Back to Journals » Journal of Inflammation Research » Volume 19
Sphingolipid-Neuroinflammation Axis in Parkinson’s Disease: Focus on S1P/SPHK1-NF‑κB Signaling
Authors Kong X, Wang H, Dong J, Cheng X, Wei J
Received 13 May 2026
Accepted for publication 18 June 2026
Published 9 July 2026 Volume 2026:19 621246
DOI https://doi.org/10.2147/JIR.S621246
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Xiangrui Kong,1,2 Hao Wang,2 Jun Dong,3 Xiangshu Cheng,1,2 Jianshe Wei1,2
1Institute for Sports and Brain Health, School of Physical Education, Henan University, Kaifeng, 475004, People’s Republic of China; 2Key Laboratory of Neurological Diseases of Kaifeng, Department of Neurology, Huaihe Hospital of Henan University, School of Life Sciences, Henan University, Kaifeng, 475004, People’s Republic of China; 3Department of Cardiovascular Medicine, Tianjin Chest Hospital, Tianjin, 300222, People’s Republic of China
Correspondence: Xiangshu Cheng, Email [email protected] Jianshe Wei, Email [email protected]
Abstract: Parkinson’s disease (PD) is the second most common neurodegenerative disorder worldwide. Its core pathological features comprise the selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), abnormal aggregation of α‑synuclein (α‑syn) into Lewy bodies, and progressive amplification of neuroinflammation, mitochondrial dysfunction, oxidative stress and lipid metabolism disorders. As a key bioactive sphingolipid, sphingosine‑1‑phosphate (S1P) is synthesized by sphingosine kinase 1 (SPHK1) and regulates immune activation, cell survival and inflammatory responses through intracellular signaling and binding to S1P receptors (S1PR1‑5). Nuclear factor‑κB (NF‑κB) is a key transcription factor closely implicated in the regulation of neuroinflammatory responses, modulating pro‑inflammatory cytokine release, microglial polarization and neuronal apoptosis. Cumulative clinical and preclinical studies have verified elevated S1P levels and activated SPHK1 in PD patients, accumulating data support a correlative link between the S1P/SPHK1‑NF‑κB signaling axis and α‑syn pathology, microglial overactivation, neuroinflammation and dopaminergic neuron degeneration, while multiple mechanistic ambiguities remain unresolved, though several mechanistic ambiguities and translational hurdles remain unaddressed. This review systematically outlines the molecular basis and cell‑specific mechanisms of this axis, its crosstalk with α‑syn aggregation, mitochondrial damage, blood‑brain barrier (BBB) leakage and gut‑brain axis disturbance during PD progression. We further objectively summarize the potential of this axis as auxiliary diagnostic biomarker and druggable therapeutic target, alongside prominent bottlenecks including insufficient detection standardization, interspecies drug response discrepancy and unsatisfactory clinical transformation of preclinical compounds, as well as pending core scientific questions restricting subsequent research advancement. Collectively, this review provides a balanced theoretical framework for exploring PD pathogenesis and developing precise therapy, with critical discussion on existing limitations to guide follow-up basic and translational investigations.
Keywords: Parkinson’s disease, sphingosine-1-phosphate, sphingosine kinase 1, nuclear factor-κB, microglia
Introduction
Parkinson’s disease (PD) is a highly prevalent movement disorder in the middle-aged and elderly population. It is characterized by cardinal motor symptoms including resting tremor, rigidity, bradykinesia and postural balance impairment, together with a wide spectrum of non-motor manifestations such as cognitive decline, depression, sleep disturbances and autonomic nervous system dysfunction. With the progressive aging of the global population, the incidence of PD continues to rise, placing a heavy burden on social healthcare and family caregiving.1 To date, the exact etiology and pathogenesis of PD have not been fully elucidated, and the traditional single-factor hypothesis focusing merely on dopaminergic neuron loss fails to explain the multisystem involvement, progressive deterioration and heterogeneous characteristics of this disease. Current academic consensus indicates that PD arises from a complex pathological network driven by the interaction of multiple risk factors, including genetic susceptibility, environmental toxin exposure, aging, gut microbiota dysbiosis, neuroinflammation, protein misfolding, mitochondrial defects and lipid metabolism imbalance. Among them, chronic neuroinflammation-related neurodegenerative cascade is hypothesized to serve as one important linking factor across diverse pathogenic contributors and promotes the spread of pathological lesions from the local substantia nigra to the whole brain.2,3
Neuroinflammation exerts a dual-edged sword effect in PD. Under physiological conditions, moderate activation of microglia and astrocytes contributes to clearing α-syn aggregates, repairing damaged neurons and maintaining central nervous system (CNS) homeostasis. However, in the pathological microenvironment of PD, persistent stimulation by pathological α-synuclein preformed fibrils (α-syn PFFs), damage-associated molecular patterns (DAMPs) released from injured mitochondria, and gut microbiota translocation products induces the emergence of pro-inflammatory reactive microglia and the transformation of astrocytes into reactive A1 subtype. These activated glial cells secrete abundant pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6 and interferon-γ (IFN-γ), and trigger the activation of core inflammatory cascades including nuclear factor-κB (NF-κB), mitogen-activated protein kinase (MAPK) and NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome, ultimately accelerating dopaminergic neuronal apoptosis, synaptic loss and Lewy body propagation.4–6
Importantly, an unresolved critical debate persists regarding whether neuroinflammation acts as an initiating causal trigger or a secondary downstream consequence in PD pathogenesis. A body of preclinical evidence supports that isolated sustained microglial overactivation is capable of initiating nigral dopaminergic degeneration and de novo α-syn aggregation, favoring the causal role of inflammation.7,8 In contrast, multiple human postmortem analyses and longitudinal clinical cohort studies observe obvious neuroinflammatory infiltration occurs subsequent to neuronal injury and abnormal α-syn deposition, suggesting inflammation develops secondarily in response to pre-existing cellular damage.9 Such conflicting findings hinder the clarification of PD pathological sequence and highlight the necessity to dissect key signaling bridges linking metabolic disturbance and neuroinflammation. In this context, identifying the pivotal signaling axis connecting lipid metabolism disturbance and neuroinflammation has become a core breakthrough to decipher the complex pathological network of PD and develop neuroprotective as well as disease-modifying therapies.
As an essential regulatory system for cell membrane structure and signal transduction, sphingolipid metabolism plays a fundamental role in CNS development, immune modulation, neuronal survival and synaptic plasticity. Dysfunction of sphingolipid homeostasis is closely implicated in multiple neurodegenerative diseases, such as Alzheimer’s disease, PD and multiple sclerosis.10,11 As a core bioactive sphingolipid metabolite, S1P is produced via the phosphorylation of sphingosine catalyzed by sphingosine kinase 1 (SPHK1)/SPHK2. SPHK1 and SPHK2 exert distinctly divergent physiological and pathological functions: physiologically, membrane-localized SPHK1 responds rapidly to extracellular stimuli to mediate inducible signaling, while nucleus- and mitochondria-enriched SPHK2 maintains constitutive basal S1P levels to regulate regular cell cycle and mitochondrial homeostasis; pathologically, only SPHK1 undergoes dramatic upregulation and phosphorylation upon inflammatory and toxic stimulation to produce excessive proinflammatory S1P, whereas SPHK2 expression and enzymatic activity remain nearly unchanged under PD-related pathological conditions and barely participate in neuroinflammatory amplification.12,13
Through intracellular direct actions and extracellular binding to sphingosine-1-phosphate (S1P) receptors (S1PR1-5), S1P regulates cell proliferation, migration, apoptosis, inflammatory responses and vascular permeability.14 SPHK1 is mainly localized on the plasma membrane and can be rapidly activated in response to inflammation, oxidative stress and growth factors, acting as a crucial inflammation-driven enzyme for S1P synthesis. In contrast, SPHK2 is primarily distributed in the nucleus and mitochondria and participates in cell cycle regulation and apoptosis. The two isoforms exhibit distinct differences in cellular distribution, functional modulation and pathological roles in the CNS.15,16 NF-κB is an evolutionarily conserved transcription factor family composed of heterodimers such as p50/p65 and p52/RelB. Under resting conditions, NF-κB is sequestered in the cytoplasm by binding to IκB proteins. Upon stimulation by pro-inflammatory cytokines, DAMPs and lipid signals, the IκB kinase (IKK) complex induces IκB phosphorylation and degradation, allowing NF-κB dimers to translocate into the nucleus and bind to the κB sequences on target gene promoters, thereby initiating the transcription of pro-inflammatory cytokines, chemokines, adhesion molecules and anti-apoptotic proteins. As a master switch of central neuroinflammation, excessive NF-κB activation is directly correlated with nigral dopaminergic neuron death and α-syn pathological propagation in PD.17
In recent years, numerous landmark studies have demonstrated significantly elevated S1P levels in the cerebrospinal fluid of PD patients. The protein expression and enzymatic activity of SPHK1 are markedly upregulated in the substantia nigra and striatum in a strictly microglia-specific manner.17 Pathological α-syn PFFs specifically induce SPHK1 phosphorylation at the Ser225 site in microglia, with no obvious regulatory effects on SPHK1 in neurons and astrocytes, nor significant alterations in SPHK2 expression across different cell types. Further mechanistic studies based largely on cellular and rodent models have validated that S1P generated by SPHK1 acts as a co-activator of TNF receptor-associated factor 2 (TRAF2), enhances its E3 ubiquitin ligase activity, facilitates IKK complex activation and IκB degradation, and promotes NF-κB nuclear translocation and pro-inflammatory transcriptional activation, which contributes to the formation of the S1P/SPHK1-NF-κB inflammatory signaling axis.18 This axis participates in the shift of microglia toward pro-inflammatory reactive status and can amplify neuronal oxidative stress via paracrine effects under experimental conditions, facilitate α-syn phosphorylation at the Ser129 site and insoluble aggregation, impair blood–brain barrier (BBB) integrity, and aggravate peripheral immune cell infiltration. Collectively, these events may constitute a pathological closed loop of “microglial activation-neuroinflammation-neuronal damage-α-syn propagation” in preclinical systems, which is regarded as one plausible nodal connection linking lipid metabolism disorder, neuroinflammation and proteinopathy in PD pathogenesis, while sufficient human pathological evidence to confirm such a central role is still lacking.17,19,20
Based on the cutting-edge advances in PD research, this review focuses on the S1P/SPHK1-NF-κB axis and elaborates from five dimensions: molecular foundation, cell-specific regulation, pathological network crosstalk, clinical transformation and therapeutic prospects. By integrating the latest evidence from clinical samples, animal models, cell experiments and multi-omics data, we construct a three-dimensional regulatory network of this axis in PD onset and progression, and analyze its crosstalk with classic PD pathological mechanisms, including α-syn pathology, mitochondrial dysfunction, oxidative stress, gut-brain axis disorder and BBB damage. Meanwhile, we summarize the research progress of targeted drugs and the biomarker potential of this axis, as well as current bottlenecks in clinical application. This review provides comprehensive and frontier theoretical references for mechanistic innovation and precise treatment of PD (Figure 1).
 |
Figure 1 Mechanism of S1P/SPHK1-NF-κB signaling axis regulation of pathological progression in PD. Multiple pathogenic stimuli specifically activate the microglial SPHK1-S1P pathway, which mediates NF-κB activation via TRAF2, drives glial transition toward pro-inflammatory reactive status, amplifies neuroinflammatory cascades, induces dopaminergic neuron damage, α-syn pathology propagation and BBB disruption, forming a pathological loop that ultimately leads to motor and non-motor symptoms of PD. Of note, resting homeostatic microglia present ramified morphology, while pro-inflammatory reactive microglia shift to amoeboid morphology upon pathological stimulation, consistent with typical morphological characteristics of microglial transformation. |
Molecular Basis and Regulatory Features of the S1P/SPHK1-NF-κB Axis
S1P Metabolic Network and the Structure, Activation, and Functional Differentiation of SPHK1
S1P is a monophosphorylated sphingosine (Sph) derivative generated through the hydrolysis of sphingomyelin (SM) by sphingomyelinase (SMase) to produce ceramide (Cer), followed by catalysis via ceramidase (CDase) to form Sph, and final phosphorylation by SPHK1/SPHK2. Its metabolic balance is maintained by synthases (SPHK1/2), degrading enzymes (S1P lyase [S1PL], S1P phosphatase [SGPP1/2]), and transporters (SPNS lysolipid transporter 2, sphingosine-1-phosphate transporter 2 [Spns2], major facilitator superfamily domain containing 2B [Mfsd2b]), forming a sphingolipid biostat of “ceramide-Sph-S1P”. Imbalance in this homeostasis directly determines cell survival and inflammatory phenotypes.21–23 Ceramide tends to induce apoptosis, oxidative stress, and inflammatory activation, whereas S1P mainly mediates cell survival, migration, and bidirectional anti-inflammatory/pro-inflammatory regulation, serving as a key lipid marker of pathological states in the CNS.
