Back to Journals » ImmunoTargets and Therapy » Volume 9
Time to Develop Therapeutic Antibodies Against Harmless Proteins Colluding with Sepsis Mediators?
Received 14 May 2020
Accepted for publication 22 July 2020
Published 5 October 2020 Volume 2020:9 Pages 157—166
DOI https://doi.org/10.2147/ITT.S262605
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Jianhua Li,1,* Guoqiang Bao,2,* Haichao Wang1,3
1The Feinstein Institutes for Medical Research, Northwell Health, Manhasset, NY 11030, USA; 2Department of General Surgery, Tangdu Hospital, Xi’an, Shaanxi 710032, People’s Republic of China; 3Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, Hempstead, NY 11549, USA
*These authors contributed equally to this work
Correspondence: Haichao Wang Tel +1 516 562-2823
Email [email protected]
Abstract: Sepsis refers to a systemic inflammatory response syndrome resulting from microbial infections, and is partly attributable to dysregulated inflammation and associated immunosuppression. A ubiquitous nuclear protein, HMGB1, is secreted by activated leukocytes to orchestrate inflammatory responses during early stages of sepsis. When it is released by injured somatic cells at overwhelmingly higher quantities, HMGB1 may induce macrophage pyroptosis and immunosuppression, thereby impairing the host’s ability to eradicate microbial infections. A number of endogenous proteins have been shown to bind HMGB1 to modulate its extracellular functions. Here, we discuss an emerging possibility to develop therapeutic antibodies against harmless proteins that collude with pathogenic mediators for the clinical management of human sepsis and other inflammatory diseases.
Keywords: antibody, HMGB1, immunosuppression, inflammation, innate immune cells, pyroptosis, sepsis
Introduction
Cohabitating with microbes, mammals employ epithelial barriers as the first layer of defense to limit the access of many pathogens. If they are breached, the host mounts an immediate immune response termed “inflammation” (“set on fire” in Greek) to eliminate these invading pathogens.1 For instance, circulating monocytes are ceaselessly patrolling the body to search for invading pathogens, and immediately infiltrate into the infected tissues upon detecting microbial products.2 Once reaching extravascular tissues, these monocytes are terminally differentiated into tissue-specific resident macrophages, which ingest and eradicate pathogens together with neutrophils and other phagocytes.3
Meanwhile, macrophages/monocytes are also equipped with Pattern Recognition Receptors [PRR, such as the Toll-like Receptor 2 (TLR2), TLR3, TLR4 and TLR9] that can bind distinct Pathogen-Associated Molecular Patterns molecules (PAMPs, such as bacterial peptidoglycan, double-stranded RNA, endotoxin and CpG-DNA).4,5 The engagement of PAMPs with respective PRRs triggers the immediate release of tumor necrosis factor (TNF),6 interleukin (IL)-1,7 and interferon (IFN)-α,8 which collectively facilitate pathogen elimination. If unsuccessful, invading pathogens can leak into the bloodstream to trigger a systemic inflammatory response and life-threatening organ dysfunction termed “sepsis”.9 The pathogenesis of sepsis is complex but attributable to dysregulated inflammatory responses and immunosuppression. 10−12 For instance, neutralizing antibodies against TNF, the first cytokine elaborated in the inflammatory cascade, were protective in animal models of endotoxemic/bacteremic shock.6 However, the early release of TNF makes it difficult to target in clinical settings,13 prompting the search for other late mediators with wider therapeutic windows.
High Mobility Group Box 1 (HMGB1)
High mobility group 1 (HMG-1) was initially identified as a 30-kDa protein with a high mobility on electrophoresis gels,14 and recently renamed as the high mobility group box-1 (HMGB1).15 It contains a continuous stretch of negatively charged residues in the C-terminus, and two internal repeats of positively charged domains (“HMG boxes” known as “A box” and “B box”) in the N-terminus (Figure 1).16 These HMG boxes enable HMGB1 to bind chromosomal DNA to fulfill its nuclear functions in stabilizing nucleosomal structure and stability, and facilitating the binding of transcription factors to their cognate DNA sequences during gene expression.16,17 Conditional knockout of HMGB1 expression renders animals more susceptible to both infectious18 and injurious insults,19,20 supporting a beneficial role of intracellular HMGB1 in health.
