J, To KKW, Chen ZS, et al. ABC transporters affects tumor immune microenvironment to regulate cancer immunotherapy and multidrug resistance. Drug Resist Updat. 2023;66:100905. doi:10.1016/j.drup.2022.100905
35. Shi Y, Li Y, Wu B, et al. Normalization of tumor vasculature: a potential strategy to increase the efficiency of immune checkpoint blockades in cancers. Int Immunopharmacol. 2022;110:108968. doi:10.1016/j.intimp.2022.108968
36. Shapira-Netanelov I, Furman O, Rogachevsky D, et al. Patient-derived gastric cancer assembloid model integrating matched tumor organoids and stromal cell subpopulations. Cancers. 2025;17(14):2287. doi:10.3390/cancers17142287
37. Yan HHN, Chan AS, Lai FP, et al. Organoid cultures for cancer modeling. Cell Stem Cell. 2023;30(7):917–937. doi:10.1016/j.stem.2023.05.012
38. Vitale C, Marzagalli M, Scaglione S, et al. Tumor microenvironment and hydrogel-based 3D cancer models for in vitro testing immunotherapies. Cancers (Basel). 2022;14(4):1013. doi:10.3390/cancers14041013
39. Kastenschmidt JM, Schroers-Martin JG, Sworder BJ, et al. A human lymphoma organoid model for evaluating and targeting the follicular lymphoma tumor immune microenvironment. Cell Stem Cell. 2024;31(3):410–420.e4. doi:10.1016/j.stem.2024.01.012
40. Salavati H, Debbaut C, Pullens P, et al. Interstitial fluid pressure as an emerging biomarker in solid tumors. Biochim Biophys Acta Rev Cancer. 2022;1877(5):188792. doi:10.1016/j.bbcan.2022.188792
41. Chen L, Liu J, Chen Q, et al. Modulating tumor interstitial fluid pressure using ultrasound and microbubble therapy: a preclinical study for enhanced drug delivery in cancer treatment. BMC Cancer. 2025;26(1):40. doi:10.1186/s12885-025-15218-1
42. Offeddu GS, Cambria E, Shelton SE, et al. Personalized vascularized models of breast cancer desmoplasia reveal biomechanical determinants of drug delivery to the tumor. Adv Sci. 2024;11(38):e2402757. doi:10.1002/advs.202402757
43. Jiang X, Xu S, Miao Y, et al. Curvature-mediated rapid extravasation and penetration of nanoparticles against interstitial fluid pressure for improved drug delivery. Proc Natl Acad Sci U S A. 2024;121(22):e2319880121. doi:10.1073/pnas.2319880121
44. Wasson EM, He W, Ahlquist J, et al. A perfused multi-well bioreactor platform to assess tumor organoid response to a chemotherapeutic gradient. Front Bioeng Biotechnol. 2023;11:1193430. doi:10.3389/fbioe.2023.1193430
45. Kim S, Min S, Choi YS, et al. Tissue extracellular matrix hydrogels as alternatives to Matrigel for culturing gastrointestinal organoids. Nat Commun. 2022;13(1):1692.
46. Hajal C, Offeddu GS, Shin Y, et al. Engineered human blood-brain barrier microfluidic model for vascular permeability analyses. Nat Protoc. 2022;17(1):95–128.
47. Jang Y, Kang S, Han H, et al. Fibrosis-encapsulated tumoroid, a solid cancer assembloid model for cancer research and drug screening. Adv Healthc Mater. 2024;13(31):e2402391.
48. Hanna EA, Crawford AJ, Du W, et al. A 3D multi-compartment assembloid to study combined immune cell infiltration and cytotoxicity. Cell Rep Methods. 2026;6(2):101307. doi:10.1016/j.crmeth.2026.101307
49. Xu X, Gao Y, Dai J, et al. Gastric cancer assembloids derived from patient-derived xenografts: a preclinical model for therapeutic drug screening. Small Methods. 2024;8(9):e2400204.
50. Zhang Y, Hu Q, Pei Y, et al. A patient-specific lung cancer assembloid model with heterogeneous tumor microenvironments. Nat Commun. 2024;15(1):3382.
51. Van Zundert I, Fortuni B, Rocha S. From 2D to 3D cancer cell models-the enigmas of drug delivery research. Nanomaterials. 2020;10(11).
52. Ashworth JC, Cox TR. The importance of 3D fibre architecture in cancer and implications for biomaterial model design. Nat Rev Cancer. 2024;24(7):461–479.
53. Neufeld L, Yeini E, Reisman N, et al. Microengineered perfusable 3D-bioprinted glioblastoma model for in vivo mimicry of tumor microenvironment. Sci Adv. 2021;7(34). doi:10.1126/sciadv.abi9119
54. Breslin S, O’Driscoll L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discov Today. 2013;18(5–6):240–249. doi:10.1016/j.drudis.2012.10.003
55. Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321–346. doi:10.1146/annurev-bioeng-071813-105259
56. Hachey SJ, Hughes CCW. Applications of tumor chip technology. Lab Chip. 2018;18(19):2893–2912.
57. Saorin G, Caligiuri I, Rizzolio F. Microfluidic organoids-on-a-chip: the future of human models. Semin Cell Dev Biol. 2023;144:41–54. doi:10.1016/j.semcdb.2022.10.001
58. Zhou Q, Dong C, Fan W, et al. Tumor extravasation and infiltration as barriers of nanomedicine for high efficacy: the current status and transcytosis strategy. Biomaterials. 2020;240:119902.
59. Sharpe BP, Nazlamova LA, Tse C, et al. Patient-derived tumor organoid and fibroblast assembloid models for interrogation of the tumor microenvironment in esophageal adenocarcinoma. Cell Rep Methods. 2024;4(12):100909.
