Back to Journals » Cancer Management and Research » Volume 18

TNFAIP3 Reprograms T Cell Exhaustion and Restores Anti-Leukemia Immunity in Acute Myeloid Leukemia
Authors Li Y, Wu H, Bai J, Peng X, Chen Y, Zhang J, Zhang L, Li L
Received 28 January 2026
Accepted for publication 7 May 2026
Published 13 May 2026 Volume 2026:18 592664
DOI https://doi.org/10.2147/CMAR.S592664
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Kattesh Katti
Yanhong Li,1,2,* Hongxia Wu,3,* Jun Bai,1,2 Xiaohuan Peng,1,2 Yue Chen,1 Jinping Zhang,1,2 Liansheng Zhang,1,2 Lijuan Li1,2
1Department of Hematology, Lanzhou University Second Hospital, Lanzhou, 730000, People’s Republic of China; 2Gansu Province Hematologic Disease Clinical Medical Research Center, Lanzhou University Second Hospital, Lanzhou, 730000, People’s Republic of China; 3Department of Nuclear Medicine, Lanzhou University Second Hospital, Lanzhou, 730000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liansheng Zhang, Email [email protected] Lijuan Li, Email [email protected]
Purpose: Acute myeloid leukemia (AML) is an aggressive hematological malignancy that is associated with profound immune dysfunction. T-cell exhaustion is a major mechanism of immune evasion in AML; however, the key molecular regulators driving this process remain unclear. This study aimed to identify immunoregulatory genes associated with T cell exhaustion in AML and define their functional roles.
Patients and Methods: Single-cell RNA sequencing (scRNA-seq) data from patients with AML and healthy donors were integrated to identify genes associated with T-cell exhaustion. TNFAIP3 was identified as a candidate hub gene and subsequently validated in peripheral T cells from 24 AML patients and 12 healthy donors using qRT-PCR and Western blotting. Functional studies were performed using lentivirus-mediated TNFAIP3 overexpression in patient-derived CD4+ and CD8+ T-cells. Apoptosis, immune checkpoint expression, cytokine production, and NF-κB signaling activity were assessed using pathway enrichment analyses.
Results: scRNA-seq analysis revealed widespread immune remodeling in AML, with TNFAIP3 consistently downregulated in AML T cells. Reduced TNFAIP3 expression was confirmed at both the mRNA and protein levels in patients with AML. TNFAIP3 overexpression in CD4+ and CD8+ T cells attenuates apoptosis, reduces the expression of the immune checkpoint molecules PD-L1 and TIM-3, and promotes a pro-inflammatory cytokine profile characterized by increased IFN-γ and TNF-α production. Mechanistically, these effects were associated with the suppression of NF-κB signaling and enrichment of pathways related to immune synapse formation and T-cell receptor signaling.
Conclusion: Our findings demonstrate that TNFAIP3 is a critical regulator of T cell dysfunction and exhaustion in AML. Downregulation of TNFAIP3 contributes to impaired anti-leukemic immunity, whereas its restoration enhances T-cell survival and effector function. TNFAIP3 therefore represents a promising therapeutic target for immune-based interventions aimed at restoring T-cell-mediated immunity in AML.
Keywords: acute myeloid leukemia, TNFAIP3, T cell, NF-κB, immune checkpoint
Introduction
Acute myeloid leukemia (AML) is a clonal hematopoietic malignancy characterized by the accumulation of immature myeloid blasts in bone marrow and peripheral blood. Despite progress in chemotherapy and targeted therapies, outcomes for AML remain unsatisfactory, particularly in patients harboring high-risk genetic alterations, such as FLT3-ITD mutations or relapsed/refractory diseases.1,2 In addition to intrinsic oncogenic drivers, disease progression is profoundly influenced by extrinsic immunosuppressive mechanisms.
T cells serve as central effectors of anti-tumor immunity; however, their activity in AML is frequently compromised by exhaustion. Exhausted T-cells are characterized by diminished cytokine secretion and elevated expression of co-inhibitory receptors, including PD-1, TIM-3, and LAG-3.3 This dysfunctional state weakens the immune surveillance and facilitates leukemic escape. Increasing evidence indicates that immune evasion is a hallmark of AML progression and therapeutic resistance. Leukemia cells evade host immunity by impairing antigen presentation, upregulating immune checkpoints such as PD-L1 and TIM-3, and suppressing T cell effector function.4,5 Although both CD4+ and CD8+ subsets are essential for tumor control, they frequently display reduced cytokine output and enhanced inhibitory receptor expression in the AML microenvironment.
Tumor necrosis factor alpha-induced protein 3 (TNFAIP3, also known as A20) is a ubiquitin-editing enzyme that negatively regulates NF-κB signaling and contributes to immune tolerance and inflammation control. TNFAIP3 mutations, deletions, and reduced expression have been reported.6,7 Importantly, TNFAIP3 deficiency can promote T cell dysfunction, increase immune checkpoint expression, and foster an immunosuppressive milieu.8,9 Mechanistically, TNFAIP3 inhibits NF-κB activity by removing K63-linked ubiquitin chains and promoting degradation of signaling intermediates such as RIP1 and TRAF6, thereby preventing uncontrolled immune activation.10,11 In T cells, TNFAIP3 regulates apoptosis, cytokine production, and metabolic fitness, thereby limiting chronic activation and shaping cell fate decisions.12 Murine studies have shown that T cell–specific loss of TNFAIP3 increases susceptibility to activation-induced cell death and elevates exhaustion markers such as PD-1 and TIM-3-features typical of tumor-associated dysfunctional T cells. Within the tumor microenvironment, TNFAIP3 downregulation has been linked to impaired CD8+ T cell cytotoxicity and heightened exhaustion, particularly in solid tumors and chronic infections.13 However, its function in hematologic malignancies, particularly AML, in which immune evasion is a major barrier to effective therapy, remains poorly understood. Elucidating how TNFAIP3 shapes the immune phenotype of AML-infiltrating T-cells may provide new strategies for improving immune surveillance and therapeutic responses.
In this study, we applied single-cell transcriptomic profiling of peripheral immune cells from AML patients and healthy donors and identified TNFAIP3 as a central regulator within the T-cell dysfunction network. Functional experiments demonstrated that the restoration of TNFAIP3 expression in AML-derived T cells reduced immune checkpoint expression, rebalanced cytokine secretion, and attenuated NF-κB activation. Together, these findings highlight TNFAIP3 as a promising target for reinvigorating T cell immunity in AML.
Materials and Methods
Single-Cell RNA Sequencing Data Acquisition and Analysis
Single-cell RNA sequencing (scRNA-seq) data of bone marrow cells from four patients with AML and two healthy donors were retrieved from the Gene Expression Omnibus (GEO) database. For this study, we focused on newly diagnosed (D0), treatment-naïve AML samples. Within this cohort, samples with available FLT3-ITD mutation annotation and high-quality Seq-Well scRNA-seq data were selected for downstream analysis after quality control filtering. Raw UMI count matrices were processed using the Seurat (v4.0.6). Low-quality cells (<200 detected genes or >20% mitochondrial transcripts) and genes expressed in fewer than three cells were excluded. Datasets were integrated using canonical correlation analysis (CCA), followed by principal component analysis (PCA), and Uniform Manifold Approximation and Projection (UMAP) based on the top 30 PCs. The clustering was performed at a resolution of 0.5. Cell type annotation relied on canonical marker genes and was verified using the SingleR software. T cells were defined based on CD3D and CD3E expression and were subdivided into CD4+ and CD8+ subsets. Differentially expressed genes (DEGs) between AML and healthy donors were identified using the Wilcoxon rank-sum test (|log2FC| > 0.25, adjusted p < 0.05). Gene Ontology (GO) and KEGG enrichment were performed with clusterProfiler, and gene set enrichment analysis (GSEA) used immune-related signatures from MSigDB.