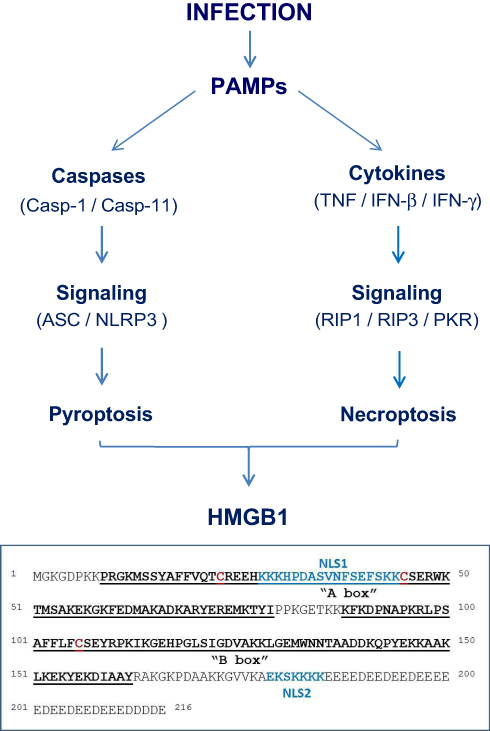 |
Figure 1 Pathogen-Associated Molecular Pattern molecules (PAMPs) trigger HMGB1 release through inducing pyroptosis or necroptosis. |
Secretion by Activated Macrophages/Monocytes
Two decades ago, we initiated an effort to search for late mediators that could contribute to the pathogenesis of lethal sepsis. Specifically, we stimulated macrophage cultures with an early cytokine (eg, TNF) and screened the cell-conditioned medium for proteins that were released relatively late. This effort led to the identification of 30-kDa protein with an N-terminal amino acid sequence identical to HMG-1 (HMGB1).15,21 Subsequently, we and others demonstrated that many exogenous PAMPs (eg, ds-RNA, CpG-DNA and endotoxins)21,22 and endogenous cytokines [eg, interferon (IFN)-γ, IFN-β, serum amyloid A (SAA), and Cold-inducible RNA-binding protein (CIRP)]23–26 similarly induced HMGB1 translocation to cytoplasmic vesicles.23,27–30 Consequently, these activated macrophages/monocytes secrete cytoplasmic HMGB1 vesicles via non-classical endoplasmic reticulum-Golgi exocytotic pathways.21,23,27–30
Release by Injured Somatic Cells
In addition, HMGB1 can be passively released by somatic cells undergoing cytoplasmic membrane destruction due to accidental (mechanical or chemical) events or regulated processes governed by specific caspases or kinases. For instance, many PAMPs induce a form of programmed necrosis, pyroptosis (Figure 1),31 that is characterized by the oligomerization of the apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) and its integration with pro-caspase-1 and a NOD-Like Receptor (eg, NLRP3) to form a large inflammasome complex (pyroptosome) that eventually disrupts cytoplasmic membranes.32,33 Similarly, many proinflammatory cytokines (eg, TNF and IFN-γ) induce another form of programmed necrosis (necroptosis) via activating protein kinase receptor-interacting protein 3 (RIP3) and the interferon-induced double-stranded RNA-activated protein kinase R (PKR) (Figure 1).34–36 Collectively, pyroptosis33,37 and necroptosis38 allow passive HMGB1 release following ischemia/reperfusion,39,40 non-penetrating trauma,41,42 chemical toxemia,43 or radiation,44 leading to massive HMGB1 release during lethal infections and injuries.
Extracellular HMGB1 and Innate Immunity
Extracellular HMGB1 binds many cell surface receptors such as the TLR445–47 and the receptor for advanced glycation end products (RAGE) (Figure 2).48–51 Due to its dramatically (4–30-fold) different affinities to TLR4/MD2 (KD = 22.0 nM)47 and RAGE (KD = 97.7–710 nM),48,49 HMGB1 may first bind TLR4/MD2 when it is actively secreted by innate immune cells at relatively lower amount.52 However, when it is passively released by somatic cells at relatively higher levels, HMGB1 may also bind RAGE48,50,53 and other low-affinity receptors. Like TLR4,4,45–47 RAGE also recognizes many other ligands including the advanced glycation end-products (AGEs), complement component (eg, C1q, KD = 5.6 µM) or endotoxins (KD = 2–35 nM).51 Thus, HMGB1 may orchestrate divergent inflammatory responses through activating different PRRs interacting with a wide array of inflammatory ligands.
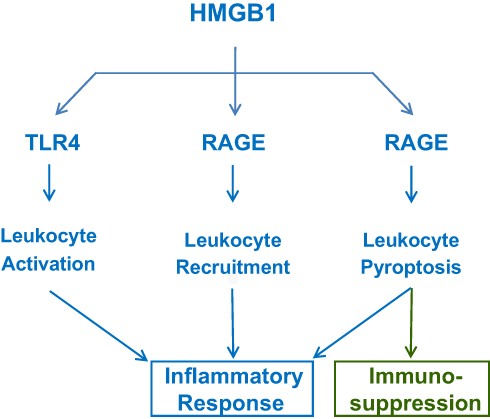 |
Figure 2 Extracellular HMGB1 induces divergent inflammatory responses through different receptors. Note that RAGE is involved in HMGB1 endocytosis and induction of leukocyte pyroptosis and possible immunosuppression due to immune cell depletion. |
Extracellular HMGB1 Amplifies Inflammation
First, as a highly charged molecule, HMGB1 binds and facilitates the cellular uptake of negatively charged PAMPS (eg, CpG-DNA and LPS) via RAGE-receptor-mediated endocytosis.53 Upon reaching acidic endosomal and lysosomal compartments near HMGB1’s isoelectric pH, HMGB1 becomes neutrally charged and sets free its cargos,53 thereby facilitating their recognition by respective receptors such as TLR954 or caspase-11 53. Second, the engagement of RAGE with HMGB1 also induces chemotaxis55 and the migration of monocytes, dendritic cells56,57 and neutrophils,58 thereby facilitating the recruitment of innate immune cells to the sites of infection and injury (Figure 2).55 Third, HMGB1 can even directly activate macrophages59 and endothelial cells60 to produce various cytokines and chemokines,45,46,60–63 thereby sustaining a dysregulated inflammatory responses during infections (Figure 2).64 Thus, extracellular HMGB1 functions as an alarmin signal to alert, recruit and activate immune cells, thereby amplifying inflammatory responses.
Following traumatic injury, HMGB1 is detected in the circulation within a few hours,41,42,65 and its systemic levels correlated with post-traumatic inflammatory responses41,66 and worsening clinical scores.67 Accordingly, HMGB1-neutralizing antibodies have been proven protective in animal models of ischemia/reperfusion,39,68,69 trauma,70,71 chemical toxemia,72–74 atherosclerosis,75 gastric ulcer,76 and hyperoxia,77 supporting a pathogenic role of HMGB1 in injury-elicited inflammatory diseases.
