")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 15
Therapeutic Advantage of Targeting PRMT5 in Combination with Chemotherapies or EGFR/HER2 Inhibitors in Triple-Negative Breast Cancers
Authors Dakroub R, Huard S, Hajj-Younes Y, Suresh S, Badran B, Fayyad-Kazan H, Dubois T
Received 13 July 2023
Accepted for publication 4 October 2023
Published 6 November 2023 Volume 2023:15 Pages 785—799
DOI https://doi.org/10.2147/BCTT.S430513
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Rayan Dakroub,1,2 Solène Huard,1 Yara Hajj-Younes,1 Samyuktha Suresh,1 Bassam Badran,2 Hussein Fayyad-Kazan,2 Thierry Dubois1
1Breast Cancer Biology Group, Translational Research Department, Institut Curie-PSL Research University, Paris, 75005, France; 2Laboratory of Cancer Biology and Molecular Immunology, Faculty of Sciences-I, Lebanese University, Hadath, 1003, Lebanon
Correspondence: Thierry Dubois, Breast Cancer Biology Group, Translational Research Department, Institut Curie-PSL Research University, Paris, 75005, France, Tel +33 156246250, Email [email protected]
Purpose: Triple-negative breast cancer (TNBC) is the most aggressive breast cancer subgroup characterized by a high risk of resistance to chemotherapies and high relapse potential. TNBC shows inter-and intra-tumoral heterogeneity; more than half expresses high EGFR levels and about 30% are classified as HER2-low breast cancers. High PRMT5 mRNA levels are associated with poor prognosis in TNBC and inhibiting PRMT5 impairs the viability of subsets of TNBC cell lines and delays tumor growth in TNBC mice models. TNBC patients may therefore benefit from a treatment targeting PRMT5. The aim of this study was to assess the therapeutic benefit of combining a PRMT5 inhibitor with different chemotherapies used in the clinics to treat TNBC patients, or with FDA-approved inhibitors targeting the HER family members.
Methods: The drug combinations were performed using proliferation and colony formation assays on TNBC cell lines that were sensitive or resistant to EPZ015938, a PRMT5 inhibitor that has been evaluated in clinical trials. The chemotherapies analyzed were cisplatin, doxorubicin, camptothecin, and paclitaxel. The targeted therapies tested were erlotinib (EGFR inhibitor), neratinib (EGFR/HER2/HER4 inhibitor) and tucatinib (HER2 inhibitor).
Results: We found that PRMT5 inhibition synergized mostly with cisplatin, and to a lesser extent with doxorubicin or camptothecin, but not with paclitaxel, to impair TNBC cell proliferation. PRMT5 inhibition also synergized with erlotinib and neratinib in TNBC cell lines, especially in those overexpressing EGFR. Additionally, a synergistic interaction was observed with neratinib and tucatinib in a HER2-low TNBC cell line as well as in a HER2-positive breast cancer cell line. We noticed that synergy can be obtained in TNBC cell lines that were resistant to PRMT5 inhibition alone.
Conclusion: Altogether, our data highlight the therapeutic potential of targeting PRMT5 using combinatorial strategies for the treatment of subsets of TNBC patients.
Keywords: TNBC, erlotinib, neratinib, cisplatin, drug combination
Introduction
Breast cancer is a heterogeneous disease with distinct subgroups categorized according to the expression level of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). Luminal breast cancers express ER and PR; HER2-positive tumors carry an amplification of the HER2 gene; and triple-negative breast cancers (TNBC) do not express ER and PR and have no HER2 gene amplification.1,2 Recently, about 65% of luminal breast cancers and 35% of TNBC were found to be HER2-low breast tumors,3,4 defined as tumors expressing HER2 with immunohistochemical (IHC) scores of 1+ and 2+ without HER2 gene amplification. Breast cancer subgroups differ in their grade and prognosis, with TNBC being the most aggressive. TNBC is an invasive tumor usually associated with drug resistance, high metastatic potential, and poor prognosis.5–8 A high percentage of TNBC patients experience relapse within 3–5 years following treatment.7 Compared to the other breast cancer subgroups, TNBC is enriched in a subpopulation of cells with self-renewal ability, termed breast cancer stem cells (BCSC) or tumor-initiating cells (TIC) that are drug-resistant and thought to be involved in the high relapse rate of TNBC patients.7,9,10 Another major concern in TNBC is its inherent inter-tumoral heterogeneity with different TNBC subtypes: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), and luminal androgen receptor (LAR).11,12 Interestingly, HER2-low breast cancer is associated with the expression of androgen receptor (AR) in luminal and in TNBC.3 The high inter- and intra-tumoral heterogeneity poses a considerable challenge in TNBC treatment options as one specific target/drug may not be beneficial to all TNBC but rather to a subset of them. Although chemotherapies such as platinum agents (cisplatin, carboplatin), taxanes (paclitaxel and docetaxel), and anthracyclines (doxorubicin, epirubicin), remain the standard treatment option,2,13,14 a few new treatments have been recently approved by the Food and Drug Administration (FDA) for selected TNBC patients: poly (ADP-ribose) polymerase (PARP) inhibitors for patients with BRCA1/2 mutations,6,15,16 anti-PDL-1 antibodies for metastatic and early disease,7,17 and the antibody-drug conjugate sacituzumab govitecan.18 Although more than half of TNBC overexpress the epidermal growth factor receptor (EGFR),19–21 early clinical trials failed to demonstrate the clinical benefit of its inhibition.5 It is possible that the patients were not selected for high EGFR expression, or the pathway was not stimulated or was activated by downstream effectors such as AKT.5,20 By screening an FDA-approved drug library in TNBC cells, we have reported that erlotinib, a reversible EGFR inhibitor, acts in synergy with the first described PRMT5 inhibitor (EPZ01566622), to impair the proliferation of TNBC cells.23
Arginine methylation is a common post-translational modification catalyzed by nine protein arginine methyltransferases (PRMT1-9), regulating transcription, pre-mRNA splicing, cell signaling, DNA repair, and stem cell maintenance.24–28 Several PRMTs are overexpressed in different cancer types.27,29–33 High PRMT5 mRNA expression is associated with poor prognosis in TNBC.23,34–36 Inhibitors specifically targeting PRMT5 have been developed and are currently being evaluated in clinical trials.26,27,37 Using the first described PRMT5 inhibitor (EPZ015666), we showed that inhibiting PRMT5 impairs cell proliferation of TNBC cell lines, reduces mammosphere formation, and slows tumor growth in a TNBC patient-derived xenograft (PDX) mice model.23 By methylating and stabilizing KLF4 and KLF5, PRMT5 sustains stemness in TNBC, and its inhibition reduces tumor growth in xenograft models derived from TNBC cell lines.35,38 These data suggest that PRMT5 could be an attractive therapeutic target for TNBC.23,38
In this study, we used a more potent PRMT5 inhibitor, EPZ015938, optimized from EPZ015666, to improve pharmacokinetic properties, yielding a more drug-like molecule that has been evaluated in a clinical trial.37,39 We assessed its antiproliferative effects on several breast cancer cell lines and non-cancerous breast cells. We evaluated the therapeutic benefit of combining it with different chemotherapies currently used in the clinic to treat TNBC patients.14 We also combined EPZ015938 with different FDA-approved inhibitors targeting the HER family members: erlotinib,40,41 neratinib (EGFR, HER2, and HER4 inhibitor),42 and tucatinib (HER2 inhibitor).43 Our results showed that inhibiting PRMT5 in combination with some chemotherapies or with inhibitors targeting the HER family could be a promising therapeutic strategy to treat subsets of TNBC patients.
Materials and Methods
Cell Culture
The MDA-MB-231 cell line was a kind gift from Dr. Mina Bissell (University of California, Berkeley, CA, USA) and its use was approved by our institute with the establishment of a material transfer agreement. All other cell lines were purchased from the American Type Culture Collection (ATCC, LGC Promochem). All cell lines were authenticated by short tandem repeat profiling in 2021 and tested for mycoplasma by the MycoAlert Mycoplasma Detection Kit (Lonza Biosciences, Durham, NC, USA). MDA-MB-468, BT474, and T47D cells were cultured in RPMI-1640 GlutaMAX™ (LifeTechnologies) supplemented with 10% (vol/vol) fetal bovine serum (FBS, LifeTechnologies), 100 U/mL penicillin, and 100 µg/mL streptomycin (P/S, LifeTechnologies). HCC38, HCC70, and HCC1954 cells were cultured in RPMI-1640 GlutaMAX™ supplemented with 10% (vol/vol) FBS, 100 U/mL P/S, 1.5 g/L sodium bicarbonate (LifeTechnologies), 10 mmol/L Hepes (LifeTechnologies), and 1 mmol/L sodium pyruvate (LifeTechnologies). MCF10A cells were cultured in DMEM-F12 (LifeTechnologies) supplemented with 0.01 mg/mL insulin, 100 ng/mL cholera toxin (Sigma), 500 ng/mL hydrocortisone (SERB Laboratories), and 20 ng/mL epidermal growth factor (Sigma). MDA-MB-453 and MDA-MB-231 cells were cultured in DMEM-F12 (LifeTechnologies) supplemented with 10% FBS and 1% P/S. BT20 and MCF7 cells were cultured in MEM (Sigma-Aldrich) containing 10% FBS, 1% P/S, 1.5 g/L sodium bicarbonate, 0.1 mmol/L non-essential amino-acids (NEAA, LifeTechnologies) and 1 mmol/L sodium pyruvate. All cell lines were maintained at 37°C with 5% CO2.