As a critical rate-limiting enzyme for S1P synthesis, SPHK1 belongs to the dihydrosphingosine kinase family, with its encoding gene located on chromosome 17 and a protein molecular weight of approximately 43 kDa. It contains an N-terminal catalytic domain, a C-terminal regulatory domain, and a membrane-binding region, and its activation mechanism is characterized by multi-signal integration: under resting conditions, SPHK1 exists in an inactive form in the cytoplasm. Upon stimulation by inflammatory factors (TNF-α, IL-1β), growth factors (platelet-derived growth factor [PDGF], epidermal growth factor [EGF]), oxidative stress, pathological α-syn, lipopolysaccharide (LPS), etc., SPHK1 undergoes conformational change and translocates to the inner side of the cell membrane via phosphorylation at Ser225 mediated by upstream kinases such as protein kinase C (PKC), extracellular regulated protein kinases (ERK), and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), then binds to Sph substrate to rapidly catalyze S1P production. In addition, SPHK1 stability and activity are regulated by post-translational modifications including ubiquitination, acetylation, and SUMOylation, among which TRAF2-mediated K63 ubiquitination is a key step for its activation and binding to the NF-κB signaling complex.24,25 In contrast to SPHK1, SPHK2 is mainly localized in the nucleus, mitochondria, and endoplasmic reticulum; its activation depends on cell cycle and metabolic signals, and the S1P it catalyzes is primarily involved in nuclear receptor regulation, mitochondrial respiration, and apoptosis. SPHK2 plays a weak role in inflammatory regulation in the CNS and shows no significant expression changes in PD pathology. The distinct cellular localization, activation mechanisms, and functional differentiation of SPHK1/SPHK2 determine the specific regulatory role of the S1P/SPHK1-NF-κB axis in PD neuroinflammation.15,26
Signal transduction of S1P in the CNS occurs via intracellular and extracellular pathways: in the intracellular pathway, S1P acts directly as a second messenger, binding to intracellular proteins such as TRAF2, protein phosphatase 2A (PP2A), and histone deacetylase 1 (HDAC1) to regulate signaling pathways including NF-κB, MAPK, and mammalian target of rapamycin (mTOR), among which the S1P-TRAF2 interaction represents one important intracellular pathway facilitating NF-κB activation. In the extracellular pathway, S1P is secreted extracellularly via the Spns2 transporter and binds to S1PR1-5 G protein-coupled receptors on the cell membrane,21,22 activating downstream signals through G protein subtypes such as Gi, Gq, and G12/13. S1PR1 is mainly expressed in microglia, vascular endothelial cells, and lymphocytes, mediating immune cell migration, BBB integrity, and microglial activation; S1PR3 is primarily involved in pro-inflammatory polarization of microglia. These two are the main receptor subtypes through which S1P regulates central neuroinflammation. Notably, S1P in the CNS is mainly synthesized and secreted by microglia and astrocytes, with weak synthetic capacity in neurons. This cell-source specificity further establishes microglia as the core regulatory cells of the S1P/SPHK1-NF-κB axis.21,27–29
Canonical and Non-Canonical Activation of the NF-κB Signaling Pathway and Central Neural Regulation
The NF-κB family comprises five members: RelA (p65), RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2), which exert transcriptional regulatory functions by forming homodimers/heterodimers. Among them, the p50/p65 heterodimer is the most predominant active form in the CNS, responsible for regulating the expression of most pro-inflammatory genes.30 NF-κB activation is divided into the canonical pathway and the non-canonical pathway: the canonical pathway is the core activation mode of PD neuroinflammation, initiated by stimuli such as TNF-α, IL-1β, LPS, α-syn preformed fibrils (α-syn PFFs), and S1P. It recruits TRAF2/6 via cell membrane receptors (TNF receptor 1 [TNFR1], IL-1 receptor [IL-1R)], Toll-like receptor 4 [TLR4]) to activate the inhibitor of NF-κB kinase (IKK) complex (IKKα, IKKβ, NF-κB essential modulator [NEMO]). IKKβ phosphorylates IκBα at Ser32/36, leading to IκBα degradation via the ubiquitin-proteasome pathway. The released p50/p65 dimer rapidly translocates into the nucleus, binds to the 5′-GGGRNNYYCC-3′ κB motif in target gene promoters, and initiates transcription of pro-inflammatory factors including TNF-α, IL-1β, IL-6, inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and chemokines (C-C motif chemokine ligand 2 [CCL2], C-X-C motif chemokine ligand 10 [CXCL10]). It also upregulates anti-apoptotic proteins (B-cell lymphoma-2 [Bcl-2], X-linked inhibitor of apoptosis protein [XIAP]) to maintain microglial survival and establish a persistent inflammatory state.31,32 The non-canonical pathway relies on NF-κB-inducing kinase (NIK) and IKKα, mainly regulating lymphocyte development and secondary lymphoid organ formation, with limited roles in PD neuroinflammation.33
In the CNS, NF-κB activation exhibits strict cell specificity and spatiotemporal characteristics: under physiological conditions, NF-κB in neurons maintains low-level basal activity, participating in synaptic plasticity, learning and memory, and neuronal survival regulation;34 as central innate immune cells, microglia have an NF-κB pathway highly sensitive to pathological stimuli, serving as the main source of neuroinflammation;35 NF-κB activation in astrocytes mediates reactive gliosis and pro-inflammatory factor release, amplifying inflammatory signals.36,37 In the pathological microenvironment of PD, excessive NF-κB activation displays dual toxicity: sustained NF-κB activation in microglia drives the transition toward a pro-inflammatory neurotoxic reactive state, releasing large amounts of neurotoxic factors that directly damage dopaminergic neurons; abnormal NF-κB activation in neurons induces the expression of pro-apoptotic proteins (Bcl-2-associated X protein [Bax], cysteinyl aspartate-specific proteinase 3 [Caspase-3]), exacerbating mitochondrial dysfunction and oxidative stress, while promoting α-syn phosphorylation and aggregation.38,39 Furthermore, NF-κB participates in PD pathological processes by regulating autophagy, mitophagy, endoplasmic reticulum stress, and other pathways. Its activity imbalance is a key molecular switch for central neuroinflammation shifting from physiological defense to pathological damage.
Core Molecular Mechanism of the S1P/SPHK1-NF-κB Axis
The core regulatory mechanism of the S1P/SPHK1-NF-κB axis is that S1P generated by SPHK1 activation directly mediates the cascade activation of the NF-κB canonical pathway, constructing a complete signal transduction chain from upstream pathological stimuli to downstream inflammatory output. This process shows high specificity and efficiency in microglia. As a typical damage-associated molecular pattern (DAMP), α-syn PFFs serve as the core pathological molecule of PD, which can be recognized and bound by multiple receptors on the microglial membrane, such as TLR4, cluster of differentiation 36 (CD36), P2X7 and Fc gamma receptor (FcγR), thereby further activating intracellular signaling pathways including PKC, ERK and PI3K/Akt. These pathways directly catalyze SPHK1 phosphorylation at Ser225, promoting the translocation of inactive cytoplasmic SPHK1 to the inner cell membrane and synchronously upregulating SPHK1 gene transcription and protein expression.17,40–44 This activation process has strict cell selectivity: α-syn PFFs only induce SPHK1 activation in microglia, with no obvious regulatory effect on SPHK1 in neurons and astrocytes, nor do they alter SPHK2 expression levels. This specificity stems from the high expression of α-syn recognition receptors and SPHK1 upstream activating kinases in microglia, laying the foundation for the cell-specific regulation of this axis.17
Activated membrane-bound SPHK1 rapidly catalyzes Sph to produce S1P. Intracellular S1P directly binds to the C-terminal RING domain of TRAF2, enhancing TRAF2 E3 ubiquitin ligase activity via allosteric activation, and promoting the association of TRAF2 with the IKK complex and NEMO to form the TRAF2-S1P-IKK signaling complex, which constitutes the core intracellular mechanism underlying S1P-mediated NF-κB activation.18,20 Meanwhile, microglia secrete excess S1P extracellularly via the Spns2 transporter, which acts on S1PR1/S1PR3 on the surface of themselves and adjacent microglia, astrocytes, and vascular endothelial cells. This further amplifies SPHK1 activation and inflammatory signals through the Gi/ERK and Gq/nuclear factor of activated T cells (NFAT) pathways, forming dual positive feedback loops of autocrine and paracrine signaling.45,46 The S1P-TRAF2 complex significantly improves the phosphorylation activation efficiency of IKKβ, accelerates the phosphorylation and ubiquitin-dependent degradation of IκBα at Ser32/36, and releases the p50/p65 dimer from cytoplasmic inhibition for rapid nuclear translocation.18,47,48 During this process, S1P also directly inhibits PP2A activity, reducing p65 dephosphorylation, prolonging the nuclear retention time and transcriptional activity of NF-κB, preventing its rapid inactivation, and thus sustaining persistent pro-inflammatory gene expression.49
Nuclear p50/p65 dimers bind to κB binding sites in the promoter regions of pro-inflammatory genes, upregulating the transcription and expression of key molecules including TNF-α, IL-1β, IL-6, iNOS, COX-2, and CCL2.50 These pro-inflammatory factors and chemokines further activate surrounding glial cells and infiltrating peripheral immune cells, and continuously activate the SPHK1-NF-κB axis through positive feedback, ultimately driving the progression of chronic neuroinflammation and progressive loss of dopaminergic neurons.51 Meanwhile, S1P activates the PI3K/Akt pathway via S1PR1, catalyzing p65 phosphorylation at Ser536 to enhance its transcriptional activity and further amplify inflammatory signals.52,53 As a metabolic precursor of S1P, ceramide exerts an antagonistic effect on the S1P/SPHK1-NF-κB axis by inhibiting SPHK1 activity, activating PP2A, and stabilizing IκB, indicating that the balance of sphingolipid metabolic homeostasis directly determines the activation intensity and duration of this axis.54,55
Cell Specificity and Regional Distribution Characteristics of the S1P/SPHK1-NF-κB Axis in the CNS
The regulation of the S1P/SPHK1-NF-κB axis in the CNS is not uniformly distributed, but exhibits cell-type specificity (Table 1), brain region specificity, and age dependence. This distributional difference directly determines the selective vulnerability of PD pathology. The substantia nigra pars compacta (SNpc), the core brain region affected in PD, shows significantly higher axis activation intensity in microglia and dopaminergic neurons than the cortex, hippocampus, and other brain regions, serving as an important molecular basis for the selective damage of dopaminergic neurons.17
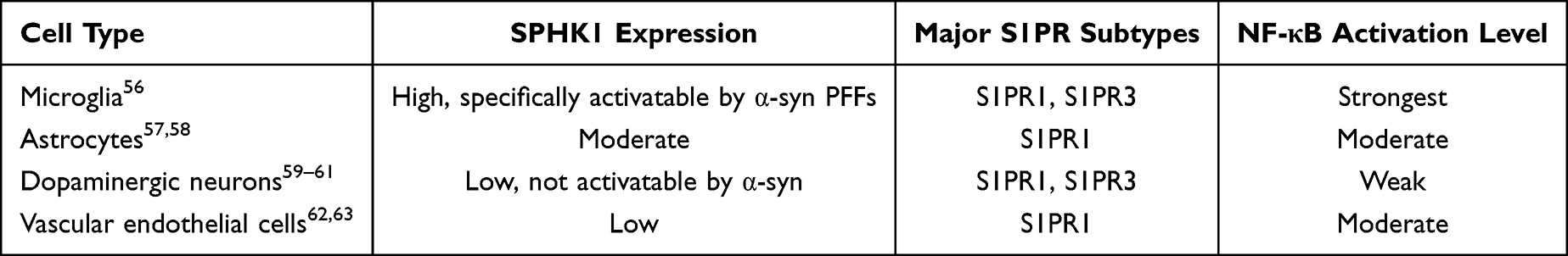 |
Table 1 Cell-Specificity and Functional Characteristics of the S1P/SPHK1-NF-κB Axis |
In terms of cell types, microglia are prominent cellular contributors to axis function and constitute a major source of S1P synthesis and secretion under pathological conditions based on preclinical findings: microglia highly express axis-related molecules such as SPHK1, S1PR1/S1PR3, and TLR4/CD36, respond rapidly to stimuli including α-syn PFFs and pro-inflammatory factors, and have the highest efficiency of SPHK1 activation and NF-κB nuclear translocation, acting as the primary initiators and amplifiers of neuroinflammation.56 Astrocytes express moderate levels of SPHK1 and S1PR, mainly receiving S1P and pro-inflammatory factors released by microglia via paracrine signaling to activate NF-κB and transform into reactive A1 subtypes. They release neurotoxic factors such as IL-1α, C1q, and TNF-α to exacerbate neuronal damage, and secrete chemokines to recruit peripheral immune cells and expand the inflammatory scope.57,58 Neurons express low levels of SPHK1, and α-syn PFFs cannot induce SPHK1 activation in neurons; however, neuronal cell membranes express S1PR1/S1PR3, which can receive S1P and pro-inflammatory factors released by microglia to activate the intracellular NF-κB pathway, inducing oxidative stress, mitochondrial damage, and apoptosis, while promoting α-syn Ser129 phosphorylation and aggregation, making neurons direct target cells of inflammatory damage.59–61 Vascular endothelial cells express S1PR1 and NF-κB; activation of the S1P/SPHK1-NF-κB axis disrupts the expression of tight junction proteins (zonula occludens-1 [ZO-1], occludin) in the BBB, increases vascular permeability, and promotes the infiltration of peripheral monocytes and T cells into the brain parenchyma, further exacerbating neuroinflammation.62,63
In terms of brain region distribution, PD-vulnerable regions such as the SNpc, ventral tegmental area (VTA), and striatum have higher microglial density, stronger basal SPHK1 expression and activation potential, and dopaminergic neurons are significantly more sensitive to oxidative stress and inflammatory factors than other neuron types. Thus, it is speculated that the activation intensity of this axis in the substantia nigra is much higher than that in non-vulnerable brain regions such as the cortex and cerebellum.64–66 Meanwhile, dopaminergic neurons in the substantia nigra are rich in iron ions, prone to mitochondrial dysfunction, and have higher oxidative stress levels; oxidative stress further activates SPHK1 and NF-κB, ultimately leading to the selective loss of dopaminergic neurons in the substantia nigra.67 In addition, as a major risk factor for PD, aging markedly enhances the activation sensitivity of the S1P/SPHK1‑NF‑κB axis by upregulating SPHK1 expression in microglia, reducing the activity of sphingolipid-degrading enzymes, and elevating basal NF-κB activity, thereby accelerating age-related neuroinflammation and neurodegeneration.68,69
Current understanding of signal transmission of the S1P/SPHK1-NF-κB axis between glial cells and neurons is dominated by the microglia-centered paracrine amplification model. Recent studies confirm that α-syn PFFs treatment specifically increases SPHK1 expression and activates SPHK1 in microglia, while SPHK2 shows no significant changes in any cell type. This cell specificity provides a molecular basis for the spatial heterogeneity of the axis.17,56 Using spatial transcriptomics, researchers have found significantly elevated S1P levels in the substantia nigra and striatum, with SPHK1 being the most significantly upregulated gene related to the “Sph metabolic process”. Furthermore, microglial SPHK1 knockdown or treatment with the specific inhibitor PF543 significantly alleviates α-syn PFFs-induced neuropathological changes, indicating that microglia are the core cell type for the axis to function in the substantia nigra.17,62,70 Deciphering this spatiotemporally dependent glia-neuron interaction heterogeneity will provide a theoretical basis for precise interventions targeting specific cells and brain regions (Figure 2).
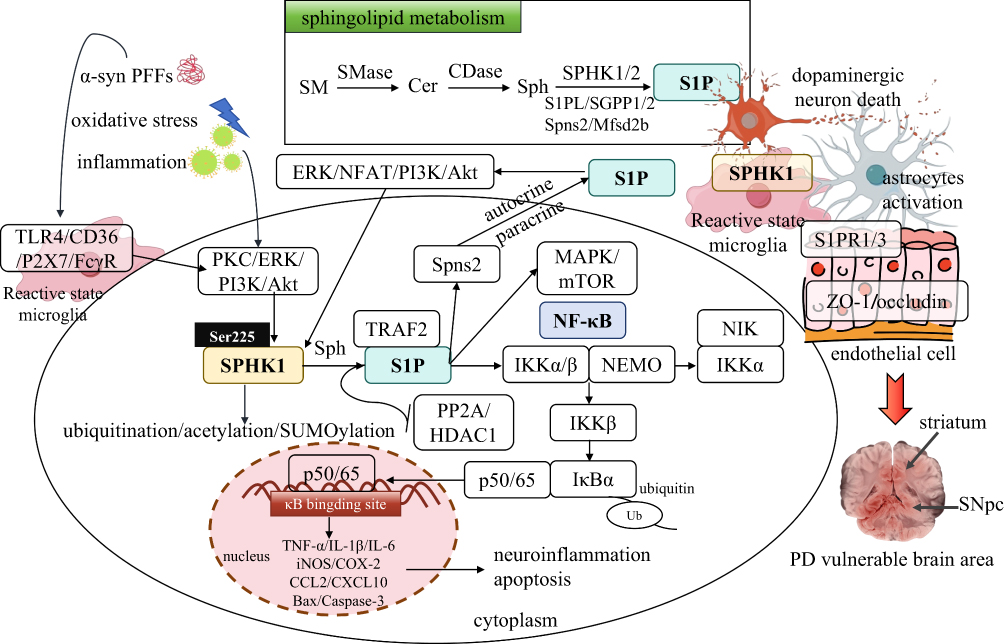 |
Figure 2 The core regulatory network of the S1P/SPHK1-NF-κB axis in PD. α-syn PFFs, oxidative stress, and inflammatory stimuli activate the PKC/ERK/PI3K/Akt signaling pathway via microglial surface receptors such as TLR4/CD36/P2X7/FcγR, mediating Ser225 phosphorylation and membrane translocation of SPHK1, which catalyzes the conversion of Sph to S1P. Intracellular S1P directly activates the canonical NF-κB pathway by binding to TRAF2 and inhibiting PP2A/HDAC1: the S1P-TRAF2 complex promotes the activation of the IKK complex (IKKα/β/NEMO), accelerates the ubiquitination and degradation of IκBα, and allows p50/p65 dimers to translocate into the nucleus, bind to κB binding sites, and initiate the transcription of pro-inflammatory genes including TNF-α, IL-1β, IL-6, iNOS, and COX-2, triggering neuroinflammation and neuronal apoptosis. Meanwhile, S1P is transported extracellularly via Spns2, and further amplifies SPHK1-NF-κB signaling through autocrine/paracrine actions by binding to S1PR1/3. Sphingolipid metabolic homeostasis (SM→Cer→Sph→S1P) serves as the upstream basis for axis activation, and the functional differentiation of SPHK1/2 determines the regulatory specificity of the axis. This axis exhibits cell- and brain region-specificity in the CNS: microglia are the core regulatory cells, astrocytes and vascular endothelial cells are involved in inflammatory amplification, and dopaminergic neurons are the ultimate damaged target cells. The excessive activation of this axis in PD-vulnerable brain regions such as the SNpc and striatum is a key molecular mechanism for the selective death of dopaminergic neurons. |
Commonly adopted PD models including toxin-induced MPTP models and transgenic animals have inherent drawbacks: these artificial experimental systems cannot fully recapitulate the slow, multifactorial progression of spontaneous human PD, which partly accounts for conflicting experimental conclusions regarding the pathogenic weight of S1P/SPHK1-NF-κB cascade. Most target compounds exert favorable neuroprotective effects in preclinical tests, while numerous candidates end up with ineffective or negative outcomes in early clinical trials, reflecting prominent translational obstacles from preclinical research to clinical medication development.