Patient Samples and T Cell Isolation
Peripheral blood samples were collected from 24 patients with newly diagnosed AML and 12 healthy donors at Lanzhou University Second Hospital (Lanzhou, China). All participants provided informed consent and the study was approved by the institutional ethics committee. PBMCs were isolated using Ficoll-Paque density gradient centrifugation. CD4+ and CD8+ T cells were purified using magnetic-activated cell sorting (MACS; Miltenyi Biotec). Cell purity (>90%) was confirmed by BD FACSAria cell sorter (BD Biosciences) flow cytometry. Total RNA was extracted using TRIzol (Invitrogen), and RNA integrity was assessed using NanoDrop and agarose gel electrophoresis. cDNA was synthesized from 1 μg RNA using the PrimeScript RT kit (Takara).
qRT-PCR and Western Blotting
TNFAIP3 mRNA levels in CD4+ and CD8+ T cells were measured by qRT-PCR. RNA was reverse-transcribed using PrimeScript RT reagent (Takara) and qRT-PCR was performed using SYBR Green Master Mix (Roche) on a Roche LightCycler 480. GAPDH served as the internal control, and relative expression was calculated using the 2^−ΔΔCt method. For protein detection, cells were lysed with RIPA buffer containing protease and phosphatase inhibitors (Beyotime). Proteins (30 μg/lane) were separated by SDS-PAGE, transferred to PVDF membranes (Millipore), blocked with 5% non-fat milk, and incubated overnight at 4°C with antibodies against TNFAIP3 (Abcam #EPR2663), phospho-NF-κB p65 (Ser536) (Proteintech #6N1), and β-actin (Proteintech #66009-1-ig) HRP-conjugated secondary antibodies were applied and signals were detected using enhanced chemiluminescence (Thermo Fisher). Primer sequences for TNFAIP3: forward 5’-CACGCTCAAGGAAACAGACA-3’, reverse 5’-CATGGGTGTGTCTGTGGAAG-3’. Primer sequences for GAPDH: forward 5’-ATCTTCCAGGAGCGAGATCC-3’; reverse 5’-ACCACTGACACGTTGGCAGT-3’.
Lentiviral Overexpression of TNFAIP3
Full-length human TNFAIP3 was cloned into the pLVX lentiviral vector. Lentiviruses were produced in HEK293T cells by co-transfection with psPAX2 and pMD2.G (Addgene), using Lipofectamine 3000 (Thermo Fisher Scientific). Viral supernatants were harvested at 48 h and 72 h, filtered (0.45 μm), and concentrated by ultracentrifugation. Patient-derived CD4+ and CD8+ T cells were cultured in RPMI-1640 with 10% FBS and 100 IU/mL IL-2 and transduced at an MOI of 20 with 8 μg/mL polybrene (Sigma). Spinoculations were performed at 800 × g for 90 min at 32°C, followed by an overnight incubation. The medium was refreshed after 24 h, and the transduction efficiency was assessed by GFP expression. Samples with >70% efficiency were used for downstream experiments. TNFAIP3 overexpression was confirmed by qRT-PCR and Western blotting. Functional assays were performed 48 hours after lentiviral transduction, a time point at which stable overexpression of TNFAIP3 at both mRNA and protein levels was confirmed.
Protein Profiling and Cytokine Assays
To evaluate immunomodulatory effects of TNFAIP3, immune checkpoint proteins and cytokines were profiled. For checkpoint analysis, the Human Immune Checkpoint Array 1 (RayBiotech) was used. Lysates were incubated with array membranes, detected with biotinylated antibodies and HRP-streptavidin, and visualized by chemiluminescence. Densitometry was performed using ImageJ software. Cytokine levels in the supernatants (collected 48 h post-transduction) were quantified using the 13-plex Human Th1/Th2 Panel (LEGENDplex, BioLegend), including IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12, IFN-α, IFN-γ, TNF-α, IL-17, and VEGF. Supernatants from 1 × 106 T cells (triplicate cultures) were cleared by centrifugation, incubated with capture beads and detection antibodies, and analyzed using a BD FACSCanto II Cytometer. Cytokine concentrations were determined using LEGENDplex™ software using a five-parameter logistic (5-PL) curve.
Flow Cytometry
Apoptosis and checkpoint expression were assessed by flow cytometry. For apoptosis, CD4+ and CD8+ T cells were stained with Annexin V-FITC and propidium iodide (PI) (BioLegend), according to the manufacturer’s instructions. Cells (1 × 10 < II >) were analyzed using a BD FACSCanto II instrument. Populations were classified as early apoptotic (Annexin V⁺/PI−), late apoptotic (Annexin V⁺/PI⁺), or necrotic (Annexin V/PI ⁺). For checkpoint analysis, cells were stained with fluorochrome-conjugated antibodies against TIM-3 (APC), PD-L1 (PE), and ICOS (FITC), with isotype controls. After 30 min at 4°C, the cells were washed and analyzed. Single-stained cells and FMO controls were used for compensation and gating, respectively. Data were processed using the FACSDiva v10.8.1, recording ≥10,000 events per sample. The antibodies used were BV421 anti-human TIM-3 (BioLegend), BV510 anti-human CD8 (BioLegend), and fluorescein isothiocyanate (FITC) anti-human CD4 (BD Biosciences).
Statistical Analysis
Data are presented as mean ± standard deviation (SD). Statistical significance was assessed using the Student’s t-test or one-way ANOVA, as appropriate. Statistical significance was set at P < 0.05. For bioinformatics analyses, multiple testing corrections were performed using the Benjamini–Hochberg method.
Result
Single-Cell Transcriptomic Profiling Reveals Immune Dysregulation in AML
To characterize the immune alterations in AML, we analyzed single-cell RNA sequencing (scRNA-seq) data from peripheral blood mononuclear cells (PBMCs) from four patients with AML and two healthy donors (Table 1). After stringent quality control and dataset integration, unsupervised clustering was performed using principal component analysis (PCA), uniform manifold approximation, and projection (UMAP). Cell types were annotated according to canonical marker expression. A heatmap of the top 10 marker genes per cluster demonstrated distinct transcriptional signatures, confirming the accurate segregation of cell populations across AML and control samples (Figure 1A). We identified 12 major immune and hematopoietic cell populations: CD14⁺ monocytes, hematopoietic stem and progenitor cells (HSPCs), T cells, promonocytes, erythroid cells, natural killer (NK) cells, granulocyte–monocyte progenitors (GMPs), B cells, monocytes, plasma cells, and plasmacytoid dendritic cells (PDCs) (Figure 1B). Dimensionality reduction by UMAP and t-SNE showed a clear clustering of these subsets across all samples.
