Back to Journals » Journal of Inflammation Research » Volume 15
Sodium Tanshinone IIA Sulfonate Inhibits Vascular Endothelial Cell Pyroptosis via the AMPK Signaling Pathway in Atherosclerosis
Authors Zhu J, Chen H, Guo J, Zha C, Lu D ![]()
Received 23 August 2022
Accepted for publication 3 November 2022
Published 14 November 2022 Volume 2022:15 Pages 6293—6306
DOI https://doi.org/10.2147/JIR.S386470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Adam Bachstetter
Ji Zhu,1,* Hang Chen,2,* Jianan Guo,2 Chen Zha,1 Dezhao Lu2
1The Third Affiliated Hospital of Zhejiang Chinese Medical University (Zhongshan Hospital of Zhejiang Province), Hangzhou, People’s Republic of China; 2School of Life Sciences, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dezhao Lu, Email [email protected]
Introduction: Atherosclerosis (AS) is the underlying cause of cardiovascular events. Endothelial cell mitochondrial damage and pyroptosis are important factors contributing to AS. Changes in internal mitochondrial conformation and increase in reactive oxygen species (ROS) lead to the disruption of mitochondrial energy metabolism, activation of the NLRP3 inflammasome and pyroptosis, which in turn affect atherogenesis by impairing endothelial function. AMPK is a core player in the regulation of cellular metabolism, not only by regulating mitochondrial homeostasis but also by regulating cellular inflammatory responses. Sodium tanshinone IIA sulfonate (STS), a water-soluble derivative of tanshinone IIA, has significant antioxidant and anti-inflammatory effects, and roles in cardiovascular protection.
Purpose: In this study, we investigated whether STS plays a protective role in AS by regulating endothelial cell mitochondrial function and pyroptosis through an AMPK-dependent mitochondrial pathway.
Methods and Results: Male ApoE−/− mice and HUVECs were used for the experiments. We found that STS treatment largely abrogated the upregulation of key proteins in aortic vessel wall plaques and typical pyroptosis signaling in ApoE−/− mice fed a western diet, consequently enhancing pAMPK expression, plaque stabilization, and anti-inflammatory responses. Consistently, STS pretreatment inhibited cholesterol crystallization (CC) -induced cell pyroptosis and activated pAMPK expression. In vitro, using HUVECs, we further found that STS treatment ameliorated mitochondrial ROS caused by CC, as evidenced by the finding that STS inhibited mitochondrial damage caused by CC. The improvement of endothelial cell mitochondrial function by STS is blocked by dorsomorphin (AMPK inhibitor). Consistently, the blockade of endothelial cell pyroptosis by STS is disrupted by dorsomorphin.
Conclusion: Our results suggest that STS enhances maintenance of mitochondrial homeostasis and inhibits mitochondrial ROS overproduction via AMPK, thereby improving endothelial cell pyroptosis during AS.
Keywords: atherosclerosis, pyroptosis, tanshinone IIA, AMPK, mitochondria
Introduction
Atherosclerosis (AS) is a commonly recognized metabolic and chronic inflammatory disease. And AS is accompanied by endothelial dysfunction, lipid deposition, and vascular wall thickening.1 AS is a significant cardiovascular burden and a leading cause of death worldwide, and is recognized as the basis of cardiovascular disease (CVD).2,3 ASCVD results in approximately 2.4 million deaths annually in China.4–6 Vascular endothelial cells constitute the inner wall of arterial vessels, and the morphological integrity and balance of their function significantly contribute to vascular health.7 Endothelial dysfunction, structural alterations, and chronic inflammation of the vascular wall are known to be critical steps in AS development, as well as in the instability of advanced plaques.8,9 However, the underlying mechanisms and appropriate means to clinically manage the disease require further investigation.
Endothelial cell dysfunction is characterized by proinflammatory cytokines, elevated ROS, disrupted vascular tone, and various types of programmed cell death (PCD), including apoptosis, autophagy, pyroptosis, and necroptosis.10 Pyroptosis is a newly discovered form of programmed inflammatory cell death, which is suggested to contribute to vascular endothelial cell dysfunction and the pathogenesis of AS.11,12 Pyroptosis can be driven by the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, which is composed of NLRP3, the adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase1.13,14 Once activated, NLRP3 recruits and cleaves pro-caspase1 into its active forms, which process pro-inflammatory cytokines such as IL-1β and IL-18 and HMGB1.15 Active forms of caspase-1 also cleave gasdermin D (GSDMD), separating the N-terminal pore-forming structural domain (PFD) of GSDMD from the C-terminal deterrent structural domain (RD) and the subsequent oligomerization of PFD transferred into the cell membrane. And the oligomer of GSDMD leads to massive pore formation, resulting in cell death, leakage of the cellular contents, including proinflammatory substances, and increased secondary inflammation.16 Oxidized low density lipoprotein (ox-LDL) and cholesterol crystals (CC) are the two main factors promoting atherosclerosis. In addition, ox-LDL and cholesterol crystals represent the central activators of the NLRP3 inflammasome and endothelial cell pyroptosis through excessive ROS production.17–19 Moreover, some risk factors for AS, such as hyperlipidemia, smoking, obesity, diabetes, and hypertension, can result in EC dysfunction and even death by triggering inflammasome assembly.20 Therefore, endothelial cell pyroptosis may play a prominent role in AS-interrelated inflammation, and targeted regulation of the endothelial NLRP3 inflammasome in atherosclerotic lesions may represent a novel direction for treating AS.21
Recent studies have suggested that some risk factors for AS (eg, ox-LDL and cholesterol crystals) mediate endothelial cell dysfunction primarily via mitochondrial damage.22 Mitochondrial dysfunction can lead to ROS production, mitochondrial DNA (mtDNA) damage, ATP reduction, and membrane potential decline. Mitochondria are the main source of ROS production and mtROS has also been shown to drive NLRP3 inflammasome activation.23,24 Thus, maintenance of mitochondrial homeostasis is necessary to sustain endothelial cell survival and modulate the inflammation in AS.25 AMP-activated protein kinase (AMPK) is a highly conserved serine/threonine protein kinase and a key molecule in the regulation of bioenergy metabolism.26 Importantly, activated AMPK monitors mitochondrial function and cellular energy status in AS.27 The up-regulation of the AMPK pathway has been proven to be conducive to the preservation of mitochondrial function in metabolic diseases under the overactivation of the NLRP3 inflammasome.28 Thus, the AMPK/PGC-1α pathway shows potential as a novel therapeutic target for AS.