Driving Roles of the S1P/SPHK1‑NF‑κB Axis in Core Pathological Progression of PD
Activation of the S1P/SPHK1‑NF‑κB Axis Mediates Aberrant Microglial Polarization and a Vicious Cycle of Neuroinflammation
As the innate immune sentinels of the CNS, microglia determine whether neuroinflammation exerts protective or detrimental effects. Resting microglia exhibit a ramified morphology and maintain homeostasis by phagocytosing apoptotic cells and α‑syn aggregates. Under pathological stimulation, microglia undergo reactive transformation into distinct functional states, featuring either pro‑inflammatory, neurotoxic reactive profiles or anti‑inflammatory, neurorestorative homeostatic reparative profiles. Transition toward pro-inflammatory reactive microglial status represents a hallmark of PD‑related neuroinflammation, and the S1P/SPHK1‑NF‑κB axis acts as a key molecular switch driving microglia to acquire pro-inflammatory reactive characteristics.71,72 In the pathological microenvironment of PD, persistent stimuli including α‑syn PFFs, mitochondrial DAMPs, and gut microbiota translocation products activate the microglial S1P/SPHK1‑NF‑κB axis via receptors such as TLR4 and CD36, initiating the transition toward a pro-inflammatory reactive microglial state.73,74 First, S1P generated by SPHK1 activation directly binds to TRAF2, activates the canonical NF‑κB pathway, and upregulates transcription and release of pro-inflammatory signature mediators including TNF‑α, IL‑1β, IL‑6, iNOS, cyclooxygenase‑2 (COX‑2), and CD86. Second, S1P activates the ERK/c‑Jun N‑terminal kinase (JNK) pathway via S1PR3, further enhancing NF‑κB activity while suppressing expression of key transcription factors driving homeostatic reparative reactive microglia formation including peroxisome proliferator‑activated receptor γ (PPARγ), arginase‑1 (Arg‑1), IL‑10, and transforming growth factor‑β (TGF‑β), blocking microglial transition to the anti‑inflammatory restorative phenotype.75,76 In addition, NF‑κB upregulates expression of α‑syn receptors (TLR4, P2X7) and chemokine receptors (C‑C motif chemokine receptor 2 [CCR2], C‑X‑C motif chemokine receptor 3 [CXCR3]) on microglia, strengthening their recognition of α‑syn aggregates and response to pro‑inflammatory factors.77
Studies have confirmed that in α‑syn PFF‑induced PD mouse models, microglial SPHK1 knockdown or treatment with the specific inhibitor PF543 markedly suppresses NF‑κB nuclear translocation, decreases pro-inflammatory mediators secreted by reactive microglia, and facilitates microglial transition toward homeostatic reparative reactive features, and enhances phagocytic clearance of α‑syn aggregates. In microglia overexpressing SPHK1, even without exogenous α‑syn stimulation, NF‑κB is spontaneously activated and cells shift into a pro-inflammatory reactive state, releasing abundant neurotoxic factors that directly induce apoptosis of co‑cultured dopaminergic neurons.17 Such imbalanced microglial polarization not only directly injures neurons but also activates astrocytes to transform into reactive A1 astrocytes via paracrine effects. A1 astrocytes lose their neurotrophic and synaptic repair functions and instead release neurotoxic factors including IL‑1α, complement component 1q (C1q), and TNF‑α, further exacerbating dopaminergic neuron loss and synaptic dysfunction. This represents a core pathological event in progressive neurodegeneration in PD.78–80
The S1P/SPHK1‑NF‑κB Axis Regulates Aberrant Aggregation, Phosphorylation, and Pathological Propagation of α‑Syn
Aberrant folding, phosphorylation, oligomerization, and fibrillar aggregation of α‑syn, as well as prion‑like propagation of pathological α‑syn between neurons, are core pathological features of PD.81 The S1P/SPHK1‑NF‑κB axis comprehensively regulates α‑syn pathology through both direct and indirect mechanisms.82 For direct regulation, activation of the S1P/SPHK1‑NF‑κB axis directly promotes α‑syn phosphorylation and aggregation. First, pro‑inflammatory factors (TNF‑α, IL‑1β) released by microglia activate neuronal NF‑κB via surface receptors, upregulate activities of glycogen synthase kinase 3β (GSK3β) and casein kinase 2 (CK2), and directly catalyze α‑syn phosphorylation at Ser129. Ser129 phosphorylation is a key marker of pathological α‑syn aggregation; phosphorylated α‑syn more readily forms oligomers and insoluble fibrils with markedly enhanced toxicity. Second, S1P directly binds the N‑terminal domain of α‑syn, alters its conformation, promotes conversion of α‑syn from physiological α‑helix to pathological β‑sheet, and accelerates oligomerization and fibril formation.17,83,84 In addition, NF‑κB activation inhibits the neuronal autophagy‑lysosome pathway (downregulates microtubule‑associated protein 1 light chain 3‑II [LC3‑II] and Beclin‑1, upregulates p62), reducing clearance of pathological α‑syn and causing intracellular accumulation of aggregates to form Lewy bodies.38,85
For indirect regulation, activation of the S1P/SPHK1‑NF‑κB axis disrupts neuronal homeostasis and creates a pathological microenvironment favorable for α‑syn aggregation. Axis activation induces microglia to release large amounts of reactive oxygen species (ROS) and nitric oxide (NO), triggering neuronal oxidative stress. Oxidative stress directly modifies cysteine residues of α‑syn and promotes its misfolding.17,86 Meanwhile, axis activation exacerbates neuronal mitochondrial dysfunction (reduces mitochondrial membrane potential, inhibits respiratory-chain complex activity, and increases mitochondrial ROS release); DAMPs released from damaged mitochondria further activate the axis and α‑syn aggregation. Furthermore, axis activation disrupts BBB integrity, allowing peripheral toxins, iron ions, and inflammatory cells to enter the brain parenchyma; iron overload directly catalyzes α‑syn aggregation.17,82,87
Critically, activation of the S1P/SPHK1‑NF‑κB axis markedly facilitates intercellular propagation of pathological α‑syn. Pro‑inflammatory factors released by pro-inflammatory reactive microglia upregulate expression of neuronal α‑syn uptake receptors such as lymphocyte activation gene 3 (LAG3) and prion protein C (PrPC), enhancing endocytosis of extracellular α‑syn PFFs by neurons. Meanwhile, axis activation damages neuronal membrane integrity and synaptic connections, promoting release of α‑syn aggregates into the extracellular space via synaptic clefts and exosomes; these aggregates are taken up by neighboring neurons, triggering a new round of aggregation and injury, thereby enabling whole-brain propagation of α-syn pathology from the substantia nigra to the locus coeruleus, dorsal motor nucleus of the vagus, cerebral cortex, and other brain regions. This is consistent with the progressive worsening of clinical PD symptoms. Animal experiments confirm that inhibition of the SPHK1‑NF‑κB axis significantly reduces α‑syn Ser129 phosphorylation, decreases insoluble α‑syn aggregates, blocks α‑syn pathological propagation, and alleviates motor deficits and dopaminergic neuron loss in PD model mice, directly validating the axis as a key driver of α‑syn pathology.17,87
The S1P/SPHK1‑NF‑κB Axis Mediates Selective Injury of Dopaminergic Neurons
Selective and progressive loss of dopaminergic neurons in the SNpc underlies PD motor symptoms. Their vulnerability stems from high metabolic demand, dopamine‑induced oxidative stress, iron overload, and fragile mitochondrial function. The S1P/SPHK1‑NF‑κB axis directly triggers oxidative stress, mitochondrial damage, and apoptotic cascades by targeting core vulnerable nodes of dopaminergic neurons, which is proposed as one important molecular contributor to neuronal death mainly from preclinical observations.86 TNF‑α, IL‑1β, iNOS, and ROS released by microglia upon S1P/SPHK1‑NF‑κB axis activation act on dopaminergic neurons via paracrine signaling, activate the intracellular NF‑κB pathway, upregulate expression of NADPH oxidase 2 (NOX2) and iNOS, generate massive superoxide anions, hydrogen peroxide, and peroxynitrite, and induce oxidative stress.88–90 Meanwhile, dopamine in dopaminergic neurons is metabolized by monoamine oxidase (MAO) to produce hydrogen peroxide, which generates hydroxyl radicals under iron catalysis. Axis activation upregulates expression of divalent metal transporter 1 (DMT1) and downregulates ferritin, worsening iron overload. Oxidative stress further modifies lipids, proteins, and DNA, causing membrane lipid peroxidation, protein denaturation, and DNA damage, directly destroying neuronal structure and function.91–93
Mitochondria are the energy core of dopaminergic neurons and a key regulatory hub for oxidative stress and apoptosis. The S1P/SPHK1‑NF‑κB axis disrupts mitochondrial homeostasis through multiple mechanisms. On one hand, pro‑inflammatory factors activate neuronal NF‑κB, downregulate expression and activity of mitochondrial respiratory-chain complexes (Complex I, III, IV), reduce ATP synthesis efficiency, and cause energy metabolic failure.60,94,95 On the other hand, S1P directly binds mitochondrial membrane proteins and triggers opening of the mitochondrial permeability transition pore (mPTP), leading to reduced mitochondrial membrane potential and release of cytochrome c into the cytoplasm. In addition, axis activation inhibits mitophagy, reducing clearance of damaged mitochondria and causing accumulation of dysfunctional mitochondria that further release ROS and pro‑apoptotic factors.14,94,96 Studies reveal reduced mitochondrial Complex I activity in the substantia nigra of PD patients, while inhibition of the SPHK1‑NF‑κB axis significantly restores mitochondrial function, reduces ROS release, and protects dopaminergic neurons.17,97,98
Oxidative stress and mitochondrial damage jointly activate the intrinsic apoptotic pathway, and the S1P/SPHK1‑NF‑κB axis accelerates neuronal death by regulating expression of apoptosis‑related proteins. NF‑κB activation upregulates pro‑apoptotic proteins including Bcl‑2‑associated X protein (Bax), Bcl‑2‑associated death promoter (Bad), Caspase‑3, and Caspase‑9, and downregulates anti‑apoptotic proteins including Bcl‑2, Bcl‑2‑extra large (Bcl‑xL), and XIAP, disrupting the balance of Bcl‑2 family proteins and promoting mitochondrial cytochrome c release and apoptotic body formation. Meanwhile, axis activation stimulates the p53 pathway to further amplify apoptotic signals.17,99 In addition, glutamate and excitotoxins released by microglia mediate neuronal calcium overload via N‑methyl‑D‑aspartate (NMDA) receptors, activate calcium‑dependent proteases and nucleases, and directly degrade neuronal cytoskeletal proteins and DNA, resulting in concurrent neuronal necrosis and apoptosis.100 Cell co‑culture experiments confirm that conditioned medium from microglia with activated S1P/SPHK1‑NF‑κB axis induces dopaminergic neuron apoptosis, while inhibition of SPHK1 or NF‑κB reduces apoptosis rate, directly proving the axis as a core driver of dopaminergic neuron death.101
The S1P/SPHK1‑NF‑κB Axis Contributes to Blood‑Brain Barrier Disruption, Peripheral Immune Infiltration, and Gut‑Brain Axis Disturbance
PD is not a pure CNS disorder but a systemic disease involving the CNS, peripheral immunity, gastrointestinal tract, cardiovascular system, and other systems. Its pathological progression is accompanied by BBB leakage, peripheral immune cell infiltration, gut microbiota dysbiosis, and intestinal inflammation. As a key signaling hub linking the CNS and periphery, the S1P/SPHK1‑NF‑κB axis comprehensively mediates integrated regulation of multisystem pathology in PD, providing molecular support for the “systemic‑brain axis” hypothesis of PD pathogenesis.102,103 The BBB is a critical structure maintaining CNS homeostasis, composed of cerebral microvascular endothelial cells, tight junctions, pericytes, and astrocytic endfeet; its disruption is a key event aggravating PD neuroinflammation.104 The S1P/SPHK1‑NF‑κB axis directly targets cerebral microvascular endothelial cells to damage BBB structure and function. First, S1P released by microglia binds endothelial S1PR1, activates the NF‑κB pathway, downregulates expression and membrane localization of tight junction proteins including ZO‑1, occludin, and claudin‑5, loosening tight junctions and increasing permeability. Second, NF‑κB upregulates expression of endothelial adhesion molecules including intercellular cell adhesion molecule-1 (ICAM‑1) and vascular cell adhesion molecule‑1 (VCAM‑1), promoting adhesion and transmigration of peripheral monocytes, CD4+ T cells, and CD8+ T cells into the brain parenchyma. Infiltrating peripheral immune cells further activate the microglial S1P/SPHK1‑NF‑κB axis, releasing more pro‑inflammatory factors and accelerating PD pathological progression.105,106 Clinical studies confirm a significant positive correlation between cerebrospinal fluid (CSF) S1P levels and BBB permeability markers (albumin quotient) in PD patients, and SPHK1 inhibition markedly improves BBB integrity and reduces peripheral immune cell infiltration.107,108
The gut‑brain axis is an emerging hotspot in PD pathogenesis. PD patients frequently present with gastrointestinal symptoms including constipation, intestinal inflammation, and gut microbiota dysbiosis, and intestinal α‑syn pathology precedes central involvement, suggesting intestinal inflammation may transmit pathological signals to the CNS via the vagus nerve.109 The S1P/SPHK1‑NF‑κB axis plays a pivotal role in bidirectional regulation between intestinal and central neuroinflammation. On one hand, gut microbiota dysbiosis (eg., reduced Bifidobacterium, increased Proteobacteria) causes intestinal barrier leakage; lipopolysaccharide (LPS), bacterial metabolites, and inflammatory factors enter the circulation, activate the peripheral immune cell SPHK1‑NF‑κB axis, release pro‑inflammatory factors and S1P, and transmit signals to the CNS via the bloodstream and vagus nerve, activating the microglial axis and initiating neuroinflammation.110,111 On the other hand, central S1P/SPHK1‑NF‑κB axis activation reversely regulates intestinal inflammation and microbiota composition via the hypothalamic‑pituitary‑adrenal (HPA) axis and vagus nerve, worsening intestinal barrier damage and α‑syn aggregation to form a systemic‑brain pathological cycle.110 Animal experiments confirm elevated intestinal SPHK1 expression and NF‑κB activity and increased intestinal inflammatory factor release in 1‑methyl‑4‑phenyl‑1,2,3,6‑tetrahydropyridine (MPTP)‑induced PD mice, while intestinal SPHK1 inhibition simultaneously alleviates intestinal and central neuroinflammation and reduces substantia nigra dopaminergic neuron loss,112,113 directly validating the S1P/SPHK1‑NF‑κB axis as a key molecular bridge for pathological transmission along the gut‑brain axis.