Inhibitors
EPZ015938 (PRMT5 inhibitor; ref: HY-101563), neratinib (HER1/2/4 inhibitor; ref: HY-32721), and tucatinib (HER2 inhibitor; ref: HY-16069) were purchased from MedChemExpress. Erlotinib (EGFR inhibitor; ref: S7786), cisplatin (ref: S1166), and paclitaxel (ref: S1150) were obtained from Selleckchem. Camptothecin (ref: C9911) and doxorubicin (ref: D1515) were purchased from Sigma-Aldrich. Cisplatin was resuspended in water and the other inhibitors in DMSO.
Proliferation Assay
To determine the half-maximal inhibitory concentration (IC50) of the PRMT5 inhibitor on the eleven breast cell lines, cells were seeded in 96 well plates and then treated after 48 hours with 0.05% DMSO or 1 µM maximal concentration of EPZ015938 followed by two-fold serial dilutions. Cell proliferation was then assessed using MTT (M2128-1G, Sigma-Aldrich, St. Louis, MO, USA), WST-1 (11,644,807,001, Sigma-Aldrich), or CellTiterGlo (G7572, Promega, Madison, WI, USA) after four doubling times (3–7 days depending on the cell line) (Table S1).
For combination analyses, four TNBC cell lines (BT20, MDA-MB-231, MDA-MB-453, and MDA-MB-468) were seeded in 96-well plates and treated with vehicle or varying concentrations of inhibitors, either on their own or in combination. For combinations, the two inhibitors were added simultaneously. Cell proliferation was measured after four doubling times by MTT or CellTiterGlo assays (Table S1). Inhibitors were used at a maximal concentration of ~2xIC50 (or 5 µM if the cell line was resistant to the inhibitor) (Tables S2 and S3), followed by two-fold serial dilutions. Drug interactions were assessed using the Loewe model and calculated on the Combenefit software.44 The combination index (CI) was calculated using the formula 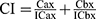 , where Cax and Cbx are the concentrations of drugs A and B that produce an effect “x” (such as 50% cell death), and ICax and ICbx are the concentrations of drugs A and B that yield the same effect “x” when used alone.45 CI = 1 indicates additivity, CI > 1 indicates antagonism and CI < 1 indicates synergism. Combination experiments were done in triplicates, and a minimum of three independent experiments were performed.
, where Cax and Cbx are the concentrations of drugs A and B that produce an effect “x” (such as 50% cell death), and ICax and ICbx are the concentrations of drugs A and B that yield the same effect “x” when used alone.45 CI = 1 indicates additivity, CI > 1 indicates antagonism and CI < 1 indicates synergism. Combination experiments were done in triplicates, and a minimum of three independent experiments were performed.
Colony Formation Assay
For colony formation assay (CFA), two TNBC cell lines were seeded at low density in 6-well plates and treated 24 hours later with vehicle or one single concentration of each drug alone or in combination and incubated until colonies formed (~9 days for MDA-MB-468 and ~12 days for BT20). For combinations, the two inhibitors were added simultaneously. As a vehicle, 0.05% DMSO was used for all the combinations except when cisplatin was examined (0.05% DMSO + H2O). Different doses of inhibitors (cisplatin, EPZ015938, erlotinib, neratinib) were first tested on these cell lines, and concentrations that decreased colony number to a maximum of 50% were chosen. The colonies were then fixed and stained with 0.05% Coomassie Brilliant Blue in 50% methanol and 10% acetic acid solution for 20 minutes then rinsed with ultrapure water. Colonies were imaged using the ChemiDoc MP imager (Bio-rad Laboratories, Hercules, CA, USA) and quantified by ImageJ 1.43u software.46
Western Blotting
Western blotting was performed as previously described47 with few modifications. The cells were lysed in Laemmli buffer containing 50 mmol/L Tris pH 6.8, 2% sodium dodecyl sulfate (SDS), 5% glycerol, 2 mmol/L 1.4-dithio-dl-threitol (DTT), 2.5 mmol/L ethylenediaminetetraacetic acid (EDTA), 2.5 mmol/L ethylene glycol tetraacetic acid (EGTA), 2 mmol/L sodium orthovanadate, and 10 mmol/L sodium fluoride (Sigma-Aldrich), a cocktail of protease (Roche) and phosphatase (Thermo Scientific) inhibitors, and then boiled at 100°C for 10 minute. The protein concentration in each sample was determined with the reducing agent-compatible version of the BCA Protein Assay kit (Thermo Scientific, 23,227). Equal amounts of total protein (20 µg) were fractionated by SDS-PAGE under reducing conditions (4–12% TGX gels, Bio-Rad, Marnes la Coquette, France) and blotted onto nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% BSA in TBS containing 0.1% Tween 20 (TBS-T) and hybridized with anti-PRMT5 (Cell Signaling Technology #79998) or anti-actin (Sigma-Aldrich #A5441) antibodies overnight at 4°C. Membranes were washed in TBS-T and then hybridized with the secondary antibody for 1 hour at room temperature. Antibodies were diluted in TBS-T containing 5% BSA. The membranes were washed with TBS-T, and immune complexes were revealed by enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific) and imaged using a ChemiDoc™ XRS+ System (Bio-Rad Laboratories, Inc.).
Transcriptomic Microarray
Total RNA was purified with miRNeasy kit from Qiagen (Qiagen, Courtaboeuf, France) according to the supplier recommendations. A quality control of total RNA was carried out with a Nanodrop ND1000 spectrophotometer (Thermo Fisher) to monitor the concentration and purity of samples, and integrity of total RNA was controlled using RNA6000 Lab-on-a-chip with a Bioanalyzer (Agilent Technologies) as described.48 Samples were then hybridized with the GeneChip Human Exon 1.0 ST arrays (Affymetrix) following the manufacturer’s instructions. Data quality control was performed using Affymetrix Expression Console. The data were analyzed as described elsewhere.48
Statistical Analysis
GraphPad Prism 8.4.3 was used for statistical analysis. Data are presented as mean ± standard deviation (SD) and p-values were calculated using the Student’s t-test. P<0.05 was considered statistically significant.
Results
Breast Cancer Cell Lines Show Distinct Sensitivity to PRMT5 Inhibition
We treated ten breast cancer cell lines from the different breast cancer subgroups (six TNBC, two HER2-positive, and two luminal) and one non-cancerous breast cell line with nanomolar concentrations (3.9 nM – 1000 nM) of EPZ015938 (Table S2). To take into account the well-established TNBC inter-tumoral heterogeneity,11,12 cell lines representing different TNBC subtypes were tested (Table S2). Among the TNBC cell lines, HCC38 cells were the most sensitive (IC50 = 21.9 nM ± 8.7 nM) to PRMT5 inhibition, followed by MDA-MB-453 (IC50 = 109.4 nM ± 13.4 nM) and MDA-MB-468 (IC50 = 319.3 nM ± 226.2 nM) cells (Figure 1 and Table S2). The three other TNBC cell lines (BT20, HCC70, and MDA-MB-231) were resistant to PRMT5 inhibition as the treatment did not permit the calculation of an IC50 (Figure 1 and Table S2). EPZ015938 also impaired the proliferation of the two luminal cell lines T47D (IC50 = 303.9 nM ± 244.8 nM) and MCF7 (IC50 = 191.5 nM ± 47 nM) (Figure 1 and Table S2). Out of the two HER2-positive cell lines, HCC1954 (IC50 = 54.2 nM ± 19.4 nM) was more sensitive to PRMT5 inhibition than BT474 (IC50 = 625.5 nM ± 217.6 nM) (Figure 1 and Table S2). Importantly, EPZ015938 was less effective in inhibiting the proliferation of the non-cancerous cell line MCF10A (IC50 = 722.8 nM ± 122 nM) compared to the sensitive breast cancer cell lines (Figure 1 and Table S2). These results show that the cells respond differently to PRMT5 inhibition, and this is independent of the PRMT5 expression level (Supplementary Figure 1).
