Back to Journals » Cancer Management and Research » Volume 15
TP63 Functions as a Tumor Suppressor Regulated by GAS5/miR-221-3p Signaling Axis in Human Non-Small Cell Lung Cancer Cells
Authors Shen Q, Wang H, Zhang L
Received 8 September 2022
Accepted for publication 4 February 2023
Published 24 February 2023 Volume 2023:15 Pages 217—231
DOI https://doi.org/10.2147/CMAR.S387781
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Qiming Shen, Haoyou Wang, Lin Zhang
Department of Thoracic Surgery, The First Hospital of China Medical University, Shenyang, 110001, Liaoning, People’s Republic of China
Correspondence: Qiming Shen, Department of Thoracic Surgery, The First Hospital of China Medical University, No. 155 North Nanjing Road, Heping Area, Shenyang, 110001, Liaoning, People’s Republic of China, Tel +86-15804019855, Email [email protected]
Background: Tumor protein p63 (TP63) has been proven to play a role as a tumor suppressor in some human cancers, including non-small cell lung cancer (NSCLC). This study aimed to investigate the mechanism of TP63 and analyze the underlying pathway dysregulating TP63 in NSCLC.
Methods: RT-qPCR and Western blotting assays were used to determine gene expression in NSCLC cells. The luciferase reporter assay was performed to explore the transcriptional regulation. Flow cytometry was used to analyze the cell cycle and cell apoptosis. Transwell and CCK-8 assays were performed to test cell invasion and cell proliferation, respectively.
Results: GAS5 interacted with miR-221-3p, and its expression was significantly reduced in NSCLC. GAS5, as a molecular sponge, upregulated the mRNA and protein levels of TP63 by inhibiting miR-221-3p in NSCLC cells. The upregulation of GAS5 inhibited cell proliferation, apoptosis, and invasion, which was partially reversed by the knockdown of TP63. Interestingly, we found that GAS5-induced TP63 upregulation promoted tumor chemotherapeutic sensitivity to cisplatin therapy in vivo and in vitro.
Conclusion: Our results revealed the mechanism by which GAS5 interacts with miR-221-3p to regulate TP63, and targeting GAS5/miR-221-3p/TP63 may be a potential therapeutic strategy for NSCLC cells.
Keywords: TP63, miR-221-3p, GAS5, NSCLC, apoptosis, invasion
Graphical Abstract:

Introduction
As the major type of lung cancer, non-small cell lung cancer (NSCLC) accounts for 85% of the new diagnoses.1,2 Despite surgical therapies, chemotherapy, radiation therapy, and molecular targeted therapies being promoted, the potential for continued proliferation and metastasis of NSCLC results in a 5-year survival rate of approximately 16%.3,4 Therefore, further exploring the molecular mechanisms of NSCLC is necessary, which may provide more suitable strategies to treat NSCLC patients.
Several studies have demonstrated that tumor protein p63 (TP63), as a p53 homologous, activates several p53 target genes to mediate increased anti-proliferative effects and acts a tumor suppressor in cancer.5–8 A common deletion of TP63 is observed in squamous cell carcinoma and contributed to oncogenesis.8 In addition, TP63 is essential for epithelial development. TP63-null mice show epithelial dysplasia.9 Although TP63 has been already recognized as a structural homologue of the p53 with antitumor activity in colorectal cancer,10–12 the molecular mechanism of the regulation of TP63 as an anticancer molecular target remains to be explored.
Micro-RNAs (miRNAs) are widely accepted to play an important role in the development and metastasis of NSCLC.13 They typically bind to mRNA 3′-untranslated regions (3′-UTR), which leads to degradation of the target mRNA or inhibition of the translation process.14,15 A549 cell proliferation and invasion are inhibited by miR-206 by acting on CORO1C in NSCLC.13 As a tumor suppressor, miR-204 represses the invasion of NSCLC tumors by directly decreasing NUAK1 expression in NSCLC.16 We had predicted three miRNAs that targeted TP63 that upregulated in NSCLC. The role of miR-221-3p as an oncogene or tumor suppressor depends on the tumor type.17 The overexpression of miR-221 is related to the malignant potential and metastatic activity of colorectal cancer.18 However, the role of miR-221-3p in NSCLC is complex and needs to be further explored.
TP63 has many different transcriptional isoforms and it has two subclasses of N-terminal isoforms (TA and DeltaN).19 DeltaNp63 isoforms induce cell proliferation20 and suppress cell apoptosis,21 while TAp63 isoforms repress cell-cycle progression22 and enhance cell apoptosis.23 The DeltaN isoforms and TA isoforms exert oncogenic and tumor-suppressive role, respectively. We had predicted that miR-221-3p targeted transcript variant 2 of TP63 (NM_001114978.2, TAp63beta) by RNA22 v2. In addition, we did not find any literature that reported the regulation of miR-221-3p on TAp63beta in NSCLC. Therefore, the regulation of miR-221-3p on TAp63beta in NSCLC was further studied.
Furthermore, increasing works demonstrated that long noncoding RNAs (lncRNAs) are associated with many diseases, including cancer, by influencing the suppressive effect of miRNAs. For example, lncRNA growth arrest-specific 5 (GAS5)-AS1 decreases cell proliferation and metastasis by sponging miR-106b-5p to promote TUSC2 level in glioma.24 MALAT1 exerts a suppressive role on cellular proliferation by increasing miR-124 and reducing STAT3 level in lung cancer.25 GAS5 is involved in carcinogenesis, and some studies have reported that GAS5, as an endogenous sponge, competitively binds miR-221-3p to regulate the target genes of miR-221-3p.26,27 However, we had not found the literatures that reported the regulation of GAS5 on Tp63 in NSCLC. Therefore, the aim of our study is to state the mechanism of GAS5 and miR-221-3p that are responsible for the dysregulation of TP63 in NSCLC.
In this study, we observed that miR-221-3p could directly target TP63, affecting TP63 expression in NSCLC. In vitro functional analysis confirmed that TP63 overexpression can regulate cell cycle progression to reduce cell proliferation and invasion, and further induce cell apoptosis, which can be deteriorated by miR-221-3p. Upon further study, we predicted and validated GAS5 as a ceRNA of miR-221-3p. Suppression of GAS5 attenuated the effect of the miR-221-3p inhibitor on apoptosis, proliferation, and invasion. Additionally, knockdown of GAS5 facilitated tumor growth, which was eliminated by TP63 overexpression.
Methods
Cell Culture
Normal lung epithelial bronchus BEAS-2B cells were purchased from Saibaikang Biotechnology Co., Ltd. (Shanghai, China). Human NSCLC cell lines A549 and H1299 were purchased from the Cell Biology of the Chinese Academy of Sciences (Shanghai, China). All NSCLC cell lines were maintained in RPMI-1640 medium (Life Technologies, USA), containing 10% fetal bovine serum (FBS, Life Technologies, USA), 100 U/mL penicillin/streptomycin, in a wet chamber at 37℃ with 5% CO2.
Clinical Samples and Ethic Statement
Ten pairs of NSCLC and nontumor tissues were gained from NSCLC patients in our hospital during 2011 and 2012 (Table 1). All the protocols related to patient organizations in the present study were performed in accordance with the principles stated in the Declaration of Helsinki and were approved by the ethics committee of The First Hospital of China Medical University. The written informed consent before surgery was obtained from each patient. After obtaining relevant tumor and adjacent lung tissue samples, they were stored at −80℃ for a long time. The tissue samples were thawed at 4℃ before being used for RT-qPCR.