Potential Roles of the S1P/SPHK1‑NF‑κB Axis in PD Motor and Non‑Motor Symptoms
Motor complications induced by long‑term levodopa therapy are a major challenge in PD clinical management. Studies show elevated CSF S1P levels in PD patients are closely associated with pathological α‑syn aggregation, and S1P triggers microglial activation via NF‑κB signaling to exacerbate neuroinflammation. Notably, SPHK1 activation in microglia induced by α‑syn PFFs is a key initiating event of neuroinflammatory cascades, and persistent inflammation may impair plastic adaptation of striatal medium spiny neurons to dopaminergic input. Furthermore, the SPHK1 inhibitor PF543 markedly alleviates PD‑like neuropathological abnormalities in hA53T α‑syn transgenic mice, indicating axis modulation may positively affect levodopa responsiveness.17,114 Incorporating the S1P/SPHK1‑NF‑κB axis into the mechanistic network of motor complications, evaluating whether axis targeting delays or mitigates levodopa‑induced dyskinesia, and exploring axis markers as predictors of drug response hold important clinical value.
In addition, the S1P/SPHK1‑NF‑κB axis participates in peripheral immune dysregulation, neuroendocrine disturbance, and non‑motor symptom modulation in PD. Axis activation causes aberrant activation of peripheral lymphocytes and monocytes, releasing pro‑inflammatory factors and aggravating systemic chronic low‑grade inflammation, which further worsens central neuroinflammation. Meanwhile, axis activation disrupts HPA axis function and cortisol rhythm, triggering non‑motor symptoms including depression, anxiety, and sleep disturbances, consistent with the broad non‑motor manifestations of PD patients.17 During the prodromal phase of PD, upregulated SPHK1, elevated S1P, and activated NF‑κB occur in the intestine, olfactory bulb, and dorsal motor nucleus of the vagus, inducing typical prodromal symptoms including olfactory dysfunction, constipation, depression, anxiety, and sleep disorders. These manifestations are closely linked to axis‑mediated local neuroinflammation, early α‑syn aggregation, and intestinal and olfactory mucosal barrier damage.111,115,116 As disease progresses, sustained central axis activation further affects non‑nigral brain regions including the locus coeruleus, raphe nucleus, and hypothalamus, worsening non‑motor symptoms such as cognitive decline, orthostatic hypotension, and urinary dysfunction by regulating metabolism of norepinephrine, serotonin, dopamine, and other neurotransmitters. Research indicates that in pathological α‑syn‑induced neuroinflammation, the SPHK1/S1P signaling pathway triggers microglial activation via NF‑κB‑dependent mechanisms; this process is not restricted to the substantia nigra and may also regulate neurotransmitter metabolism across wider brain regions. Clinical data show that CSF levels of S1P are significantly elevated in patients with PD, approximately 16-fold higher than in control subjects, and exhibit a significant positive correlation with the severity of behavioral deficits.17,23 This suggests that peripheral blood S1P levels may also correlate with non-motor symptom scores. This axis could serve as an important target for prodromal screening and targeted intervention of non-motor symptoms, addressing the limitation that current PD treatments focus solely on motor symptoms (Figure 3 and Table 2).
 |
Table 2 The S1P/SPHK1‑NF‑κB Axis Mediates Core Pathological Events in PD |
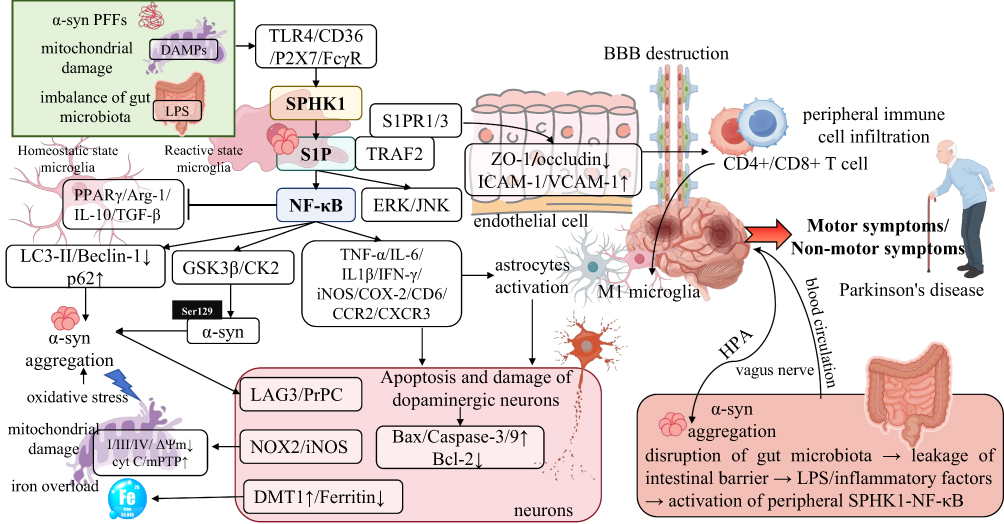 |
Figure 3 Multidimensional driving mechanism of the S1P/SPHK1‑NF‑κB axis in the core pathological progression of PD. In the PD pathological microenvironment, upstream stimuli including α‑syn PFFs, mitochondrial DAMPs, and LPS mediated by gut microbiota dysbiosis activate SPHK1 in microglia via receptors such as TLR4/CD36/P2X7/FcγR, catalyzing the production of S1P; S1P dually activates the canonical NF‑κB pathway and ERK/JNK signaling by binding to TRAF2 and S1PR1/3, forming a core regulatory axis for PD pathological progression. This axis drives the entire PD disease course through multilevel cascade effects: it drives microglial reactive transformation toward a pro-inflammatory neurotoxic state and restricts the development of homeostatic reparative microglial features, while activating A1‑type reactive astrocytes, forming a vicious cycle of neuroinflammation; it directly promotes α‑syn Ser129 phosphorylation and β‑sheet conformational conversion, inhibits the autophagy‑lysosome pathway, exacerbates abnormal α‑syn aggregation, and mediates prion‑like pathological propagation of α‑syn by upregulating receptors such as LAG3/PrPC; it triggers oxidative stress bursts, mitochondrial dysfunction, and iron overload in dopaminergic neurons, initiates neuronal apoptotic cascades by regulating apoptosis‑related proteins, and mediates the selective loss of nigral dopaminergic neurons; it targets cerebral microvascular endothelial cells, downregulates tight junction proteins (ZO‑1/occludin/claudin‑5), upregulates adhesion molecules (ICAM‑1/VCAM‑1), disrupts BBB integrity, and promotes the infiltration of immune cells such as peripheral CD4+/CD8+ T cells into the brain parenchyma; as a key molecular bridge of the gut‑brain axis, LPS and inflammatory factors released from intestinal barrier leakage activate the peripheral SPHK1‑NF‑κB axis, which transmits signals to the CNS via the vagus nerve and blood circulation, and meanwhile, central axis activation reversely aggravates intestinal inflammation and α‑syn aggregation through the HPA axis, forming a systemic‑cerebral pathological cycle. Ultimately, sustained activation of this axis not only drives the progressive worsening of PD motor symptoms but also participates in the development of non‑motor symptoms during the prodromal phase, including olfactory dysfunction, constipation, depression, and anxiety, which provides a core molecular explanation for the multisystem pathological mechanism of PD and offers key regulatory targets for targeted intervention in PD. |
Crosstalk Network Between the S1P/SPHK1‑NF‑κB Axis and Other Core Pathological Mechanisms in PD
Synergistic Amplification Loop Between the S1P/SPHK1‑NF‑κB Axis, Oxidative Stress, and Endoplasmic Reticulum Stress
Oxidative stress and endoplasmic reticulum stress (ERS) are important auxiliary mechanisms of neurodegeneration in PD. Both form a tight synergistic amplification loop with the sphingosine-1-phosphate/sphingosine kinase 1-nuclear factor-kappa B (S1P/SPHK1‑NF‑κB) axis to jointly drive pathological progression.117 As both an upstream stimulus and a downstream effect of axis activation, oxidative stress regulates axis activity through a dual mechanism. On one hand, oxidative products such as reactive oxygen species (ROS) and peroxynitrite can directly activate SPHK1 (Ser225 phosphorylation) and NF‑κB (p65 oxidative modification), initiating axis activation. On the other hand, iNOS and NOX2 released upon axis activation further aggravate oxidative stress.118,119
ERS regulates the axis through the unfolded protein response (UPR): α‑syn aggregation and oxidative stress in PD cause endoplasmic reticulum dysfunction and activate the protein kinase R-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) branches of UPR. Among them, the IRE1 pathway directly activates tumor necrosis factor receptor-associated factor 2 (TRAF2), which in turn activates NF‑κB and SPHK1. Meanwhile, UPR upregulates the expression of pro‑inflammatory factors and synergizes with the axis to amplify neuroinflammation. In turn, axis activation exacerbates endoplasmic reticulum calcium imbalance and protein misfolding, further aggravating ERS and forming a cross‑regulatory network.117,120,121
Bidirectional Regulation Between the S1P/SPHK1‑NF‑κB Axis and Autophagy/Mitophagy
Autophagy and mitophagy are core mechanisms for clearing misfolded proteins and damaged mitochondria. Their dysfunction is a major cause of α‑syn aggregation and mitochondrial damage in PD, and the S1P/SPHK1‑NF‑κB axis is bidirectionally regulated with autophagy pathways. On one hand, activation of the S1P/SPHK1‑NF‑κB axis downregulates the expression of key autophagic proteins (LC3, Beclin‑1, autophagy-related gene 5 [ATG5]) via NF‑κB, inhibits both mTOR-dependent and ‑independent autophagic pathways, reduces the clearance of α‑syn aggregates and damaged mitochondria, and leads to pathological accumulation. On the other hand, defective autophagy results in the accumulation of α‑syn and DAMPs, further activating the S1P/SPHK1‑NF‑κB axis. As an important form of selective autophagy, mitophagy is more significantly regulated by the axis: activation of the S1P/SPHK1‑NF‑κB axis downregulates PTEN-induced kinase 1 (PINK1)/Parkin pathway activity, impairs the recognition and removal of damaged mitochondria, causes mitochondrial dysfunction and ROS release, and further amplifies axis activation and neuroinflammation.122,123
Synergistic Activation Between the S1P/SPHK1‑NF‑κB Axis and the NLRP3 Inflammasome
The NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome is another central regulatory platform for central neuroinflammation, composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and Caspase-1. Upon activation, it cleaves pro-interleukin-1β (pro-IL-1β) and pro-interleukin-18 (pro-IL-18) into mature pro‑inflammatory cytokines, forming a “dual switch” for neuroinflammation together with NF‑κB. The S1P/SPHK1‑NF‑κB axis closely synergizes with the NLRP3 inflammasome.124 First, activation of the S1P/SPHK1‑NF‑κB axis upregulates the expression of NLRP3 and pro-IL-1β via NF‑κB, providing the first signal for NLRP3 activation. Second, ROS, potassium ion (K+) efflux, and lysosomal rupture induced by axis activation supply the second signal for NLRP3 activation, promoting inflammasome assembly and Caspase‑1 activation. Finally, mature IL‑1β released by NLRP3 activation further activates the SPHK1‑NF‑κB axis, and the two synergistically mediate the persistence and exacerbation of chronic neuroinflammation in PD.125,126
Interaction Between the S1P/SPHK1‑NF‑κB Axis and PD Genetic Risk Factors
PD genetic risk factors (such as leucine-rich repeat kinase 2 (LRRK2), PINK1, Parkin, and mutant α‑syn) modulate disease susceptibility and pathological progression by regulating the activity of the S1P/SPHK1‑NF‑κB axis. The LRRK2 G2019S mutant directly phosphorylates SPHK1 and inhibitor of IKKβ, enhancing axis activation and NF‑κB activity, aggravating microglial inflammation and α‑syn aggregation.127 Mutations in PINK1/Parkin lead to mitophagy defects, and damaged mitochondria release DAMPs to activate the S1P/SPHK1‑NF‑κB axis.128 α‑syn A53T and A30P mutants are more prone to form oligomers and more strongly activate the microglial S1P/SPHK1‑NF‑κB axis.129 In contrast, protective genetic factors such as nuclear factor erythroid 2-related factor 2 (Nrf2) can attenuate axis activation and neuroinflammation by upregulating antioxidant proteins and inhibiting SPHK1 and NF‑κB activity. These interactions between genetic factors and the axis provide a molecular explanation for the genetic susceptibility and heterogeneity of PD.130
The S1P/SPHK1‑NF‑κB Axis and Epigenetic Regulation
The S1P/SPHK1‑NF‑κB axis also deeply participates in PD pathogenesis through epigenetic mechanisms, constituting another important layer of gene‑environment interaction. S1P can directly inhibit the activity of histone deacetylases (HDAC1/2), alter chromatin accessibility, and thereby upregulate the transcription efficiency of NF‑κB target genes and α‑syn‑related genes.131 The expression and activation of SPHK1 are also finely regulated by DNA methylation and non‑coding RNAs (microRNAs [miRNAs], long non-coding RNAs [lncRNAs]). For example, miR‑124 and miR‑146a directly target SPHK1 mRNA and inhibit its translation. In PD models, the expression of these microRNAs is significantly downregulated, thus relieving the repression of SPHK1 and promoting sustained axis activation.132–134
In addition, NF‑κB can recruit the histone acetyltransferase p300/CREB-binding protein (CBP) to acetylate the promoter regions of pro‑inflammatory genes, forming long‑term inflammatory memory and rendering microglia hypersensitive to subsequent pathological stimuli, further amplifying neuroinflammation and neuronal damage.135 Such epigenetic regulation not only explains how aging and environmental toxins long‑term remodel inflammatory gene expression, but also provides novel intervention strategies for targeting epigenetic regulation to reverse aberrant activation of the S1P/SPHK1‑NF‑κB axis (Figure 4).