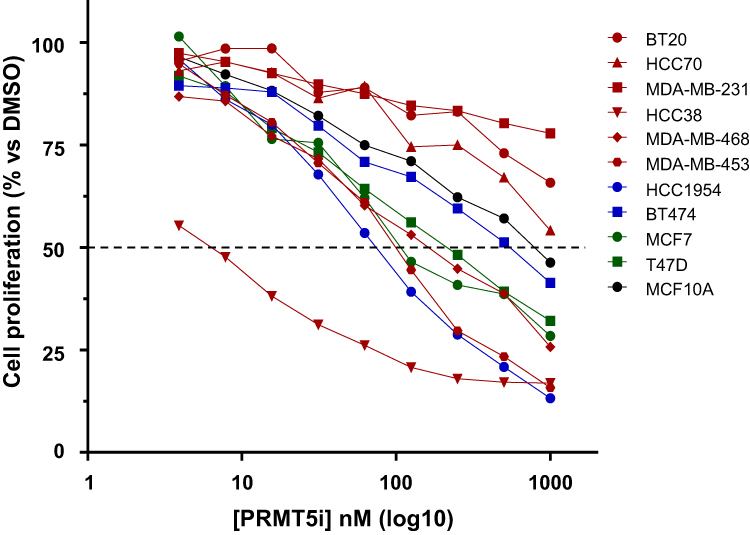 |
Figure 1 Evaluation of the sensitivity of various breast cell lines to PRMT5 inhibition (EPZ015938). Six TNBC (red), two HER2-positive (blue), two luminal (green) breast cancer cell lines, and one non-tumorigenic breast cell line (black) were treated with nanomolar doses (3.9 nM – 1000 nM) of EPZ015938 (PRMT5i). Cell proliferation was determined after four mitotic cycles. The percentage of viable cells was normalized to DMSO-treated cells. The mean of at least three independent experiments is presented for each cell line (error bars are not shown to better visualize the different cell lines but IC50 ± SD are indicated in Table S2). |
PRMT5 Inhibition Synergizes with Cisplatin, Doxorubicin, and Camptothecin, but Not with Paclitaxel, to Impair TNBC Cell Proliferation
Drug combinations have gained increasing interest as a means to overcome resistance, increase treatment efficacy, and reduce relapse, all representing concerns in TNBC management. Therefore, we tested the antiproliferative effects of inhibiting PRMT5 in combination with different chemotherapies used in the clinics to treat TNBC patients.14 The drug combinations were assessed in four TNBC cell lines: two sensitive (MDA-MB-453 and MDA-MB-468) and two resistant (BT20 and MDA-MB-231) to PRMT5 inhibition with EPZ015938 (Figure 1 and Table S2). Cells were treated with varying concentrations of both drugs starting with a dose corresponding to ~2xIC50 for sensitive cells and to 5 µM for resistant cells, then viability was quantified after four mitotic cycles. To assess the nature of drug interactions (synergy, additivity, antagonism), we employed the widely used Loewe additivity model which assumes there is no interaction when a compound is combined with itself.49,50
PRMT5 inhibition acted synergistically with cisplatin to inhibit the proliferation of BT20, MDA-MB-453 and MDA-MB-468 (Figure 2A) cells; additivity was observed in MDA-MB-231 cells (Supplementary Figure 2). The highest synergy scores (>30) and lowest CI values were obtained in BT20 and MDA-MB-468 cells, and importantly, at doses lower than the IC50 for both PRMT5 inhibitor and cisplatin in MDA-MB-468 cells. Remarkably, synergy was observed at low doses (125–250 nM) of EPZ015938 in BT20 cells (Figure 2A), a cell line resistant to PRMT5 inhibition alone (Figure 1). Next, we examined whether the EPZ015938 plus cisplatin combination affected the ability of TNBC cell lines to form colonies. At the tested doses, the EPZ015938 plus cisplatin combination decreased the colony number by 63.7% ± 1.9% in BT20 (Figure 2B) and 77.8% ± 3.9% in MDA-MB-468 (Figure 2C) cells. This reduction is more pronounced compared to cisplatin alone in BT20 (20.4% ± 2.7%) and in MDA-MB-468 (35.4% ± 2.1%) or compared to EPZ015938 alone in BT20 (46.7% ± 2.9%) and in MDA-MB-468 (45.6% ± 8.8%) cells (Figure 2B and C). These results suggest that targeting TNBC with PRMT5 inhibitors in combination with cisplatin may offer a promising therapeutic option by using lower doses of each drug, hence potentially lowering toxicity in patients.
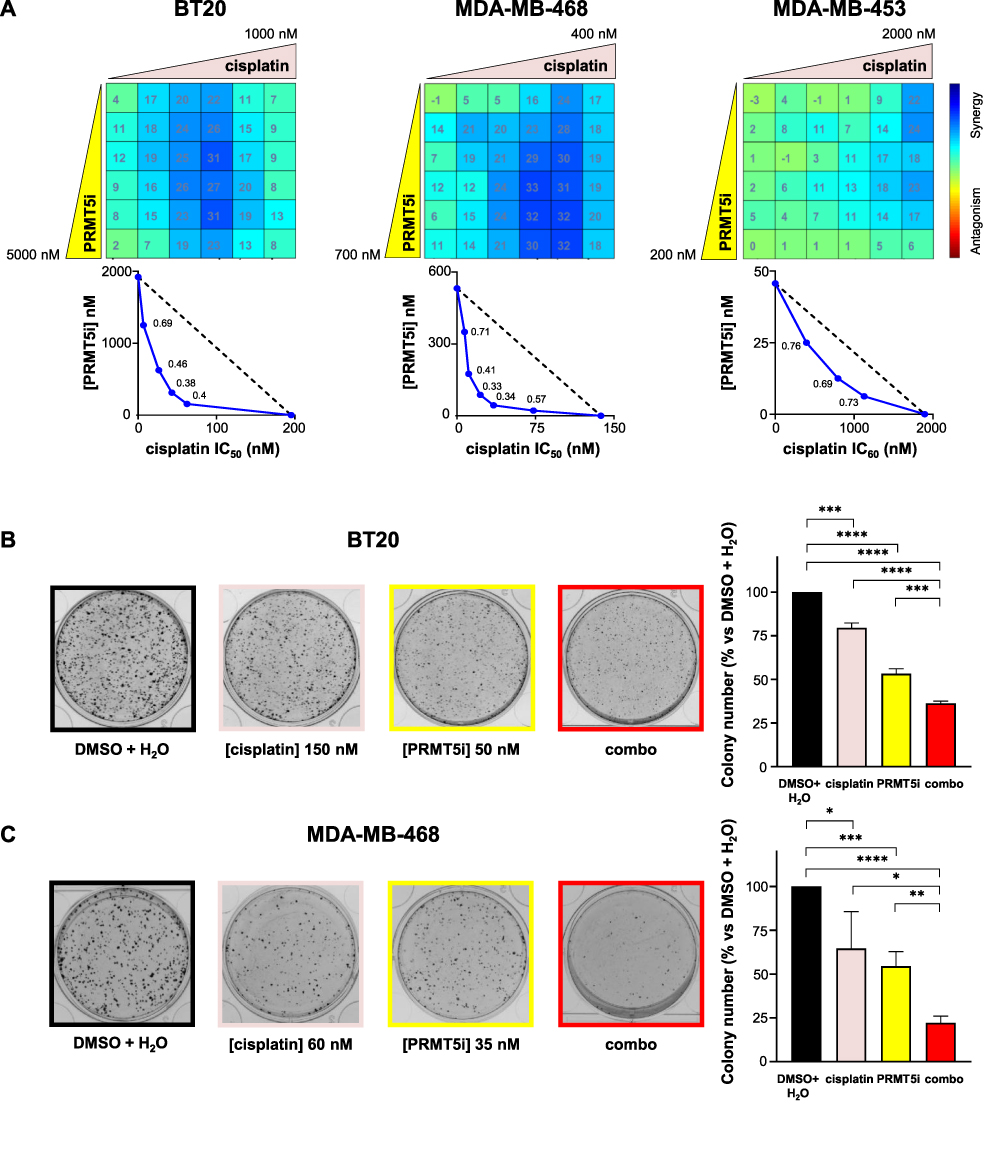 |
Figure 2 Effect of the inhibition of PRMT5 in combination with cisplatin on the proliferation (A) and colony formation (B and C) of TNBC cell lines. (A) BT20, MDA-MB-468, and MDA-MB-453 TNBC cells were seeded in 96-well plates and treated with varying concentrations of EPZ015938 (PRMT5i) and/or cisplatin, then cell proliferation was measured after four mitotic cycles (7 days). The percentage of viable cells was normalized to (DMSO + H2O)-treated cells. Each drug was used at a maximal concentration of 2xIC50 for sensitive cell lines (5 µM maximum for resistant cells), followed by two-fold serial dilutions. The nature of drug interaction between EPZ015938 and cisplatin was assessed using the Loewe model on the Combenefit software. The synergy matrix (upper panel) and isobologram (bottom panel) for each cell line are shown. Isobolograms represent the IC50 (BT20, MDA-MB-468) or IC60 (MDA-MB-453) of cisplatin (X-axis) obtained at various EPZ015938 concentrations (Y-axis). CI were calculated at the different EPZ015938 concentrations used and are shown on the isobolograms. Data are representative of at least three independent experiments. (B and C) BT20 (B) and MDA-MB-468 (C) cells were seeded at low densities and then treated with DMSO + H2O, EPZ015938 (PRMT5i), cisplatin, or a combination (combo) of the two drugs. The colony number was quantified using ImageJ software. An image for each condition is shown and is representative of three independent experiments. Quantification of colony number is expressed as a percentage relative to (DMSO + H2O)-treated cells and represented as the mean ± SD from at least three independent experiments (right panels). P values were calculated using a Student’s t-test and presented as: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. |
Moreover, we found that doxorubicin synergized with PRMT5 inhibition to impair cell proliferation in BT20 and MDA-MB-453 cell lines (Figure 3A); additivity was observed in MDA-MB-468 (Figure 3A) cells and weak antagonism in MDA-MB-231 cells (Supplementary Figure 2). Camptothecin synergized with PRMT5 inhibition in BT20 and MDA-MB-468 cell lines (Figure 3B); additivity was observed in MDA-MB-453 (Figure 3B) and weak antagonism in MDA-MB-231 (Supplementary Figure 2). The paclitaxel/EPZ015938 combination had an additive effect on all the tested cell lines except on MDA-MB-453 cells in which the drug combination exhibited antagonism (Supplementary Figures 2 and 3). These results showed that a particular drug combination could show synergy, additivity, or antagonism depending on the TNBC cell line.