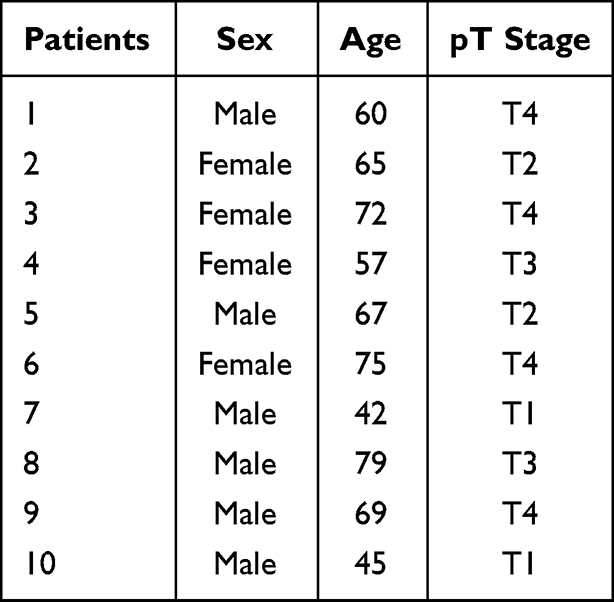 |
Table 1 The Clinicopathological Variables in NSCLC Patients (n=10) |
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Trizol reagent (Life Technologies, USA) or the mirVana miRNA Isolation Kit (Applied Biosystems, Foster City, CA, USA) was used to isolate total RNA from cells and tissues. Complementary DNA of mRNA and miRNA was generated by the PrimeScript RT reagent Kit (Takara, Japan) and the SYBR PrimeScript miRNA RT-PCR Kit, respectively. The TP63, lncRNA GAS5 and miR-221-3p expression was measured by SYBR Green Master Mix (Takara, Japan), and GAPDH and U6 were used as the internal control of mRNA and miRNA, respectively. The PCR primers were as follows: GAS5 forward, 5’-GCAAGCCTAACTCAAGCCATTG-3’ and reverse, 5’-CTTGCTCCACACAGTGTAGTC-3’; TP63 forward, 5’-GGCTGCTGTTG CTTAAACCAC-3’ and reverse, 5’-CACATTACCTTTTAGAGCCACGC-3’; miR-221-3p forward, 5’-AGCTAGTCGATGTAACAGACG-3’ and reverse, 5’-CTCAACTGGTGTCGTGGA-3’; GAPDH forward, 5’-GTTCGTCATGGGTGTGAACC-3’ and reverse, 5’-CATCCACAGTCTTCTGGGTG-3’; U6 forward, 5’-CTCGCTTCGGCAGCACA-3’ and reverse, 5’-AACGCTTCACGAATTTGCGT-3’. Quantitative analysis was calculated using the 2−ΔΔCt method.
Cell Transfection
Lipofectamine 3000 was performed for cell transfection (Life Technologies, USA). A TP63 overexpression plasmid was generated by subcloning TP63 cDNA into the expression plasmid pcDNA3.1 (Invitrogen, USA) and named pc-TP63. The empty vectors were used as controls and named pc-Ctrl. The miR-221-3p inhibitor, inhibitor negative control (inhibitor-NC), mimic negative control (mimic-NC), and miR-221-3p mimic were purchased from Gene-Chem (Shanghai, China). The specific interference siRNA of GAS5 (si-GAS5) (si-GAS5-1: 5’-TATGGTGCTGGGTGCAGATG-3’; si-GAS5-2: 5’-TGAAGTCCTAAAGAGCAAGCCT-3’; si-GAS5-3: 5’-CTTGCCTGGACCAGCTTAATT-3’) were used to knockdown GAS5, and the control siRNA was named si-NC. For against TP63, we designed 3 shRNA that targeted Tap63beta. The shRNA against TP63 (sh-TP63) (sh-TP63-1: 5’- GATCCGTATCCCACAGATTGCAGCTCGAGCTGCAATCTGTGGGATACGTTTTTG-3’; sh-TP63-2: 5’- GATCGATAGCATTTGACCCTATTCTCGAGAATAGGGTCAAATGCTATCTTTTTG-3’; sh-TP63-3: 5’- GATCCCTCCGTATCCCACAGATTCTCGAGAATCTGTGGGATACGGAGGTTTTTG-3’) was cloned into the Pgpu6 vector. Empty vectors were used as control and named sh-NC. Geneticin (G418; Sigma, St Louis, MO, USA) was used to select stable cell lines and used in xenograft tumor analysis.
Construction of Non-Small Lung Cancer Model in Nude Mice
To construct subcutaneous xenograft models, the right shoulder blade of female BALB/c nude mice (aged 4 weeks) was subcutaneously inoculated with NSCLC cells (106). Then, we continue to feed for 2 weeks, measured the size of tumor every day, and carried out the follow-up experiment when the tumor grew to about 400 mm3 in each group. In five parallel groups, four mice with similar tumor size were selected for the follow-up treatment experiment in each group. After about 28 days of treatment, the mice were killed with 40 mg/kg pentobarbital sodium. After the tumor was removed, it is weighed sequentially, and then the tumor was photographed.
Immunohistochemistry (IHC)
The tumor tissues of mice were processed into 4-μm paraffin-embedded tissues after being fixed with formalin to detect the level of proliferation biomarker Ki67. The sections were de-paraffinized and rehydrated, and then endogenous peroxidase was blocked by hydrogen peroxide solution (3%). After antigen retrieval, the silences were incubated with primary antibody for Ki67 (ab16667, Abcam, 1:200 dilution), at 4℃ overnight. Then, the sections were incubated with an HRP-conjugated goat anti-rabbit IgG H&L (ab6721, Abcam) at a dilution of 1:1000. The sections were stained with diaminobenzidine and counterstained with hematoxylin. All the animal experiments were conducted as per the protocols approved by the Animal Care and Use Committee of The First Hospital of China Medical University and were in accordance with the ethical guidelines by the International Council for Laboratory Animal Science (ICLAS).
Dual Luciferase Assay
The wild-type (WT) or mutant (MUT) binding site of miR-221-3p in TP63 or GAS5 3’UTR was cloned into a pmirGLO dual-luciferase vector after linearizing the pmirGLO vector with the restriction enzymes. Cells were co-transfected with miR-221-3p mimics (or mimic-NC) and Pglo-GAS5-WT, Pglo-GAS5-MUT, Pglo-TP63-WT or Pglo-TP63-MUT by Lipofectamine 3000 reagent (Invitrogen, USA). Then, the Dual-Luciferase® Reporter Assay System kit (E1910, Promega) was used to determine luciferase activity after transfection 48 h. The experiments were implemented in triplicate.
Cell Viability Assay
Cells at exponential growth stage were placed (4000 cells/well) in 96-well plates containing 100 μL complete medium for 24 h. CCK-8 solution (10 μL, Beyotime, China) was added after different treatments, and the samples were incubated at 37℃ for 4 h. The plates were read with microplate reader (SmartSpec 3000; CA Bio-Rad, USA), and the absorbance was measured at 450 nm.