Tanshinone IIA, a diterpene quinone that originates from the roots of the traditional Chinese herb Salvia miltiorrhiza, is used as a treatment for CVD and columnar ectopy.29 Sodium tanshinone IIA sulfonate (STS) is a water-soluble derivative of tanshinone IIA,30 and sodium tanshinone IIA sulfonate injection has been approved by the Chinese State Food and Drug Administration (CFDA) for the treatment of cardiovascular diseases.31,32 Recently, several studies have explored other potential therapeutic effects of tanshinone IIA and its derivatives in various diseases, including central nervous system disorders,33 diabetes,34 cancer,35 depression36 and liver diseases.37,38 Tanshinone IIA and its derivatives also displays a potent beneficial effect in AS by improving endothelial function via multiple processes, including by stabilizing vulnerable AS plaques via the TLR4/MyD88/NF-κB signaling pathway,39 improving mitochondrial function,40 alleviating oxidative stress,41 inhibiting cell death42,43 and reducing the inflammatory response.41 Nonetheless, whether STS impacts NLRP3 inflammasome activation and subsequent pyroptosis in endothelial cells, and what the underlying mechanisms could be, remain unclear. Considering the NLRP3 inflammasome and pyroptosis are important in endothelial dysfunction and AS, these factors warrant investigation. Therefore, we examined the protective effects of STS, particularly on NLRP3 inflammasome activation and pyroptosis, and explored the potential mechanism by which these occur. In the present study, we found that STS inhibits NLRP3 inflammasome activation and subsequent pyroptosis, and may be mediated by the AMPK/mitochondria axis in HUVECs treated with cholesterol crystals.
Materials and Methods
Materials
Dorsomorphin (Compound C, S7840), and Sodium tanshinone IIA sulfonate (STS, purity 98.61%, S3766) were obtained from Selleck (Shanghai, China). LPS (Escherichia coli O11:B4) and cholesterol were purchased from Sigma (St Louis, USA). And cholesterol crystals as were prepared as previously described.44,45
Animals and Establishment of as Model
Male ApoE−/− mice (18–22 g, 6 weeks of age) were provided by GemPharmatech Co., Ltd (Nanjing, China). The animals were acclimated for 7 days with a 12 h light-dark cycle with water and food ad libitum. Animal care and experiments were performed according to the Guide of Chinese Regulation for the Use and Care of Laboratory Animals. The experiments were approved by the Medical Code and Ethics Committee of Zhejiang Chinese Medical University (approval number: 20200720-06). ApoE−/− mice were fed with a western diet [containing 0.5% (wt/wt) cholesterol, fat 42% kcal, carbohydrate 42.7% kcal, protein 15.3% kcal, #TP26304, Trophi Feed High-tech Co., Ltd., Nantong, China] for 12 weeks to establish the AS model. Then the ApoE−/− mice were with a western diet were randomly assigned to three groups as follows: (1) the western diet (WD) AS model group, in which the mice were administrated intragastrically saline every day and continually fed a western diet for 4 weeks; (2) the low STS (AS+LT) group, in which the mice were administrated intragastrically 10 mg/kg STS every day and continually fed a western diet for 4 weeks; and (3) the high STS (AS+HT)group, in which the mice were administrated intragastrically 20 mg/kg STS every day and continually fed a western diet for 4 weeks. Each group has four mice. And in normal control group, four male ApoE−/− mice were fed with a normal control diet [containing fat 12.5% kcal, carbohydrate 68.1% kcal, protein 19.4% kcal, #TP26352, Trophi Feed High-tech Co., Ltd., Nantong, China] for 16 weeks.
Analysis of Atherosclerotic Lesions
Mouse aortas were opened longitudinally from the ascending aorta to the abdominal aorta, and fixed in 4% paraformaldehyde for 36 h. Hematoxylin and eosin (HE) staining assay was used to detect atherosclerotic lesions, and the Oil Red O (Solarbio, China) staining kit was used to evaluate lipid accumulation. Masson staining was used to evaluate the content of collagen fibers in atherosclerotic plaques. Immunofluorescence staining was performed to stain the location of endothelial cells in atherosclerotic lesions using CD31 antibody (1:100, Abcam, USA), and to detect the expression of GSDMD (1:50, Santa Cruz, USA) and the co-localization of endothelial NLRP3 (1:100, AdipoGen, USA) and ASC (1:100, AdipoGen, USA) by visualizing using a fluorescence microscope (Olympus, Japan). Immunohistochemistry staining was performed to stain the level of IL-1β and IL-18 in atherosclerotic lesions using IL-1β antibody (1:100, Proteintech, China) and IL-18 antibody (1:100, Proteintech, China) by visualizing using a Nano Zoomer (Hamamatsu, Japan). Quantitative analysis of lesions was performed using Fiji software (NIH, USA).