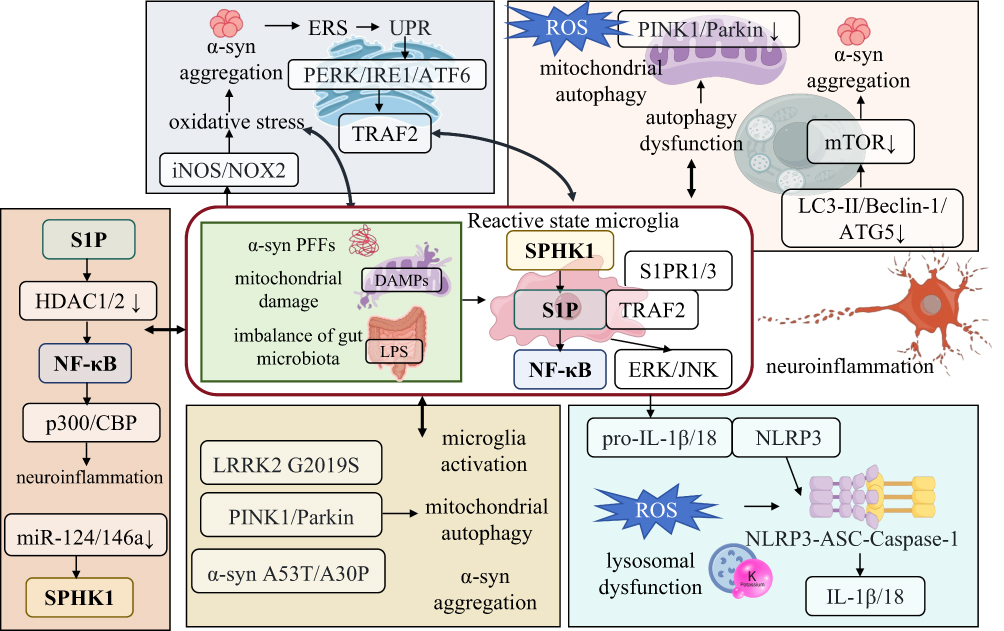 |
Figure 4 Cross-interaction network between the S1P/SPHK1‑NF‑κB axis and core pathological mechanisms of PD. The S1P/SPHK1‑NF‑κB axis serves as a central regulatory hub in PD pathogenesis, forming a multi-dimensional cross-network with oxidative stress/endoplasmic reticulum stress, autophagy/mitophagy, the NLRP3 inflammasome, genetic risk factors, and epigenetic regulation to drive disease progression. Upstream stimuli such as α‑syn PFFs, mitochondrial DAMPs, and gut microbiota dysbiosis activate SPHK1 in microglia, leading to S1P production and amplification of neuroinflammation via the NF‑κB/ERK/JNK pathways. This axis establishes a positive feedback loop with oxidative/endoplasmic reticulum stress and autophagic deficiency, exacerbating the accumulation of pathological substances. Meanwhile, it synergizes with the NLRP3 inflammasome to sustain chronic inflammation, while genetic factors including LRRK2, PINK1/Parkin, and epigenetic regulation mediated by HDACs and microRNAs further enhance the axis activity. These interactions collectively constitute the core pathological circuit of neuroinflammation, α‑syn aggregation, mitochondrial dysfunction, and neuronal degeneration in PD. |
Clinical Translation Potential of the S1P/SPHK1-NF-κB Axis as a Biomarker for PD
Studies on clinical samples have confirmed that molecules related to the S1P/SPHK1-NF-κB axis can serve as potential biomarkers for early diagnosis, disease monitoring, and prognosis evaluation of PD: the level of S1P in the CSF of PD patients is 12–16 times higher than that of healthy controls, and it is significantly positively correlated with the Hoehn-Yahr scale and the Unified Parkinson’s Disease Rating Scale (UPDRS) score, while negatively correlated with the degree of dopaminergic neuron loss;17,136,137 the activity of SPHK1 and the phosphorylation level of p65 in the CSF are also significantly increased, which can be detected in the early stage of PD (Hoehn-Yahr stage 1–2), earlier than the appearance of classic biomarkers (α-syn oligomers, DJ-1);17,138,139 the level of S1P in peripheral blood, the expression of SPHK1 in peripheral blood mononuclear cells (PBMCs), and the activity of NF-κB are also significantly increased, and positively correlated with the levels in CSF, which can be used as non-invasive biomarkers for early screening and disease monitoring of PD.39,136,140
The diagnostic specificity of a single axis biomarker is limited. Combined detection with classic PD biomarkers (CSF α-syn oligomers, total α-syn, phosphorylated α-syn, DJ-1, peripheral blood neurofilament light chain [NfL]) can significantly improve the accuracy and specificity of PD diagnosis. The combined detection of CSF S1P + α-syn oligomers has precise diagnostic sensitivity and high specificity, which is significantly superior to a single biomarker; the combined detection of blood S1P + NfL can be used as a non-invasive screening tool for early risk assessment of PD in middle-aged and elderly people in communities; at the same time, the level of axis biomarkers can reflect neuroinflammatory activity, predict the rate of disease progression and treatment response, and provide a basis for precise stratification and individualized treatment of PD.141–143
Existing studies have mostly focused on the activation characteristics of the S1P/SPHK1-NF-κB axis in typical PD (mainly motor symptoms, good response to levodopa), but the clinical manifestations of PD are highly heterogeneous, including non-tremor type mainly characterized by postural instability and gait disturbance, aggressive type characterized by rapid progression, and diffuse type accompanied by significant cognitive impairment. Clinical studies have found that the level of S1P in the CSF of PD patients is higher than that of healthy controls, and this increase is closely related to the abnormal activation of SPHK1, while the expression of SPHK2 has no significant change. It is worth noting that α-syn PFFs can selectively induce the phosphorylative activation of SPHK1 (Ser225 site) in microglia but have no significant effect on neurons and astrocytes, and this cell selectivity may vary among different PD subtypes.17 Although large-scale subtype stratification studies are currently lacking, existing evidence suggests that patients with aggressive PD may exhibit a stronger axis activation state, accompanied by more prominent shift of microglia toward pro-inflammatory reactive status and BBB damage. Clarifying the differential expression and dynamic evolution of this axis in different PD subtypes will help realize disease classification based on inflammatory phenotypes and promote the clinical implementation of individualized anti-inflammatory treatment strategies.
There are still three major bottlenecks in the clinical translation of current axis biomarkers. The standardization of detection methods is insufficient, and there is no unified kit and quality control standard for the detection of S1P and SPHK1 activity in CSF/blood; the specificity needs to be improved, as elevated S1P levels can also be seen in diseases such as multiple sclerosis (MS), cerebral infarction, and inflammatory bowel disease (IBD); the sensitivity of non-invasive detection is insufficient, and the level of blood S1P fluctuates greatly, affected by diet, drugs, and peripheral inflammation. Future optimization directions include: developing high-specificity and high-sensitivity monoclonal antibody detection kits for S1P, SPHK1, and phosphorylated p65 (p-p65); establishing normal reference ranges and PD diagnostic thresholds for axis biomarkers in CSF/blood; constructing a combined diagnostic model of axis biomarkers, clinical characteristics, and imaging (dopamine transporter [DAT] scan) combined with machine learning algorithms to improve diagnostic efficiency.
Therapeutic Strategies Targeting the S1P/SPHK1-NF-κB Axis and Progress in Drug Development for PD
Specific Inhibitors Targeting SPHK1
As the upstream rate-limiting enzyme of the S1P/SPHK1-NF-κB axis, SPHK1 is a core target for PD treatment. Its specific inhibitors can directly block S1P synthesis, inhibit NF-κB activation, and simultaneously not interfere with the normal physiological functions of SPHK2, making it a key direction in current drug development. Currently, the main types of SPHK1 inhibitors that have entered preclinical research include the following: PF543 is the first highly selective SPHK1 inhibitor with a selectivity for SPHK2 exceeding 1000-fold, which can effectively cross the BBB. In PD mouse models induced by α-syn PFFs and MPTP, it can significantly reduce brain S1P levels, inhibit NF-κB nuclear translocation, restrain microglial transition into pro-inflammatory reactive status and lower the secretion of pro-inflammatory mediators, while decreasing abnormal α-syn aggregation, protecting dopaminergic neurons, improving motor disorders, and showing no obvious peripheral toxicity;144 SKI-II is a non-selective dual SPHK1/2 inhibitor, which can inhibit the activation of this signaling axis but may cause certain side effects due to simultaneous inhibition of SPHK2, and is currently mainly used for basic mechanism research;145 ABC294640 is a selective SPHK2 inhibitor with weak effect on SPHK1, which is mostly used in metabolism regulation and tumor-related research,146 and has few studies in the field of PD.
Modulators Targeting S1P Receptors
Modulators targeting S1P receptors can effectively block S1P-mediated autocrine and paracrine loops, thereby inhibiting neuroinflammation. Currently, several S1PR modulators have been approved for the clinical treatment of multiple sclerosis (MS)147 and have shown good application prospects in preclinical PD models. Fingolimod (FTY720), as a non-selective S1PR1/3/4/5 modulator, is the first oral disease-modifying drug approved for relapsing-remitting MS (RRMS). After phosphorylation in vivo, it exerts dual immunomodulatory and neuroprotective effects by promoting lymphocyte homing, reducing peripheral immune cell infiltration, and directly acting on astrocytes in the CNS.44,148 Siponimod and Ozanimod are both dual S1PR1/5 modulators; the former has been approved for RRMS and active secondary progressive MS (SPMS), while the latter is simultaneously approved for RRMS, ulcerative colitis (UC), and Crohn’s disease (CD). Ponesimod, as a highly selective S1PR1 agonist, has also been approved for RRMS treatment. In addition, several new-generation S1PR modulators are in clinical development: Amiselimod (MT-1303) and Etrasimod (APD334) are both S1PR1/4/5 modulators; the former has completed Phase II clinical trials for RRMS, CD, and UC, while the latter has entered Phase III studies in UC and CD; Mocravimod (KRP-203), as a dual S1PR1/3 modulator, is being explored in Phase II trials for UC, systemic lupus erythematosus (SLE), and organ transplant rejection. The successful application of these drugs in peripheral immune-mediated diseases such as MS provides important theoretical basis and preclinical support for expanding their indications to PD, which is characterized by chronic neuroinflammation.149,150
Inhibitors Targeting the NF-κB Pathway
As the core downstream transcription factor of the S1P/SPHK1-NF-κB axis, NF-κB inhibitors can directly block the transcriptional expression of pro-inflammatory genes, but in application, it is crucial to solve the problems of central targeting selectivity and systemic side effects, avoiding excessive inhibition of NF-κB activity under physiological conditions. Selective IKKβ inhibitors such as BMS-345541 and IMD-0354 can specifically act on IKKβ, prevent IκB degradation and NF-κB pathway activation, and can significantly reduce pro-inflammatory factor release and exert neuroprotective effects in PD models. However, some compounds have defects such as poor BBB penetration and easy induction of peripheral immunosuppression.151,152 p65 acetylation or phosphorylation inhibitors reduce its transcriptional activity by interfering with the post-translational modification of p65. Natural products represented by curcumin and resveratrol can effectively cross the BBB, and while inhibiting NF-κB, they also have antioxidant and anti-inflammatory effects. Preclinical studies have confirmed that they can improve the pathological and behavioral symptoms of PD models.153,154 Microglia-specific NF-κB inhibitors construct targeted delivery systems relying on microglial surface markers such as CD11b and CX3CR1, which can accurately deliver the inhibitors to microglia, reduce non-specific effects on neurons and peripheral immune cells, and significantly improve the safety and targeting of intervention.
Sphingolipid Metabolism Homeostasis Modulators
Regulating the sphingolipid metabolism network, increasing the ceramide/sphingosine ratio, and reducing S1P levels to restore sphingolipid biological homeostasis can also indirectly inhibit the excessive activation of the S1P/SPHK1-NF-κB axis. Studies have shown that double knockout of sphingosine kinase can lead to a significant increase in intracellular sphingosine and ceramide levels (ceramide increased by more than 10-fold) and induce the constitutive expression of autophagy markers, suggesting that sphingolipid metabolic reprogramming may exert anti-inflammatory effects by restoring the ceramide-S1P balance. The glucosylceramide synthase (GCS) inhibitor BZ1 can significantly reduce glycosphingolipid levels in neurons, reduce pathological aggregation of α-synuclein, rescue lysosomal dysfunction associated with GBA1 mutations, and alleviate α-synuclein-induced dopaminergic neurodegeneration. In GBA-related PD mouse models, long-term administration of the brain-penetrant GCS inhibitor Venglustat can significantly reduce glycosphingolipid accumulation in the CNS, decrease pathological α-synuclein aggregates, and improve related memory deficits.155,156 In addition, ceramidase inhibitors (such as NOE-SA, D-erythro-MAPP) can block the conversion of ceramide to sphingosine, reduce the synthetic precursor of S1P, and preclinical studies have confirmed that they can reduce brain S1P content and inhibit central neuroinflammation. S1P lyase activators can accelerate the degradation of S1P into hexanal and phosphoethanolamine, thereby reducing S1P levels. The marketed lipid-lowering drug bezafibrate, which has the effect of activating S1P lyase, has shown clear neuroprotective potential in PD models.157 Sphingomyelinase activators can promote the conversion of sphingomyelin to ceramide, antagonizing the pro-inflammatory effect of S1P at the metabolic level. Recombinant acid sphingomyelinase has been shown to improve neuroinflammation and neuronal damage in PD models.158 The above strategies provide a new intervention idea for indirectly inhibiting the excessive activation of the S1P/SPHK1-NF-κB axis by restoring sphingolipid metabolism homeostasis.
Progress in Natural Products and Traditional Chinese Medicine Intervention
In addition to small chemical molecules and biological agents, natural products and traditional Chinese medicine (TCM) have shown unique advantages in intervening PD by targeting the S1P/SPHK1-NF-κB axis, becoming an important supplement to disease-modifying therapy. Multiple studies have confirmed that polyphenolic compounds such as berberine, puerarin, resveratrol, and curcumin can achieve multi-node regulation of the axis by inhibiting SPHK1 phosphorylation, reducing S1P production, and blocking IκB degradation and p65 nuclear translocation, while also having multiple neuroprotective effects such as antioxidation, mitochondrial protection, and promotion of α-syn clearance.159,160 TCM compounds and monomer components can also regulate gut microbiota and sphingolipid metabolism homeostasis, indirectly inhibit the activation of the central S1P/SPHK1-NF-κB axis through the gut-brain axis, and improve motor and non-motor symptoms in PD models.161,162 Such naturally derived interventions usually have good BBB permeability and low toxicity and side effects, making them suitable for long-term intervention. However, the accuracy of their targets, dose–effect relationship, and standardized preparations are still the main bottlenecks in clinical translation. In the future, combining network pharmacology and molecular docking technology can further explore natural axis modulators with high selectivity and multi-target synergy.
Combined Treatment Strategy: Multi-Target Synergistic Blockade of Pathological Loops
Single-target intervention is difficult to block the complex and interlaced pathological network of PD. Combining the S1P/SPHK1-NF-κB axis targeting strategy with other core pathological mechanism intervention methods such as α-synuclein clearance, mitochondrial protection, antioxidation, and NLRP3 inflammasome inhibition can achieve multi-pathway synergy, blocking disease progression more efficiently. The combination of SPHK1 inhibitors and α-syn antibodies, such as PF543 combined with α-syn monoclonal antibodies, can inhibit neuroinflammation while promoting the clearance of pathological α-syn, and the therapeutic effect is significantly better than single-drug regimens; the combination of S1PR modulators and mitochondrial protectants, such as FTY720 combined with idebenone, can synergistically improve mitochondrial function and reduce oxidative stress levels, thereby exerting stable neuroprotective effects; the combination of NF-κB inhibitors and NLRP3 inhibitors, such as curcumin combined with MCC950, can dual-block the core pathways of neuroinflammation, significantly reducing pathological damage in PD models and providing reliable experimental basis for clinical combined treatment.163–165
Bottlenecks in Clinical Translation and Breakthrough Pathways
Although a large number of preclinical studies support the therapeutic potential of targeting the S1P/SPHK1-NF-κB axis for PD, candidate drugs entering clinical trials still face multiple translational obstacles. The use of the specific SPHK1 inhibitor PF543 in α-syn PFFs-induced PD mouse models can significantly reduce brain S1P levels, inhibit NF-κB nuclear translocation, restrict microglial transition toward pro-inflammatory reactive features, protect dopaminergic neurons, and improve motor disorders. However, the effective dose of this inhibitor in non-human primates and human cell systems is significantly increased, which may be related to the difference in binding affinity of the catalytic domain of SPHK1 between species. In addition, long-term inhibition of SPHK1 may affect peripheral sphingolipid metabolism homeostasis, and its safety needs further evaluation. The key pathways to break through the above bottlenecks include: developing next-generation SPHK1 inhibitors based on the combination of orthosteric conformation and allosteric regulation to improve the selectivity for human SPHK1 and BBB penetration; adopting local administration or cell-specific nanocarriers to reduce systemic exposure and off-target toxicity; and using biomarkers (such as baseline CSF S1P levels, SPHK1 activity) for patient enrichment enrollment to improve the success rate of clinical trials.