Flow Cytometry
According to experimental groups, A549 and H1299 cells were transfected and cultured in a cell incubator. We used apoptosis inducers (TNF-α+SM-164 from Beyotime, Shanghai, China; #C0006S) in this experiment. Next, the cells were digested with trypsin and collected. After removing supernatant, the cells were resuspended with 1mL pre-cooled PBS. After centrifugation, the cells were resuspended with 1×binding buffer. Each sample was treated with 10 μL RNase A (Sigma) and 25 μL propidium iodide (PI) solution and incubated at 37℃ for 30 min (in dark). The red fluorescence was detected by flow cytometry at 488 nm. In the drug resistance test, cells were stained with Annexin-V/propidium iodide detection kit (Key GEN, China), and apoptosis rates were measured by flow cytometry.
Transwell Assay
The Transwell system was used to detect the cell invasion. After digestion with trypsin, NSCLC cells were centrifugated, resuspended and counted. Then the cells (200 μL, 5×104 cells/well) were inoculated into the upper chambers. In the lower chamber, 500 μL medium with 10% serum was filled. After incubation for 24 h at 37℃, the migrated cells were fixed for 15 min with 4% paraformaldehyde, then cells were stained with 1% crystal violet for 10 min. The cell number was observed and photographed.
Western Blotting
After collecting the cell samples, the cell lysate buffer (P0013, Beyotime) that mainly contained Tris, NaCl, Triton X-100, sodium pyrophosphate, β-glycerophosphate, and so on, was added to prepare the samples. After lysis, cells were centrifugated. The total protein solution was quantitatively analyzed with BCA kit. Then, sodium dodecyl sulfate polyacrylamide gel (10%) was used to separate the samples by electrophoresis. Cellulose acetate membrane was used to transfer the separated protein. The silence was blocked with 5% non-fat milk, and the membrane was incubated with TP63 antibody (1:1000, Affinity, AF0233) overnight at 4℃. Then, the samples were incubated with anti-rabbit IgG secondary antibody (0.2 μg/mL, Jackson) at 37℃ for 1.5 h. The experiment was exerted in triplicate.
Statistical Analysis
Data were expressed as the mean ± SEM from at least three independent experiments. The expression levels of TP63, miR-221-3p, and GAS5 in tumors and nontumor tissues were compared using the paired-samples t-test. P < 0.05 was considered statistically significant.
Results
TP63 Was Inversely Correlated with miR-221-3p in NSCLC Tissues
We analyzed TP63 and miR-221-3p expression in NSCLC clinical samples. TP63 mRNA expression was greatly downregulated in tumor tissues (Figure 1A). Compared with adjacent tissues, miR-221-3p expression was greatly higher in tumor tissues (Figure 1B). As shown in Figure S1, compared with BEAS-2B cells, the expression of TP63 and miR-221-3p was also decreased and increased, respectively, in A549 and H1299. The correlation analysis results showed that, between the expression of TP63 and miR-221-3p, a significant negative correlation was observed (Pearson r = −0.8271, P = 0.0032; Figure 1C), which suggested that miR-221-3p regulated TP63 expression during the development of NSCLC.
 |
Figure 1 TP63 was the direct target of miR-221-3p and the expression levels of TP63 and miR-221-3p was negatively correlated in NSCLC cells. (A) The mRNA level of TP63 was detected by RT-qPCR and it greatly downregulated in tumors. (B) miR-221-3p expression was significantly increased in tumor tissues compared with adjacent nontumor tissues. (C) TP63 mRNA level was negatively related with miR-221-3p level. NT, nontumor tissues. (D) The putative miR-221-3p binding site in TP63 3’-UTR region. (E) A549 cells were co-transfected with miR-221-3p (or mimic-NC) and TP63-WT (or TP63-MUT) 3’-UTR constructs. (F) TP63 mRNA levels were detected by RT-qPCR. (G) Western blotting was used to measure TP63 protein levels in cells transfected with the miR-221-3p mimic. mimic-NC, mimic negative control; **P<0.01, ***P<0.001 versus the control group. |
Some putative miRNAs that may target the mRNA of TP63 were predicted by bioinformatics databases (ENCORI, miRWalk and TargetSpy) (Supplementary Table 1). The miRNAs expression data in GSE29248 that was selected after retrieval in GEO database (https://www.ncbi.nlm.nih.gov/geo/), and the upregulated differential expression miRNAs were gained by the online GEO2R. The overlapped upregulated miRNAs that targeted TP63 were selected by the Venny tool (http://www.ehbio.com/test/venn/#/), and three miRNAs, including miR-221-3p, miR-483-3p and miR-28-3p, were revealed to be involved in NSCLC (Figure S2A). Among them, miR-221-3p targeted transcript variant 2 of TP63 (TAp63beta), which was predicted by RNA22 v2 (Figure 1D). Then, the TAp63beta was chose for further study.
As shown in Figure S3A, miR-221-3p mimic significantly increased the expression of miR-221-3p. The luciferase assay results certificated that miR-221-3p directly bound to the 3′-UTR of TP63 (Figure 1E) and miR-221-3p mimic strongly decreased the 3’-UTR reporter gene activity of wild-type TP63 compared with the mimic negative control, but there was no inhibitory effect on these of mutant TP63. The expression of TP63 was significantly repressed by miR-221-3p mimic, compared with cells treated with mimic negative control, at the mRNA and protein levels of A549 and H1299 cells (Figure 1F and G). Therefore, these results indicate that miR-221-3p reduced TP63 expression by binding to TP63 3’-UTR.
The Promoting Effect of miR-221-3p on Cancer Cells Was Reversed by Overexpression of TP63
Next, we investigated the possibility that miR-221-3p could exert a regulatory function in NSCLC by targeting TP63. We detected the expression of miR-221-3p in A549 and H1299 cells that were co-transfected with miR-221-3p mimic (or mimic negative control) and TP63 plasmids (or empty vectors), and miR-221-3p mimic significantly increased the expression of miR-221-3p (Figure S3B). The TP63 expression had been markedly reduced in A549 and H1299 cells that were transfected with miR-221-3p mimic, while co-transfection with the mimic and TP63 plasmid had restored TP63 expression (Figure 2A and B). As shown in Figure 2C, compared with the mimic negative control, miR-221-3p mimic greatly promoted cell viability of A549 and H1299 cells. miR-221-3p overexpression increased as the cells entered into the S phase during the cell cycle of A549 and H1299 (Figure 2D and Figure S4A), and increased the expression of Cyclin D1 in A549 and H1299 cell cycles (Figure 2E). Moreover, miR-221-3p had an inhibitory effect on apoptosis in A549 and H1299 cells (Figure 2F and Figure S4B), and inhibited the expression of Caspase-3 (Figure 2G). In addition, as shown in Figure 2H and Figure S4C, Transwell assays showed that, in A549 and H1299 cells, miR-221-3p mimic increased cell invasion. Meanwhile, miR-221-3p mimic increased the expression of MMP2 in A549 and H1299 cells (Figure 2I). However, TP63 overexpression eliminated the effects of miR-221-3p mimic on proliferation, cell viability, apoptosis, and invasion of A549 and H1299 cells.