Cell Culture
HUVECs (Lonza, USA) cultured in Endothelial cell medium (ECM, Sciencell, USA) at 37°C with 5% CO2 and 95% air.
Cell Death Assay
Cell death was evaluated using a PI assay. For assay of cell death, HUVECs after treatment were detected using a Calcein-AM/PI Double Staining Kit (Dojindo Kumamoto, Japan).
Cellular Mitochondrial ROS (mtROS) and Mitochondrial Membrane Potential (MMP) Assay
After incubating with Mito-Tracker Green (100 nM, Thermo Fisher Scientific, USA) at 37°C for 30 min, HUVECs were incubated with MitoSox Red (5 μM, Thermo Fisher Scientific, USA) in phosphate-buffered saline (PBS) at 37°C for 30 min. The cells were then washed thrice with warm PBS. The MMP was determined with TMRE (100nM, MCE, USA). And the level of mtROS and MMP were assayed using the ImageXpress® Micro Confocal High-Content Imaging System (Molecular Devices, USA) as described before.46,47
Western Blot Analysis
Total protein was extracted from HUVECs using Cell lysis buffer for Western and IP [20mM Tris(pH7.5), 150mM NaCl, 1% Triton X-100, Beyotime, China]. The concentrations of protein were determined by a BCA protein assay kit (Vazime, China). 20 μg proteins were separated by BeyoGel™ Plus Precast PAGE Gel (Beyotime, China) and transferred onto PVDF membranes (Millipore, USA). After blocking with 5% dry milk, the membrane was incubated with primary antibodies against NLRP3 (1:1000, AdipoGen, USA), ASC (1:1000, AdipoGen, USA), Caspase-1 (1:1000, Abcam, UK), GSDMD (1:500, Santa, USA), and IL-1β (1:500, Santa Cruz, USA), AMPKα (1:1000, Cell Signaling Technology, USA), and Phospho-AMPKα (Thr172) (1:1000, Cell Signaling Technology, USA), β-actin (1:2000, Proteintech, China) overnight at 4°C. Following incubation, the membrane was washed thrice with TBST (0.05% Tween 20). After incubation with the corresponding secondary antibody [goat anti-rabbit (1:5000, BOSTER Biological Technology co. ltd, China) and goat anti-mouse (1:5000, BOSTER Biological Technology co. ltd, China)], the membrane was exposed to an enhanced chemiluminescence kit (Vazime, China), and observed using a Clinx ChemiScope 3500 (Clinx Science instrument Co. Ltd, China).
Measurement of TG, TC, LDL-C, and HDL-C Contents
Levels of TC, TG, LDL-C and HDL-C in serum were measured using commercial kits (Nanjing Jiancheng Bioengineering Institute, China)
Statistical Analysis
GraphPad Prism 8.0 software was used for statistical analysis. One-way ANOVA was used to analyze differences among groups. All data in this study are presented as the mean ± SD. P-values < 0.05 were considered statistically significant.
Results
Effects of STS on Aortic Lesions, Lipid Homeostasis, and Inflammation in WD-Induced ApoE−/− Mice
To confirm the protective effect of STS postconditioning on AS, we used HE and Masson staining in histological sections of the ascending aorta of the ApoE−/− mice. And Oil red O staining was performed to show aortic lesions. The serum levels of low-density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), triglyceride (TG), and total cholesterol (TC) were also determined to investigate the effect of STS on lipid profiles. Our results showed that after treatment with STS, the atherosclerotic plaque decreased significantly compared to the WD group as shown by Oil red O staining (Figure 1H). H&E staining indicated that the aortas from the WD group exhibited an increased intimal lesion area containing a necrotic core and Masson staining showed unstable plaque with cholesterol crystal and healed plaque rupture in WD group (Figure 1F and G). However, STS treatment improved this condition (Figure 1F and G). The results showed that STS significantly decreased the levels of LDL-C, TG, and TC, especially in the high-dose group (Figure 1A–D). However, the HDL-C levels were not changed in the WD group (Figure 1E).
 |
Figure 1 Sodium tanshinone IIA sulfonate alleviates atherosclerosis in ApoE−/− mice. (A) Body Weight, (B) plasma total cholesterol (TC) level, (C) plasma triglyceride (TG) level, (D) Plasma low-density lipoprotein cholesterol (LDL-c) level, and (E) plasma high density liptein cholesterol (HDL-c) level. (F) H&E staining of the aortic arches of the ApoE−/− mice. (G) Masson staining of the aortic arches. (H) Oil red O staining of the aortic arches. Data are represented as mean ± SD of three independent replicates; **P < 0.01 vs NCD group; †P < 0.05, ††P < 0.01 vs AS model group. |
STS Inhibited Pyroptosis in the Aortic Intima of WD-Induced ApoE−/− Mice
Compared to the NCD group, immunofluorescent triple staining of the aortic sinus of NLRP3, ASC, and CD31 (endothelial cell marker) revealed that the content of endothelial NLRP3-ASC colocalization was significantly increased in the ascending aorta of WD group, while STS significantly suppressed this increase (Figure 2A). Our results further showed that the expression of GSDMD in the ascending aorta of WD-fed ApoE−/− mice was significantly increased compared to that of the NCD group, while STS significantly suppressed the enhanced expression of GSDMD (Figure 2B). Furthermore, the positive area of IL-1β and IL-18 was increased in plaques of mice fed with Western diet, but decreased in plaques of mice administrated with STS (Figure 2C and D). Our findings suggested that STS suppressed the pyroptosis of endothelial cells in the aortic intima of WD-fed ApoE−/− mice.