Extracellular HMGB1 Induces Immunosuppression
It is well known that antecedent traumatic injury often dampens subsequent innate immunity against secondary infections, suggesting a possible pathogenic role of DAMP in immune tolerance or immunosuppression. As aforementioned, when HMGB1 is passively released by damaged tissues at overwhelmingly high levels, it can bind RAGE48–50,78 to induce TLR4 internalization and desensitization to subsequent stimulation with inflammatory ligands (eg, endotoxin). Furthermore, at relatively higher doses (eg, 10 µg/mL), HMGB1 could also binds RAGE to trigger macrophage pyroptosis,53,79 apoptosis80 and necrosis,80 resulting in possible depletion of innate immune cells and immunosuppression (Figure 2). Therefore, excessive HMGB1 accumulation may induce immune tolerance81,82 as well as immunosuppression83 that compromises the host’s ability to eradicate microbial infections during lethal injuries and infections.84,85
HMGB1 as a Late Mediator of Lethal Sepsis
In preclinical settings, sepsis is routinely induced by the infusion of bacterial endotoxins (endotoxemia) or the disruption of host epithelial barrier to induce microbial translocation by a surgical procedure termed cecal ligation and puncture (CLP).86 In murine models of endotoxemia and CLP-sepsis, HMGB1 is first detected in the circulation 8 hours after the disease onset, and subsequently increased to plateau levels from 16 to 32 hours,21,87 distinguishing HMGB1 from TNF and other early proinflammatory cytokines.13 The pathogenesis of HMGB1 in endotoxemia was inferred from studies using HMGB1-neutralizing antibodies, which conferred a dose-dependent protection against endotoxin-induced tissue injury and lethality.21,88 Intriguingly, anti-HMGB1 antibodies did not affect endotoxin-induced cytokine production in the lung inflammation model, suggesting that HMGB1 may contribute to lung injury through additional inflammation-independent mechanisms.88 Indeed, it has been shown that excessive HMGB1 accumulation in the bronchoalveolar space adversely compromised bacteria-killing capacities of alveolar macrophages,77 which were similarly reversed by HMGB1-neutralizing antibodies.89
In a more clinically relevant animal model of sepsis (induced by CLP), delayed administration of HMGB1-specific neutralizing antibodies, beginning 24 h after CLP, dose-dependently rescued rodents from lethal sepsis.87,90,91 Moreover, targeted inhibition of HMGB1 expression in macrophages and dendritic cells reduced systemic HMGB1 accumulation, and similarly rescued mice from sepsis.92 Taken together, these experimental data establish extracellular HMGB1 as a critical late mediator of experimental sepsis, which can be therapeutically targeted with wider therapeutic windows than other early cytokines.1,13,93,94
Endogenous HMGB1-Binding Proteins
To prevent potentially harmful inflammatory responses, mammals have also developed many strategies to counter-regulate HMGB1-mediated cytokine productions. For instance, an endothelial anticoagulant cofactor, thrombomodulin (TM), could bind HMGB1 to prevent its interaction with macrophage cell surface receptors,95 thereby preventing HMGB1-induced inflammatory response.96,97 Similarly, a liver-derived acute-phase protein, haptoglobin (Hp), could capture HMGB1 to trigger CD163-dependent endocytosis of HMGB1/Hp complexes, and instead induced the production of anti-inflammatory enzymes (heme oxygenase-1) and cytokines (eg, IL-10).98 Moreover, a complement factor C1q also interacted with HMGB1 (KD = 200 nM) and formed a tetramolecular complex with RAGE and LAIR-1, resulting in the production of anti-inflammatory cytokines (eg, IL-10) and pro-resolution lipid mediators.99,100 Thus, in a sharp contrast to exogenous PAMPs (eg, CpG-DNA and LPS), many endogenous proteins can bind HMGB1 to tilt the balance towards anti-inflammatory responses via distinct signaling pathways.95,98–100
Development of Antibodies Against an Endogenous Protein (Tetranectin, TN) Colluding with HMGB1
Tetranectin (TN) was initially characterized as an oligomeric plasminogen-binding protein101 exhibiting >75% amino acid sequence identity between humans and rodents.102 It is expressed abundantly in the lung103 but also present at relatively high levels (10–12 µg/mL) in the bloodstream of healthy humans 104. In rodents, enhanced expression or genetic depletion of TN led to abnormal osteogenesis 105, excessive curvature of the thoracic spine-spinal deformity,106 deficient motor function (such as limb rigidity),107 or impaired wound healing,108,109 suggesting a generally beneficial role of TN in health.