 |
Figure 2 The oncogenic effect of miR-221-3p was through targeting TP63 in NSCLC cells. (A and B) RT-qPCR and Western blotting were used to measure TP63 mRNA expression and was used to detect its protein level in A549 and H1299 cells. (C) Increased miR-221-3p expression upregulated cell viability detecting by CCK8, and this promotion was reversed by overexpression of TP63. (D) Cell cycle was promoted by miR-221-3p, while TP63 upregulation eliminated the promotion of progression. (E) Cyclin D1 was upregulated by miR-221-3p, while the upregulation of TP63 eliminated its upregulation. (F) miR-221-3p down-regulated the process of apoptosis of NSCLC cells and promoted the survival of A549/H1299 cells, while TP63 had the opposite effect. (G) miR-221-3p down-regulated the apoptosis-related protein Caspase-3 in NSCLC cells, while TP63 had the opposite effect. (H) After miR-221-3p mimic treatment, cell invasion was increased, and this process was weakened by the overexpression of TP63. pc-TP63, TP63 overexpression vector. (I) After miR-221-3p simulated treatment, the cell invasion associated protein MMP2 increased, and the overexpression of TP63 decreased its increase. pc-TP63, TP63 overexpression vector; **P<0.01, versus mimic-NC+pc-Ctrl (empty plasmid). |
miR-221-3p Was Targeted by lncRNA GAS5
Compared to that in adjacent tissues, the expression of GAS5 dramatically decreased in tumor tissues (Figure 3A). In addition, the expression of GAS5 was downregulated in A549 and H1299 (Figure S5A). The correlation analysis results revealed that GAS5 expression level was negatively related with miR-221-3p expression level in tumors (Pearson r = −0.747, P = 0.013; Figure 3B).
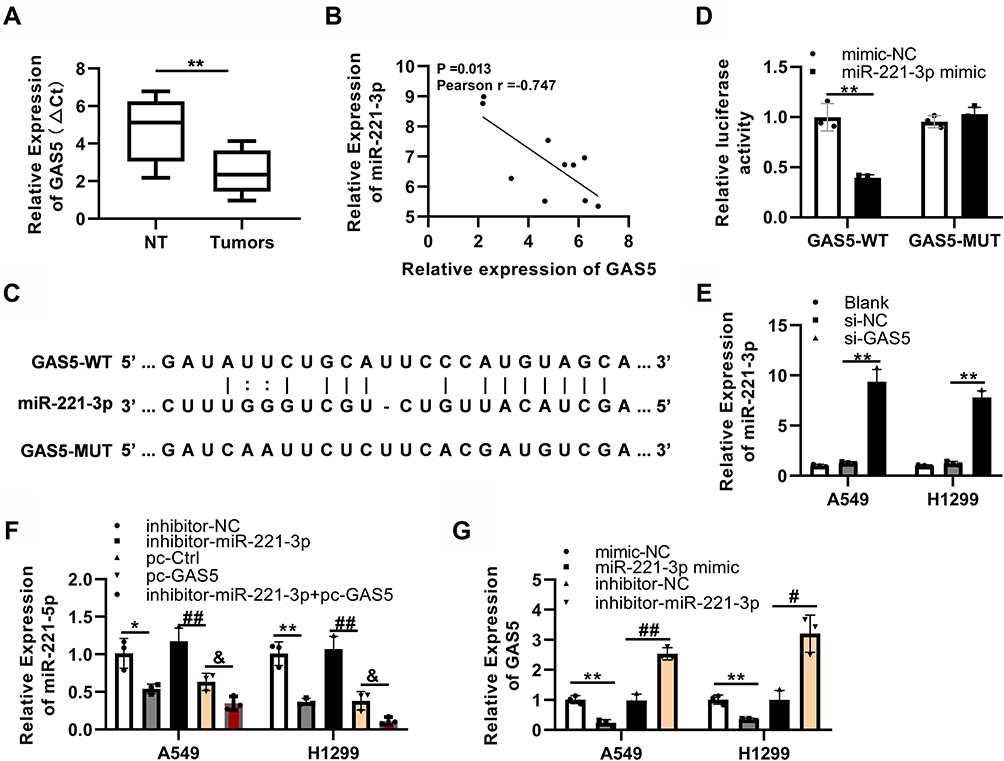 |
Figure 3 GAS5 directly targets and has an inhibitory effect on the expression level of miR-221-3p in NSCLC cells. (A) GAS5 expression level in tumor tissues was lower than that in adjacent tissues. (n=10, means ± SD). **P<0.01 versus nontumor tissues (NT). (B) Perform Pearson correlation analysis on GAS5 and miR-221-3p (P = 0.013, Pearson r = 0.747). (C) Prediction of binding sites between GAS5 and miR-221-3p. (D) A549 cells were co-transfected miR-221-3p mimic or mimic-negative control (mimic-NC) with luciferase constructs containing GAS5-WT or GAS5-MUT sequences in. **P<0.01 versus mimic-NC. (E) Effect of down-regulation of GAS5 gene expression on miR-221-3p level in A549/H1299 cells. **P<0.01 versus si-NC (siRNA negative control). (F) Effect of upregulation of GAS5 gene expression on miR-221-3p level in A549/H1299 cells. *P<0.05, **P<0.01 versus inhibitor negative control. ##P<0.01 versus pc-Ctrl. &P<0.05 versus pc-GAS5 or inhibitor-miR-221-3p. (G) The GAS5 expression in A549 or H1299 cells was regulated via exogenously transfecting miR-221-3p mimic or miR-221-3p inhibitor. **P<0.01 versus mimic-NC. #P<0.05, ##P<0.01 versus inhibitor negative control. |
However, in this study, we also should verify that miR-221-3p was targeted by GAS5. Some putative miRNAs that may be as potential targets of GAS5 were predicted by bioinformatics databases (ENCORI and miRcode) (Supplementary Table 2). The overlapped upregulated miRNAs that were as potential targets of GAS5 also were selected by the Venny tool, and two miRNAs, including miR-221-3p and miR-455-5p were revealed to be involved in NSCLC (Figure S2B). According to the online database ENCORI results, we predicted GAS5 as the potential target of miR-221-3p (Figure 3C). Then, we employed the luciferase assay to validate the binding of miR-221-3p with GAS5. As shown in Figure S5B, miR-221-3p mimic greatly decreased the GAS5 expression. miR-221-3p mimic greatly inhibited luciferase activity in A549 cells that were transfected with the GAS5-WT but not that of the GAS5-MUT (Figure 3D). Furthermore, to inhibit the expression of GAS5, the 3 siRNA of GAS5 were used to reduce the expression of GAS5, and the most effective siRNA-3 was used to conduct follow-up experiments (Figure S5C). RT-qPCR results revealed that, in A549 and H1299 cells, knockdown of GAS5 lead to an increase in miR-221-3p expression (Figure 3E). Additionally, GAS5 overexpression decreased miR-221-3p expression in A549 and H1299 cells (Figure 3F). However, miR-221-3p mimic strongly decreased GAS5 expression, whereas miR-221-3p inhibitor enhanced the GAS5 expression of A549 and H1299 cells (Figure 3G).