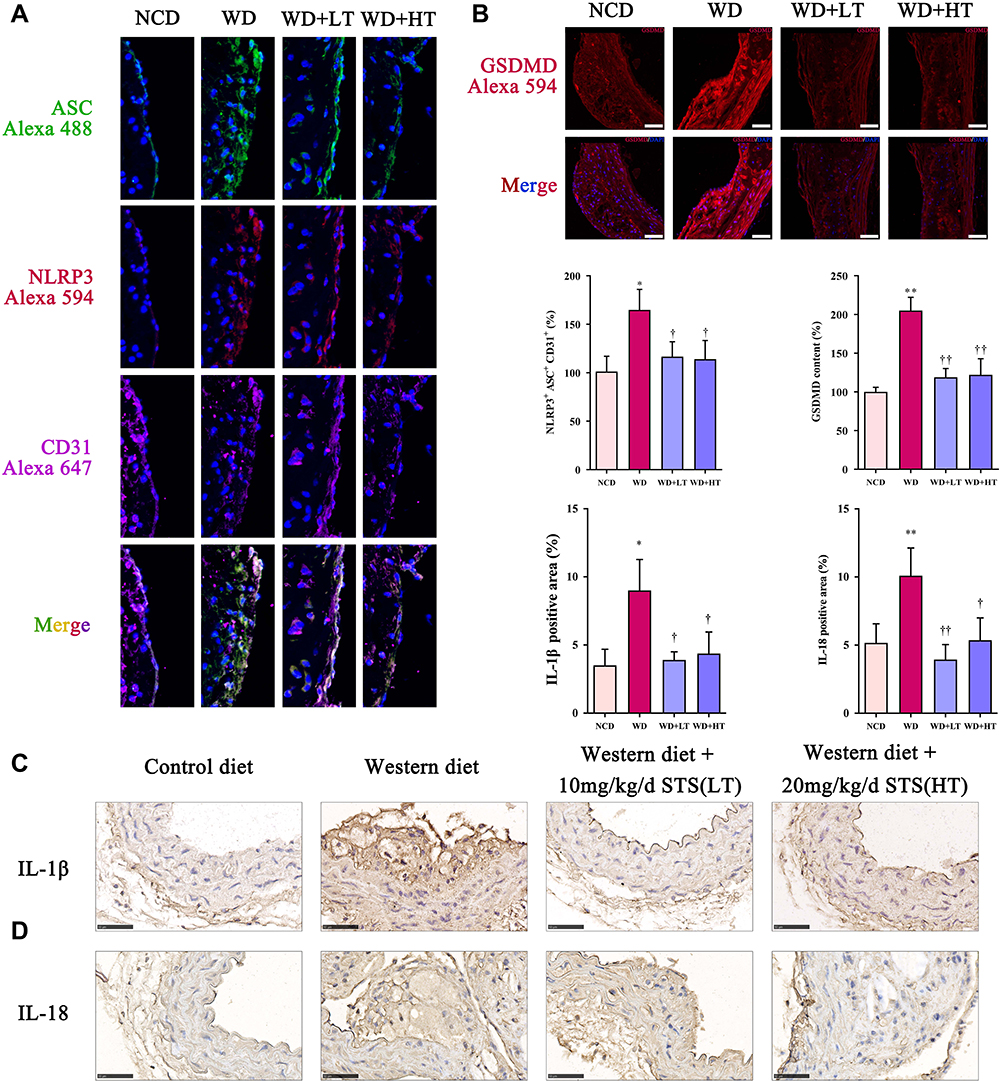 |
Figure 2 Sodium tanshinone IIA sulfonate alleviates the pyroptosis in atherosclerotic plaques. (A) NLRP3 and ASC in endothelial cells (marked by CD31) the aortic arches were detected by immunofluorescence. (B) GSDMD level in sections of the aortic arches was detected by immunofluorescence. (C) Immunohistochemistry analysis of IL-1β expression. (D) Immunohistochemistry analysis of IL-18 expression. Data are represented as mean ± SD of three independent replicates; *P < 0.05, **P < 0.01 vs NCD group; †P < 0.05, ††P < 0.01 vs AS model group; Scale bar = 50 μm. |
STS Inhibited NLRP3 Inflammasome and Pyroptosis in HUVECs
We used 500 μM cholesterol crystals to induce an inflammatory response in HUVECs. Using the PI assay, we demonstrated that treatment with STS dose-dependently decreased the cell death with stimulus of cholesterol crystals (Figure 3A). To evaluate the effects of STS on the NLRP3 inflammasome and pyroptosis, we measured the level of NLRP3, ASC, activated caspase-1, IL-1β, and GSDMD. STS treatment decreased the levels of NLRP3, ASC, cleaved caspase-1, cleaved IL-1β, and cleaved GSDMD in HUVECs compared to the cholesterol crystals group (Figure 3B). We then investigated the specific function of STS in NLRP3 inflammasome assembly. First, NLRP3 specks were detected by immunofluorescence, and our results showed that treatment with STS decreased the number of cholesterol crystals-induced NLRP3 specks (Figure 3C). These results suggest that STS regulates the formation of the NLRP3 inflammation to suppress pyroptotic cell death in HUVECs.