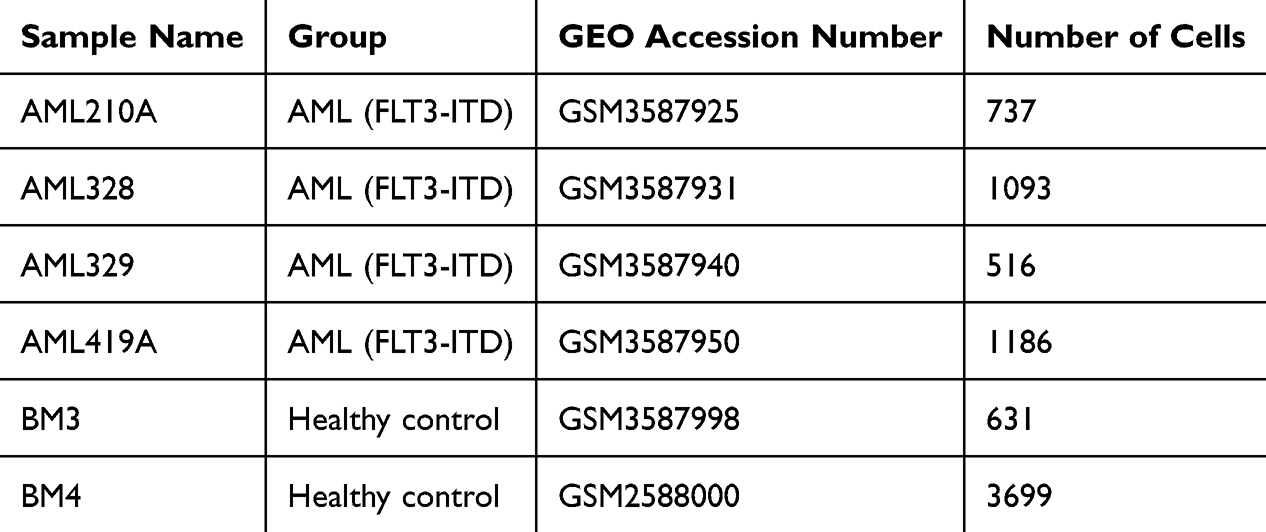 |
Table 1 Overview of the Study Design and GEO-Derived scRNA-Seq Datasets, Including Peripheral Blood Mononuclear Cells (PBMCs) from Four AML Patients and Two Healthy Donors |
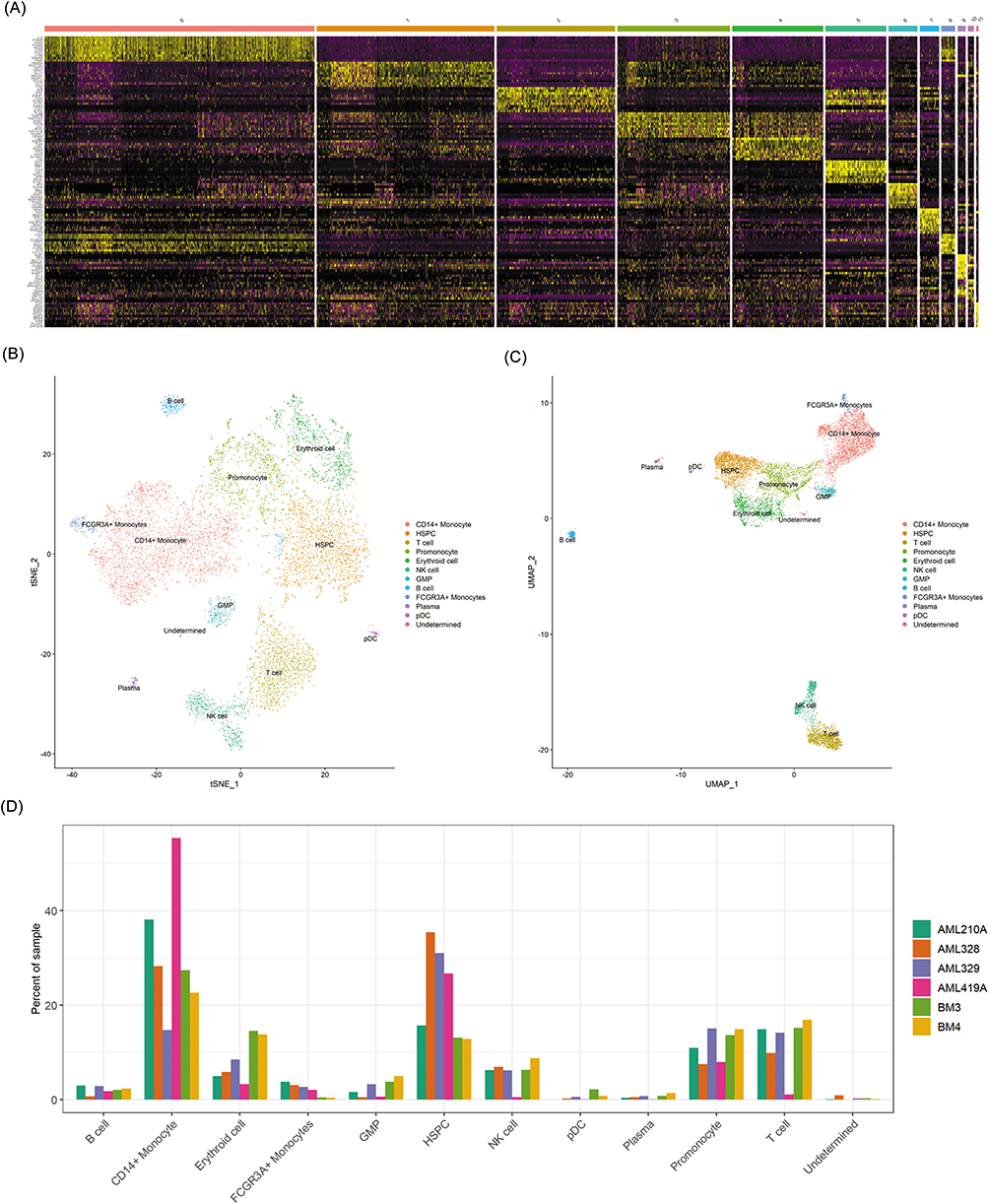 |
Figure 1 Single-cell profiling reveals immune remodeling in AML. (A) Heatmap of the top 10 cluster-specific marker genes, highlighting distinct transcriptional signatures (yellow boxes). (B) UMAP plot of integrated scRNA-seq data showing 12 major immune and hematopoietic cell types annotated by canonical markers. (C) t-SNE projection of the same dataset, illustrating the distribution of annotated immune subsets across samples. (D) Bar plot depicting the relative proportions of immune cell types, demonstrating expansion of myeloid-derived populations (e.g., monocytes, progenitor-like cells) and depletion of T cells in AML. |
Comparative analysis revealed marked shifts in immune composition between patients with AML and controls. AML samples were characterized by a pronounced expansion of myeloid-lineage cells, particularly monocytes and progenitor-like populations, accompanied by a notable reduction in T cell frequencies (Figure 1C and D). Collectively, these results demonstrate extensive immune remodeling in AML, with a strong myeloid bias and impaired T cell representation in the peripheral immune compartment.
Altered T Cell Composition and Transcriptional Profile in AML
To further dissect immune alterations, we assessed the cell-type composition at the individual sample level. Stacked bar plots illustrate the relative abundance of the 12 major immune cell subsets across donors (Figure 2A). Patients with AML display a pronounced expansion of monocytes and progenitor-like populations, accompanied by reduced T cell frequencies, highlighting the characteristic myeloid skewing and immune imbalance in AML. Within the T-cell compartment, differential gene expression analysis between AML patients and healthy controls revealed widespread transcriptional reprogramming. A heatmap of the top 50 differentially expressed genes (DEGs) demonstrated significant downregulation of several immune-regulatory genes in AML T cells, including TNFAIP3, ZFP36L2, TSC22D3, TXNIP, SRGN, SARAF, ZFP36, and GADD45 (Figure 2B). These genes are functionally linked to apoptosis, cytokine signaling, and immune regulation, suggesting impaired effector potential. Gene Ontology (GO) enrichment analysis indicated that DEGs were predominantly involved in T cell activation, cadherin binding, and cell–substrate junction organization (Figure 2C–E). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis further highlighted enrichment in pathways such as Human T-cell leukemia virus 1 infection, implicating disrupted antiviral and anti-tumor immune responses (Figure 2F). Together, these findings demonstrate that AML is characterized not only by quantitative depletion of T cells, but also by qualitative transcriptional dysfunction, underscoring the profound impairment of adaptive immunity.