The GAS5/miR-221-3p Signal Pathway Regulated Cell Proliferation, Apoptosis, and Invasion
To investigate whether the GAS5/miR-221-3p axis has a regulatory function in NSCLC cells, si-GAS5 (or si-NC) and the miR-221-3p inhibitor (or inhibitor-NC) were used to co-transfected into the A549 or H1299 cells. However, the control cells were transfected with the mutant GAS5 (GAS5-MUT) and miR-221-3p inhibitor. As shown in Figure 4A and Figure S5A, after co-transfection, RT-qPCR was performed to measure GAS5 expression. The decreased cell viability by miR-221-3p inhibitor was eliminated by GAS5 knockdown in A549 and H1299 (Figure 4B and Figure S7B). Similarly, the knockdown of GAS5 promoted the cell enter into S phase from G0/G1 phase and reversed the stagnation of cell in G0/G1 phase were induced by miR-221-3p inhibitor (Figure 4C, Figures S6A and S7C–D). Induction of knockdown of GAS5 by miR-221-3p inhibitor upregulates cellular CyclinD1 expression and reverses knockdown of GAS5 suppresses CyclinD1 expression (Figure 4D). In terms of apoptosis analysis, knockdown of GAS5 significantly inhibited apoptosis and the expression of Caspase-3, but this effect could be eliminated by miR-221-3p inhibitor (Figure 4E–F, Figures S6B and S7E). In addition, si-GAS5 promoted the invasion ability of A549 and H1299, and increased the expression of MMP2 (Figure 4G–H, Figures S6C and S7F).
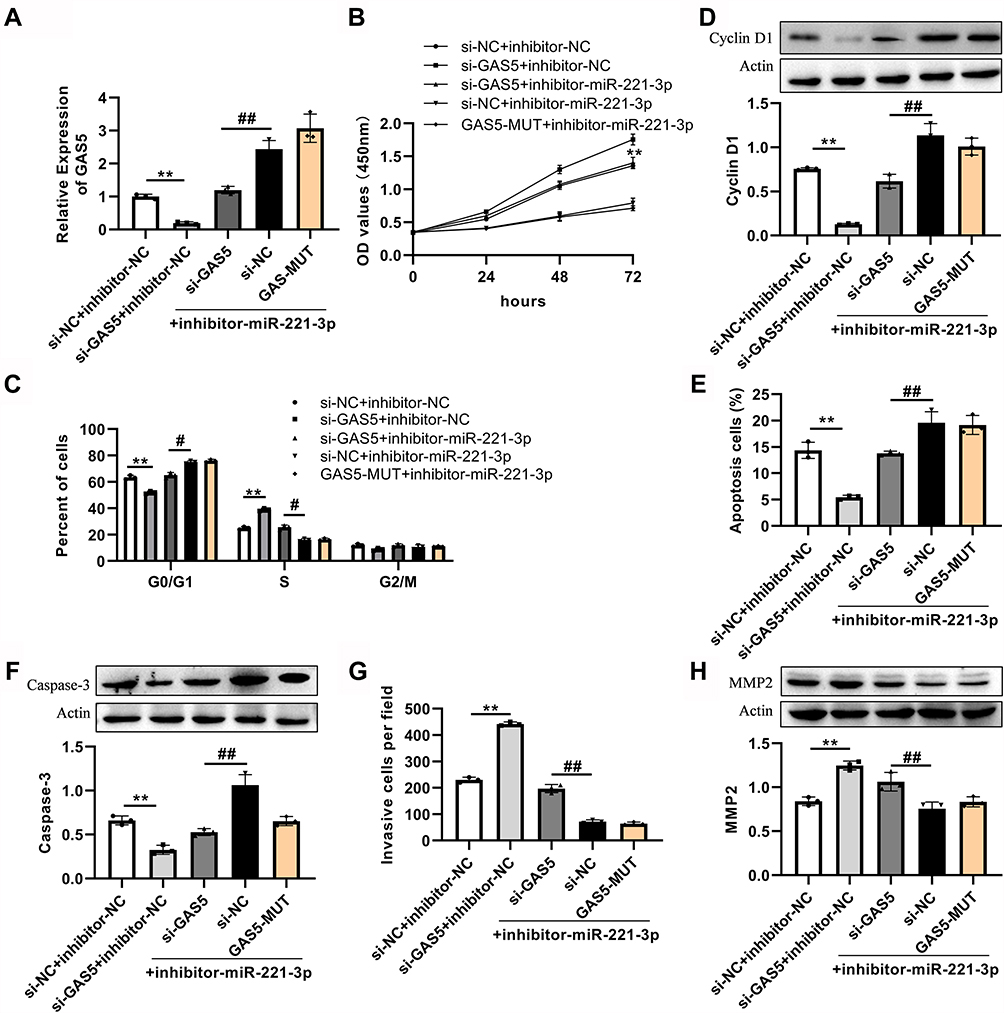 |
Figure 4 The effect of miR-221-3p inhibitor on cell process was weakened after GAS5 knockdown in A549 cells. (A) RT-qPCR was used to evaluate the GAS5 expression in cells after knockdown or addition of miR-221-3p inhibitor. (B) The cell viability was detected by CCK8. (C) si-GAS5 transfected cells affected cell cycle, reduced G0/G1 phase arrest, and increased S phase cells, while inhibitors reversed this effect. (D). si-GAS5 transfected cells reduced the expression of cyclin D1, a cyclin-related protein, but miR-221-3p inhibitor reversed this effect. (E) si-GAS5 transfected cells reduced apoptosis, but miR-221-3p inhibitor reversed this effect. (F) si-GAS5 transfected cells reduced the expression of apoptosis-related protein Caspase-3, but miR-221-3p inhibitor reversed this effect. (G) The number of invaded cells increased after GAS5 knockdown, while the opposite effect was generated by miR-221-3p inhibitor. si-NC, negative control siRNA. GAS5-MUT, mutant GAS5. (H) After GAS5 knockout, invasion related protein MMP2 increased, while miR-221-3p inhibitor had the opposite effect; **P < 0.01 versus si-NC+inhibitor-NC. #P<0.05 versus si-GAS5+inhibitor. ##P < 0.01 versus si-GAS5+inhibitor-miR-221-3p. |
Then, the pc-GAS5 (or pc-Ctrl) and the miR-221-3p mimic (or mimic-NC) were used to co-transfected into the A549 or H1299 cells, and after co-transfection, RT-qPCR was performed to measure GAS5 expression (Figures S8A and S9A). In A549 and H1299 cells, the GAS5 overexpression inhibited the cell viability and proliferation (Figure S8B–8D and S9B–D), and the GAS5 overexpression increased the apoptosis (Figures S8E and S9E). In addition, in A549 and H1299 cells, the invasion ability was also inhibited by GAS5 overexpression (Figures S8F and S9F). However, miR-221-3p mimic weakened these effects.
The Positive Correlation Exists Between GAS5 and TP63
We intended to analyze whether GAS5 further regulates the expression of TP63 by inhibiting miR-221-3p. The results are shown in Figure 5A and 5B, both at mRNA and protein levels, the miR-221-3p inhibition resulted in upregulation of TP63 expression in A549 and H1299 cells, which was eliminated by si-GAS5. Compared with the negative control group (co-transfection with si-NC and inhibitor-NC), TP63 expression downregulated in knockdown group of GAS5 (co-transfection with si-GAS5 and inhibitor-NC). Additionally, in NSCLC tissues, GAS5 expression was positively related with TP63 expression level (Pearson r = 0.7846, P = 0.0072; Figure 5C). Collectively, the above results suggest that GAS5 promoted TP63 expression by sponging miR-221-3p.