60. Ding S, Hsu C, Wang Z, et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell. 2022;29(6):905–917.e6. doi:10.1016/j.stem.2022.04.006
61. Hachey SJ, Sobrino A, Lee JG, et al. A human vascularized microtumor model of patient-derived colorectal cancer recapitulates clinical disease. Transl Res. 2023;255:97–108. doi:10.1016/j.trsl.2022.11.011
62. De Vita A, Liverani C, Molinaro R, et al. Lysyl oxidase engineered lipid nanovesicles for the treatment of triple negative breast cancer. Sci Rep. 2021;11(1):5107. doi:10.1038/s41598-021-84492-3
63. Ding Y, Wang Y, Hu Q. Recent advances in overcoming barriers to cell-based delivery systems for cancer immunotherapy. Exploration. 2022;2(3):20210106. doi:10.1002/EXP.20210106
64. Han X, Alu A, Liu H, et al. Biomaterial-assisted biotherapy: a brief review of biomaterials used in drug delivery, vaccine development, gene therapy, and stem cell therapy. Bioact Mater. 2022;17:29–48. doi:10.1016/j.bioactmat.2022.01.011
65. Lopez-Vince E, Wilhelm C, Simon-Yarza T. Vascularized tumor models for the evaluation of drug delivery systems: a paradigm shift. Drug Deliv Transl Res. 2024;14(8):2216–2241. doi:10.1007/s13346-024-01580-3
66. Micalet A, Upadhyay A, Javanmardi Y, et al. Patient-specific colorectal-cancer-associated fibroblasts modulate tumor microenvironment mechanics. iScience. 2024;27(6):110060. doi:10.1016/j.isci.2024.110060
67. Schuth S, Le Blanc S, Krieger TG, et al. Patient-specific modeling of stroma-mediated chemoresistance of pancreatic cancer using a three-dimensional organoid-fibroblast co-culture system. J Exp Clin Cancer Res. 2022;41(1):312. doi:10.1186/s13046-022-02519-7
68. Malik DA, Schmieder EAS, Genova G, et al. Organoids in translation: a bench-to-bedside framework for pancreatic cancer precision medicine. J Transl Med. 2026;24(1):136. doi:10.1186/s12967-025-07596-8
69. Lim J, Ching H, Yoon JK, et al. Microvascularized tumor organoids-on-chips: advancing preclinical drug screening with pathophysiological relevance. Nano Converg. 2021;8(1):12. doi:10.1186/s40580-021-00261-y
70. Eble JA, Niland S. The extracellular matrix in tumor progression and metastasis. Clin Exp Metastasis. 2019;36(3):171–198.
71. Zushin PH, Mukherjee S, Wu JC. FDA Modernization Act 2.0: transitioning beyond animal models with human cells, organoids, and AI/ML-based approaches. J Clin Invest. 2023;133(21). doi:10.1172/JCI175824
72. Liu W, Pang PD, Wu CA, et al. New approach methodologies for drug discovery. Cell. 2026;189(7):1877–1903.
73. Ingber DE. Challenges and opportunities for human Organ Chips in FDA assessments and pharma pipelines. Cell Stem Cell. 2026;33(2):176–183. doi:10.1016/j.stem.2025.12.022
74. Schmeisser S, Miccoli A, von Bergen M, et al. New approach methodologies in human regulatory toxicology - Not if, but how and when! Environ Int. 2023;178:108082. doi:10.1016/j.envint.2023.108082
75. Stokar-Regenscheit N, Bell L, Berridge B, et al. Complex in vitro model characterization for context of use in toxicologic pathology: use cases by collaborative teams of biologists, bioengineers, and pathologists. Toxicol Pathol. 2024;52(2–3):123–137. doi:10.1177/01926233241253811
76. Kopec AK, Yokokawa R, Khan N, et al. Microphysiological systems in early stage drug development: perspectives on current applications and future impact. J Toxicol Sci. 2021;46(3):99–114. doi:10.2131/jts.46.99
77. Hargrove-Grimes P, Low LA, Tagle DA. Microphysiological systems: stakeholder challenges to adoption in drug development. Cells Tissues Organs. 2022;211(3):269–281. doi:10.1159/000517422
78. Baker TK, Van vleet TR, Mahalingaiah PK, et al. The current status and use of microphysiological systems by the pharmaceutical industry: the International Consortium for Innovation and quality microphysiological systems affiliate survey and commentary. Drug Metab Dispos. 2024;52(3):198–209. doi:10.1124/dmd.123.001510
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Engineering Extracellular Vesicles for Tumor Targeted Therapy: Source Optimization, Modification, and Clinical Application
Sui J, Qin H, Zhang Z, Lv X, Lin X, Liu Z, Zhao X, Liu X, Zhang H
International Journal of Nanomedicine 2026, 21:592579
Published Date: 9 April 2026
Precision Thyroid Oncology: A Review of Multi-Omics Biomarkers and Spatiotemporal Technologies
Huang L, Deng X, Xi Z, Huan X, Mao J, Li X
International Journal of General Medicine 2026, 19:602509
Published Date: 18 May 2026
Clinical Outcomes and Treatment-Related Complications in Triple-Negative Breast Cancer: A Review of 24 Case Reports from 2020 to 2025
Febriyanti RM, Hakim MLN, Rofiidatul I, Islami MS, Utami PS, Qurrotuaini SP, Halimah E, Mohd Hashim N, Diantini A
Therapeutics and Clinical Risk Management 2026, 22:619823
Published Date: 30 June 2026