Diverse pharmacological agents targeting SPHK1, S1PR and NF-κB have displayed protective effects across cell and animal PD models, yet these agents differ greatly in target selectivity, blood–brain barrier penetration capability and adverse reaction profiles. SPHK1 selective inhibitors efficiently suppress microglial pro-inflammatory activation in vitro but are restricted by poor central nervous system bioavailability in vivo; distinct S1PR subtypes are unevenly expressed in brain tissue, leading to disparate even opposite biological outcomes after receptor modulation; pan-NF-κB inhibitors produce non-specific systemic immunosuppression, which largely impedes their long-term clinical usage. Such inherent defects partially explain why few relevant candidates have successfully advanced into late-stage clinical research for PD treatment.
Challenges and Perspectives: Future Directions of Research on the S1P/SPHK1‑NF‑κB Axis
Key Scientific Questions and Research Challenges
Although the central role of the S1P/SPHK1‑NF‑κB axis in PD has been established, numerous critical scientific questions remain to be addressed. The dynamic activation pattern and cell‑specific regulatory mechanisms of the S1P/SPHK1‑NF‑κB axis at different PD stages (prodromal, early, and progressive stages) are unclear. The bidirectional regulatory network linking the S1P/SPHK1‑NF‑κB axis with the gut‑brain axis, peripheral immunity, and neuroendocrine system lacks systematic investigation. The functional divergence and synergistic mechanisms of SPHK1/SPHK2 and S1PR subtypes require further elucidation. Drugs targeting the S1P/SPHK1‑NF‑κB axis face clinical translation bottlenecks including blood‑brain barrier permeability, central selectivity, and long‑term safety. Standardized clinical detection of S1P/SPHK1‑NF‑κB axis‑related biomarkers and integrated diagnostic models have not yet been established.
Current research on the S1P/SPHK1‑NF‑κB axis heavily relies on experimental animal models such as mice and rats. However, significant species differences exist between rodents and humans in microglial phenotypes, sphingolipid metabolic profiles, NF‑κB regulatory networks, and blood‑brain barrier composition, which severely limit efficient translation from basic research to clinical practice. For instance, the basal expression and inflammatory response patterns of SPHK1 in human microglia differ from those in mice, and the distribution ratio and signaling coupling efficiency of human S1PR subtypes in the brain also diverge from those in model animals. Moreover, most PD patients are elderly with multiple comorbidities, and their systemic metabolism and peripheral inflammation differ substantially from the standardized backgrounds of experimental animals. Consequently, several SPHK1 inhibitors and S1PR modulators effective in animal models have failed to achieve expected neuroprotective outcomes in clinical trials. Therefore, future studies should strengthen research based on human induced pluripotent stem cell‑derived dopaminergic neurons and microglia, brain organoids, and non‑human primate PD models to more realistically mimic human pathological conditions and improve the clinical translation rate of axis‑targeted drug development.
Future Research Directions and Cutting‑Edge Perspectives
Future research may leverage cutting‑edge technologies such as single‑cell RNA sequencing, spatial transcriptomics, and in situ hybridization to conduct spatiotemporal dynamic single‑cell analyses of the S1P/SPHK1‑NF‑κB axis. This will enable systematic dissection of its spatiotemporal activation patterns across different brain regions, microglial subsets, and neuronal subsets in PD, and precisely clarify its dynamic regulatory role in the pathological propagation of α‑syn. Meanwhile, in‑depth investigations into the molecular mechanisms of the gut‑brain axis are warranted, focusing on the bidirectional regulation between the intestinal SPHK1-NF‑κB axis and its central counterpart, defining the key roles of gut microbiota, intestinal inflammation, and the vagus nerve in axis activation and pathological signal transmission, and developing targeted intestinal intervention strategies accordingly.
In drug development, based on the structural biology of SPHK1, S1PR, and NF‑κB, small‑molecule inhibitors with high selectivity, favorable blood‑brain barrier permeability, and microglia specificity, as well as targeted delivery systems, should be designed, and Phase I–II clinical trials of relevant candidate drugs steadily advanced. For clinical translation, a standardized detection system for axis‑related biomarkers in cerebrospinal fluid and blood should be established, and a multi‑marker integrated diagnostic model for early PD diagnosis, dynamic disease monitoring, and prognostic evaluation constructed in combination with dopamine transporter imaging and clinical functional scores. Furthermore, stratified therapy should be explored according to differences in patients’ genetic backgrounds, axis activation levels, and inflammatory phenotypes, ultimately achieving individualized precision application of drugs targeting the S1P/SPHK1‑NF‑κB axis.
Conclusion
Collectively, abundant preclinical studies indicate aberrant S1P/SPHK1-NF-κB signaling is closely involved in PD-associated neuroinflammation and dopaminergic neuronal loss. Despite encouraging pharmacological outcomes observed in cell and animal models, multiple unresolved challenges restrict its clinical transformation, including inherent defects of experimental animal systems, inconsistent research findings across different laboratories, prominent interspecies gaps between rodents and human patients, and unsatisfactory results of related candidate drugs in early clinical trials. Current evidence cannot rule out that abnormal activation of this signaling cascade may serve as a secondary pathological response rather than an initial pathogenic trigger in sporadic PD. Therefore, further human tissue-based investigation and well-designed clinical researches are required to validate its therapeutic potential, and targeted medications derived from this axis remain only preliminary preclinical candidates pending more rigorous verification before clinical application.
Data Sharing Statement
All data and material generated or analyzed during this study are included in this published article.
Author Contributions
Xiangrui Kong; Conceptualization, Supervision, Visualization, Writing – original draft. Hao Wang; Conceptualization, Supervision, Visualization, Writing – original draft. Jun Dong; Conceptualization, Supervision, Visualization, Writing – original draft. Xiangshu Cheng; Conceptualization, Supervision, Funding acquisition, Writing – review and editing. Jianshe Wei; Conceptualization, Supervision, Funding acquisition, Writing – review and editing. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
This work was supported partly by National Natural Science Foundation of China (81271410, 32161143021), Henan Provincial Science and Technology Key Projects Program (252301420027, 252102311160), and Henan Provincial Medical Science and Technology Key Projects Program (SBGJ202402075).
Disclosure
The authors declare no competing interests in this work.
References
1. Li Y, Lv Z, Dai Y, et al. The global, regional, and National burden of Parkinson’s disease in 204 countries and territories, 1990-2021: a systematic analysis for the global burden of disease study 2021. BMC Public Health. 2025;25(1):3047. doi:10.1186/s12889-025-23492-8
2. Zhang S, Wang T, Peng Y, et al. Parkinson’s disease: pathogenesis and therapeutic strategies. Mol Biomed. 2026;7(1):46. doi:10.1186/s43556-026-00445-0
3. Liu T, Guo D, Wei J. The pathogenesis of Parkinson’s disease and crosstalk with other diseases. Biocell. 2024;48(8):1155–25. doi:10.32604/biocell.2024.051518
4. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458–3483. doi:10.1016/j.neuron.2022.10.020
5. Lawrence JM, Schardien K, Wigdahl B, Nonnemacher MR. Roles of neuropathology-associated reactive astrocytes: a systematic review. Acta Neuropathol Commun. 2023;11(1):42. doi:10.1186/s40478-023-01526-9
6. Li Y, Xia Y, Yin S, et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front Immunol. 2021;12:719807. doi:10.3389/fimmu.2021.719807
7. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J Immunol Res. 2018;2018:4784268. doi:10.1155/2018/4784268
8. Mendes-Oliveira J, Campos FL, Ferreira SA, Tomé D, Fonseca CP, Baltazar G. Endogenous GDNF is unable to halt dopaminergic injury triggered by microglial activation. Cells. 2023;13(1):74. doi:10.3390/cells13010074
9. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi:10.1212/WNL.38.8.1285
10. Islam MN, Mandal N, Khanom NI, et al. Sphingolipid signaling and metabolism in neuronal and glial cells: implications for cerebrovascular and neurodegenerative disorders. Aging Dis. 2025. PMID: 41400579. doi:10.14336/AD.2025.1024
11. Qin B, Fu Y, Raulin AC, et al. Lipid metabolism in health and disease: mechanistic and therapeutic insights for Parkinson’s disease. Chin Med J. 2025;138(12):1411–1423. doi:10.1097/CM9.0000000000003627
12. Schneider JS, Singh G. Altered expression of glycobiology-related genes in Parkinson’s disease brain. Front Mol Neurosci. 2022;15:1078854. doi:10.3389/fnmol.2022.1078854
13. Shin KO, Kim KP, Cho Y, et al. Both sphingosine kinase 1 and 2 coordinately regulate cathelicidin antimicrobial peptide production during keratinocyte differentiation. J Invest Dermatol. 2019;139(2):492–494. doi:10.1016/j.jid.2018.08.015
14. Pyszko JA, Strosznajder JB. The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol. 2014;52(3):260–269. doi:10.5114/fn.2014.45567
15. Diaz Escarcega R, McCullough LD, Tsvetkov AS. The functional role of sphingosine kinase 2. Front Mol Biosci. 2021;8:683767. doi:10.3389/fmolb.2021.683767
16. Diaz Escarcega R, Murambadoro K, Valencia R, et al. Sphingosine kinase 2 regulates protein ubiquitination networks in neurons. Mol Cell Neurosci. 2024;130:103948. doi:10.1016/j.mcn.2024.103948
17. Li Y, Xia Y, Yin S, et al. Discovery of S1P/SPHK1 dysregulation in Parkinson’s disease and its role in pathological α-synuclein-induced neurodegeneration. The Innovation Life. 2025;3:100164. doi:10.59717/j.xinn-life.2025.100164
18. Alvarez SE, Harikumar KB, Hait NC, et al. Sphingosine-1-phosphate is a missing cofactor for the E3ubiquitin ligase TRAF2. Nature. 2010;465(7301):1084–1088. doi:10.1038/nature09128
19. Bisht M, Kadian JP, Hooda T, Jain N, Lather A, Aggarwal N. Explore the role of the Sphingosine-1-Phosphate signalling as a novel promising therapeutic target for the management of parkinson’s disease. Drug Res. 2024;74(8):365–378. doi:10.1055/a-2401-4578
20. Napolitano G, Karin M. Sphingolipids: the oil on the TRAFire that promotes inflammation. Sci Signal. 2010;3(141):pe34. doi:10.1126/scisignal.3141pe34
21. Xiao J. Sphingosine 1-Phosphate lyase in the developing and injured nervous system: a dichotomy? Mol Neurobiol. 2023;60(12):6869–6882. doi:10.1007/s12035-023-03524-3
22. Ishay Y, Rotnemer-Golinkin D, Ilan Y. The role of the sphingosine axis in immune regulation: a dichotomy in the anti-inflammatory effects between sphingosine kinase 1 and sphingosine kinase 2-dependent pathways. Int J Immunopathol Pharmacol. 2021;35:20587384211053274. doi:10.1177/20587384211053274
23. Van Echten-Deckert G, Van Echten-Deckert G. The role of sphingosine 1-phosphate metabolism in brain health and disease. Pharmacol Ther. 2023;244:108381. doi:10.1016/j.pharmthera.2023.108381
24. Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer Zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374(5–6):413–428. doi:10.1007/s00210-007-0132-3
25. Pyne NJ, Tonelli F, Lim KG, Long JS, Edwards J, Pyne S. Sphingosine 1-phosphate signalling in cancer. Biochem Soc Trans. 2012;40(1):94–100. doi:10.1042/BST20110602
26. Song DD, Zhou JH, Sheng R. Regulation and function of sphingosine kinase 2 in diseases. Histol Histopathol. 2018;33(5):433–445. doi:10.14670/HH-11-939
27. Lee JY, Jin HK, Bae JS. Sphingolipids in neuroinflammation: a potential target for diagnosis and therapy. BMB Rep. 2020;53(1):28–34. doi:10.5483/BMBRep.2020.53.1.278
28. Ziegler AC, Müller T, Gräler MH. Sphingosine 1-phosphate in sepsis and beyond: its role in disease tolerance and host defense and the impact of carrier molecules. Cell Signal. 2021;78:109849. doi:10.1016/j.cellsig.2020.109849
29. Tang HB, Jiang XJ, Wang C, Liu SC. S1P/S1PR3 signaling mediated proliferation of pericytes via Ras/pERK pathway and CAY10444 had beneficial effects on spinal cord injury. Biochem Biophys Res Commun. 2018;498(4):830–836. doi:10.1016/j.bbrc.2018.03.065
30. Babkina II, Sergeeva SP, Gorbacheva LR. The role of NF-κB in neuroinflammation. Neurochem J. 2021;15:114–128. doi:10.1134/S1819712421020045
31. Singh SS, Rai SN, Birla H, Zahra W, Rathore AS, Singh SP. NF-κB-Mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox Res. 2020;37(3):491–507. doi:10.1007/s12640-019-00147-2
32. Guo Q, Jin Y, Chen X, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther. 2024;9(1):53. doi:10.1038/s41392-024-01757-9
33. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–558. doi:10.1038/nri.2017.52
34. Engelmann C, Haenold R. Transcriptional control of synaptic plasticity by transcription factor NF-κB. Neural Plast. 2016;2016:7027949. doi:10.1155/2016/7027949
35. Xue F, Zhang M, Zhao RY, et al. Dectin-1 participates in neuroinflammation and dopaminergic neurodegeneration through synergistic signaling crosstalk with TLR4. Brain Behav Immun. 2025;126:260–273. doi:10.1016/j.bbi.2025.02.013
36. Villarreal A, Seoane R, González Torres A, et al. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: implications for its role in the propagation of reactive gliosis. J Neurochem. 2014;131(2):190–205. doi:10.1111/jnc.12790
37. Chu PH, Chen SC, Chen HY, Wu CB, Huang WT, Chiang HY. Astrocyte-associated fibronectin promotes the proinflammatory phenotype of astrocytes through β1 integrin activation. Mol Cell Neurosci. 2023;125:103848. doi:10.1016/j.mcn.2023.103848
38. Rocha SM, Kirkley KS, Chatterjee D, Aboellail TA, Smeyne RJ, Tjalkens RB. Microglia-specific knock-out of NF-κB/IKK2 increases the accumulation of misfolded α-synuclein through the inhibition of p62/sequestosome-1-dependent autophagy in the rotenone model of Parkinson’s disease. Glia. 2023;71(9):2154–2179. doi:10.1002/glia.24385
39. Lin S, Shu Y, Shen R, et al. The regulation of NFKB1 on CD200R1 expression and their potential roles in Parkinson’s disease. J Neuroinflammation. 2024;21(1):229. doi:10.1186/s12974-024-03231-3