 |
Figure 5 The expression of TP63 is influenced by GAS5 in NSCLC cells. (A) RT-qPCR was used to detect TP63 mRNA levels. (B) TP63 protein level in the si-GAS5 and miR-221-3p inhibitor groups was lower compared with si-NC and miR-221-3p inhibitor group. (C) The expression of GAS5 and TP63 in tumor tissues was positively correlated. **P<0.01 versus si-NC+inhibitor-NC. |
Function of GAS5 Overexpression in NSCLC in vitro and in vivo
In the final part of this study, we evaluated whether GAS5 overexpression altered the sensitivity of A549 and H1299 cells to cisplatin (CIS) in vivo and in vitro. Furthermore, to inhibit the expression of TP63, the shRNA-3 of TP63 was used to inhibit the expression of TP63, and the most effective shRNA-3 was used to conduct follow-up experiments (Figure S10). As expected, compared with the control group, GAS5 overexpression after CIS treatment of A549 (Figure 6A and C) and H1299 cells (Figure S11A and B) significantly promoted cell apoptosis and reduced the number of invasive cells, as well as increased Caspase-3 expression and decreased MMP2 expression (Figure 6B and D). This effect of GAS5 overexpression was eliminated by TP63 knockdown. In addition, GAS5 silencing decreased the apoptosis and increased the invasion of H1299 and A549 to CIS, and TP63 overexpression declined these effects (Figure S12A and B). The following experiments in nude mice showed that GAS5 overexpression decreased tumor growth in vivo to a large extent compared to CIS treatment alone (Figure 6E). However, tumor growth in mice after TP63 knockdown was more significant, and this effect was offset and eliminated by co-transfection with the plasmid expressing GAS5 (pc-GAS5) (Figure 6E). The tumor volume and weight, that derived from GAS5 overexpression group, were greatly smaller than the tumor volume and weight of blank group, while co-transfection with sh-TP63 abolished this effect (Figure 6F and G). In addition, the IHC results of Ki67 indicated that GAS5 overexpression significantly inhibited NSCLC cell proliferation after CIS treatment, and TP63 silencing abolished this effect (Figure S13). Based on the above results, it can be concluded that the efficacy of CIS can be improved after overexpression of lncRNA GAS5 in vivo.
 |
Figure 6 The effect of GAS5 and TP63 on the tumor ‘s chemotherapeutic sensitivity to CIS in vitro and in vivo. (A) The percentage of apoptosis induced by CIS increased significantly after the overexpression of GAS5, but sh-TP63 decreased this effect. (B) After overexpression of GAS5, the expression of caspase-3, an apoptosis-related protein induced by CIS, was significantly increased, but sh-TP63 reduced this effect. (C) After overexpression of GAS5, the invasion induced by CIS was significantly reduced, but sh-TP63 reduced this effect. (D) After overexpression of GAS5, the expression of invasion related protein MMP2 induced by CIS was significantly reduced, but sh-TP63 reduced this effect; **P<0.01 versus pc-Ctrl+si-NC+CIS. #P<0.01 versus pc-Ctrl+si-TP63+CIS. (E) Representative photographs of tumor formation (n=4, mean ± SD) were selected and displayed. (F and G) Tumor growth was significantly inhibited after the downregulation of TP63, and the decrease of TP63 expression also eliminated the promotion effect of GAS5 overexpression on tumor weight (F) and tumor volume (G). (n=6, mean ± SD); **P<0.01, versus pc-Ctrl+sh-NC+CIS. |
Discussion
The anti-proliferation effect of TP63 in human gastric, lung, and pancreatic tumor cells has been demonstrated in previous studies.5 Similar in function to p53 in its family, TP63 can induce and promote tumor cell apoptosis in vivo and in vitro, and ultimately significantly inhibit tumor growth.5 In the present study, we investigated the underlying mechanism responsible for the dysregulation of TP63 in NSCLC progression. TAp63 suppresses tumorigenesis and metastasis, and the complete inactivation TAp63 promotes tumour progression.28 In the present study, we predicted and verified that miR-221-3p was the upstream regulator of TAp63beta. Our results indicated that TP63 mRNA level downregulated in NSCLC tissues, and TAp63beta was not only related to tumor cell apoptosis but also related to loss of tumorigenicity in nude mice. TAp63beta also appears as a tumor suppressor in other cancer, such as cervical carcinoma.29 TAp63beta may be an important tumor-related gene, which may be regulated by a variety of miRNAs in various tumors. A previous study demonstrated that miR-375 overexpression promoted TAp63beta expression, and TAp63beta overexpression repressed the migration of cervical cancer cell.30
In NSCLC tumor tissues, miR-221-3p expression was upregulated, and it was negatively related with TP63, revealing that miR-221-3p/TP63 may be relevant to lung cancer occurrence and development. miR-221-3p is also highly expressed in other cancers, such as liver cancer,31 thyroid cancer,32 and pancreatic cancer.33 We only used 10 pairs of NSCLC and nontumor tissues to detect the miR-221-3p expression, but it is difficult to collect samples of NSCLC and nontumor tissues, and we will try our best to collect samples of NSCLC and nontumor tissues to further support the results in the future. The mechanism of miR-221-3p in tumor progression is complex. miR-221-3p reduces the first-line chemotherapy drug paclitaxel resistance of NSCLC and promotes cell apoptosis.34 In pancreatic cancer, miR-221-3p targeted RB transcriptional corepressor 1 to enhance the 5-fluorouracil resistance of cells.35
lncRNA plays a variety of regulatory roles in the behaviours of various tumors, including the proliferation, multi-organ metastasis and drug resistance.36,37 GAS5, as a lncRNA that is relation with cell proliferation, has low expression in various malignant tumors. Here, the downregulation of GAS5 expression in NSCLC organizations was found and verified. Consistent with NSCLC, previous studies proved that GAS5 expression reduced in clear cell renal cell carcinoma tissues and gastric cancer tissues,38 especially in gastric cancer tissues, showing poor overall survival rate and poor disease-free survival.39 In the present study, GAS5 performed function as an inducer in the process of NSCLC cell apoptosis by targeting miR-221-3p/TP63 signal transduction. The binding of miR-221-3p with GAS5 was observed. Moreover, GAS5 overexpression inhibited miR-221-3p expression, but miR-221-3p mimic also suppressed GAS5 expression. Further, contrary to low GAS5 expression, high miR-221-3p expression also supports the negative relation between GAS5 and miR-221-3p to some extent in tumors. Knockdown of GAS5 lead to downregulation of TP63 expression in NSCLC cells. GAS5 expression is positively related with TP63 expression. These findings suggested that GAS5 promoted TP63 expression by sponging miR-221-3p. GAS5 represses miR-221-3p to increase tumor suppressor IRF2 expression in NSCLC cells, which blocks the progression of NSCLC.40 Some literatures explored GAS5 as sponge of miR-221-3p to affect the regulation of miR-221-3p on other genes expression. In follicular thyroid carcinoma, GAS5 targeted miR-221-3p/CDKN2B axis to induce G0/G1 phase arrest and inhibit cell proliferation.41 A previous study demonstrated that GAS5 directly interact with miR-221-3p by dual-luciferase reporter assay and RNA immunoprecipitation assay.26 Therefore, the possibility of competition of GAS5 and TP63 for the target sequences in miR-221-3p is extremely high. However, in the future, we should further demonstrate the possibility of direct interaction between miR-221-3p and GAS5 or TP63 by RNA immunoprecipitation assay. Additionally, GAS5 inhibits growth, invasion and migration via the miR-205/PTEN pathway in NSCLC.42 GAS5 has lower expression in exosomes from the serum of NSCLC patients.43 The above data certificated that GAS5 may be as a mark for predicting the prognosis of cancer patients.