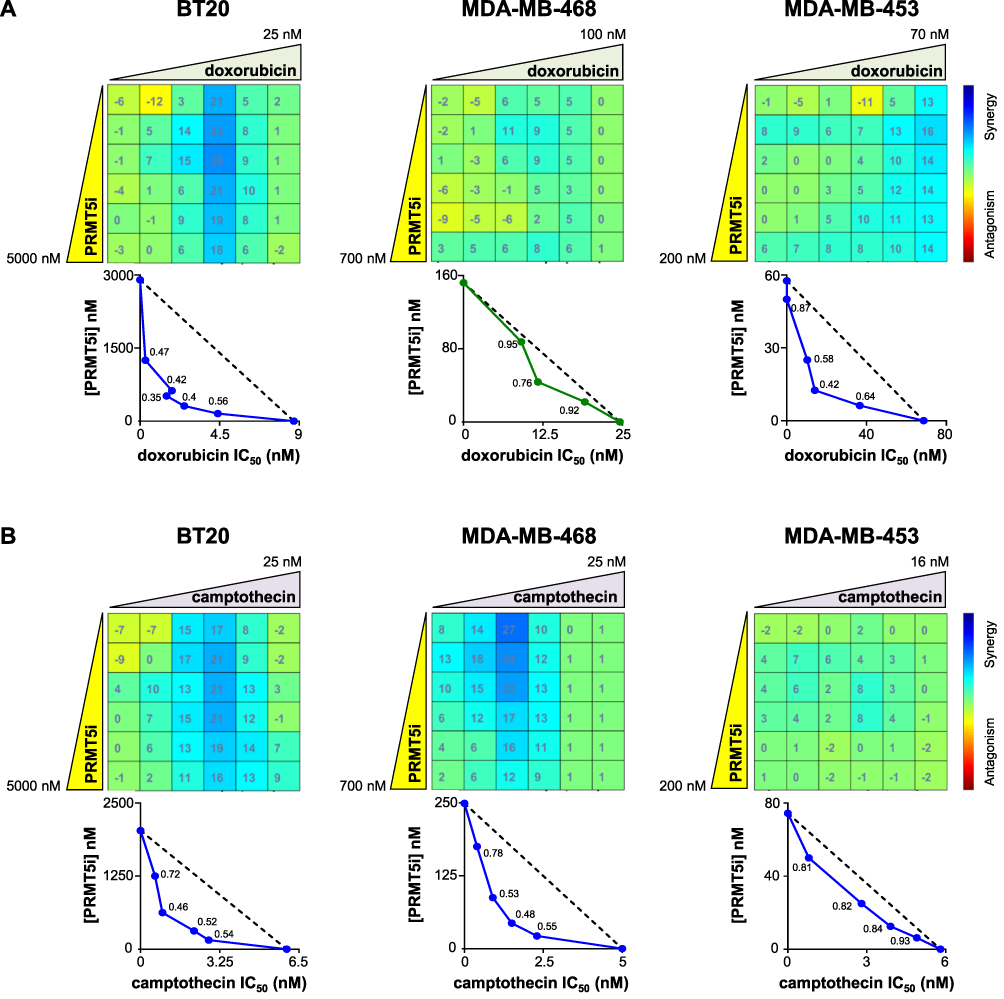 |
Figure 3 Effect of the inhibition of PRMT5 in combination with doxorubicin (A) or camptothecin (B) on TNBC cell proliferation. BT20, MDA-MB-468, and MDA-MB-453 TNBC cells were seeded in 96-well plates and treated with varying concentrations of EPZ015938 (PRMT5i) and/or doxorubicin (A) or camptothecin (B), then cell proliferation was measured after four mitotic cycles (7 days). The percentage of viable cells was normalized to DMSO-treated cells. Each drug was used at a maximal concentration of 2xIC50 for sensitive cell lines (5 µM maximum for resistant cells), followed by two-fold serial dilutions. The nature of drug interaction between EPZ015938 and doxorubicin (A) or camptothecin (B) was assessed using the Loewe model on the Combenefit software. The synergy matrix (upper panel) and isobologram (bottom panel) for each cell line are shown. Isobolograms represent the IC50 of doxorubicin (A) or camptothecin (B) (X-axis) obtained at various EPZ015938 concentrations (Y-axis). CI were calculated at the different EPZ015938 concentrations used and are shown on the isobolograms. Data are representative of at least three independent experiments. |
Taken together, analyzing the effects on cell viability of PRMT5 inhibition combined with different clinically relevant chemotherapies to treat TNBC patients revealed that (i) there is a heterogeneity of response to a particular combination between TNBC cell lines, (ii) the most effective synergistic interaction was obtained with cisplatin, and (iii) the response of a cell line to a particular combination is independent of its sensitivity to PRMT5 inhibitor when used alone.
PRMT5 Inhibition Synergizes with Inhibitors Targeting HER Family Members to Impair the Proliferation of TNBC Cells
As more than half of TNBC tumors overexpress EGFR and approximately a third of TNBC are classified as HER2-low breast cancers, we tested the combination between EPZ015938 and FDA-approved inhibitors that target different HER family members (EGFR/HER1, HER2, and HER4). We performed the analyses in the same four TNBC cell lines used for the combination with chemotherapies. Among them, MDA-MB-468 and BT20 express high levels of EGFR (Supplementary Figure 4). MDA-MB-453 expresses AR as well as higher levels of HER2 (with no HER2 amplification) compared to other TNBC cell lines51 (Supplementary Figure 4) and could be considered as a HER2-low breast cancer cell line.
Erlotinib synergized with EPZ015938 in the two EGFR-high expressing cell lines, BT20 and MDA-MB-468, to impair their proliferation (Figure 4A). BT20 cells were the most responsive to the combination with synergistic scores reaching 36 (Figure 4A). This combination was additive in MDA-MB-453 cells (Figure 4A) and varied between additivity or weak antagonism in MDA-MB-231 cells (Supplementary Figure 2). The erlotinib/EPZ015938 combination also significantly decreased the ability of BT20 and MDA-MB-468 cells to form colonies by 62.2% ± 3.7% and 69.4% ± 13.4%, respectively (Figure 4B and C), compared to an 8.7% ± 0.2% (BT20) and 25.1% ± 6.7% (MDA-MB-468) reduction caused by erlotinib treatment alone and a 49.9% ± 3.2% (BT20) and 40.5% ± 9.7% (MDA-MB-468) reduction by EPZ015938 treatment alone (Figure 4B and C).