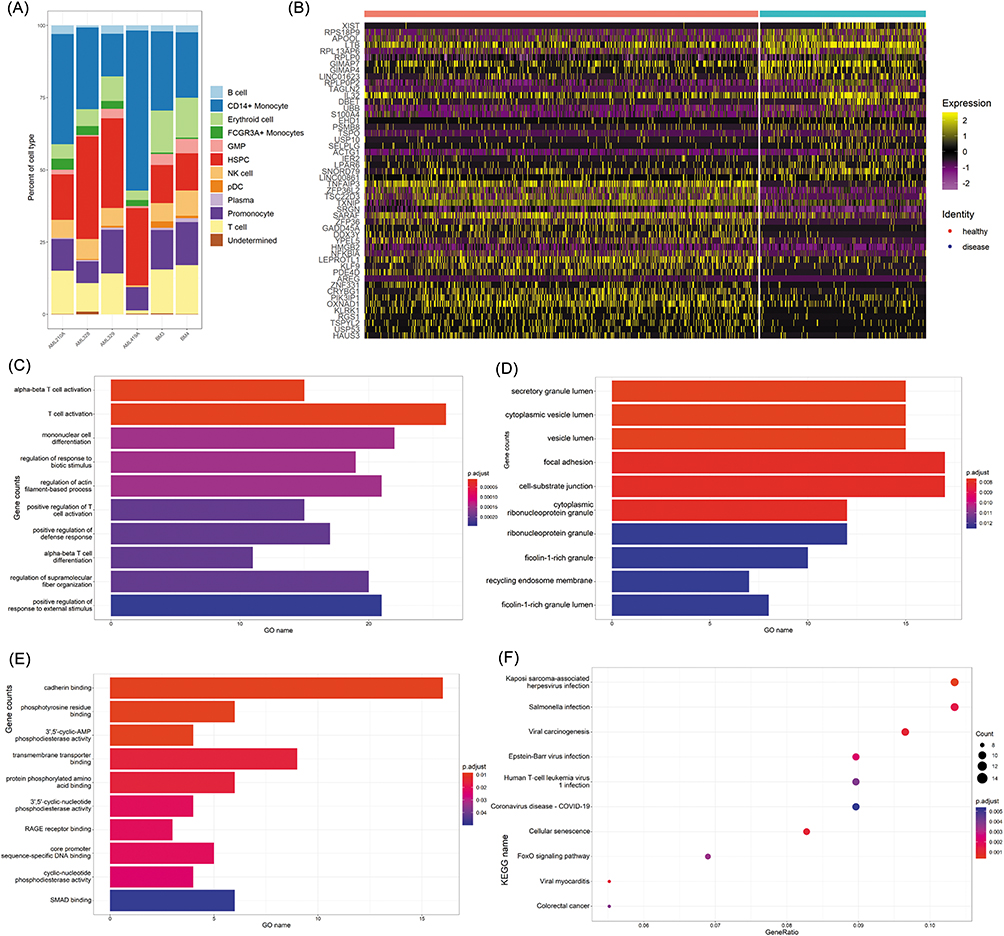 |
Figure 2 Transcriptional alterations and functional enrichment of AML T cells. (A) Stacked bar plots showing the distribution of 12 immune cell subsets across individual AML patients and healthy donors. (B) Heatmap of the top 50 differentially expressed genes (DEGs) in T cells, highlighting downregulation of immune-regulatory and stress-response genes such as TNFAIP3, ZFP36L2, TSC22D3, and TXNIP in AML. (C–E) Gene Ontology (GO) enrichment of DEGs, categorized by biological process (BP), cellular component (CC), and molecular function (MF), with significant terms related to T cell activation, cadherin binding, and immune regulation. (F) KEGG pathway enrichment demonstrating association of DEGs with immune dysfunction pathways, including Human T-cell leukemia virus 1 infection, suggesting disruption of antiviral and anti-tumor signaling in AML T cells. |
Co-Expression Network Analysis Identifies TNFAIP3 as a Central Immunoregulatory Hub
To identify the key drivers of T cell dysfunction in AML, we performed a co-expression network analysis of T cell–specific DEGs. This approach revealed distinct gene modules with coordinated expression patterns (Figure 3A and B). Among these modules, TNFAIP3 exhibited the highest connectivity (degree = 60), indicating its pivotal role in shaping transcriptional networks in T cells (Table 2). TNFAIP3, which encodes the ubiquitin-editing enzyme A20, inhibits NF-κB signaling via dual E3 ligase and deubiquitinase activity. Its dysregulation has been linked to impaired cytokine signaling, aberrant activation thresholds, and immune dysfunction in various malignancies. Functional enrichment analysis of TNFAIP3-associated modules highlighted GO terms related to T-cell activation, lymphocyte differentiation, and effector functions, whereas KEGG analysis showed enrichment of the FoxO signaling pathway (Figure 3C and D), which governs apoptosis, oxidative stress responses, and metabolic homeostasis. Experimental validation by qRT-PCR in peripheral T cells from 24 AML patients and 12 healthy donors confirmed the significant downregulation of TNFAIP3 in AML-derived T cells (P < 0.05) (Figure 3E), reinforcing its role as a central immunoregulatory node in AML.
 |
Table 2 Network Centrality Analysis Ranking Genes by Connectivity; TNFAIP3 Shows the Highest Degree (60), Designating It as the Top Hub Gene |
 |
Figure 3 TNFAIP3 acts as a central immunoregulatory hub in AML T cells. (A) Co-expression network of T cell–specific DEGs, displaying modular architecture with tightly clustered gene groups. (B) Core gene modules identified by topological overlap and hierarchical clustering, highlighting potential regulatory nodes. (C and D) GO and KEGG enrichment of the TNFAIP3-associated module, enriched in pathways related to T cell activation, lymphocyte differentiation, and FoxO signaling. (E) qRT-PCR validation of TNFAIP3 expression in peripheral T cells from 24 AML patients and 12 healthy donors, confirming significant downregulation in AML, consistent with scRNA-seq findings. ***P < 0.001. |
TNFAIP3 Modulates Immune Checkpoint Expression in CD4+ T Cells
To explore the functional impact of TNFAIP3 on AML-derived T cells, CD4+ and CD8+ subsets were isolated from patient PBMCs using MACS, achieving purities >92% and 94.6%, respectively (Figure 4A). Immune checkpoint profiling using RayBio® Human Immune Checkpoint Array 1 revealed that TNFAIP3 overexpression in CD4+ T cells led to a significant downregulation of PD-L1 (B7-H1) and TIM-3 compared to controls (Figure 4B and Table 3). Overall, six immune checkpoint molecules were suppressed, indicating broad immunomodulatory effects. Given TIM-3’s established role in T cell exhaustion, its decreased expression suggests that TNFAIP3 may partially restore effector function in CD4+ T cells. GO enrichment analysis of differentially expressed proteins highlighted terms related to mononuclear cell proliferation, lymphocyte activation, and protein deacetylation, indicating the involvement of TNFAIP3 in chromatin remodeling and immune activation (Figure 4C–E). Collectively, these findings indicated that TNFAIP3 reprograms CD4+ T cells by downregulating inhibitory checkpoints and enhancing immune responsiveness.