The complex mechanism of tumor resistance is an obvious challenge to current therapies. It is a valid approach to increase the chemotherapy efficacy to find and predict drug-sensitive marker molecules and explore new mechanisms and measures to reverse drug resistance.44 To quest the effect of GAS5 on NSCLC cell drug resistance in vitro and in vivo, CIS was used as chemotherapy drug. As a common drug used in chemotherapy, CIS significantly represses the tumor growth; however, drug resistance always lead to the chemotherapy flunk.45,46 In this study, GAS5 targeted miR-221-3p/TP63 signal pathway to increase the NSCLC cells’ sensitivity to CIS. Meanwhile, in the subcutaneous tumor bearing nude model, we observed that after CIS treatment, the tumor volume of tumor-bearing mice planted with A549 cells of GAS5 overexpression was significantly lower than that of the control group, indicating that GAS5 overexpression greatly enhanced the antitumor activity of CIS in vivo. Besides, GAS5 competes with PTEN for miR-21 binding to regulate the NSCLC chemo-sensitivity to CIS.47 GAS5 also enhances CIS sensitivity by suppressing autophagy of NSCLC cells.48
Conclusion
In summary, we demonstrate that reduced expression of TP63 performs important functions in NSCLC development. The subsequent analysis of the possible mechanisms causing its low expression revealed for the first time that GAS5 could play a tumor repressive gene in the development of NSCLC by regulating the miR-221-3p/TP63 axis, thus improving the sensitivity of NSCLC cell to chemotherapy and cisplatin to a certain extent.
Abbreviations
TP63, tumor protein p63; miRNAs, micro-RNAs; mRNA, messenger RNA; NSCLC, non-small cell lung cancer; lncRNA, long noncoding RNA; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; PI, propidium iodide; CIS, cisplatin.
Data Sharing Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
This study was approved by the ethics committee of The First Hospital of China Medical University.
Consent to Participate
Written informed consent before surgery was obtained from each patient.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Martinez P, Martinez-Marti A, Navarro A, Cedrés S, Felip E. Molecular targeted therapy for early-stage non-small-cell lung cancer: will it increase the cure rate? Lung Cancer. 2014;84(2):97–100. doi:10.1016/j.lungcan.2014.01.018
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.21654
3. Reungwetwattana T, Weroha SJ, Molina JR. Oncogenic pathways, molecularly targeted therapies, and highlighted clinical trials in non-small-cell lung cancer (NSCLC). Clin Lung Cancer. 2012;13(4):252–266. doi:10.1016/j.cllc.2011.09.004
4. Zhao M, Xu P, Liu Z, et al. Dual roles of miR-374a by modulated c-Jun respectively targets CCND1-inducing PI3K/AKT signal and PTEN-suppressing Wnt/β-catenin signaling in non-small-cell lung cancer. Cell Death Dis. 2018;9(2):78. doi:10.1038/s41419-017-0103-7
5. Kunisaki R, Ikawa S, Maeda T, et al. p51/p63, a novel p53 homologue, potentiates p53 activity and is a human cancer gene therapy candidate. J Gene Med. 2006;8(9):1121–1130. doi:10.1002/jgm.945
6. Ishida S, Yamashita T, Nakaya U, Tokino T. Adenovirus-mediated transfer of p53-related genes induces apoptosis of human cancer cells. Jpn J Cancer Res. 2000;91(2):174–180. doi:10.1111/j.1349-7006.2000.tb00929.x
7. Katoh I, Aisaki KI, Kurata SI, Ikawa S, Ikawa Y. p51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene. 2000;19(27):3126–3130. doi:10.1038/sj.onc.1203644
8. Senoo M, Tsuchiya I, Matsumura Y, et al. Transcriptional dysregulation of the p73L / p63 / p51 / p40 / KET gene in human squamous cell carcinomas: expression of Delta Np73L, a novel dominant-negative isoform, and loss of expression of the potential tumour suppressor p51. Br J Cancer. 2001;84(9):1235–1241. doi:10.1054/bjoc.2000.1735
9. Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–718. doi:10.1038/19539
10. Shimada A, Kato S, Enjo K, et al. The transcriptional activities of p53 and its homologue p51/p63: similarities and differences. Cancer Res. 1999;59(12):2781–2786.
11. Ikawa S, Nakagawara A, Ikawa Y. p53 family genes: structural comparison, expression and mutation. Cell Death Differ. 1999;6(12):1154–1161. doi:10.1038/sj.cdd.4400631
12. Sasaki Y, Morimoto I, Ishida S, Yamashita T, Imai K, Tokino T. Adenovirus-mediated transfer of the p53 family genes, p73 and p51/p63 induces cell cycle arrest and apoptosis in colorectal cancer cell lines: potential application to gene therapy of colorectal cancer. Gene Ther. 2001;8(18):1401–1408. doi:10.1038/sj.gt.3301538
13. Liao M, Peng L. MiR-206 may suppress non-small lung cancer metastasis by targeting CORO1C. Cell Mol Biol Lett. 2020;25:22. doi:10.1186/s11658-020-00216-x
14. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–531. doi:10.1038/nrg1379
15. Sun W, Julie LYS, Huang HD, Shyy JY, Chien S. microRNA: a master regulator of cellular processes for bioengineering systems. Annu Rev Biomed Eng. 2010;12:1–27. doi:10.1146/annurev-bioeng-070909-105314
16. Shi L, Zhang B, Sun X, et al. MiR-204 inhibits human NSCLC metastasis through suppression of NUAK1. Br J Cancer. 2014;111(12):2316–2327. doi:10.1038/bjc.2014.580
17. Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med. 2012;12(1):27–33. doi:10.2174/156652412798376170
18. Iida M, Hazama S, Tsunedomi R, et al. Overexpression of miR‑221 and miR‑222 in the cancer stroma is associated with malignant potential in colorectal cancer. Oncol Rep. 2018;40(3):1621–1631. doi:10.3892/or.2018.6575
19. Mangiulli M, Valletti A, Caratozzolo MF, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009;37(18):6092–6104. doi:10.1093/nar/gkp674
20. Patturajan M, Nomoto S, Sommer M, et al. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell. 2002;1(4):369–379. doi:10.1016/s1535-6108(02)00057-0
21. Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9(1):45–56. doi:10.1016/j.ccr.2005.12.013
22. Gallegos JR, Litersky J, Lee H, et al. SCF TrCP1 activates and ubiquitylates TAp63gamma. J Biol Chem. 2008;283(1):66–75. doi:10.1074/jbc.M704686200
23. Suh EK, Yang A, Kettenbach A, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444(7119):624–628. doi:10.1038/nature05337
24. Huang W, Shi Y, Han B, et al. LncRNA GAS5-AS1 inhibits glioma proliferation, migration, and invasion via miR-106b-5p/TUSC2 axis. Hum Cell. 2020;33(2):416–426. doi:10.1007/s13577-020-00331-z