TN Depletion in Sepsis
While searching for endogenous proteins modulating HMGB1 release, we noticed that the blood level of a 20-kDa protein was almost completely depleted in a patient who died of sepsis. This 20-kDa protein was identified as human TN by mass spectrometry and immunoblotting assays.110 Further analysis of a cohort of 44–45 age-matched healthy controls and critically ill patients revealed a 60–70% reduction of plasma TN levels in patients with sepsis or septic shock. In accordance with these clinical findings, circulating TN levels were similarly decreased in experimental sepsis, with a >70% reduction at 24 h after disease onset— a time point when some septic animals started to lose survival.110
Surprising Discovery of TN-Specific Protective Antibodies
To understand the role of TN in sepsis, we would normally perform three sets of experiments to find out what would happen if we: 1) genetically knocked out its expression; 2) pharmacologically supplemented septic animals with recombinant TN protein; and 3) treated septic animals with TN-specific antibodies? First, genetic disruption of TN expression rendered animals more susceptible to lethal sepsis with exacerbated lung inflammation and injury, suggesting a protective role of TN in sepsis.110 Conversely, supplementation of sub-physiological dose of TN at 2 and 24 h after the onset of lethal sepsis conferred a dose-dependent protection, further confirming its protective role in disease. Surprisingly, when we treated septic animals with polyclonal antibodies (pAbs) generated in four rabbits, we found that total IgGs purified from two rabbits reproducibly reduced septic lethality even when the first dose was given 22 h after CLP.110
Screening a library of peptides spanning the entire sequence of human TN revealed that these two protective pAbs uniquely recognized a peptide (P5) harboring an epitope sequence (NDALYEYLRQ, “P5-5”) exhibiting 60–70% identity between humans and rodents, but a 100% identity between humans and many other mammalian species (ranging from pigs to monkeys). We thus strategically immunized mice with human TN antigen and generated several hybridoma clones producing P5-5-reactive monoclonal antibodies (mAbs). When administered to septic mice 24 h after CLP, three P5-5-reacting mAbs similarly and significantly rescued mice from lethal sepsis by attenuating sepsis-induced TN depletion, tissue injury, as well as bacteremia.110 It suggests a possibility that TN domain-specific mAbs conferred protection against lethal sepsis partly by facilitating pathogen elimination.
TN Interacts with HMGB1 to Facilitate HMGB1 Endocytosis and Macrophage Pyroptosis
Intriguingly, highly purified TN protein selectively inhibited the LPS- and SAA-induced HMGB1 release without affecting the parallel release of other cytokines and chemokines. This selective inhibition of HMGB1 release was attributable to TN’s capacity in capturing HMGB1 and facilitating the endocytosis of TN/HMGB1 complexes by macrophage cultures (Figure 3).110 Consistent with previous findings that HMGB1 endocytosis triggered macrophage pyroptosis,53,79 we found that TN enhanced HMGB1-induced translocation of nuclear ASC to cytoplasmic regions, where ASC either aggregated into minute puncta that appeared to be secreted through microvesicle shedding, or aggregated into a larger focus or speck (pyroptosome) that would trigger pyroptosis.110 Consequently, TN significantly enhanced HMGB1-induced uptake of trypan blue dye and parallel release of LDH and ASC, a marker for macrophage pyroptosis.111 Taken together, these findings suggest that TN could capture HMGB1 released into the circulation during sepsis, thereby promoting HMGB1 endocytosis and macrophage pyroptosis, leading to possible immunosuppression that may compromise effective pathogen elimination (Figure 3).
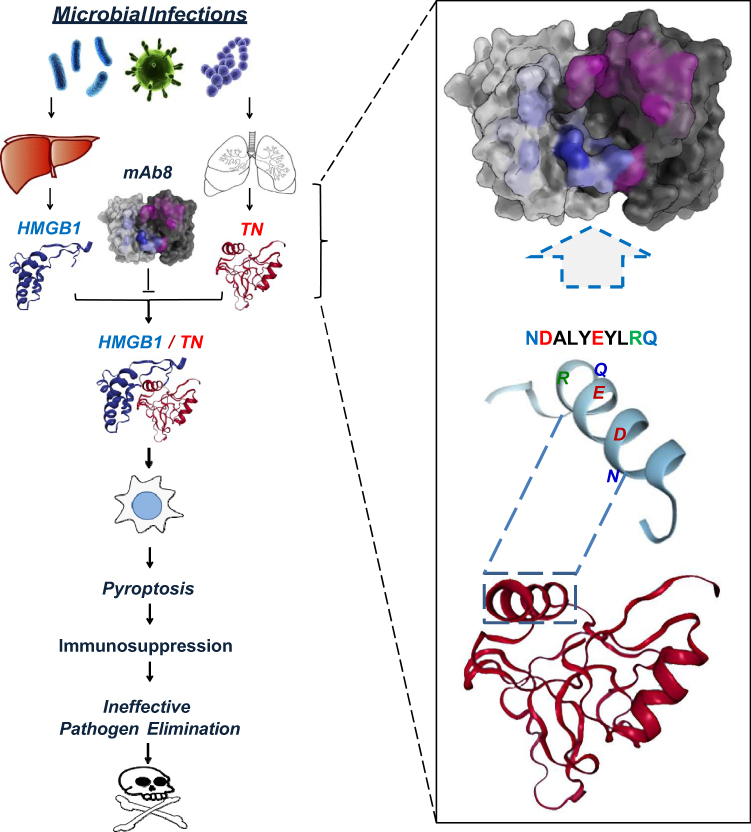 |
Figure 3 Proposed model for tetranectin domain-specific monoclonal antibody (mAb)-mediated protection against lethal sepsis. Severe microbial infections cause systemic accumulation of HMGB1 and concurrent depletion of TN, partly because circulating HMGB1 binds to TN to facilitate the endocytosis and degradation of TN/HMGB1 complexes by innate immune cells. The endocytosis of TN/HMGB1 complex also adversely triggers macrophage pyroptosis and immunosuppression that may compromise effective pathogen elimination and animal survival. Some TN-reactive mAbs, such as mAb8, could bind to specific epitope sequence (NDALYEYLRQ) of TN to interrupt its interaction with HMGB1, thereby impairing HMGB1 endocytosis and macrophage pyroptosis, and consequently reversing the sepsis-induced immunosuppression and animal lethality. |
TN-Specific mAbs Prevent Harmful TN/HMGB1 Interaction and Macrophage Pyroptosis
To understand the protective mechanism of TN domain-specific mAbs, we tested their possibility in disrupting TN/HMGB1 interaction using the Surface Plasmon Resonance technique. When the TN-conjugated sensor chip was pre-treated with mAb, the SPR response signal for subsequent HMGB1 application was reduced by >85%, which was paralleled by an almost 6-fold increase of KD, indicating that these TN domain-specific protective mAbs effectively interrupted TN/HMGB1 interaction (Figure 3).110 Furthermore, these protective mAb markedly prevented the reciprocal enhancement of cellular uptake of HMGB1 and TN, and prevented the TN/HMGB1-induced cytoplasmic ASC translocation or aggregation into large ASC specks in macrophage cultures. It suggests a possibility that TN domain-specific mAbs may confer protection against lethal sepsis partly by preventing harmful TN/HMGB1 interaction that may adversely trigger macrophage pyroptosis and immunosuppression (Figure 3).