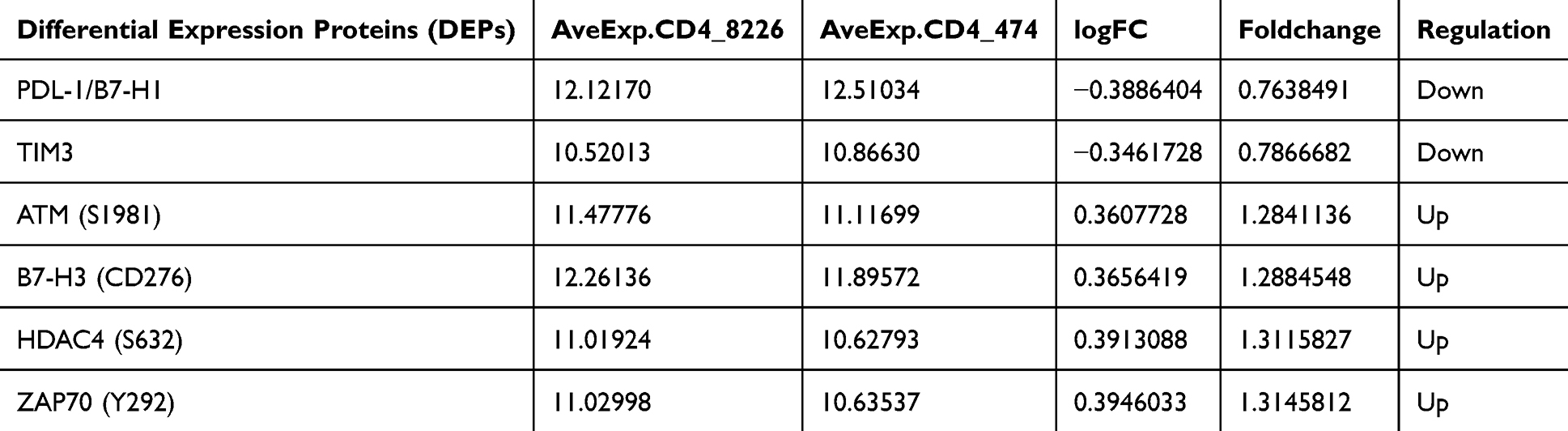 |
Table 3 Quantification of Six Checkpoint Proteins Significantly Downregulated Following TNFAIP3 Overexpression |
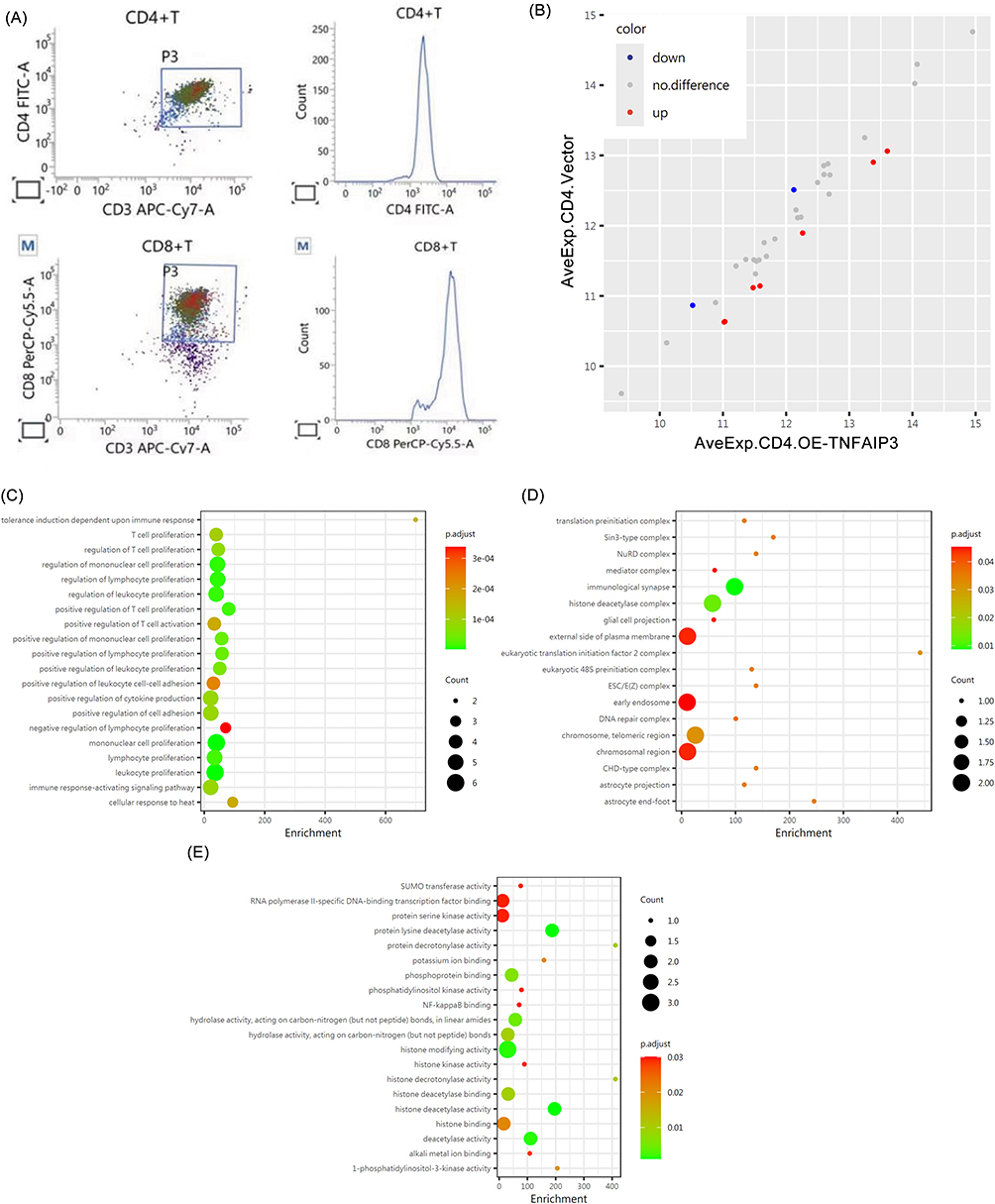 |
Figure 4 TNFAIP3 modulates immune checkpoint expression in CD4+ T cells. (A) Flow cytometry plots confirming high purity of MACS-sorted T cell subsets (>92% for CD4+, >94.6% for CD8+) used in functional assays. (B) Immune checkpoint protein profiling using RayBio® Human Immune Checkpoint Array 1, comparing TNFAIP3-overexpressing CD4+ T cells (OE-TNFAIP3) with vector controls, showing reduced PD-L1 and TIM-3 levels. (C–E) GO enrichment of differentially expressed checkpoint-associated proteins, highlighting TNFAIP3-mediated effects on mononuclear cell proliferation, lymphocyte activation, and chromatin remodeling. |
TNFAIP3 Modulates Immune Checkpoint Expression and Signaling in CD8+ T Cells
To assess the effect of TNFAIP3 on cytotoxic CD8+ T cells, we used the same immune checkpoint protein array. Twenty-one proteins were differentially expressed between the TNFAIP3-overexpressing and control cells, with notable changes in ICOS, eIF1α, CD200, B7-1 (CD80), HDAC4, and PD-L1 expression (Table 4, Figure 5A). Downregulation of immunosuppressive ligands (PD-L1 and CD200) coupled with upregulation of costimulatory molecules (ICOS and CD80) suggests enhanced activation potential and reduced exhaustion in TNFAIP3-overexpressing CD8+ T cells. GO and KEGG enrichment analyses supported these observations, highlighting the terms associated with T cell activation, receptor complex organization, and immune synapse formation (Figure 5B–D). KEGG pathways included cytokine–cytokine receptor interactions and T cell receptor signaling (Figure 5E), indicating that TNFAIP3 orchestrates the broad modulation of key signaling cascades essential for CD8+ T cell effector functions. Collectively, these results reinforce TNFAIP3’s role in promoting a more immunocompetent T cell phenotype in AML.
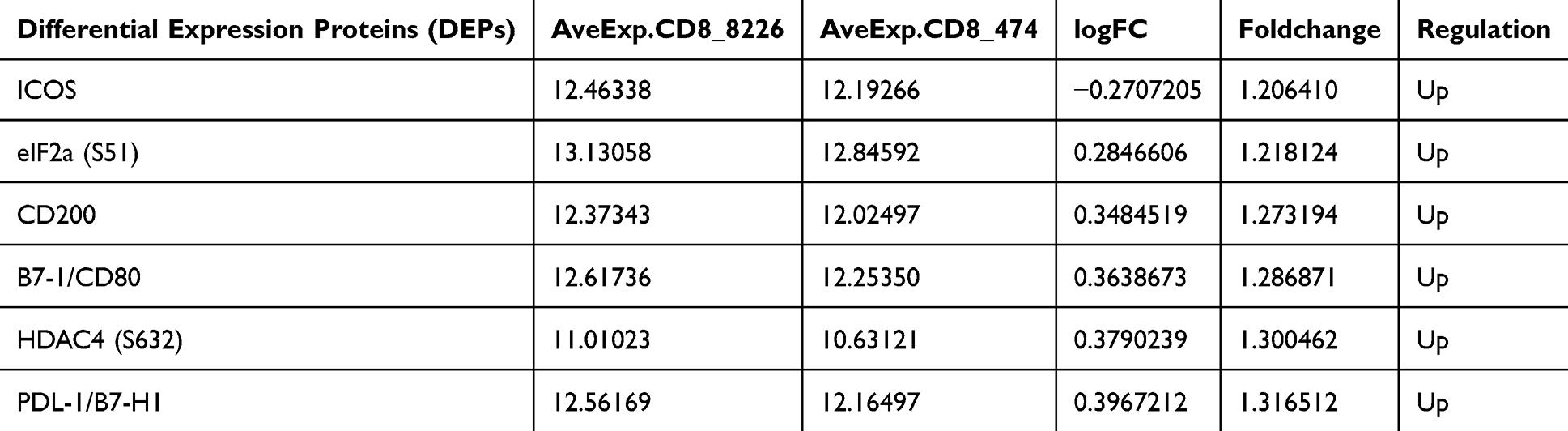 |
Table 4 Differentially Expressed Immune Checkpoint and Signaling Proteins in CD8+ T Cells with or Without TNFAIP3 Overexpression, Highlighting Significant Changes in ICOS, eIF1α, CD200, B7-1 (CD80), HDAC4, and PD-L1 |
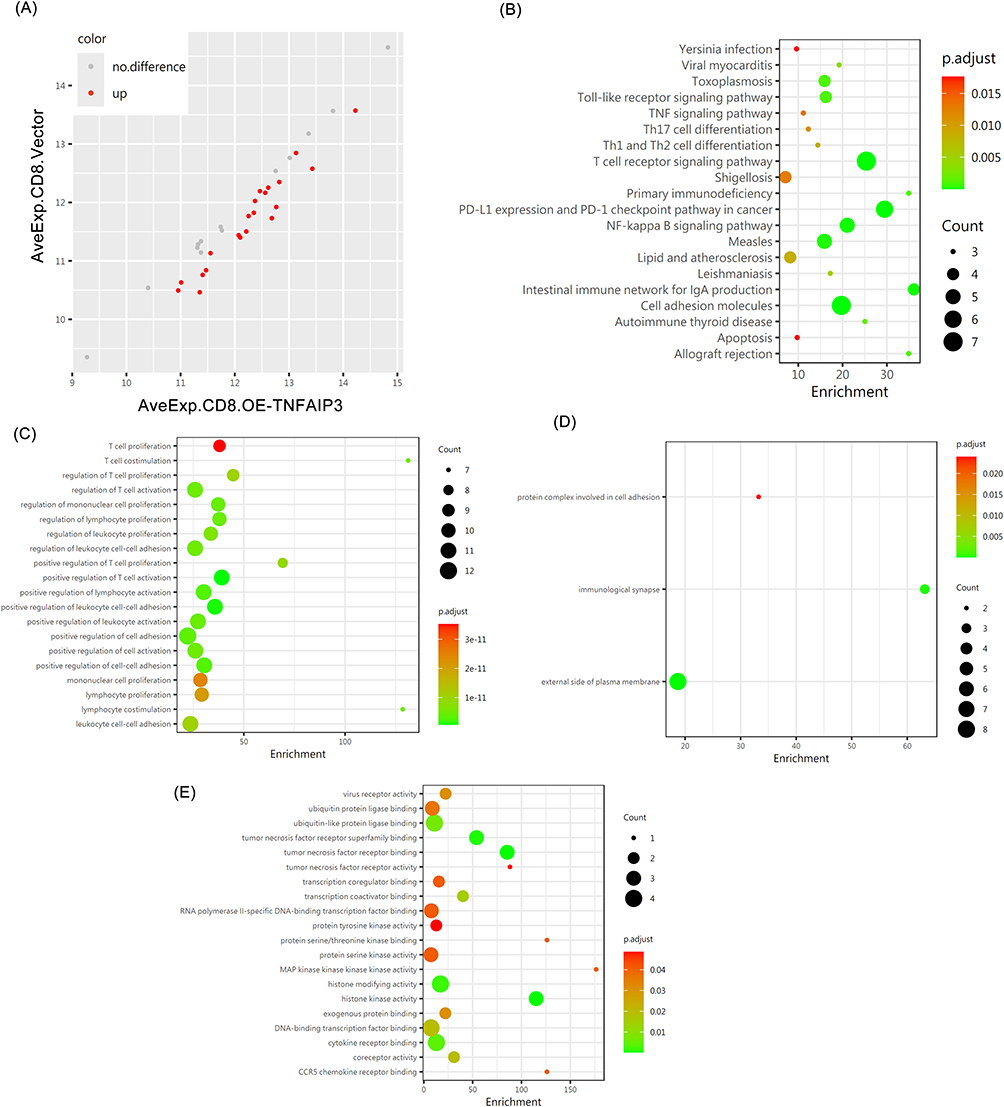 |
Figure 5 TNFAIP3 reprograms immune checkpoint expression and signaling in CD8+ T cells. (A) Dot plot summarizing fold changes and statistical significance, indicating a shift from an exhausted to an activated T cell phenotype. (B–D) GO enrichment of differentially expressed proteins, showing terms related to T cell activation, immunological synapse formation, and receptor complex organization. (E) KEGG pathway enrichment, revealing significant involvement of cytokine–cytokine receptor interactions and T cell receptor (TCR) signaling, underscoring TNFAIP3’s regulatory role in CD8+ T cell effector function. |
TNFAIP3 Reprograms T Cells by Regulating Apoptosis, Immune Checkpoints, and Cytokine Profiles
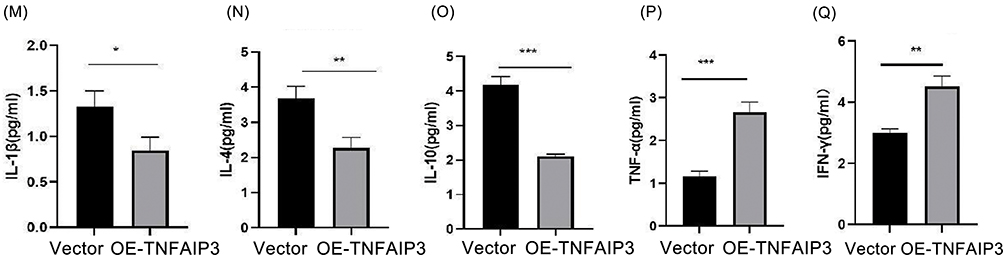