40. Pitson SM, Moretti PA, Zebol JR, et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22(20):5491–5500. doi:10.1093/emboj/cdg540
41. Benakanakere MR, Zhao J, Galicia JC, Martin M, Kinane DF. Sphingosine kinase-1 is required for toll mediated beta-defensin 2 induction in human oral keratinocytes. PLoS One. 2010;5(7):e11512. doi:10.1371/journal.pone.0011512
42. Stahelin RV, Hwang JH, Kim JH, et al. The mechanism of membrane targeting of human sphingosine kinase 1. J Biol Chem. 2005;280(52):43030–43038. doi:10.1074/jbc.M507574200
43. Luo Y, Xue H, Gao Y, Ji G, Wu T. Sphingosine kinase 2 in cancer: a review of its expression, function, and inhibitor development. Int J Biol Macromol. 2025;306(Pt 1):141392. doi:10.1016/j.ijbiomac.2025.141392
44. Motyl J, Przykaza Ł, Boguszewski PM, Kosson P, Strosznajder JB. Pramipexole and Fingolimod exert neuroprotection in a mouse model of Parkinson’s disease by activation of sphingosine kinase 1 and Akt kinase. Neuropharmacology. 2018;135:139–150. doi:10.1016/j.neuropharm.2018.02.023
45. Zhong L, Jiang X, Zhu Z, et al. Lipid transporter Spns2 promotes microglia pro-inflammatory activation in response to amyloid-beta peptide. Glia. 2019;67(3):498–511. doi:10.1002/glia.23558
46. Donoviel MS, Hait NC, Ramachandran S, et al. Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J. 2015;29(12):5018–5028. doi:10.1096/fj.15-274936
47. Chen Z, Hagler J, Palombella VJ, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9(13):1586–1597. doi:10.1101/gad.9.13.1586
48. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267(5203):1485–1488. doi:10.1126/science.7878466
49. Watterson K, Sankala H, Milstien S, Spiegel S. Pleiotropic actions of sphingosine-1-phosphate. Prog Lipid Res. 2003;42(4):344–357. doi:10.1016/S0163-7827(03)00015-8
50. Giridharan S, Srinivasan M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J Inflamm Res. 2018;11:407–419. doi:10.2147/JIR.S140188
51. Li W, Cai H, Ren L, et al. Sphingosine kinase 1 promotes growth of glioblastoma by increasing inflammation mediated by the NF-κB /IL-6/STAT3 and JNK/PTX3 pathways. Acta Pharm Sin B. 2022;12(12):4390–4406. doi:10.1016/j.apsb.2022.09.012
52. Rostami N, Nikkhoo A, Ajjoolabady A, et al. S1PR1 as a novel promising therapeutic target in cancer therapy. Mol Diagn Ther. 2019;23(4):467–487. doi:10.1007/s40291-019-00401-5
53. Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int, J, Cancer. 2009;125(12):2863–2870. doi:10.1002/ijc.24748
54. Cornell TT, Hinkovska-Galcheva V, Sun L, et al. Ceramide-dependent PP2A regulation of TNFalpha-induced IL-8 production in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296(5):L849–56. doi:10.1152/ajplung.90516.2008
55. Loh KC, Baldwin D, Saba JD. Sphingolipid signaling and hematopoietic malignancies: to the rheostat and beyond. Anticancer Agents Med Chem. 2011;11(9):782–793. doi:10.2174/187152011797655159
56. Yu Z, Zhang H, Li L, et al. Microglia-mediated pericytes migration and fibroblast transition via S1P/S1P3/YAP signaling pathway after spinal cord injury. Exp Neurol. 2024;379:114864. doi:10.1016/j.expneurol.2024.114864
57. Liu SH, Lai YL, Chen BL, Yang FY. Ultrasound enhances the expression of brain-derived neurotrophic factor in astrocyte through activation of TrkB-Akt and Calcium-CaMK Signaling Pathways. Cereb Cortex. 2017;27(6):3152–3160. doi:10.1093/cercor/bhw169
58. Zhang L, Guo K, Zhou J, et al. Ponesimod protects against neuronal death by suppressing the activation of A1 astrocytes in early brain injury after experimental subarachnoid hemorrhage. J Neurochem. 2021;158(4):880–897. doi:10.1111/jnc.15457
59. Wieczorek I, Strosznajder RP. S1P receptor modulators affect the toxicity of amyloid β oligomers in microglial and neuronal cells. Folia Neuropathol. 2024;62(4):348–361. doi:10.5114/fn.2024.145758
60. Ghasemi R, Dargahi L, Ahmadiani A. Integrated sphingosine-1 phosphate signaling in the central nervous system: from physiological equilibrium to pathological damage. Pharmacol Res. 2016;104:156–164. doi:10.1016/j.phrs.2015.11.006
61. Ramalingam N, Jin SX, Moors TE, et al. Dynamic physiological α-synuclein S129 phosphorylation is driven by neuronal activity. NPJ Parkinsons Dis. 2023;9(1):4. doi:10.1038/s41531-023-00444-w
62. Feng M, An Y, Qin Q, et al. Sphk1/S1P pathway promotes blood-brain barrier breakdown after intracerebral hemorrhage through inducing Nlrp3-mediated endothelial cell pyroptosis. Cell Death Dis. 2024;15(12):926. doi:10.1038/s41419-024-07310-4
63. Oukka M, Bettelli E. Regulation of lymphocyte trafficking in central nervous system autoimmunity. Curr Opin Immunol. 2018;55:38–43. doi:10.1016/j.coi.2018.09.008
64. Liu T, Li J, Wu H, Qiao J, Wei J. Unraveling Parkinson’s disease: the mystery of mitochondria and the role of aging. Genes Dis. 2025;13(2):101719. doi:10.1016/j.gendis.2025.101719
65. Masato A, Plotegher N, Boassa D, Bubacco L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol Neurodegener. 2019;14(1):35. doi:10.1186/s13024-019-0332-6
66. Marchetti B, Giachino C, Tirolo C, Serapide MF. “Reframing” dopamine signaling at the intersection of glial networks in the aged Parkinsonian brain as innate Nrf2/Wnt driver: therapeutical implications. Aging Cell. 2022;21(4):e13575. doi:10.1111/acel.13575
67. Buoso C, Seifert M, Lang M, et al. Dopamine‑iron homeostasis interaction rescues mitochondrial fitness in Parkinson’s disease. Neurobiol Dis. 2024;196:106506. doi:10.1016/j.nbd.2024.106506
68. Tchekalarova J, Georgieva I, Vukova T, Apostolova S, Tzoneva R. Pinealectomy-Induced melatonin deficiency exerts age-specific effects on sphingolipid turnover in rats. Int J Mol Sci. 2025;26(4):1694. doi:10.3390/ijms26041694
69. Wei L, Yang X, Wang J, et al. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J Neuroinflammation. 2023;20(1):208. doi:10.1186/s12974-023-02879-7
70. Zeng Y, Zhang W, Xue T, Zhang D, Lv M, Jiang Y. Sphk1-induced autophagy in microglia promotes neuronal injury following cerebral ischaemia-reperfusion. Eur J Neurosci. 2022;56(4):4287–4303. doi:10.1111/ejn.15749
71. L’Episcopo F, Tirolo C, Serapide MF, et al. Microglia polarization, gene-environment interactions and Wnt/β-Catenin signaling: emerging roles of glia-neuron and glia-stem/neuroprogenitor crosstalk for dopaminergic neurorestoration in aged parkinsonian brain. Front Aging Neurosci. 2018;10:12. doi:10.3389/fnagi.2018.00012
72. Mohan UP, Filipeanu CM, Lazartigues E. Microglial activation and RAS signaling: a dual-edged sword in neuroinflammation. Am J Physiol Regul Integr Comp Physiol. 2026;330(2):R151–R165. doi:10.1152/ajpregu.00268.2025
73. Stoll AC, Kemp CJ, Patterson JR, et al. Neuroinflammatory gene expression profiles of reactive glia in the substantia nigra suggest a multidimensional immune response to alpha synuclein inclusions. Neurobiol Dis. 2024;191:106411. doi:10.1016/j.nbd.2024.106411
74. Usman S, Mondal AC. Menopause triggers microglia-associated neuroinflammation in Parkinson’s disease. Brain Res. 2025;1859:149649. doi:10.1016/j.brainres.2025.149649
75. Marfia G, Navone SE, Hadi LA, et al. The adipose mesenchymal stem cell secretome inhibits inflammatory responses of microglia: evidence for an involvement of Sphingosine-1-Phosphate signalling. Stem Cells Dev. 2016;25(14):1095–1107. doi:10.1089/scd.2015.0268
76. Frontiers Editorial Office. Retraction: the intra-nuclear SphK2-S1P axis facilitates M1-to-M2 shift of microglia via suppressing HDAC1-mediated KLF4 deacetylation. Front Immunol. 2026;17:1809852. doi:10.3389/fimmu.2026.1809852
77. Niskanen J, Peltonen S, Ohtonen S, et al. Uptake of alpha-synuclein preformed fibrils is suppressed by inflammation and induces an aberrant phenotype in human microglia. Glia. 2025;73(1):159–174. doi:10.1002/glia.24626
78. Guo M, Guan A, Zhang M, et al. Exosome-mediated microglia-astrocyte interactions drive neuroinflammation in Parkinson’s disease with Peli1 as a potential therapeutic target. Pharmacol Res. 2025;219:107908. doi:10.1016/j.phrs.2025.107908
79. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi:10.1038/nature21029
80. Parente A, Borzacchiello L, Giaccio M, et al. Progressive activation of the astrocyte A1 phenotype underlies microglia-astroglia crosstalk and contributes to neuroinflammation in neuronopathic MPS. Mol Genet Metab. 2025;146(1–2):109224. doi:10.1016/j.ymgme.2025.109224
81. Kawahata I, Finkelstein DI, Fukunaga K. Pathogenic impact of α-synuclein phosphorylation and its kinases in α-Synucleinopathies. Int J Mol Sci. 2022;23(11):6216. doi:10.3390/ijms23116216
82. Motyl JA, Strosznajder JB, Wencel A, Strosznajder RP. Recent insights into the interplay of alpha-synuclein and sphingolipid signaling in Parkinson’s disease. Int J Mol Sci. 2021;22(12):6277. doi:10.3390/ijms22126277
83. Takahashi M, Ko LW, Kulathingal J, Jiang P, Sevlever D, Yen SH. Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur J Neurosci. 2007;26(4):863–874. doi:10.1111/j.1460-9568.2007.05736.x
84. Hara S, Arawaka S, Sato H, et al. Serine 129 phosphorylation of membrane-associated α-synuclein modulates dopamine transporter function in a G protein-coupled receptor kinase-dependent manner. Mol Biol Cell. 2013;24(11):
85. Choi I, Zhang Y, Seegobin SP, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11(1):1386. doi:10.1038/s41467-020-15119-w
86. Ren G, Jiao P, Yan Y, Ma X, Qin G. Baicalin exerts a protective effect in diabetic nephropathy by repressing inflammation and oxidative stress through the SphK1/S1P/NF-κB signaling pathway. Diabetes Metab Syndr Obes. 2023;16:1193–1205. doi:10.2147/DMSO.S407177
87. Chen Y, Luo X, Yin Y, et al. The interplay of iron, oxidative stress, and α-synuclein in Parkinson’s disease progression. Mol Med. 2025;31(1):154. doi:10.1186/s10020-025-01208-3
88. Stefanova N. Microglia in Parkinson’s disease. J Parkinsons Dis. 2022;12(s1):S105–S112. doi:10.3233/JPD-223237
89. Vilhardt F, Haslund-Vinding J, Jaquet V, McBean G. Microglia antioxidant systems and redox signalling. Br J Pharmacol. 2017;174(12):1719–1732. doi:10.1111/bph.13426
90. Jin MM, Wang F, Qi D, et al. A critical role of autophagy in regulating microglia polarization in neurodegeneration. Front Aging Neurosci. 2018;10:378. doi:10.3389/fnagi.2018.00378
91. Hermida-Ameijeiras A, Méndez-Alvarez E, Sánchez-Iglesias S, Sanmartín-Suárez C, Soto-Otero R. Autoxidation and MAO-mediated metabolism of dopamine as a potential cause of oxidative stress: role of ferrous and ferric ions. Neurochem Int. 2004;45(1):103–116. doi:10.1016/j.neuint.2003.11.018
92. Lee HP, Zhu X, Liu G, et al. Divalent metal transporter, iron, and Parkinson’s disease: a pathological relationship. Cell Res. 2010;20(4):397–399. doi:10.1038/cr.2010.39
93. Sperlich CL, Stockwell BR, Farina M. When pathways converge: iron, lipid peroxidation, and α-Synuclein in ferroptosis-driven dopaminergic neurodegeneration. J Neurochem. 2026;170(4):e70438. doi:10.1111/jnc.70438
94. Strub GM, Paillard M, Liang J, et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011;25(2):600–612. doi:10.1096/fj.10-167502
95. Sivasubramanian M, Kanagaraj N, Dheen ST, Tay SS. Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson’s disease and in MPP+ -treated MN9D cells in vitro. Neuroscience. 2015;290:636–648. doi:10.1016/j.neuroscience.2015.01.032
96. Shen Z, Liu C, Liu P, Zhao J, Xu W. Sphingosine 1-phosphate (S1P) promotes mitochondrial biogenesis in Hep G2 cells by activating Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Cell Stress Chaperones. 2014;19(4):541–548. doi:10.1007/s12192-013-0480-5
97. Keogh MJ, Chinnery PF. Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta. 2015;1847(11):1401–1411. doi:10.1016/j.bbabio.2015.05.015
98. Gurrea-Rubio M, Wang Q, Mills EA, et al. Siponimod attenuates neuronal cell death triggered by neuroinflammation via NFκB and mitochondrial pathways. Int J Mol Sci. 2024;25(5):2454. doi:10.3390/ijms25052454
99. Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:111–119. doi:10.1111/j.1749-6632.2003.tb07469.x
100. Zhang X, Wang D, Zhang B, Zhu J, Zhou Z, Cui L. Regulation of microglia by glutamate and its signal pathway in neurodegenerative diseases. Drug Discov Today. 2020;25(6):1074–1085. doi:10.1016/j.drudis.2020.04.001
101. Su D, Cheng Y, Li S, Dai D, Zhang W, Lv M. Sphk1 mediates neuroinflammation and neuronal injury via TRAF2/NF-κB pathways in activated microglia in cerebral ischemia reperfusion. J Neuroimmunol. 2017;305:35–41. doi:10.1016/j.jneuroim.2017.01.015
102. Dorsey ER, De Miranda BR, Horsager J, Borghammer P. The body, the brain, the environment, and Parkinson’s disease. J Parkinsons Dis. 2024;14(3):363–381. doi:10.3233/JPD-240019
103. Chen SJ, Lin CH. Gut microenvironmental changes as a potential trigger in Parkinson’s disease through the gut-brain axis. J Biomed Sci. 2022;29(1):54. doi:10.1186/s12929-022-00839-6
104. Liu T, Wei J. FGF21 confers neuroprotection in Parkinson’s disease by activating the FGFR1-sirt1 pathway. Cell Mol Biol Lett. 2025;30(1):127. doi:10.1186/s11658-025-00807-6
105. Li YJ, Shi SX, Liu Q, Shi FD, Gonzales RJ. Targeted role for sphingosine-1-phosphate receptor 1 in cerebrovascular integrity and inflammation during acute ischemic stroke. Neurosci Lett. 2020;735:135160. doi:10.1016/j.neulet.2020.135160
106. Johann L, Soldati S, Müller K, et al. A20 regulates lymphocyte adhesion in murine neuroinflammation by restricting endothelial ICOSL expression in the CNS. J Clin Invest. 2023;133(24):e168314. doi:10.1172/JCI168314
107. Stewart WG, Hejl CD, Guleria RS, Gupta S. Thymosin β4 stabilizes hypoxia induced brain microvascular endothelial cell dysfunction through S1PR1 dependent mechanisms. Sci Rep. 2025;15(1):45764. doi:10.1038/s41598-025-28435-2
108. Lu Z, Ren S, Wang B, et al. Intranasally administered muse cells attenuate neurodegeneration in Parkinson’s disease. J Transl Med. 2025;23(1):1421.
109. Chen C, Wang GQ, Li DD, Zhang F. Microbiota-gut-brain axis in neurodegenerative diseases: molecular mechanisms and therapeutic targets. Mol Biomed. 2025;6(1):64. doi:10.1186/s43556-025-00307-1
110. Chatzikonstantinou S, Poulidou V, Arnaoutoglou M, et al. Signaling through the S1P-S1PR axis in the gut, the immune and the central nervous system in multiple sclerosis: implication for pathogenesis and treatment. Cells. 2021;10(11):3217. doi:10.3390/cells10113217
111. Martín-Hernández D, Muñoz-López M, Tendilla-Beltrán H, et al. Immune system and brain/intestinal barrier functions in psychiatric diseases: is Sphingosine-1-Phosphate at the helm? Int J Mol Sci. 2023;24(16):12634.