Conclusion
Sepsis remains a major clinical problem that accounts for approximately 20% of total deaths worldwide,112 and annually costs more than $62 billion in the US alone.113 Despite a robust increase in the understanding of the pathophysiology of sepsis, many antibody-based strategies targeting early pro-inflammatory cytokines (such as TNF or IL-1) failed in clinical settings. Currently, there is still no effective therapy114 other than adjunctive use of antibiotics, fluid resuscitation, and supportive care.113 By using a bedside to bench approach, we have identified additional unexpected biological targets with potential translational promise. The interaction of a harmless protein (TN) with a pathogenic mediator (HMGB1) released into the circulation during sepsis adversely promoted HMGB1 endocytosis and macrophage pyroptosis (Figure 3), which triggers both excessive inflammation and ineffective pathogen elimination in a feed-forward mechanism ultimately leading to host lethality.114 Although pyroptosis normally serves as a host defense mechanism against infection,115 it could also allow the excessive release of HMGB1 and other proinflammatory cytokines (eg, IL-1β) that adversely drive a life-threatening inflammatory response.114 Meanwhile, excessive pyroptosis also depletes the number of innate immune cells necessary for pathogen clearance, resulting in a possible immunosuppression (Figure 3).
An antibody that interferes with TN/HMGB1 interaction thus represents a rational therapeutic approach to prevent macrophage cell death and resultant sequelae. Our recent discovery of several TN-specific mAbs capable of recognizing a highly conserved epitope sequence (NDALYEYLRQ) in humans and many mammalian species (including baboon, bovine, cougar, elephant, monkey, pig and tiger) has suggested a promising therapy for sepsis in clinical settings. At present, it remains elusive whether these TN-specific mAbs also attenuate HMGB1-induced apoptosis or necrosis that are similarly associated with poor outcomes in preclinical settings. It is also not yet known whether TN-specific mAbs similarly affect TN interaction with other proteins (for example, plasminogen) that may affect sepsis-induced dysregulated coagulopathy. Finally, given the pathogenic role of HMGB1 in injury-elicited inflammatory diseases, the role of TN in other inflammatory diseases should also be explored.114
Thus, it would be exciting to translate these pre-clinical findings into clinical applications through the use of humanized TN-specific mAbs capable of preventing its undesired interaction with pathogenic mediators that could cause macrophage pyroptosis and immunosuppression during lethal infections or injuries.110 Moreover, the discovery of mAbs capable of disrupting TN/HMGB1 interaction and endocytosis and rescuing animals from lethal sepsis has suggested an exciting possibility to develop therapeutic antibodies against harmless proteins colluding with disease mediators.
Copyright Statement
All three figures are original, and not reproduced from previously published sources.
Acknowledgments
The authors’ laboratory was supported in part by the National Institutes of Health (NIH) grants R01GM063075 and R01AT005076.
Author Contributions
All authors have made a significant contribution to the work reported, including the conception and interpretation of relevant published literature. J.L. and G.B. generated the first draft, and H.W. made significant revisions and finalized the manuscript. All authors have agreed on the journal to which the article was submitted. All authors have reviewed and agreed on all versions of the article before submission. All authors have agreed to take responsibility and be accountable for the contents of this review article.
Disclosure
H.W. and J L are co-inventor of two provisional patent applications entitled “Use of tetranectin and peptide agonists to treat inflammatory diseases” and “Tetranectin-targeting monoclonal antibodies to fight against lethal sepsis and other pathologies”. The authors report no other conflicts of interest in this work.
References
1. Wang H, Zhu S, Zhou R, Li W, Sama AE. Therapeutic potential of HMGB1-targeting agents in sepsis. Expert Rev Mol Med. 2008;10:e32. doi:10.1017/S1462399408000884
2. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6(12):1182–1190. doi:10.1038/ni1275
3. Liles WC. Immunomodulatory approaches to augment phagocyte-mediated host defense for treatment of infectious diseases. Semin Respir Infect. 2001;16(1):11–17. doi:10.1053/srin.2001.22724
4. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi:10.1038/nri1391
5. Brightbill HD, Libraty DH, Krutzik SR, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285(5428):732–736. doi:10.1126/science.285.5428.732
6. Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. doi:10.1038/330662a0
7. Dinarello CA, Thompson RC. Blocking IL-1: interleukin 1 receptor antagonist in vivo and in vitro. Immunol Today. 1991;12(11):404–410. doi:10.1016/0167-5699(91)90142-G
8. Heinzel FP. The role of IFN-gamma in the pathology of experimental endotoxemia. J Immunol. 1990;145(9):2920–2924.
9. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–810. doi:10.1001/jama.2016.0287
10. Cohen J, Vincent JL, Adhikari NK, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15:581–614. doi:10.1016/S1473-3099(15)70112-X
11. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi:10.1056/NEJMra021333
12. Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–524. doi:10.1038/nm0503-517
13. Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164:1768–1773.