To evaluate the functional impact of TNFAIP3 overexpression, AML-derived CD4+ and CD8+ T cells were transduced with the TNFAIP3-encoding lentiviral vectors. Apoptosis assays revealed a significant reduction in apoptotic cells from 11.14% to 6.64%, indicating enhanced T-cell survival (Figure 6A–C). Flow cytometry showed decreased TIM-3 expression in both CD4+ and CD8+ T cells (Figure 6D–I), suggesting partial reversal of the exhausted phenotype. Western blot analysis confirmed that increased TNFAIP3 expression was accompanied by reduced phosphorylation of NF-κB p65 (p-p65), which was consistent with the inhibition of NF-κB signaling (Figure 6J). Multiplex cytokine profiling demonstrated a functional shift: anti-inflammatory cytokines IL-1β, IL-4, and IL-10 were reduced, whereas pro-inflammatory cytokines TNF-α and IFN-γ were elevated in TNFAIP3-overexpressing T-cells (Figure 6K–Q). Together, these findings indicate that TNFAIP3 overexpression reprograms T cells toward a pro-inflammatory and anti-tumor phenotype by modulating apoptosis, immune checkpoints, and cytokine secretion, highlighting its potential to restore immune surveillance in the AML microenvironment. Figure 6 Continued. Figure 6 TNFAIP3 modulates apoptosis, immune exhaustion, NF-κB signaling, and cytokine secretion in T cells. (A) Representative flow cytometry plot of Annexin V/PI staining in control CD4+ and CD8+ T cells.(B) Representative flow cytometry plot of Annexin V/PI staining in TNFAIP3-overexpressing CD4+ and CD8+ T cells, showing reduced apoptotic cell populations compared with the control group. (C) Quantification of apoptotic cells based on Annexin V/PI staining, demonstrating a significant reduction in apoptosis from 22.5% to 14.6% upon TNFAIP3 overexpression. (D) Flow cytometry analysis of TIM-3 expression in CD4+ T cells under blank control conditions. (E) Flow cytometry analysis of TIM-3 expression in CD4+ T cells under vector control conditions. (F) Flow cytometry analysis of TIM-3 expression in CD4+ T cells with TNFAIP3 overexpression, showing decreased TIM-3-positive cells compared with controls. (G) Flow cytometry analysis of TIM-3 expression in CD8+ T cells under blank control conditions. (H) Flow cytometry analysis of TIM-3 expression in CD8+ T cells under vector control conditions. (I) Flow cytometry analysis of TIM-3 expression in CD8+ T cells with TNFAIP3 overexpression, showing reduced TIM-3 expression relative to control groups. Boxed regions indicate TIM-3-positive cells; red and grey boxes represent different T cell populations (e.g., CD4+ vs CD8+), but all denote TIM-3-positive cells. (J) Western blot analysis of A20 (TNFAIP3) and phosphorylated NF-κB p65 (p-p65) in T cell lysates. TNFAIP3 overexpression resulted in increased A20 protein levels and decreased p-p65 levels, indicating inhibition of NF-κB signaling. (K) Cytokine levels in supernatants of control CD4+ T cells. (L) Cytokine levels in supernatants of TNFAIP3-overexpressing CD4+ T cells. (M) Quantification of IL-1β levels, showing reduced expression upon TNFAIP3 overexpression. (N) Quantification of IL-4 levels, showing reduced expression upon TNFAIP3 overexpression. (O) Quantification of IL-10 levels, showing reduced expression upon TNFAIP3 overexpression. (P) Quantification of TNF-α levels, showing increased expression upon TNFAIP3 overexpression. (Q) Quantification of IFN-γ levels, showing increased expression upon TNFAIP3 overexpression.*P < 0.05, **P < 0.01, ***P < 0.001.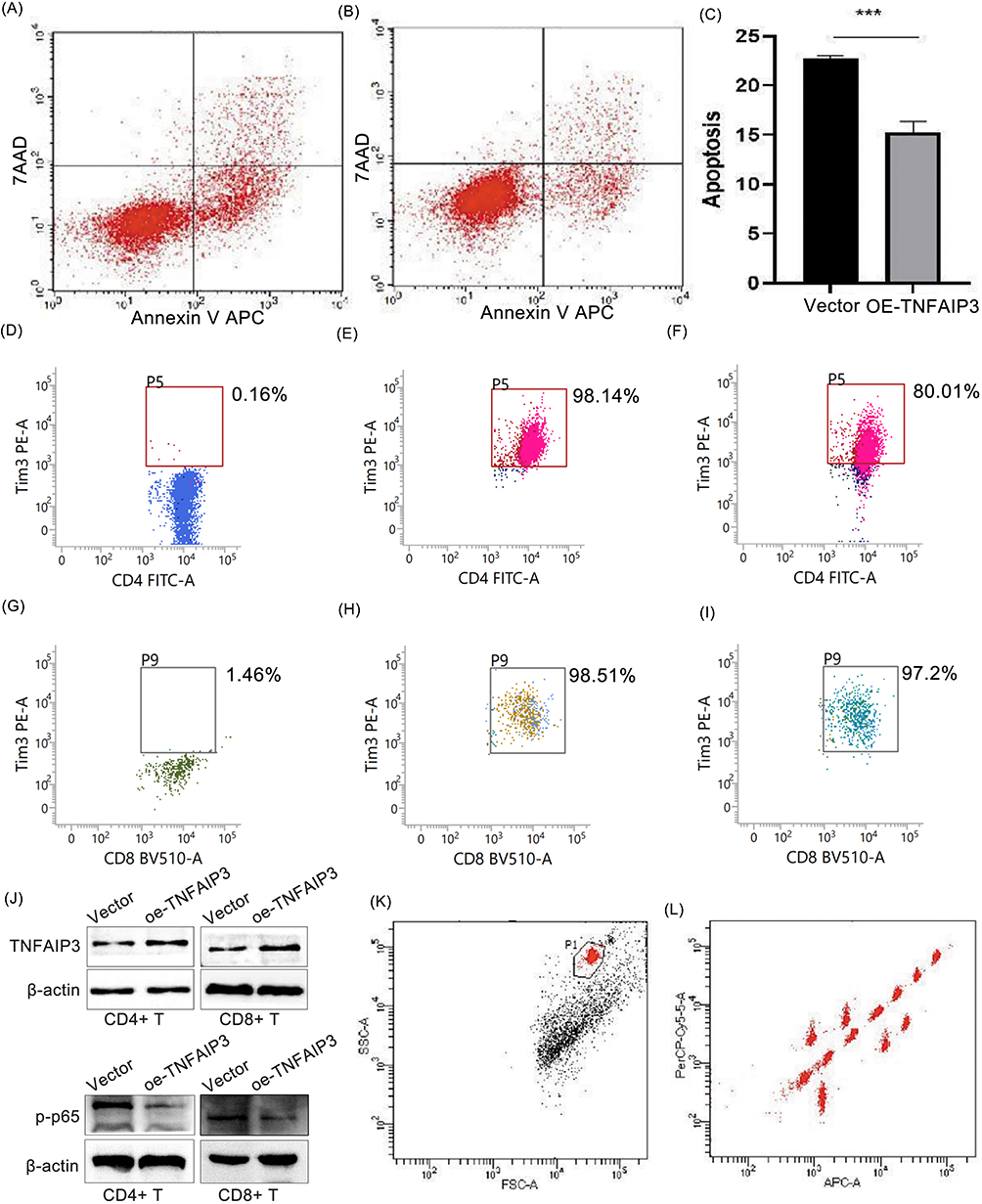
Discussion