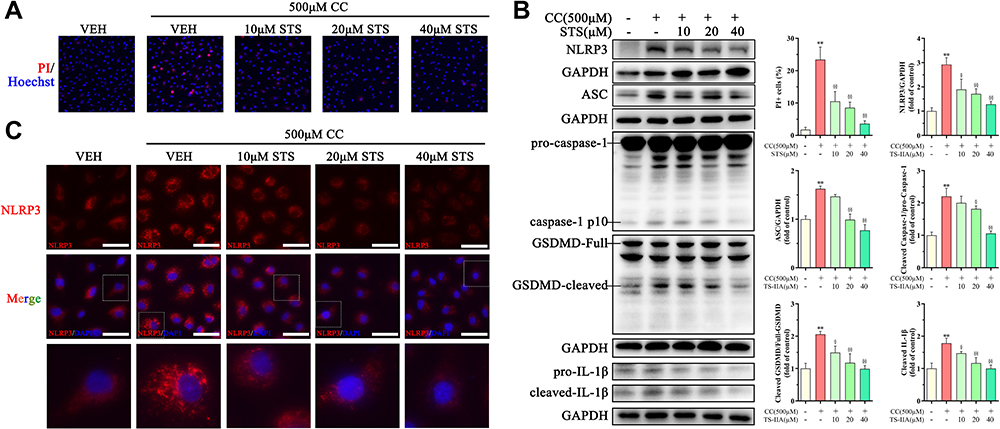 |
Figure 3 Sodium tanshinone IIA sulfonate alleviates cholesterol crystals-induced pyroptotic damage in HUVECs. HUVECs were incubated with 500 μM cholesterol crystals (CC) for 16 h. 10, 20, 40 μM Sodium tanshinone IIA sulfonate (STS) was added 1 h before CC treatment. (A) Cell death was determined using PI staining. (B) NLRP3, ASC, caspase-1, GSDMD and IL-1β protein levels were determined by Western blot. (C) NLRP3 specks were detected by immunofluorescence. Data are represented as mean ± SD of three independent replicates; **P < 0.01 vs VEH group; §P < 0.05, §§P < 0.01 vs cholesterol crystals group; Scale bar = 10 μm. |
STS Improved Mitochondrial Function in HUVECs
Mitochondrial dysfunction is one of the upstream signaling events for NLRP3 activation.24 Cholesterol crystals-induced mitochondrial damage and mtROS over-production were detected in HUVECs. Treatment with STS inhibited the cholesterol crystals-induced decrease in mitochondrial membrane potential (MMP) and elevated mtROS (Figure 4A and B). These results indicate that STS suppressed damage to mitochondria and mtROS.
 |
Figure 4 Sodium tanshinone IIA sulfonate alleviates cholesterol crystals-induced mitochondrial damage in HUVECs. HUVECs were incubated with 500 μM cholesterol crystals (CC) for 16 h. 10, 20, 40 μM Sodium tanshinone IIA sulfonate (STS) was added 1 h before CC treatment. (A) Representative images of TMRE staining in HUVECs that were stained with TMRE (red, to label MMP) and Hoechst 33342 (blue, to label nuclear). (B) HUVECs were stained with Mito-SOX probe (red, to label mtROS), Mito-tracker Green (green, to label mitochondrial) and Hoechst 33342 (blue, to label nuclear) and images were detected under Confocal High-Content Imaging System. Data are represented as mean ± SD of three independent replicates;**P < 0.01 vs VEH group; §P < 0.05, §§P < 0.01 vs cholesterol crystals group; Scale bar = 10 μm. |
STS Inhibited Mitochondrial Dysfunction, the NLRP3 Inflammasome, and Pyroptosis in HUVECs by Activating the AMPK Pathway
AMPK is an essential regulator bridging energy metabolism, mitochondria and inflammation.48,49 We found that STS treatment increased AMPK phosphorylation in HUVECs, which was also confirmed in mice aortic sinus (Figure 5). Subsequently, dorsomorphin, an AMPK inhibitor,50 was used to confirm whether STS could suppress mtROS over-production through increasing AMPKThr172 phosphorylation. Our results showed that the inhibitory effects of STS on cell death (Figure 6A) and mtROS (Figure 7B) were blocked by treatment with dorsomorphin. With regard to mitochondrial homeostasis, our results showed that the beneficial effects of STS on mitochondrial membrane potential (MMP) (Figure 7A) was disrupted by dorsomorphin. With regard to NLRP3 activation, we found that the effects of STS on inhibiting cholesterol crystals-induced activation of NLRP3 and pyroptosis were blocked by dorsomorphin (Figure 6B). Dorsomorphin also increased the specks of NLRP3 foci in the cell membrane (Figure 6C). These results indicated that STS could increase AMPK phosphorylation to suppress mtROS production, resulting in inhibition of NLRP3 inflammasome activation. These results suggest that STS can increase AMPK phosphorylation to increase mitochondrial biogenesis, thereby restoring mitochondrial function to inhibit mtROS production and thus inhibit NLRP3 activation induced-pyroptosis.