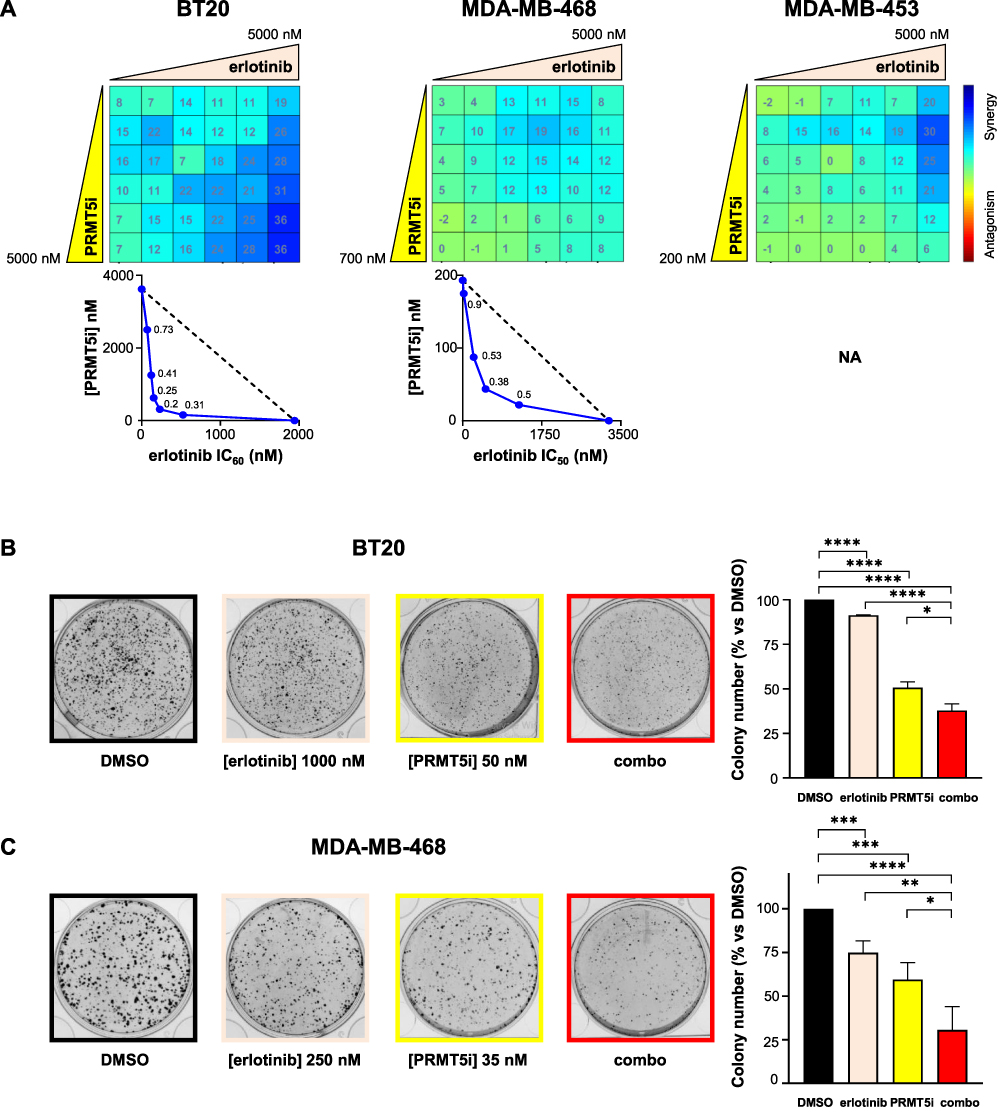 |
Figure 4 Effect of PRMT5i/erlotinib combination on the proliferation (A) and colony formation (B and C) of TNBC cell lines. (A) BT20, MDA-MB-453, and MDA-MB-468 cells were seeded in 96-well plates and treated with varying concentrations of EPZ015938 (PRMT5i) and/or erlotinib, then cell proliferation was measured after four mitotic cycles (7 days). The percentage of viable cells was normalized to DMSO-treated cells. Cells were treated with a 5 µM maximal concentration of erlotinib, and EPZ015938 was used at a maximal concentration of 2xIC50 for sensitive cell lines (5 µM for resistant cells). Both drugs were then two-fold serially diluted. The nature of drug interaction between EPZ015938 and erlotinib was assessed using the Loewe model on the Combenefit software. The synergy matrix (upper panel) and isobologram (bottom panel) for each cell line are shown. Isobolograms represent the IC50 of erlotinib (X-axis) obtained at various EPZ015938 concentrations (Y-axis). CI were calculated at the different EPZ015938 concentrations used and are shown on the isobolograms. Isobologram for MDA-MB-453 cells (A) was not plotted as erlotinib alone did not impair cell viability by more than 20% and is indicated as NA (not applicable). Data are representative of at least three independent experiments. (B and C) BT20 (B) and MDA-MB-468 (C) cells were seeded at low densities and then treated with DMSO, EPZ015938 (PRMT5i), erlotinib, or a combination (combo) of the two inhibitors. Colonies were quantified using ImageJ software. An image for each condition is shown and is representative of three independent experiments (left panel). Quantification of colony number is expressed as a percentage relative to DMSO-treated cells and represented as mean ± SD of three independent experiments (right panel). P values were calculated using a Student’s t-test and presented as: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. |
Neratinib synergized with EPZ015938 to impair the proliferation of both MDA-MB-468 and BT20 cells (Figure 5) expressing high EGFR levels (Supplementary Figure 4) and in MDA-MB-453 cells (Figure 5) which express HER2 (Supplementary Figure 4). The highest synergistic scores (> 30) were observed at low PRMT5 doses (~ 25–50 nM) in MDA-MB-453 cells (Figure 5). Of note, the highest synergistic scores in BT20 cells were obtained at the lowest concentrations of both neratinib (120 nM) and PRMT5 inhibitor (150 nM) (Figure 5). Compared to the drugs used alone, the neratinib/EPZ015938 combination did not further reduce the colony number in BT20 (Supplementary Figure 5). Although not significant, this combination tended to decrease colony formation in MDA-MB-468 cells compared to EPZ015938 alone (Supplementary Figure 5).
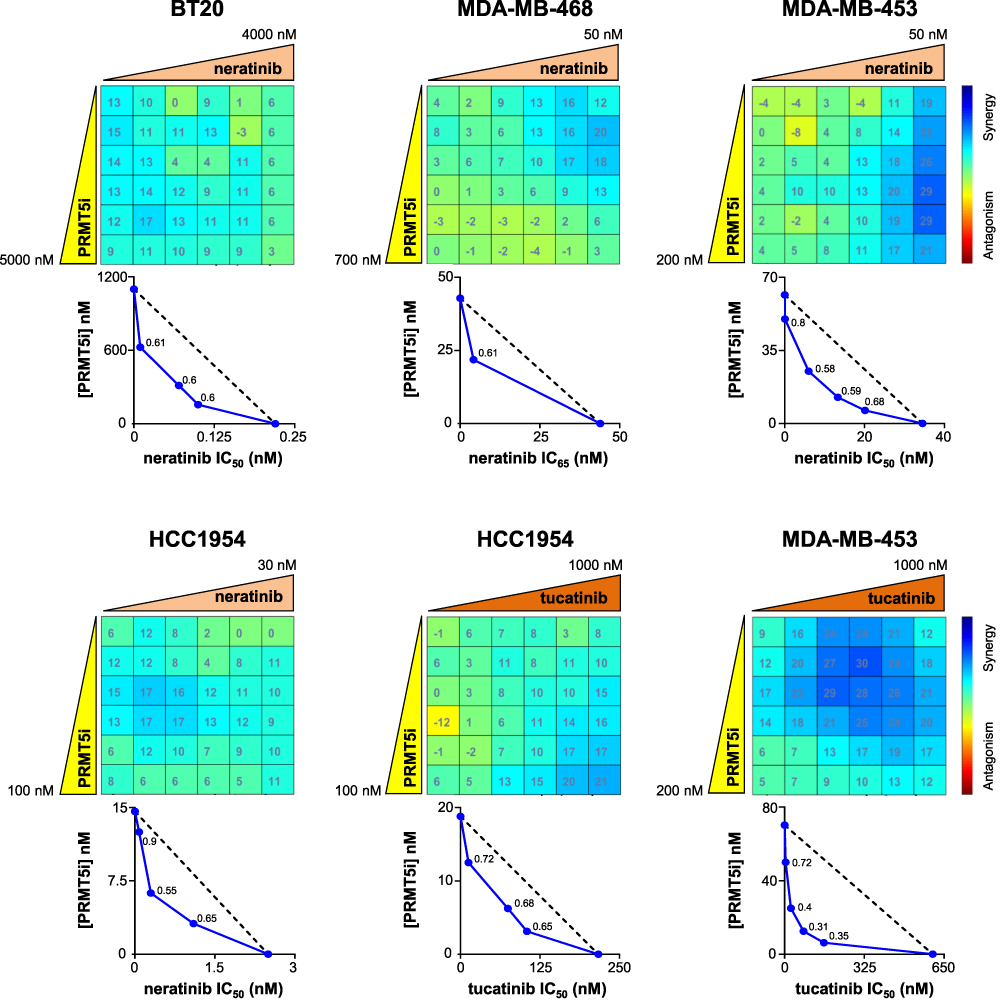 |
Figure 5 Effect of PRMT5/neratinib and PRMT5/tucatinib combinations on the proliferation of TNBC and HER2-positive breast cancer cell lines. BT20, MDA-MB-468, MDA-MB-453, and HCC1954 cells were seeded in 96-well plates and treated with varying concentrations of EPZ015938 (PRMT5i) and/or neratinib or tucatinib as indicated, then cell proliferation was measured after four mitotic cycles (7 days). The percentage of viable cells was normalized to DMSO-treated cells. Each drug was used at a maximal concentration of 2xIC50 for sensitive cell lines (5 µM maximum for resistant cells), followed by two-fold serial dilutions. The nature of drug interaction between EPZ015938 and neratinib or tucatinib was assessed using the Loewe model on the Combenefit software. The synergy matrix (upper panel) and isobologram (bottom panel) for each cell line are shown. Isobolograms represent the IC50 (BT20, MDA-MB-453, and HCC1954) or IC65 (MDA-MB-468) of neratinib or the IC50 of tucatinib (X-axis) obtained at various EPZ015938 concentrations (Y-axis). CI were calculated at the different EPZ015938 concentrations used and are shown on the isobolograms. Data are representative of at least three independent experiments. |
We then tested the combination between EPZ015938 and tucatinib or neratinib on the HER2-positive breast cancer cell line HCC1954 and found synergistic interactions between EPZ015938 and both inhibitors (Figure 5). The tucatinib/EPZ015938 combination also yielded a synergistic effect in reducing MDA-MB-453 cell proliferation (Figure 5).