112. Xie W, Gao J, Jiang R, et al. Twice subacute MPTP administrations induced time-dependent dopaminergic neurodegeneration and inflammation in midbrain and ileum, as well as gut microbiota disorders in PD mice. Neurotoxicology. 2020;76:200–212. doi:10.1016/j.neuro.2019.11.009
113. He Y, Zhao J, Ma Y, et al. Citrobacter rodentium infection impairs dopamine metabolism and exacerbates the pathology of Parkinson’s disease in mice. J Neuroinflammation. 2024;21(1):153. doi:10.1186/s12974-024-03145-0
114. Wang Y, Zhang GJ, Sun YN, et al. Identification of metabolite biomarkers for L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease by metabolomic technology. Behav Brain Res. 2018;347:175–183. doi:10.1016/j.bbr.2018.03.020
115. Bao X, Xu X, Wu Q, et al. Sphingosine 1-phosphate promotes the proliferation of olfactory ensheathing cells through YAP signaling and participates in the formation of olfactory nerve layer. Glia. 2020;68(9):1757–1774. doi:10.1002/glia.23803
116. Meng H, Lee VM. Differential expression of sphingosine-1-phosphate receptors 1-5 in the developing nervous system. Dev Dyn. 2009;238(2):487–500. doi:10.1002/dvdy.21852
117. Kong X, Liu T, Wei J. Parkinson’s Disease: the neurodegenerative enigma under the “Undercurrent” of endoplasmic reticulum stress. Int J Mol Sci. 2025;26(7):3367. doi:10.3390/ijms26073367
118. Liao KH, Liao KL. Interference with sphingosine kinase-1 reduces the hydrogen peroxide-induced oxidative stress damage in melanocytes through four and a half LIM domains 2. Gen Physiol Biophys. 2024;43(4):321–333. doi:10.4149/gpb_2024011
119. Park SW, Huq MD, Hu X, Wei LN. Tyrosine nitration on p65: a novel mechanism to rapidly inactivate nuclear factor-kappaB. Mol Cell Proteomics. 2005;4(3):300–309. doi:10.1074/mcp.M400195-MCP200
120. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26(8):3071–3084. doi:10.1128/MCB.26.8.3071-3084.2006
121. Ahumada-Montalva P, Muñoz-Carvajal F, Bórquez-Macaya S, et al. TMBIM6 enhances dopaminergic neuron survival by modulating the IRE1a pathway in Parkinson’s disease. Cell Death Dis. 2026;17(1):385. doi:10.1038/s41419-025-08391-5
122. Rajan S, Sood A, Jain R, Kamatham PT, Khatri DK. Fingolimod exerts neuroprotection by regulating S1PR1 mediated BNIP3-PINK1-Parkin dependent mitophagy in rotenone induced mouse model of Parkinson’s disease. Neurosci Lett. 2024;820:137596. doi:10.1016/j.neulet.2023.137596
123. Wang G, Bieberich E. Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv Biol Regul. 2018;70:51–64. doi:10.1016/j.jbior.2018.09.013
124. Syed SN, Weigert A, Brüne B. Sphingosine kinases are involved in macrophage NLRP3 inflammasome transcriptional induction. Int J Mol Sci. 2020;21(13):4733. doi:10.3390/ijms21134733
125. Wang Y, Wang C, He Q, et al. Inhibition of sphingosine-1-phosphate receptor 3 suppresses ATP-induced NLRP3 inflammasome activation in macrophages via TWIK2-mediated potassium efflux. Front Immunol. 2023;14:1090202. doi:10.3389/fimmu.2023.1090202
126. Soraci L, Gambuzza ME, Biscetti L, et al. Toll-like receptors and NLRP3 inflammasome-dependent pathways in Parkinson’s disease: mechanisms and therapeutic implications. J Neurol. 2023;270(3):1346–1360. doi:10.1007/s00415-022-11491-3
127. Parisiadou L, Cai H. LRRK2 function on actin and microtubule dynamics in Parkinson disease. Commun Integr Biol. 2010;3(5):396–400. doi:10.4161/cib.3.5.12286
128. Narendra DP, Youle RJ. The role of PINK1-Parkin in mitochondrial quality control. Nat Cell Biol. 2024;26(10):1639–1651. doi:10.1038/s41556-024-01513-9
129. Leandrou E, Chalatsa I, Anagnostou D, et al. α-Synuclein oligomers potentiate neuroinflammatory NF-κB activity and induce Cav3.2 calcium signaling in astrocytes. Transl Neurodegener. 2024;13(1):11. doi:10.1186/s40035-024-00401-4
130. Teng K, Ma H, Gai P, Zhao X, Qi G. SPHK1 enhances olaparib resistance in ovarian cancer through the NFκB/NRF2/ferroptosis pathway. Cell Death Discov. 2025;11(1):29. doi:10.1038/s41420-025-02309-y
131. Hait NC, Allegood J, Maceyka M, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325(5945):1254–1257. doi:10.1126/science.1176709
132. Xia J, Wu Z, Yu C, et al. miR-124 inhibits cell proliferation in gastric cancer through down-regulation of SPHK1. J Pathol. 2012;227(4):470–480. doi:10.1002/path.4030
133. Liu BF, Chen Q, Zhang M, Zhu YK. MiR-124 promotes ischemia-reperfusion induced cardiomyocyte apoptosis by targeting sphingosine kinase 1. Eur Rev Med Pharmacol Sci. 2019;23(16):7049–7058. doi:10.26355/eurrev_201908_18747
134. Shaheen N, Shaheen A, Osama M, Nashwan AJ, Bharmauria V, Flouty O. MicroRNAs regulation in Parkinson’s disease, and their potential role as diagnostic and therapeutic targets. NPJ Parkinsons Dis. 2024;10(1):186. doi:10.1038/s41531-024-00791-2
135. Kikuchi M, Morita S, Wakamori M, et al. Epigenetic mechanisms to propagate histone acetylation by p300/CBP. Nat Commun. 2023;14(1):4103. doi:10.1038/s41467-023-39735-4
136. Schwedhelm E, Englisch C, Niemann L, et al. Sphingosine-1-Phosphate, motor severity, and progression in Parkinson’s Disease (MARK-PD). Mov Disord. 2021;36(9):2178–2182. doi:10.1002/mds.28652
137. Wang N, Li X, Li B, et al. Multi-omics study identifies diagnostic metabolic signatures of early Parkinson’s disease associated with dysregulated glutathione and TCA cycle metabolism. Parkinsonism Relat Disord. 2026;144:108230. doi:10.1016/j.parkreldis.2026.108230
138. Waragai M, Sekiyama K, Sekigawa A, Takamatsu Y, Fujita M, Hashimoto M. α-Synuclein and DJ-1 as potential biological fluid biomarkers for Parkinson’s Disease. Int J Mol Sci. 2010;11(11):4257–4266. doi:10.3390/ijms11114257
139. Bereczki E, Bogstedt A, Höglund K, et al. Synaptic proteins in CSF relate to Parkinson’s disease stage markers. NPJ Parkinsons Dis. 2017;3:7. doi:10.1038/s41531-017-0008-2
140. Taneva SG, Todinova S, Andreeva T. Morphometric and nanomechanical screening of peripheral blood cells with atomic force microscopy for label-free assessment of alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Int J Mol Sci. 2023;24(18):14296. doi:10.3390/ijms241814296
141. Zhou B, Wen M, Yu WF, Zhang CL, Jiao L. The diagnostic and differential diagnosis utility of cerebrospinal fluid α -Synuclein levels in Parkinson’s disease: a meta-analysis. Parkinsons Dis. 2015;2015:567386. doi:10.1155/2015/567386
142. Bianco MG, Cristiani CM, Scaramuzzino L, et al. Combined blood neurofilament light chain and third ventricle width to differentiate progressive supranuclear palsy from Parkinson’s Disease: a machine learning study. Parkinsonism Relat Disord. 2024;123:106978. doi:10.1016/j.parkreldis.2024.106978
143. Malaty GR, Decourt B, Shill HA, Sabbagh MN. Biomarker assessment in Parkinson’s disease dementia and dementia with lewy bodies by the immunomagnetic reduction assay and clinical measures. J Alzheimers Dis Rep. 2024;8(1):1361–1371. doi:10.3233/ADR-240110
144. Yi X, Tang X, Li T, et al. Therapeutic potential of the sphingosine kinase 1 inhibitor, PF-543. Biomed Pharmacother. 2023;163:114401. doi:10.1016/j.biopha.2023.114401
145. Antoon JW, White MD, Burow ME, Beckman BS. Dual inhibition of sphingosine kinase isoforms ablates TNF-induced drug resistance. Oncol Rep. 2012;27(6):1779–1786. doi:10.3892/or.2012.1743
146. French KJ, Zhuang Y, Maines LW, et al. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010;333(1):129–139. doi:10.1124/jpet.109.163444
147. Haldar R, De A. Clinical status and CNS adverse drug report of Sphingosine-1-Phosphate receptor modulators in multiple sclerosis. Drug Discov Today. 2025;30(10):104469. doi:10.1016/j.drudis.2025.104469
148. Pépin É, Jalinier T, Lemieux GL, Massicotte G, Cyr M. Sphingosine-1-Phosphate receptors modulators decrease signs of neuroinflammation and prevent Parkinson’s disease symptoms in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine mouse model. Front Pharmacol. 2020;11:77. doi:10.3389/fphar.2020.00077
149. Maniscalco GT, Sparaco M, Di Gregorio M, et al. Real-world comparison of lymphopenia profiles in S1P receptor modulators for multiple sclerosis: a multicenter retrospective study. J Neurol. 2025;272(9):559. doi:10.1007/s00415-025-13300-z
150. Coyle PK, Freedman MS, Cohen BA, Cree BAC, Markowitz CE. Sphingosine 1-phosphate receptor modulators in multiple sclerosis treatment: a practical review. Ann Clin Transl Neurol. 2024;11(4):842–855. doi:10.1002/acn3.52017
151. Han X, Lu M, Wang S, Lv D, Liu H. Targeting IKK/NF-κB pathway reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci Lett. 2012;511(1):28–32. doi:10.1016/j.neulet.2012.01.030
152. Hikage F, Lennikov A, Mukwaya A, et al. NF-κB activation in retinal microglia is involved in the inflammatory and neovascularization signaling in laser-induced choroidal neovascularization in mice. Exp Cell Res. 2021;403(1):112581. doi:10.1016/j.yexcr.2021.112581
153. Esmaealzadeh N, Miri MS, Mavaddat H, et al. The regulating effect of curcumin on NF-κB pathway in neurodegenerative diseases: a review of the underlying mechanisms. Inflammopharmacology. 2024;32(4):2125–2151. doi:10.1007/s10787-024-01492-1
154. Wu J, Tian Z, Wang B, et al. Exploring resveratrol against Alzheimer’s disease and Parkinson’s disease through integrating network pharmacology, bioinformatics, and experimental validation strategy in vitro. Heliyon. 2024;10(18):e37908. doi:10.1016/j.heliyon.2024.e37908
155. Cosden M, Jinn S, Yao L, et al. A novel glucosylceramide synthase inhibitor attenuates alpha synuclein pathology and lysosomal dysfunction in preclinical models of synucleinopathy. Neurobiol Dis. 2021;159:105507. doi:10.1016/j.nbd.2021.105507
156. Viel C, Clarke J, Kayatekin C, et al. Preclinical pharmacology of glucosylceramide synthase inhibitor venglustat in a GBA-related synucleinopathy model. Sci Rep. 2021;11(1):20945. doi:10.1038/s41598-021-00404-5
157. Kreisler A, Duhamel A, Vanbesien-Mailliot C, Destée A, Bordet R. Differing short-term neuroprotective effects of the fibrates fenofibrate and bezafibrate in MPTP and 6-OHDA experimental models of Parkinson’s disease. Behav Pharmacol. 2010;21(3):194–205. doi:10.1097/FBP.0b013e32833a5c81
158. Alcalay RN, Mallett V, Vanderperre B, et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov Disord. 2019;34(4):526–535. doi:10.1002/mds.27642
159. Uddin MS, Al Mamun A, Kabir MT, et al. Neuroprotective role of polyphenols against oxidative stress-mediated neurodegeneration. Eur J Pharmacol. 2020;886:173412. doi:10.1016/j.ejphar.2020.173412
160. Wang J, Song Y, Chen Z, Leng SX. Connection between systemic inflammation and neuroinflammation underlies neuroprotective mechanism of several phytochemicals in neurodegenerative diseases. Oxid Med Cell Longev. 2018;2018:1972714. doi:10.1155/2018/1972714
161. Kalu A, Ray SK. Epigallocatechin-3-Gallate, quercetin, and kaempferol for treatment of Parkinson’s disease through prevention of gut dysbiosis and attenuation of multiple molecular mechanisms of pathogenesis. Brain Sci. 2025;15(2):144. doi:10.3390/brainsci15020144
162. Yang X, Hu D, Cheng R, et al. Unlocking the neuroprotective secrets of natural products: a focus on the gut-brain axis. Phytochem Rev. 2025;24:5569–5612. doi:10.1007/s11101-025-10081-1
163. Katsoulaki EE, Dimopoulos D, Hadjipavlou-Litina D. Multitarget compounds designed for Alzheimer, Parkinson, and huntington neurodegeneration diseases. Pharmaceuticals. 2025;18(6):831. doi:10.3390/ph18060831
164. Unnithan D, Sartaj A, Iqubal MK, Ali J, Baboota S. A neoteric annotation on the advances in combination therapy for Parkinson’s disease: nanocarrier-based combination approach and future anticipation. Part II: nanocarrier design and development in focus. Expert Opin Drug Deliv. 2024;21(3):437–456. doi:10.1080/17425247.2024.2331216
165. Guo X, Qiao X, Li X, et al. Lactoferrin-modified organic-inorganic hybrid mesoporous silica for co-delivery of levodopa and curcumin in the synergistic treatment of Parkinson’s disease. Phytomedicine. 2025;140:156547. doi:10.1016/j.phymed.2025.156547
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Targeted Liposomal Co-Delivery Dopamine with 3-n-Butylphthalide for Effective Against Parkinson’s Disease in Mice Model

Liang Y, Feng L, Zheng Y, Gao Y, Shi R, Zhang Z, Ying X, Zeng Y
International Journal of Nanomedicine 2024, 19:12851-12870
Published Date: 30 November 2024
GPX3 as a Novel and Potential Therapeutic Target in the Shared Molecular Mechanisms of Traumatic Brain Injury and Parkinson’s Disease
Wang Y, Fang J, Yuan Q, Yu J, Hu J
Journal of Inflammation Research 2025, 18:1911-1928
Published Date: 7 February 2025
Exploring Potential Causal Links: How Parkinson’s Disease Brain Damage Impacts Liver Health–A Mendelian Randomization and Transcriptomic Study
Liu T, Chen Q, Wu H, Li J, Xian M, Lu K, Zhu C, Wei J
Journal of Inflammation Research 2025, 18:8173-8197
Published Date: 21 June 2025
Circadian Rhythm Dysfunction in Neurodegenerative Diseases: A Bidirectional Perspective and Therapeutic Potential
Namgyal D, Lim CS
Nature and Science of Sleep 2025, 17:2969-2989
Published Date: 19 November 2025
GLP-1 Receptor Agonists in Neurological Disorders: From Mechanisms to Clinical Translation
Li P, Gao Y, Liu W
Drug Design, Development and Therapy 2026, 20:613616
Published Date: 12 June 2026