 |
Figure 5 Sodium tanshinone IIA sulfonate activates AMPK. HUVECs were incubated with 500 μM cholesterol crystals (CC) for 16 h. 10, 20, 40 μM Sodium tanshinone IIA sulfonate (STS) was added 1 h before CC treatment. (A) p-AMPK level in sections of the aortic arches was detected by immunofluorescence. (B) p-AMPKThr172, AMPK protein levels were determined by Western blot. Data are represented as mean ± SD of three independent replicates; **P < 0.01 vs VEH group; §§P < 0.01 vs cholesterol crystals group; ††P < 0.01 vs STS treatment group. |
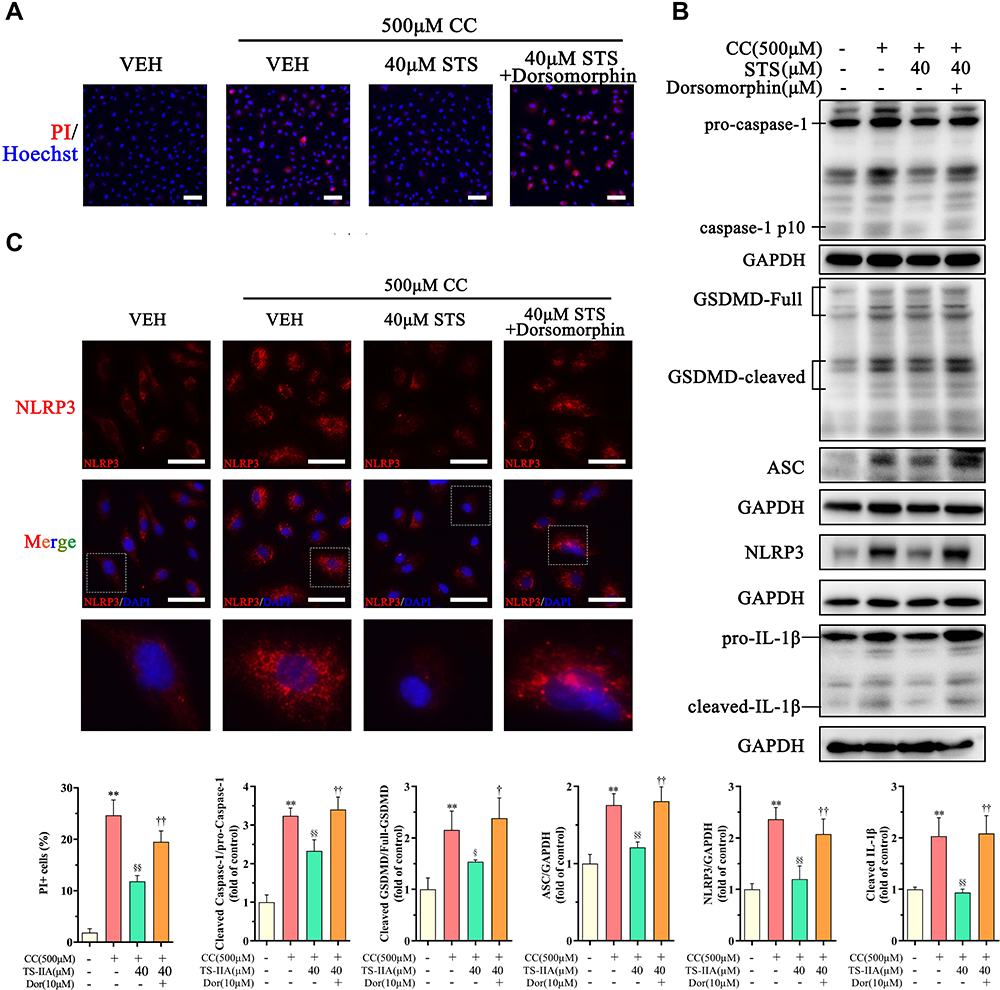 |
Figure 6 Inhibition of AMPK signaling eliminates the protective effect of Sodium tanshinone IIA sulfonate on cholesterol crystals-induced endothelial pyroptosis. HUVECs were incubated with 500 μM cholesterol crystals (CC) for 16 h and 40 μM Sodium tanshinone IIA sulfonate (STS) was added 1 h before CC treatment. 10 μM Dorsomorphin was added 2 h before STS treatment. (A) Cell death was determined using PI staining. (B) NLRP3, ASC, caspase-1, GSDMD and IL-1β protein levels were determined by Western blot. (C) NLRP3 specks were detected by immunofluorescence. Data are represented as mean ± SD of three independent replicates; **P < 0.01 vs VEH group; §P < 0.05, §§P < 0.01 vs cholesterol crystals group; †P < 0.05, ††P < 0.01 vs STS treatment group; Scale bar = 10 μm. |
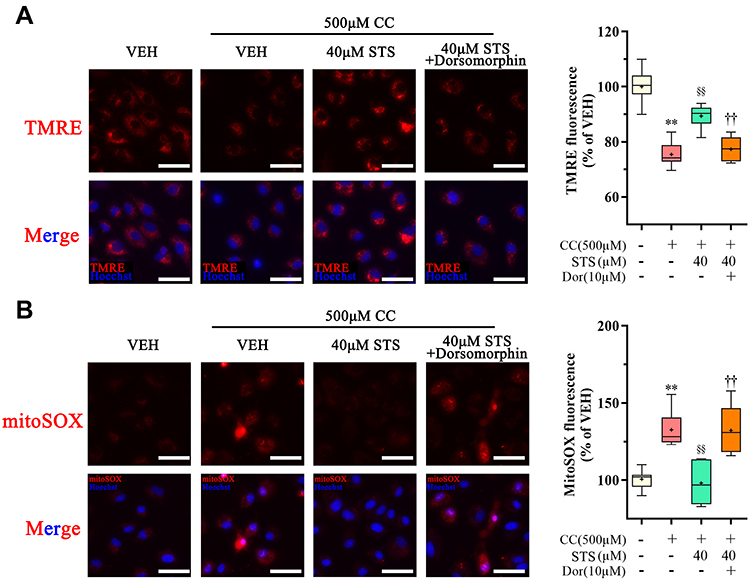 |
Figure 7 Inhibition of AMPK signaling eliminates the protective effect of Sodium tanshinone IIA sulfonate on cholesterol crystals-induced mitochondrial damage. HUVECs were incubated with 500 μM cholesterol crystals (CC) for 16 h and 40 μM Sodium tanshinone IIA sulfonate (STS) was added 1 h before CC treatment. 10 μM Dorsomorphin was added 2 h before STS treatment. (A) Representative images of TMRE staining in HUVECs that were stained with TMRE (red, to label MMP) and Hoechst 33342 (blue, to label nuclear). (B) HUVECs were stained with Mito-SOX probe (red, to label mtROS), Mito-tracker Green (green, to label mitochondrial) and Hoechst 33342 (blue, to label nuclear) and images were detected under Confocal High-Content Imaging System. Data are represented as mean ± SD of three independent replicates; **P < 0.01 vs VEH group; §§P < 0.01 vs cholesterol crystals group; ††P < 0.01 vs STS treatment group; Scale bar = 10 μm. |
Discussion
STS is a water-soluble derivative of tanshinone IIA, which is present in the roots of the traditional Chinese herb Salvia miltiorrhiza. In recent years, studies have shown beneficial effects of STS in CVD, including antioxidant, antiplatelet aggregating, and anti-inflammatory effects.51 Herein, we demonstrated that STS attenuated the formation of atherosclerotic lesions and inhibited endothelial pyroptosis in the aortic intima of WD-fed ApoE−/− mice. In addition, STS also inhibited the expression of LDL-C in serum, but the effect of HDL-C by STS is not significant. However, some studies have shown that other HDL-related biomarkers, such as HDL function or HDL particle number, may have more clinical significance than the relationship between HDL-C levels and cardiovascular disease.52–54 Moreover, in vitro, STS inhibited mitochondrial dysfunction, NLRP3 inflammasome formation, and pyroptosis in cholesterol crystals-stimulated HUVECs. We demonstrated that STS may play a role in inflammation by regulating pyroptosis in endothelial cells to prevent and treat AS. Importantly, our results showed that might prevent pyroptosis in vascular endothelial cells via up-regulation of AMPK related signaling pathway.