Altogether, our data highlight the potential of targeting PRMT5 in combination with inhibitors targeting the HER family members in EGFR-high TNBC, HER2-low breast cancer (LAR-TNBC), and HER2-positive breast cancer.
Discussion
TNBC is the most aggressive breast cancer subgroup associated with a high relapse rate and metastatic potential. Although a few targeted therapies have recently been approved for subsets of TNBC patients,15–18 chemotherapies remain the main treatment option for these patients.1,2 Due to their high expression in several cancer types, PRMTs have emerged as attractive therapeutic targets and PRMT inhibitors have been developed.27 More specifically, different PRMT5 inhibitors have been characterized and several are currently under evaluation in Phase I clinical trials.37 High levels of PRMT5 are associated with poor prognosis in TNBC23,34,35 and PRMT5 inhibition impairs tumor growth in a TNBC PDX model23 and xenograft models derived from TNBC cell lines.35,38 TNBC patients may therefore benefit from a treatment targeting PRMT5.
As expected, similar to EPZ015666,23 we found a heterogeneity of response to EPZ015938 in breast cancer cell lines (Table S2). Importantly, the cell lines that were sensitive to EPZ015938 were also sensitive to EPZ015666, but with a ~10-fold lower IC50 (Table S2). Among the six TNBC cell lines examined, only the two BL1-TNBC (HCC38 and MDA-MB-468) and the LAR-TNBC (MDA-MB-453) cell lines were sensitive to both EPZ015938 and EPZ015666 (Table S2). To generalize whether BL1- and LAR-TNBC subtypes are the most sensitive to PRMT5 inhibition remains to be examined by analyzing additional TNBC cell lines. Previous studies also found that MDA-MB-468, MDA-MB-453, MCF7, and T47D cells were sensitive to EPZ015938, whereas HCC70 (BL2-TNBC), MDA-MB-231 (M-TNBC), BT549 (M-TNBC), Hs578T (M-TNBC) and MCF10A cells were resistant.52,53 The heterogeneity of response to EPZ015938 is not restricted to breast cancer cell lines but to cell lines of different cancer types.39 The variable sensitivity of breast cancer cell lines to PRMT5 inhibition was also highlighted using GSK3203591 (later called GSK591),33 another PRMT5 inhibitor also optimized from EPZ015666.39 Understanding the reasons why some cell lines are sensitive and others resistant to PRMT5 inhibition would help to identify biomarkers of response, which then could aid in stratifying patients who could benefit from a treatment targeting PRMT5.
In this study, we assessed in four TNBC cell lines (two sensitive and two resistant to EPZ015938) whether there is a benefit of combining EPZ015938 with different chemotherapies currently given in the clinic to treat TNBC patients. We observed a heterogenous response to the different combinations among the cell lines. Synergy was observed with the EPZ015938/doxorubicin combination in BT20 and MDA-MB-453 cells, and with the EPZ015938/camptothecin combination in BT20 and MDA-MB-468 cells. Our results show that synergy can be observed in the BT20 cell line which is resistant to EPZ015938. In contrast, we did not find synergy when the PRMT5 inhibitor was combined with paclitaxel. Nevertheless, a previous study has reported a synergistic interaction between EPZ015666 and paclitaxel in MDA-MB-231 cells.54
The most striking result was observed when EPZ015938 was combined with cisplatin, with synergy seen in three out of the four TNBC cell lines. The highest synergy scores (>30) were reached in BT20 (resistant to EPZ015938) and in MDA-MB-468 (sensitive to EPZ015938) cells, importantly, at low doses of the drugs. Being resistant to PRMT5 inhibition alone does not predict that a cell line will not respond to the combination treatment. Our findings are in agreement with a study reporting that PRMT5 inhibition (EPZ015938) sensitizes breast cancer cells to cisplatin, even in BT549 cells that were resistant to PRMT5 inhibition alone.52 Altogether, these results imply that therapeutic strategies combining PRMT5 inhibition with cisplatin could be useful for a larger number of TNBC patients, regardless of their response to PRMT5 inhibition used as a monotherapy. Combining cisplatin and a PRMT5 inhibitor, at low doses, may reduce toxicity and achieve better clinical outcomes. Cisplatin also sensitizes TNBC32 and ovarian cancer55 cells to PRMT1 inhibitors. In the ovarian cancer study, it was shown that cisplatin treatment induces DNA-PK-dependent phosphorylation of PRMT1, leading to the methylation of histone H4 and the activation of genes involved in the senescence-associated secretory phenotype (SASP), protecting cells from apoptosis.55 Whether a similar PRMT1-dependent mechanism occurs in cisplatin-treated TNBC cells has not yet been reported. Moreover, whether cisplatin induces the expression of SASP genes in a PRMT5-dependent manner remains to be explored.
Although more than half of the TNBC patients overexpress EGFR, targeting EGFR as monotherapy in TNBC patients did not achieve the expected results.20,21 The screening of an FDA-approved drug library permitted us to uncover a synergistic interaction between the first described PRMT5 inhibitor (EPZ015666) and erlotinib in several TNBC cell lines.23 Here, to be more clinically relevant, we wanted to confirm these results with EPZ015938, a more drug-like PRMT5 inhibitor already being evaluated in clinical trials.37 As expected, we found that EPZ015938 synergizes with erlotinib in the two TNBC cell lines expressing high levels of EGFR (MDA-MB-468 and BT20). We further show that inhibiting PRMT5 activity sensitizes these two cell lines to erlotinib to reduce their ability to form colonies. Next, we wanted to strengthen these findings by assessing the combination between EPZ015938 and neratinib, an EGFR/HER2/HER4 inhibitor. We found that EPZ015938 also acts in synergy with neratinib in BT20 and MDA-MB-468 cell lines. Importantly, synergy with both neratinib and erlotinib was observed at low doses of EPZ015938, avoiding potential undesirable effects. Synergy with EGFR inhibitors was observed in BT20 cells which are resistant to PRMT5 inhibition alone. Together, these results suggest that combining a PRMT5 inhibitor with erlotinib or neratinib could be beneficial for TNBC patients expressing high levels of EGFR and/or having an activated EGFR signaling pathway. We also reported that PRMT1 inhibition synergized with erlotinib in MDA-MB-468 cells.32 The molecular mechanisms underlying the synergistic interaction between EGFR and PRMT inhibitors remain to be understood. Nevertheless, previous studies have reported a relationship between PRMTs and EGFR. Indeed, PRMT1 controls the expression of EGFR, and EGFR is methylated by PRMT1 and PRMT5 affecting downstream signaling pathways.32,56–61 PRMT5 methylates and activates AKT,52,62 a downstream effector of EGFR. Moreover, AKT inhibition sensitizes breast cancer cells (luminal and TNBC) to PRMT5 inhibition.52 AKT can be activated by different signaling pathways, not only by EGFR, which could explain the synergy observed when combining EGFR and PRMT5 inhibitors.
The evaluation in a clinical trial of an antibody-drug conjugate (ADC) targeting HER2 coupled to a topoisomerase I inhibitor (Trastuzumab-deruxtecan) revealed a therapeutic benefit not only in HER2-positive but also in HER2-low breast cancers (all breast cancer subgroups including TNBC).63 This ADC has recently been FDA-approved for HER2-low breast cancer patients, independently of the expression of hormone receptors.64 Interestingly, being HER2-low positively correlates with AR expression in TNBC and luminal breast cancer.3 We found that EPZ015938 synergizes with neratinib and tucatinib in MDA-MB-453, a LAR-TNBC cell line, and in HCC1954, a HER2-positive breast cancer cell line. High synergistic scores were obtained at low doses of the PRMT5 inhibitor (~10-20 nM) in MDA-MB-453 cells. These results uncover for the first time the therapeutic potential of combining PRMT5 and HER2 inhibitors in HER2-low and HER2-positive breast cancers.