In this study, we identified TNFAIP3 as a key modulator of T cell exhaustion within the immunosuppressive microenvironment of AML. By integrating single-cell transcriptomic profiling, co-expression network analysis, and functional validation, we demonstrated that TNFAIP3 is consistently downregulated in AML-derived T cells and that its restoration reprograms T cell function by attenuating immune checkpoint signaling, reshaping cytokine secretion, and suppressing NF-κB activation. Importantly, these findings extend beyond descriptive observations and provide mechanistic insight into how intrinsic regulatory pathways within T cells contribute to immune evasion in AML. T cell exhaustion in AML is a multifactorial process driven by chronic antigen exposure, persistent inflammatory signaling, and inhibitory receptor engagement. Previous studies have primarily focused on surface checkpoint molecules such as PD-L1 and TIM-3 as downstream mediators of dysfunction. Our results add an additional regulatory layer by identifying TNFAIP3 as an upstream checkpoint modulator that integrates intracellular signaling and transcriptional programs. The observed reduction in PD-L1 and TIM-3 expression following TNFAIP3 overexpression suggests that TNFAIP3 may influence exhaustion not only at the level of receptor expression but also by modulating the signaling thresholds that sustain the exhausted state. This highlights a potential shift from targeting individual checkpoints to regulating core intracellular hubs that orchestrate T cell fate decisions.
Mechanistically, TNFAIP3 functions as a ubiquitin-editing enzyme with dual E3 ligase and deubiquitinase activities, enabling precise control of NF-κB signaling dynamics. While transient NF-κB activation is required for effective T cell responses, sustained activation under chronic stimulation conditions can drive apoptosis, metabolic stress, and exhaustion. Our data demonstrate that TNFAIP3 overexpression reduces phosphorylated p65 levels, suggesting that TNFAIP3 restores signaling homeostasis rather than simply suppressing activation. This distinction is critical, as it supports a model in which TNFAIP3 acts as a rheostat that fine-tunes T cell activation intensity, preventing both insufficient activation and detrimental hyperactivation. In the AML microenvironment, where persistent antigen exposure and inflammatory cues coexist, loss of TNFAIP3 may exacerbate signaling imbalance and accelerate T cell dysfunction. Beyond checkpoint regulation, TNFAIP3 exerts broader immunomodulatory effects by reshaping cytokine networks.9 We observed a coordinated decrease in immunosuppressive cytokines (IL-1β, IL-4, IL-10) and an increase in effector cytokines (IFN-γ, TNF-α), indicating a functional reprogramming toward a pro-inflammatory, anti-tumor phenotype. This cytokine shift suggests that TNFAIP3 not only affects T cell intrinsic properties but may also remodel the surrounding immune microenvironment, potentially enhancing antigen presentation and amplifying immune responses. Consistently, pathway enrichment analyses revealed that TNFAIP3-associated networks are linked to T cell receptor signaling, cytokine–cytokine receptor interactions, and FoxO signaling, all of which are critical for maintaining T cell fitness, survival, and differentiation under stress conditions. Our co-expression network analysis further supports the central role of TNFAIP3 by identifying it as a hub gene with high connectivity to multiple exhaustion-related regulators, including ZFP36L2, TSC22D3, and GADD45. These genes are involved in transcriptional repression, stress response, and apoptosis, suggesting that TNFAIP3 participates in a coordinated regulatory module that governs T cell tolerance and dysfunction. Notably, the consistent effects observed in both CD4+ and CD8+ T cell subsets indicate that TNFAIP3-mediated regulation is conserved across functionally distinct T cell populations, reinforcing its importance as a global regulator of T cell homeostasis in AML.