In this study, we observed that STS attenuated the formation of atherosclerotic lesions in WD-fed ApoE−/− mice. The vascular endothelium plays a physiological role in maintaining vascular homeostasis and its dysfunction is one of the major causes of CVDs.7 Some studies and our previous study, have demonstrated that STS exerts anti-inflammatory effects in CVDs.41 Moreover, inflammasome-induced pyroptosis has recently been shown to be an important cause of endothelial damage.55 In line with this, several studies have confirmed that precise regulation of NLRP3 can effectively inhibit vascular endothelial injury in AS.20 The NLRP3 inflammasome is composed of NLRP3, ASC, and pro-caspase1.56 Caspase-1 is activated by the NLRP3 polyprotein complex and subsequently cleaves inflammatory factors and GSDMD, allowing the GSDMD domain to penetrate the cell membrane, induce pyroptotic cell death, and promote the release of inflammatory factors.57 In this study, the formation of the NLRP3, ASC, and GSDMD were remarkably inhibited by STS in the aortic endothelial cells of WD-fed ApoE−/− mice. And STS administration also attenuated IL-1β and IL-18 expression in the aorta of mice. Combined with the above results, the beneficial effects of STS in the prevention and reversal of AS may depend on the suppression of the activated NLRP3 inflammasome signaling pathway.
It is well-known that cholesterol crystals induce endothelial cell pyroptosis and endothelial dysfunction and cholesterol crystals markedly increase endothelial ROS production, thereby inducing NLRP3 inflammasome activation and endothelial cell pyroptosis.58,59 We observed that STS treatment of HUVECs alleviated cholesterol crystals-induced pyroptosis by decreasing the number of PI-positive cells, the enhanced expression of the NLRP3 inflammasome, mature caspase-1, and cleaved-GSDMD, and the level of activated-IL-1β. These results suggest that STS plays an anti-AS role by inhibiting endothelial cell pyroptosis.
Mitochondrial damage is thought to be one of the upstream signaling events for endothelial dysfunction, NLRP3 inflammasome activation, and pyroptosis, and excessive products such as mtROS and mtDNA released after mitochondrial damage. And these products can cause NLRP3 activation.60 We found that cholesterol crystals treatments resulted in an increase in mtROS production, as well as changes in mitochondrial membrane potential, suggesting mitochondrial dysfunction. STS can exhibit a dose-effect restoration of mitochondrial membrane potential, and reduced mtROS production. Our work shows that STS is beneficial for regulating mitochondrial function.