Combination strategies with PRMT5 inhibition appear to be promising therapeutic approaches with high translational impact. Indeed, other laboratories have also shown that PRMT5 inhibition sensitizes cancer cells to targeted therapies: anti-PDL1 in lung cancer,65 ATR inhibitor in mantle cell lymphoma (MCL),66 CDK4/6 inhibitors in MCL66 and melanoma,67 EZH2 inhibitor in colorectal cancer,68 type I PRMT inhibitors in pancreatic cancer69 and in acute myeloid leukemia (AML),70 mTOR inhibitor in glioblastoma,71 spliceosome inhibitor in AML,70 TGF-β inhibitor in pancreatic cancer,72 ULK1 inhibitor in M-TNBC cell lines,53 and PARP inhibitors in ovarian and breast cancer.73 In addition, PRMT5 inhibition in combination with gemcitabine leads to synthetic lethality in pancreatic cancer.74
Conclusion
Our study highlights the benefit of targeting PRMT5 in combination with some chemotherapies and with inhibitors targeting the HER family members. The most promising combinations with PRMT5 inhibition were obtained with (i) cisplatin in TNBC cells, (ii) EGFR inhibition in EGFR-high TNBC cells, and (iii) HER2 inhibition in HER2-low breast cancer (MDA-MB-453) and in HER2-positive breast cancer cells. In future studies, we will further evaluate the combination of inhibitors targeting PRMT5 with HER family members in additional cell lines, to strengthen our findings. These studies may identify EGFR and HER2 as biomarkers of response to these drug combinations, aiding in the stratification of patients who will most likely respond to the treatment. To translate these in vitro results to preclinical studies, we will assess the therapeutic advantage of the most promising combinations on tumor growth in various TNBC PDX models, chosen based on the expression of the appropriate biomarker of response.
Abbreviations
ADC, antibody-drug conjugate; AML, acute myeloid leukemia; AR, androgen receptor; BCSC, breast cancer stem cells; BL1, basal-like 1; BL2, basal-like 2; CFA, colony formation assay; EGFR, epidermal growth factor receptor; ER, estrogen receptor; FDA, Food and Drug Administration; HER2, human epidermal growth factor receptor 2; IC50, half-maximal inhibitory concentration; IHC, immunohistochemistry; LAR, luminal androgen receptor; M, mesenchymal; MCL, mantle cell lymphoma; PARP, poly (ADP-ribose) polymerase; PDX, patient-derived xenograft; PR, progesterone receptor; PRMT, protein arginine methyltransferase; SASP, senescence-associated secretory phenotype; SD, standard deviation; TIC, tumor-initiating cells; TNBC, Triple-negative breast cancer.
Consent for Publication
All authors read and approved the final manuscript.
Acknowledgments
We thank Dylan Payet (BCBG lab, Institut Curie) for performing some experiments and Dr. David Gentien (Genomics platform, Institut Curie) for performing the microarrays on the cell lines. We are grateful to Virginie Maire (BCBG, Institut Curie) and Dr. Ramón García-Areas (BCBG, Institut Curie) for critical reading of the manuscript. We also thank Dr. Lisa McPherson (Division of Oncology, Stanford University School of Medicine, USA) for verifying the English language of the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Institut Curie. S.S. was funded by the European Union’s Horizon 2020 Research and Innovation Program (Marie Skłodowska-Curie grant agreement No 666003). R.D. was financed by the French Embassy of Lebanon and the Lebanese University (Safar Volet 1).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397(10286):1750–1769. doi:10.1016/S0140-6736(20)32381-3
2. Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. Lancet. 2017;389(10087):2430–2442. doi:10.1016/S0140-6736(16)32454-0
3. Li Y, Tsang JY, Tam F, Loong T, Tse GM. Comprehensive characterization of HER2-low breast cancers: implications in prognosis and treatment. EBioMedicine. 2023;91:104571. doi:10.1016/j.ebiom.2023.104571
4. Popović M, Silovski T, Križić M, Dedić Plavetić N. HER2 low breast cancer: a new subtype or a trojan for cytotoxic drug delivery? Int J Mol Sci. 2023;24(9):8206. doi:10.3390/IJMS24098206
5. Zagami P, Carey LA. Triple negative breast cancer: pitfalls and progress. NPJ Breast Cancer. 2022;8(1):95. doi:10.1038/s41523-022-00468-0
6. Barchiesi G, Roberto M, Verrico M, Vici P, Tomao S, Tomao F. Emerging role of PARP inhibitors in metastatic triple negative breast cancer. Current scenario and future perspectives. Front Oncol. 2021;11:769280. doi:10.3389/fonc.2021.769280
7. Li Y, Zhang H, Merkher Y, et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol. 2022;15(1):121. doi:10.1186/s13045-022-01341-0
8. Almansour NM. Triple-negative breast cancer: a brief review about epidemiology, risk factors, signaling pathways, treatment and role of artificial intelligence. Front Mol Biosci. 2022;9:836417. doi:10.3389/fmolb.2022.836417
9. Park SY, Choi JH, Nam JS. Targeting cancer stem cells in triple-negative breast cancer. Cancers. 2019;11(7):965. doi:10.3390/cancers11070965
10. Kumar H, Gupta NV, Jain R, et al. A review of biological targets and therapeutic approaches in the management of triple-negative breast cancer. J Adv Res. 2023. doi:10.1016/j.jare.2023.02.005
11. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi:10.1172/jci45014
12. Lehmann BD, Jovanović B, Chen X, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One. 2016;11(6):e0157368. doi:10.1371/journal.pone.0157368
13. Lee J. Current treatment landscape for early triple-negative breast cancer (TNBC). J Clin Med. 2023;12(4):1524. doi:10.3390/JCM12041524
14. Denduluri N, Somerfield MR, Eisen A, et al. Selection of optimal adjuvant chemotherapy regimens for human epidermal growth factor receptor 2 (HER2) -negative and adjuvant targeted therapy for HER2-positive breast cancers: an American Society of Clinical Oncology guideline adaptation of the Cancer Care Ontario clinical practice guideline. J Clin Oncol. 2016;34(20):2416–2427. doi:10.1200/JCO.2016.67.0182
15. Singh DD, Parveen A, Yadav DK. Role of PARP in TNBC: mechanism of inhibition, clinical applications, and resistance. Biomedicines. 2021;9(11):1512. doi:10.3390/biomedicines9111512
16. Tung N, Garber JE. PARP inhibition in breast cancer: progress made and future hopes. NPJ Breast Cancer. 2022;8(1):47. doi:10.1038/s41523-022-00411-3
17. Tarantino P, Corti C, Schmid P, et al. Immunotherapy for early triple negative breast cancer: research agenda for the next decade. NPJ Breast Cancer. 2022;8(1):23. doi:10.1038/s41523-022-00386-1
18. Jeong JH, Kim SB. Antibody-drug conjugates targeting Trop-2: clinical developments in early breast cancer therapy. Breast. 2022;66:199–203. doi:10.1016/j.breast.2022.10.015
19. Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6(8):1609–1623.