14. Johns EW. History, definitions and problems. In: Johns EW, editor. The HMG Chromosomal Proteins. London: Academic Press Inc. (London) Ltd; 1982:1–8.
15. Bustin M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem Sci. 2001;26(3):152–153. doi:10.1016/S0968-0004(00)01777-1
16. Kang R, Chen R, Zhang Q, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;10.
17. Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19(8):5237–5246. doi:10.1128/MCB.19.8.5237
18. Yanai H, Matsuda A, An J, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A. 2013;110(51):20699–20704. doi:10.1073/pnas.1320808110
19. Huang H, Nace GW, McDonald KA, et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology. 2014;59:1984–1997.
20. Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146(4):1097–1107. doi:10.1053/j.gastro.2013.12.015
21. Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi:10.1126/science.285.5425.248
22. Ivanov S, Dragoi AM, Wang X, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–1981.
23. Rendon-Mitchell B, Ochani M, Li J, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170:3890–3897. doi:10.4049/jimmunol.170.7.3890
24. Kim JH, Kim SJ, Lee IS, et al. Bacterial endotoxin induces the release of high mobility group box 1 via the IFN-beta signaling pathway. J Immunol. 2009;182:2458–2466. doi:10.4049/jimmunol.0801364
25. Qiang X, Yang WL, Wu R, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–1495. doi:10.1038/nm.3368
26. Li W, Zhu S, Li J, et al. Serum Amyloid A stimulates PKR expression and HMGB1 release possibly through TLR4/RAGE receptors. Mol Med. 2015;21(1):515–525. doi:10.2119/molmed.2015.00109
27. Lu B, Antoine DJ, Kwan K, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A. 2014;111(8):3068–3073. doi:10.1073/pnas.1316925111
28. Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177(11):7889–7897. doi:10.4049/jimmunol.177.11.7889
29. Gardella S, Andrei C, Ferrera D, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3(10):955–1001. doi:10.1093/embo-reports/kvf198
30. Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi:10.1093/emboj/cdg516
31. Muendlein HI, Jetton D, Connolly WM, et al. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science. 2020;367:1379–1384. doi:10.1126/science.aay3878
32. Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun. 2016;7:11929. doi:10.1038/ncomms11929.:11929
33. Lamkanfi M, Sarkar A, Vande WL, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185(7):4385–4392. doi:10.4049/jimmunol.1000803
34. Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
35. Thapa RJ, Nogusa S, Chen P, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110(33):E3109–E3118. doi:10.1073/pnas.1301218110
36. Gunther C, Martini E, Wittkopf N, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi:10.1038/nature10400
37. Nystrom S, Antoine DJ, Lundback P, et al. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013;32:86–99. doi:10.1038/emboj.2012.328
38. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi:10.1038/nature00858
39. Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–1143. doi:10.1084/jem.20042614
40. Andrassy M, Volz HC, Igwe JC, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi:10.1161/CIRCULATIONAHA.108.769331
41. Cohen MJ, Brohi K, Calfee CS, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care. 2009;13:R174. doi:10.1186/cc8152
42. Peltz ED, Moore EE, Eckels PC, et al. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock. 2009;32:17–22. doi:10.1097/SHK.0b013e3181997173
43. Seo YS, Kwon JH, Yaqoob U, et al. HMGB1 recruits hepatic stellate cells and liver endothelial cells to sites of ethanol induced parenchymal cell injury. Am J Physiol Gastrointest Liver Physiol. 2013;305(11):G838–848. doi:10.1152/ajpgi.00151.2013
44. Bald T, Quast T, Landsberg J, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507:109–113. doi:10.1038/nature13111
45. Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi:10.1097/01.shk.0000225404.51320.82
46. Park JS, Svetkauskaite D, He Q, et al. Involvement of TLR 2 and TLR 4 in cellular activation by high mobility group box 1 protein (HMGB1). J Biol Chem. 2004;279:7370–7377. doi:10.1074/jbc.M306793200
47. Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947.
48. Ling Y, Yang ZY, Yin T, et al. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int Immunopharmacol. 2011;11:187–193. doi:10.1016/j.intimp.2010.11.014
49. Liu R, Mori S, Wake H, et al. Establishment of in vitro binding assay of high mobility group box-1 and S100A12 to receptor for advanced glycation endproducts: heparin’s effect on binding. Acta Med Okayama. 2009;63:203–211. doi:10.18926/AMO/31812