This study has several limitations. First, while in vitro overexpression demonstrates TNFAIP3’s potential to reverse T cell exhaustion, in vivo validation in AML models or patient-derived xenografts is required to confirm therapeutic efficacy. Second, although NF-κB appears to be a key downstream target, additional pathways, including metabolic regulators and chromatin modifiers, may contribute to the TNFAIP3-mediated effects and warrant further investigation. Thirdly, whether TNFAIP3 restoration synergizes with immune checkpoint blockade or T cell-based therapies remains an important translational question.14 A key point raised by previous studies is that TNFAIP3 may exert context-dependent effects on T-cell activation. In adoptive or engineered T-cell systems, including CAR-T cell models, genetic deletion or CRISPR-mediated knockout of TNFAIP3 has been reported to enhance T-cell activation, proliferation, and antitumor cytotoxicity by relieving negative regulation of NF-κB signaling. These findings suggest that TNFAIP3 can function as a molecular “brake” that limits excessive immune activation under strong artificial stimulation.8,9 In contrast, our study demonstrates that in AML-associated T cells, which are exposed to chronic antigen stimulation and an immunosuppressive tumor microenvironment, TNFAIP3 is downregulated, and its restoration reduces exhaustion-associated phenotypes, decreases inhibitory receptor expression, and improves cytokine production. These seemingly opposite observations likely reflect distinct immunological contexts. In engineered T cells, TNFAIP3 loss enhances acute activation, whereas in tumor-associated T cells, TNFAIP3 restoration may help re-establish signaling homeostasis and prevent dysfunctional exhaustion states. Therefore, TNFAIP3 should be considered a context-dependent regulator that fine-tunes T-cell activation thresholds rather than acting as a unidirectional inhibitor or activator of antitumor immunity. Although our study demonstrates that TNFAIP3 restoration enhances T-cell survival and effector function in vitro, several limitations should be acknowledged. First, all functional experiments were performed in vitro using primary AML-derived T cells, which cannot fully recapitulate the complexity of the in vivo leukemic microenvironment, including stromal interactions, cytokine gradients, and immune cell crosstalk. Second, in vivo validation using AML mouse models or patient-derived xenograft systems was not performed, which limits direct assessment of therapeutic efficacy. From a translational perspective, future strategies to modulate TNFAIP3 in vivo may include T-cell–targeted gene delivery systems, CRISPR-based epigenetic regulation, or pharmacological modulation of upstream NF-κB signaling pathways. In addition, combining TNFAIP3 modulation with immune checkpoint blockade or adoptive T-cell therapies may provide synergistic antileukemic effects and warrants further investigation. Another limitation of this study is the lack of stratified clinical analysis of AML patients, including correlations between TNFAIP3 expression and clinical outcomes such as overall survival, treatment response, or immunotherapy efficacy. This limitation is mainly due to the absence of comprehensive clinical annotation in the publicly available scRNA-seq dataset used in this study. Therefore, our findings are primarily mechanistic and require further validation in well-annotated clinical cohorts.
In summary, our study identifies TNFAIP3 as a central regulator of T cell exhaustion in AML and provides a mechanistic framework linking intracellular signaling dysregulation to impaired anti-leukemic immunity. By restoring TNFAIP3 expression, T cells can be reprogrammed toward a more functional and less exhausted state. Although further in vivo and clinical studies are required, these findings suggest that targeting TNFAIP3 or its downstream pathways may represent a promising strategy for modulating T cell immunity and improving therapeutic outcomes in AML.
Conclusion
In this study, we identified TNFAIP3 as an important regulator associated with T cell dysfunction in AML. Through integrated single-cell transcriptomic analysis and in vitro functional validation, we found that TNFAIP3 is downregulated in AML-derived T cells, and its restoration partially alleviates exhaustion-related features, including altered checkpoint expression, cytokine imbalance, and NF-κB activation. These findings provide insight into the mechanisms underlying T cell dysfunction in AML and suggest that TNFAIP3 may play a role in modulating anti-leukemic immunity. Further studies, particularly in vivo investigations, are required to validate its functional significance and therapeutic potential.
Data Sharing Statement
All data supporting the findings of this study are available from the corresponding author, Liansheng Zhang ([email protected]), upon reasonable request.
Ethics Approval and Consent to Participate
All procedures involving human participants were approved by the Ethics Committee of Lanzhou University Second Hospital, Lanzhou, China (Approval No. 2026A-052). Written informed consent was obtained from all participants prior to enrollment. This study was conducted in accordance with the Declaration of Helsinki.
Acknowledgments
The authors thank all participants and colleagues who contributed to this study.
Author Contributions
Liansheng Zhang and Lijuan Licontributed equally as corresponding authors.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of Gansu Province (21JR11RA104), National Natural Science Foundation of China (82360029), Gansu Province Science and Technology Plan Project (21JR7RA435), Cuiying Technology Innovation Project of Lanzhou University Second Hospital (2020QN-13), and the Lanzhou Science and Technology Development Plan Project (2020-ZD-99).
Disclosure
The authors declare no competing interests.
References
1. Khalifeh M, Hopewell E, Salman H. CAR-T cell therapy for treatment of acute myeloid leukemia, advances and outcomes. Mol Ther. 2025;33(6):2441–16. doi:10.1016/j.ymthe.2025.03.052
2. Cortes J. Quizartinib: a potent and selective FLT3 inhibitor for the treatment of patients with FLT3-ITD-positive AML. J Hematol Oncol. 2024;17(1):111. doi:10.1186/s13045-024-01617-7
3. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20(3):173–185. doi:10.1038/s41577-019-0224-6
4. Yang X, Ma L, Zhang X, et al. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp Hematol Oncol. 2022;11(1):11. doi:10.1186/s40164-022-00263-4
5. Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol. 2017;10(1):103. doi:10.1186/s13045-017-0474-3
6. Cheon H, Xing JC, Moosic KB, et al. Genomic landscape of TCRalphabeta and TCRgammadelta T-large granular lymphocyte leukemia. Blood. 2022;139(20):3058–3072. doi:10.1182/blood.2021013164
7. Culver-Cochran AE, Hassan A, Hueneman K, et al. Chemotherapy resistance in acute myeloid leukemia is mediated by A20 suppression of spontaneous necroptosis. Nat Commun. 2024;15(1):9189. doi:10.1038/s41467-024-53629-z
8. Matsuzawa Y, Oshima S, Takahara M, et al. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy. 2015;11(7):1052–1062. doi:10.1080/15548627.2015.1055439
9. Song XT, Evel-Kabler K, Shen L, et al. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14(3):258–265. doi:10.1038/nm1721
10. Liu S, Yu Y, Liu Q, et al. Ubiquitin-editing enzyme A20 protects the inflammatory injury of human corneal epithelial cells against lipoteichoic acid. Cell Signal. 2025;134(111962):111962. doi:10.1016/j.cellsig.2025.111962
11. Jang JH, Kim H, Jung IY, et al. A20 inhibits LPS-induced inflammation by regulating TRAF6 polyubiquitination in rainbow trout. Int J Mol Sci. 2021;22(18):9801. doi:10.3390/ijms22189801
12. Sheibani M, Hosseinzadeh A, Fatemi I, et al. Melatonin and necroptosis: therapeutic aspects based on cellular mechanisms. Mol Biol Rep. 2025;52(1):606. doi:10.1007/s11033-025-10713-x
13. Garrido AN, Machhar R, Cruz-Correa OF, et al. Single-cell RNA sequencing of circulating immune cells supports inhibition of TNFAIP3 and NFKBIA translation as psoriatic arthritis biomarkers. Front Immunol. 2025;16(1483393). doi:10.3389/fimmu.2025.1483393
14. Guo W, Ma J, Guo S, et al. A20 regulates the therapeutic effect of anti-PD-1 immunotherapy in melanoma. J Immunother Cancer. 2020;8(2):e001866. doi:10.1136/jitc-2020-001866
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Increased Co-Expression of PD-L1 and CTLA-4 Predicts Poor Overall Survival in Patients with Acute Myeloid Leukemia Following Allogeneic Hematopoietic Stem Cell Transplantation
Chen C, Qiu K, Chen J, Wang S, Zhang Y, Wang C, Li Y
ImmunoTargets and Therapy 2025, 14:25-33
Published Date: 20 January 2025