20. You KS, Yi YW, Cho J, Park JS, Seong YS. Potentiating therapeutic effects of epidermal growth factor receptor inhibition in triple-negative breast cancer. Pharmaceuticals. 2021;14(6):589. doi:10.3390/ph14060589
21. Maennling AE, Tur MK, Niebert M, et al. Molecular targeting therapy against EGFR family in breast cancer: progress and future potentials. Cancers. 2019;11(12):1826. doi:10.3390/cancers11121826
22. Chan-Penebre E, Kuplast KG, Majer CR, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11(6):432–437. doi:10.1038/nchembio.1810
23. Vinet M, Suresh S, Maire V, et al. Protein arginine methyltransferase 5: a novel therapeutic target for triple-negative breast cancers. Cancer Med. 2019;8(5):2414–2428. doi:10.1002/cam4.2114
24. Larsen SC, Sylvestersen KB, Mund A, et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal. 2016;9(443):rs9. doi:10.1126/scisignal.aaf7329
25. Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell. 2017;65(1):8–24. doi:10.1016/j.molcel.2016.11.003
26. Jarrold J, Davies CC. PRMTs and arginine methylation: cancer’s best-kept secret? Trends Mol Med. 2019;25(11):993–1009. doi:10.1016/j.molmed.2019.05.007
27. Wu Q, Schapira M, Arrowsmith CH, Barsyte-Lovejoy D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat Rev Drug Discov. 2021;20(7):509–530. doi:10.1038/s41573-021-00159-8
28. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13(1):37–50. doi:10.1038/nrc3409
29. Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. 2019;20(10):642–657. doi:10.1038/s41580-019-0155-x
30. Guccione E, Schwarz M, Di Tullio F, Mzoughi S. Cancer synthetic vulnerabilities to protein arginine methyltransferase inhibitors. Curr Opin Pharmacol. 2021;59:33–42. doi:10.1016/j.coph.2021.04.004
31. Suresh S, Huard S, Dubois T. CARM1/PRMT4: making its mark beyond its function as a transcriptional coactivator. Trends Cell Biol. 2021;31(5):402–417. doi:10.1016/j.tcb.2020.12.010
32. Suresh S, Huard S, Brisson A, et al. PRMT1 regulates EGFR and wnt signaling pathways and is a promising target for combinatorial treatment of breast cancer. Cancers. 2022;14(2):306. doi:10.3390/cancers14020306
33. Wu Q, Nie DY, Ba-alawi W, et al. PRMT inhibition induces a viral mimicry response in triple-negative breast cancer. Nat Chem Biol. 2022;18(8):821–830. doi:10.1038/s41589-022-01024-4
34. Suresh S, Vinet M, Dakroub R, et al. Expression, localization and prognosis association of MEP50 in breast cancer. Cancers. 2022;14(19):4766. doi:10.3390/cancers14194766
35. Zhou Z, Feng Z, Hu D, et al. A novel small-molecule antagonizes PRMT5-mediated KLF4 methylation for targeted therapy. EBioMedicine. 2019;44:98–111. doi:10.1016/j.ebiom.2019.05.011
36. Wu Y, Wang Z, Zhang J, Ling R. Elevated expression of protein arginine methyltransferase 5 predicts the poor prognosis of breast cancer. Tumour Biol. 2017;39(4):1010428317695917. doi:10.1177/1010428317695917
37. Feustel K, Falchook GS. Protein arginine methyltransferase 5 (PRMT5) inhibitors in oncology clinical trials: a review. J Immunother Precis Oncol. 2022;5(3):58–67. doi:10.36401/jipo-22-1
38. Wang X, Qiu T, Wu Y, et al. Arginine methyltransferase PRMT5 methylates and stabilizes KLF5 via decreasing its phosphorylation and ubiquitination to promote basal-like breast cancer. Cell Death Differ. 2021;28(10):2931–2945. doi:10.1038/s41418-021-00793-0
39. Gerhart SV, Kellner WA, Thompson C, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep. 2018;8(1):9711. doi:10.1038/s41598-018-28002-y
40. Tang PA, Tsao MS, Moore MJ. A review of erlotinib and its clinical use. Expert Opin Pharmacother. 2006;7(2):177–193. doi:10.1517/14656566.7.2.177
41. Minna JD, Dowell J. Erlotinib hydrochloride. Nat Rev Drug Discov. 2005;Suppl:S14–5. doi:10.1038/nrd1612
42. Collins DM, Conlon NT, Kannan S, et al. Preclinical characteristics of the irreversible Pan-HER kinase inhibitor neratinib compared with lapatinib: implications for the treatment of HER2-Positive and HER2-mutated breast cancer. Cancers. 2019;11(6):737. doi:10.3390/cancers11060737
43. Conlon NT, Kooijman JJ, van Gerwen SJC, et al. Comparative analysis of drug response and gene profiling of HER2-targeted tyrosine kinase inhibitors. Br J Cancer. 2021;124(7):1249–1259. doi:10.1038/s41416-020-01257-x
44. Di Veroli GY, Fornari C, Wang D, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32(18):2866–2868. doi:10.1093/bioinformatics/btw230
45. Zhao L, Au JLS, Wientjes MG. Comparison of methods for evaluating drug-drug interaction. Front Biosci. 2010;2(1):241. doi:10.2741/E86
46. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
47. Maire V, Mahmood F, Rigaill G, et al. LRP8 is overexpressed in estrogen‐negative breast cancers and a potential target for these tumors. Cancer Med. 2019;8(1):325. doi:10.1002/CAM4.1923
48. Lerebours F, Vacher S, Guinebretiere JM, et al. Hemoglobin overexpression and splice signature as new features of inflammatory breast cancer? J Adv Res. 2021;28:77. doi:10.1016/J.JARE.2020.08.009
49. Tang J, Wennerberg K, Aittokallio T. What is synergy? The Saariselkä agreement revisited. Front Pharmacol. 2015;6:181. doi:10.3389/fphar.2015.00181
50. Meyer CT, Wooten DJ, Lopez CF, Quaranta V. Charting the fragmented landscape of drug synergy. Trends Pharmacol Sci. 2020;41(4):266–280. doi:10.1016/j.tips.2020.01.011
51. Smith SE, Mellor P, Ward AK, et al. Molecular characterization of breast cancer cell lines through multiple omic approaches. Br Cancer Res. 2017;19(1):1–12. doi:10.1186/S13058-017-0855-0/FIGURES/5
52. Yin S, Liu L, Brobbey C, et al. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat Commun. 2021;12(1):3444. doi:10.1038/s41467-021-23833-2
53. Brobbey C, Yin S, Liu L, et al. Autophagy dictates sensitivity to PRMT5 inhibitor in breast cancer. Sci Rep. 2023;13(1):1–13. doi:10.1038/s41598-023-37706-9
54. Mueller HS, Fowler CE, Dalin S, et al. Acquired resistance to PRMT5 inhibition induces concomitant collateral sensitivity to paclitaxel. Proc Natl Acad Sci U S A. 2021;118(34). doi:10.1073/pnas.2024055118
55. Musiani D, Giambruno R, Massignani E, et al. PRMT1 is recruited via DNA-PK to chromatin where it sustains the senescence-associated secretory phenotype in response to cisplatin. Cell Rep. 2020;30(4):1208–1222.e9. doi:10.1016/j.celrep.2019.12.061
56. Epstein DM, Buck E. Old dog, new tricks: extracellular domain arginine methylation regulates EGFR function. J Clin Invest. 2015;125(12):4320–4322. doi:10.1172/jci85001
57. Liao HW, Hsu JM, Xia W, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125(12):4529–4543. doi:10.1172/jci82826
58. Nakai K, Xia W, Liao HW, Saito M, Hung MC, Yamaguchi H. The role of PRMT1 in EGFR methylation and signaling in MDA-MB-468 triple-negative breast cancer cells. Breast Cancer. 2018;25(1):74–80. doi:10.1007/s12282-017-0790-z
59. Wang WJ, Hsu JM, Wang YN, et al. An essential role of PRMT1-mediated EGFR methylation in EGFR activation by ribonuclease 5. Am J Cancer Res. 2019;9(1):180–185.
60. Hsu JM, Chen CT, Chou CK, et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13(2):174–181. doi:10.1038/ncb2158
61. Yao B, Gui T, Zeng X, et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. 2021;13(1):58. doi:10.1186/s13073-021-00871-5
62. Huang L, Zhang XO, Rozen EJ, et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat Commun. 2022;13(1):3955. doi:10.1038/s41467-022-31645-1
63. Modi S, Jacot W, Yamashita T, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. New England J Med. 2022;387(1):9–20. doi:10.1056/NEJMOA2203690/SUPPL_FILE/NEJMOA2203690_DATA-SHARING.PDF
64. FDA. FDA approves first targeted therapy for HER2-Low breast cancer. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-her2-low-breast-cancer.
65. Hu R, Zhou B, Chen Z, et al. PRMT5 inhibition promotes PD-L1 expression and immuno-resistance in lung cancer. Front Immunol. 2021;12:722188. doi:10.3389/fimmu.2021.722188
66. Che Y, Liu Y, Yao Y, et al. Exploiting PRMT5 as a target for combination therapy in mantle cell lymphoma characterized by frequent ATM and TP53 mutations. Blood Cancer J. 2023;13(1):27. doi:10.1038/s41408-023-00799-6
67. AbuHammad S, Cullinane C, Martin C, et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc Natl Acad Sci U S A. 2019;116(36):17990–18000. doi:10.1073/pnas.1901323116
68. Yang L, Ma DW, Cao YP, et al. PRMT5 functionally associates with EZH2 to promote colorectal cancer progression through epigenetically repressing CDKN2B expression. Theranostics. 2021;11(8):3742–3759. doi:10.7150/thno.53023
69. Fedoriw A, Rajapurkar SR, O’Brien S, et al. Anti-tumor activity of the type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell. 2019;36(1):100–114.e25. doi:10.1016/j.ccell.2019.05.014
70. Fong JY, Pignata L, Goy PA, et al. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell. 2019;36(2):194–209.e9. doi:10.1016/j.ccell.2019.07.003
71. Holmes B, Benavides-Serrato A, Saunders JT, et al. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J Neurooncol. 2019;145(1):11–22. doi:10.1007/s11060-019-03274-0
72. Hong E, Barczak W, Park S, et al. Combination treatment of T1-44, a PRMT5 inhibitor with Vactosertib, an inhibitor of TGF-β signaling, inhibits invasion and prolongs survival in a mouse model of pancreatic tumors. Cell Death Dis. 2023;14(2):93. doi:10.1038/s41419-023-05630-5
73. O’Brien S, Butticello M, Thompson C, et al. Inhibiting PRMT5 induces DNA damage and increases anti-proliferative activity of Niraparib, a PARP inhibitor, in models of breast and ovarian cancer. BMC Cancer. 2023;23(1):775. doi:10.1186/S12885-023-11260-Z
74. Wei X, Yang J, Adair SJ, et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc Natl Acad Sci U S A. 2020;117(45):28068–28079. doi:10.1073/pnas.2009899117
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.