50. Hori O, Brett J, Slattery T, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi:10.1074/jbc.270.43.25752
51. Teissier T, Boulanger E. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology. 2019;20:279–301.
52. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. 2020;11:484. doi:10.3389/fimmu.2020.00484
53. Deng M, Tang Y, Li W, et al. The endotoxin delivery protein HMGB1 Mediates Caspase-11-dependent lethality in sepsis. Immunity. 2018;49:740–753. doi:10.1016/j.immuni.2018.08.016
54. Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi:10.1038/ni1457
55. Degryse B, Bonaldi T, Scaffidi P, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–1206. doi:10.1083/jcb.152.6.1197
56. Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81:59–66. doi:10.1189/jlb.0306180
57. Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81:84–91. doi:10.1189/jlb.0306171
58. Orlova VV, Choi EY, Xie C, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–1139. doi:10.1038/sj.emboj.7601552
59. Zhu S, Ashok M, Li J, et al. Spermine protects mice against lethal sepsis partly by attenuating surrogate inflammatory markers. Mol Med. 2009;15:275–282. doi:10.2119/molmed.2009.00062
60. Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi:10.1182/blood-2002-05-1300
61. Kokkola R, Andersson A, Mullins G, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi:10.1111/j.0300-9475.2005.01534.x
62. Treutiger CJ, Mullins GE, Johansson AS, et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–385. doi:10.1046/j.1365-2796.2003.01204.x
63. Lv B, Wang H, Tang Y, Fan Z, Xiao X, Chen F. High-mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF-kappaB and Egr-1. Thromb Haemost. 2009;102:352–359. doi:10.1160/TH08-11-0759
64. Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions: time for change. Lancet. 2013;381:774–775. doi:10.1016/S0140-6736(12)61815-7
65. Levy RM, Mollen KP, Prince JM, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1538–R1544. doi:10.1152/ajpregu.00272.2007
66. Manganelli V, Signore M, Pacini I, et al. Increased HMGB1 expression and release by mononuclear cells following surgical/anesthesia trauma. Crit Care. 2010;14(6):R197. doi:10.1186/cc9316
67. Sugita A, Kinoshita K, Sakurai A, et al. Systemic impact on secondary brain aggravation due to ischemia/reperfusion injury in post-cardiac arrest syndrome: a prospective observational study using high-mobility group box 1 protein. Crit Care. 2017;21:247–1828.
68. Wu H, Ma J, Wang P, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21:1878–1890.
69. Qiu J, Nishimura M, Wang Y, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28(5):927–938. doi:10.1038/sj.jcbfm.9600582
70. Okuma Y, Liu K, Wake H, et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol. 2012;72(3):373–384. doi:10.1002/ana.23602
71. Shimazaki J, Matsumoto N, Ogura H, et al. Systemic involvement of high-mobility group box 1 protein and therapeutic effect of anti-high-mobility group box 1 protein antibody in a rat model of crush injury. Shock. 2012;37(6):634–638. doi:10.1097/SHK.0b013e31824ed6b7
72. Yang R, Zhang S, Cotoia A, Oksala N, Zhu S, Tenhunen J. High mobility group B1 impairs hepatocyte regeneration in acetaminophen hepatotoxicity. BMC Gastroenterol. 2012;12:45. doi:10.1186/1471-230X-12-45.:45-12
73. Nadatani Y, Watanabe T, Tanigawa T, et al. High mobility group box 1 promotes small intestinal damage induced by nonsteroidal anti-inflammatory drugs through Toll-like receptor 4. Am J Pathol. 2012;181(1):98–110. doi:10.1016/j.ajpath.2012.03.039
74. Zhou RR, Liu HB, Peng JP, et al. High mobility group box chromosomal protein 1 in acute-on-chronic liver failure patients and mice with ConA-induced acute liver injury. Exp Mol Pathol. 2012;93:213–219. doi:10.1016/j.yexmp.2012.05.006
75. Hirata Y, Kurobe H, Higashida M, et al. HMGB1 plays a critical role in vascular inflammation and lesion formation via toll-like receptor 9. Atherosclerosis. 2013;231:227–233. doi:10.1016/j.atherosclerosis.2013.09.010
76. Nadatani Y, Watanabe T, Tanigawa T, et al. High-mobility group box 1 inhibits gastric ulcer healing through toll-like Receptor 4 and receptor for advanced glycation end products. PLoS One. 2013;8(11):e80130. doi:10.1371/journal.pone.0080130
77. Patel VS, Sitapara RA, Gore A, et al. High Mobility Group Box-1 mediates hyperoxia-induced impairment of Pseudomonas aeruginosa clearance and inflammatory lung injury in mice. Am J Respir Cell Mol Biol. 2013;48:280–287.
78. Li Z, Scott MJ, Fan EK, et al. Tissue damage negatively regulates LPS-induced macrophage necroptosis. Cell Death Differ. 2016;23(9):1428–1447. doi:10.1038/cdd.2016.21
79. Xu J, Jiang Y, Wang J, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21(8):1229–1239. doi:10.1038/cdd.2014.40
80. Chen R, Fu S, Fan XG, et al. Nuclear DAMP complex-mediated RAGE-dependent macrophage cell death. Biochem Biophys Res Commun. 2015;458:650–655. doi:10.1016/j.bbrc.2015.01.159
81. Robert SM, Sjodin H, Fink MP, Aneja RK. Preconditioning with high mobility group box 1 (HMGB1) induces lipoteichoic acid (LTA) tolerance. J Immunother. 2010;33(7):663–671. doi:10.1097/CJI.0b013e3181dcd111
82. Aneja RK, Tsung A, Sjodin H, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. 2008;84(5):1326–1334. doi:10.1189/jlb.0108030
83. Gregoire M, Tadie J-M, Uhel F, et al. Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukoc Biol. 2017;101(6):1281–1287. doi:10.1189/jlb.5HI0316-128RR
84. Wild CA, Bergmann C, Fritz G, et al. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24(8):485–494. doi:10.1093/intimm/dxs051
85. Patel VS, Sitapara RA, Gore A, et al. HMGB1 mediates hyperoxia-induced impairment of pseudomonas aeruginosa clearance and inflammatory lung injury in mice. Am J Respir Cell Mol Biol. 2012;48:280–287.
86. Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock–a review of laboratory models and a proposal. J Surg Res. 1980;29:189–201. doi:10.1016/0022-4804(80)90037-2
87. Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi:10.1073/pnas.2434651100
88. Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi:10.4049/jimmunol.165.6.2950
89. Entezari M, Javdan M, Antoine DJ, et al. Inhibition of extracellular HMGB1 attenuates hyperoxia-induced inflammatory acute lung injury. Redox Biol. 2014;2:314–322. doi:10.1016/j.redox.2014.01.013
90. Qin S, Wang H, Yuan R, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–1642.
91. Suda K, Kitagawa Y, Ozawa S, et al. Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J Surg. 2006;30:1755–1762.
92. Ye C, Choi JG, Abraham S, et al. Human macrophage and dendritic cell-specific silencing of high-mobility group protein B1 ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci U S A. 2012;109(51):21052–21057. doi:10.1073/pnas.1216195109
93. Wang H, Yang H, Tracey KJ. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004;255(3):320–331. doi:10.1111/j.1365-2796.2003.01302.x
94. Wang H, Ward MF, Sama AE. Novel HMGB1-inhibiting therapeutic agents for experimental sepsis. Shock. 2009;32(4):348–357. doi:10.1097/SHK.0b013e3181a551bd
95. Herzog C, Lorenz A, Gillmann HJ, et al. Thrombomodulin’s lectin-like domain reduces myocardial damage by interfering with HMGB1-mediated TLR2 signalling. Cardiovasc Res. 2014;101:400–410. doi:10.1093/cvr/cvt275
96. Takehara K, Murakami T, Kuwahara-Arai K, Iba T, Nagaoka I, Sakamoto K. Evaluation of the effect of recombinant thrombomodulin on a lipopolysaccharide-induced murine sepsis model. Exp Ther Med. 2017;13(6):2969–2974. doi:10.3892/etm.2017.4308
97. Abeyama K, Stern DM, Ito Y, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115(5):1267–1274. doi:10.1172/JCI22782
98. Yang H, Wang H, Levine YA, et al. Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight. 2018;3(24):e126617. doi:10.1172/jci.insight.126617
99. Son M, Porat A, He M, et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood. 2016;128(18):2218–2228. doi:10.1182/blood-2016-05-719757
100. Liu T, Xiang A, Peng T, et al. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci U S A. 2019;116:23254–23263. doi:10.1073/pnas.1907490116
101. Clemmensen I, Petersen LC, Kluft C. Purification and characterization of a novel, oligomeric, plasminogen kringle 4 binding protein from human plasma: tetranectin. Eur J Biochem. 1986;156:327–333. doi:10.1111/j.1432-1033.1986.tb09586.x
102. Sorensen CB, Berglund L, Petersen TE. Cloning of a cDNA encoding murine tetranectin. Gene. 1995;152:243–245. doi:10.1016/0378-1119(94)00703-U
103. Wewer UM, Iba K, Durkin ME, et al. Tetranectin is a novel marker for myogenesis during embryonic development, muscle regeneration, and muscle cell differentiation in vitro. Dev Biol. 1998;200:247–259. doi:10.1006/dbio.1998.8962
104. Jensen BA, McNair P, Hyldstrup L, Clemmensen I. Plasma tetranectin in healthy male and female individuals, measured by enzyme-linked immunosorbent assay. J Lab Clin Med. 1987;110:612–617.
105. Wewer UM, Ibaraki K, Schjorring P, Durkin ME, Young MF, Albrechtsen R. A potential role for tetranectin in mineralization during osteogenesis. J Cell Biol. 1994;127:1767–1775.
106. Iba K, Durkin ME, Johnsen L, et al. Mice with a targeted deletion of the tetranectin gene exhibit a spinal deformity. Mol Cell Biol. 2001;21:7817–7825. doi:10.1128/MCB.21.22.7817-7825.2001
107. Wang ES, Zhang XP, Yao HB, et al. Tetranectin knockout mice develop features of Parkinson disease. Cell Physiol Biochem. 2014;34:277–287. doi:10.1159/000362998
108. Iba K, Hatakeyama N, Kojima T, et al. Impaired cutaneous wound healing in mice lacking tetranectin. Wound Repair Regen. 2009;17:108–112. doi:10.1111/j.1524-475X.2008.00447.x
109. Iba K, Abe Y, Chikenji T, et al. Delayed fracture healing in tetranectin-deficient mice. J Bone Miner Metab. 2013;31:399–408. doi:10.1007/s00774-013-0436-y
110. Chen W, Qiang X, Wang Y, et al. Identification of tetranectin-targeting monoclonal antibodies to treat potentially lethal sepsis. Sci Transl Med. 2020;12:12–539. doi:10.1126/scitranslmed.aaz3833
111. Franklin BS, Bossaller L, De ND, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–737. doi:10.1038/ni.2913
112. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
113. Buchman TG, Simpson SQ, Sciarretta KL, et al. Sepsis among medicare beneficiaries: 1. The burdens of sepsis, 2012-2018. Crit Care Med. 2020;48:276–288. doi:10.1097/CCM.0000000000004224
114. Paterson CW, Ford ML, Coopersmith CM. Breaking the bond between tetranectin and HMGB1 in sepsis. Sci Transl Med. 2020;12:12–539. doi:10.1126/scitranslmed.abb2575
115. Aachoui Y, Leaf IA, Hagar JA, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339(6122):975–978. doi:10.1126/science.1230751
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.