25. Li S, Mei Z, Hu HB, Zhang X. The lncRNA MALAT1 contributes to non-small cell lung cancer development via modulating miR-124/STAT3 axis. J Cell Physiol. 2018;233(9):6679–6688. doi:10.1002/jcp.26325
26. Chen Z, Pan T, Jiang D, et al. The lncRNA-GAS5/miR-221-3p/DKK2 axis modulates ABCB1-mediated adriamycin resistance of breast cancer via the Wnt/β-catenin signaling pathway. Mol Ther Nucleic Acids. 2020;19:1434–1448. doi:10.1016/j.omtn.2020.01.030
27. Chen D, Zhang M, Liu H. Downregulation of lncRNA ZFAS1 inhibits the hallmarks of thyroid carcinoma via the regulation of miR‑302‑3p on cyclin D1. Mol Med Rep. 2021;23:2. doi:10.3892/mmr.2020.11774
28. Su X, Chakravarti D, Cho MS, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467(7318):986–990. doi:10.1038/nature09459
29. Ben Khalifa Y, Teissier S, Tan MK, et al. The human papillomavirus E6 oncogene represses a cell adhesion pathway and disrupts focal adhesion through degradation of TAp63β upon transformation. PLoS Pathog. 2011;7(9):e1002256. doi:10.1371/journal.ppat.1002256
30. Lu H, Qi Z, Lin L, et al. The E6-TAp63β-Dicer feedback loop involves in miR-375 downregulation and epithelial-to-mesenchymal transition in HR-HPV+ cervical cancer cells. Tumour Biol. 2016;2016:1. doi:10.1007/s13277-016-5378-2
31. Dong Y, Zhang N, Zhao S, Chen X, Li F, Tao X. miR-221-3p and miR-15b-5p promote cell proliferation and invasion by targeting Axin2 in liver cancer. Oncol Lett. 2019;18(6):6491–6500. doi:10.3892/ol.2019.11056
32. Ye T, Zhong L, Ye X, Liu J, Li L, Yi H. miR-221-3p and miR-222-3p regulate the SOCS3/STAT3 signaling pathway to downregulate the expression of NIS and reduce radiosensitivity in thyroid cancer. Exp Ther Med. 2021;21(6):652. doi:10.3892/etm.2021.10084
33. Li F, Xu JW, Wang L, Liu H, Yan Y, Hu SY. MicroRNA-221-3p is up-regulated and serves as a potential biomarker in pancreatic cancer. Artif Cells, Nanomed Biotechnol. 2018;46(3):482–487. doi:10.1080/21691401.2017.1315429
34. Ni L, Xu J, Zhao F, et al. MiR-221-3p-mediated downregulation of MDM2 reverses the paclitaxel resistance of non-small cell lung cancer in vitro and in vivo. Eur J Pharmacol. 2021;899:174054. doi:10.1016/j.ejphar.2021.174054
35. Zhao L, Zou D, Wei X, et al. MiRNA-221-3p desensitizes pancreatic cancer cells to 5-fluorouracil by targeting RB1. Tumour Biol. 2016;37:16053–16063. doi:10.1007/s13277-016-5445-8
36. Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839(11):1097–1109. doi:10.1016/j.bbagrm.2014.08.012
37. Pickard MR, Mourtada-Maarabouni M, Williams GT. Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim Biophys Acta. 2013;1832(10):1613–1623. doi:10.1016/j.bbadis.2013.05.005
38. Qiao HP, Gao WS, Huo JX, Yang ZS. Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pac J Cancer Prev. 2013;14(2):1077–1082. doi:10.7314/apjcp.2013.14.2.1077
39. Sun M, Jin FY, Xia R, et al. Decreased expression of long noncoding RNA GAS5 indicates a poor prognosis and promotes cell proliferation in gastric cancer. BMC Cancer. 2014;14:319. doi:10.1186/1471-2407-14-319
40. Ma J, Miao H, Zhang H, et al. LncRNA GAS5 modulates the progression of non-small cell lung cancer through repressing miR-221-3p and up-regulating IRF2. Diagn Pathol. 2021;16(1):46. doi:10.1186/s13000-021-01108-0
41. Liu Y, Li YF, Liu J, Deng ZG, Zeng L, Zhou WB. Long noncoding RNA GAS5 Targeting miR-221-3p/cyclin-dependent kinase inhibitor 2B axis regulates follicular thyroid carcinoma cell cycle and proliferation. Pathobiology. 2021;88(4):289–300. doi:10.1159/000513338
42. Dong L, Li G, Li Y, Zhu Z. Upregulation of long noncoding RNA GAS5 inhibits lung cancer cell proliferation and metastasis via miR-205/PTEN axis. Med Sci Monit. 2019;25:2311–2319. doi:10.12659/msm.912581
43. Li C, Lv Y, Shao C, et al. Tumor-derived exosomal lncRNA GAS5 as a biomarker for early-stage non-small-cell lung cancer diagnosis. J Cell Physiol. 2019;234(11):20721–20727. doi:10.1002/jcp.28678
44. O’Reilly EA, Gubbins L, Sharma S, et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015;3:257–275. doi:10.1016/j.bbacli.2015.03.003
45. Ito S, Sano T, Mizusawa J, et al. A Phase II study of preoperative chemotherapy with docetaxel, cisplatin, and S-1 followed by gastrectomy with D2 plus para-aortic lymph node dissection for gastric cancer with extensive lymph node metastasis: JCOG1002. Gastric Cancer. 2017;20(2):322–331. doi:10.1007/s10120-016-0619-z
46. Coleman RL, Brady MF, Herzog TJ, et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG oncology/gynecologic oncology group study GOG-0213): a multicentre, open-label, randomised, Phase 3 trial. Lancet Oncol. 2017;18(6):779–791. doi:10.1016/s1470-2045(17)30279-6
47. Cao L, Chen J, Ou B, Liu C, Zou Y, Chen Q. GAS5 knockdown reduces the chemo-sensitivity of non-small cell lung cancer (NSCLC) cell to cisplatin (DDP) through regulating miR-21/PTEN axis. Biomed Pharmacother. 2017;93:570–579. doi:10.1016/j.biopha.2017.06.089
48. Zhang N, Yang GQ, Shao XM, Wei L. GAS5 modulated autophagy is a mechanism modulating cisplatin sensitivity in NSCLC cells. Eur Rev Med Pharmacol Sci. 2016;20(11):2271–2277.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Estrogen Regulates the Expression and Function of lncRNA-H19 in Ectopic Endometrium
Liu S, Qiu J, Tang X, Li Q, Shao W
International Journal of Women's Health 2022, 14:821-830
Published Date: 28 June 2022
A 5`-tRNA Derived Fragment NamedtiRNA-Val-CAC-001 Works as a Suppressor in Gastric Cancer
Zheng J, Li C, Zhu Z, Yang F, Wang X, Jiang P, Yan F
Cancer Management and Research 2022, 14:2323-2337
Published Date: 4 August 2022
Eriochloa villosa Alleviates Progression of Benign Prostatic Hyperplasia in vitro and in vivo
Baek EB, Hwang YH, Park S, Hong EJ, Won YS, Kwun HJ
Research and Reports in Urology 2022, 14:313-326
Published Date: 24 September 2022
Human Mesenchymal Stem Cell-Derived Exosomes Promote the Proliferation and Melanogenesis of Primary Melanocytes by Attenuating the H2O2-Related Cytotoxicity in vitro
Wang Y, He Z, Luo B, Wong H, Wu L, Zhou H
Clinical, Cosmetic and Investigational Dermatology 2024, 17:683-695
Published Date: 18 March 2024
MicroRNA-18b-5p Inhibits the Malignant Progression of Prostate Cancer Through Downregulating TRAF5

Wang X, Yu X, Wang Y, Lian J, Li Z, Dong C, Song H, Zhang L, Zhang H, Wang Y
International Journal of General Medicine 2025, 18:1831-1843
Published Date: 31 March 2025