AMPK, as an energy receptor, plays a central role in maintaining mitochondrial homeostasis, and controlling inflammatory stress and oxidative stress.61 AMPK is considered a potential target for metabolism-related CVD, including AS, coronary heart disease, and diabetic cardiomyopathy.62 AMPK inhibits inflammatory signaling and NLRP3 activation.48 In this study, we found that WD caused decreased pAMPK expression in the aortic vasculature, and cholesterol crystals decreased the expression of pAMPK in endothelial cells in vitro. STS increased AMPK expression in WD mice aortic vessels, as well as in cholesterol crystals-treated HUVECs. Moreover, dorsomorphin, an AMPK inhibitor, inhibited the activation of AMPK in endothelial cells by STS and also suppressed the inhibitory effect of STS on endothelial cell pyroptosis. AMPK not only inhibits inflammation but also promotes oxidative phosphorylation and mitochondrial homeostasis.61 The addition of dorsomorphin greatly disrupted the restoration of STS for mitochondrial membrane potential, and also further increased the production of mtROS. ROS generation was the first intermediate found to be common to various stimuli-induced NLRP3 inflammasome activation. And mitochondria are the major source of intracellular ROS. The over-production of mtROS is responsible for the activation of the NLRP3 inflammasome.56 Overall, the regulation of endothelial pyroptosis by STS is AMPK-dependent, and the regulation of mtROS plays a central role in this process.
In summary, STS ameliorates endothelial pyroptotic cell death and AS in an NLRP3-dependent manner. STS increases the phosphorylation of AMPK, where it improves mitochondrial function and blocks mtROS production, leading to suppression of NLRP3 inflammasome activation and pyroptosis in endothelial cells. Our results suggest that STS may be used for preventing AS.
Data Sharing Statement
All of the data used in the study are available from the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Animal Experimental Ethics Committee of Zhejiang Chinese Medical University (approval number: IACUC-20200720-06).
Acknowledgments
We appreciated the technical support from the Public Platform of Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University.
Author Contributions
DZ Lu and J Zhu conceived and designed the study. J Zhu, H Chen, and JN Guo performed major experiments, data analysis, and drafted the manuscript. C Zha provided technological support. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work. These authors have contributed equally to this work and share first authorship: Ji Zhu and Hang Chen.
Funding
This research was supported by the Natural Science Foundation of Zhejiang Province (LY19H280003, LY20H280007), the Opening Project of Key Laboratory of Integrative Chinese and Western Medicine for the Diagnosis and Treatment of Circulatory Diseases of Zhejiang Province (Nos. 2C32004 and 2C32101), and the Scientific Research and Innovation Fund of Zhejiang Chinese Medicine University (KC201946).
Disclosure
The authors report no conflicts of interest in relation to this work and declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ross R, Epstein FH. Atherosclerosis — an inflammatory disease. N Engl J Med. 1999;340(2):115–126. doi:10.1056/NEJM199901143400207
2. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201:S1–S7. doi:10.1016/S0167-5273(15)31026-3
3. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118(4):535–546. doi:10.1161/CIRCRESAHA.115.307611
4. Yan R, Li W, Yin L, et al. Cardiovascular diseases and risk‐factor burden in urban and rural communities in high‐, middle‐, and low‐income regions of china: a large community‐based epidemiological study. J Am Heart Assoc. 2017;6(2). doi:10.1161/JAHA.116.004445
5. Xi B, Liu F, Hao Y, Dong H, Mi J. The growing burden of cardiovascular diseases in China. Int J Cardiol. 2014;174(3):736–737. doi:10.1016/j.ijcard.2014.04.098
6. Yang G, Wang Y, Zeng Y, et al. Rapid health transition in China, 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet. 2013;381(9882):1987–2015. doi:10.1016/S0140-6736(13)61097-1
7. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636. doi:10.1161/CIRCRESAHA.115.306301
8. Ali L, Schnitzler JG, Kroon J. Metabolism. Curr Opin Lipidol. 2018;29(6):474–480. doi:10.1097/MOL.0000000000000550
9. Brown RA, Shantsila E, Varma C, Lip GYH. Current understanding of atherogenesis. Am J Med. 2017;130(3):268–282. doi:10.1016/j.amjmed.2016.10.022
10. He B, Nie Q, Wang F, et al. Role of pyroptosis in atherosclerosis and its therapeutic implications. J Cell Physiol. 2021;236(10):7159–7175. doi:10.1002/jcp.30366
11. Jia C, Chen H, Zhang J, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–318. doi:10.1016/j.intimp.2018.12.028
12. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019;52(2):e12563. doi:10.1111/cpr.12563
13. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
14. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40(11):1035–1052. doi:10.1016/j.it.2019.09.005
15. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):R568–R572. doi:10.1016/j.cub.2016.02.019
16. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233(3):2116–2132. doi:10.1002/jcp.25930
17. Bai B, Yang Y, Wang Q, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11(9):776. doi:10.1038/s41419-020-02985-x
18. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi:10.1038/nature08938
19. Christ A, Günther P, Lauterbach MAR, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172(1–2):162–175.e14. doi:10.1016/j.cell.2017.12.013
20. He X, Fan X, Bai B, Lu N, Zhang S, Zhang L. Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis. Pharmacol Res. 2021;165:105447. doi:10.1016/j.phrs.2021.105447
21. Xu YJ, Zheng L, Hu YW, Wang Q. Pyroptosis and its relationship to atherosclerosis. Clin Chim Acta. 2018;476:28–37. doi:10.1016/j.cca.2017.11.005
22. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
23. Jin H, Ko YS, Park SW, Kim HJ. P2Y2R activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free Radic Biol Med. 2019;136:109–117. doi:10.1016/j.freeradbiomed.2019.04.004
24. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
25. Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. 2018;103:115–124. doi:10.1016/j.molimm.2018.09.010
26. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135. doi:10.1038/nrm.2017.95
27. Gao F, Chen J, Zhu H. A potential strategy for treating atherosclerosis: improving endothelial function via AMP-activated protein kinase. Sci China Life Sci. 2018;61(9):1024–1029. doi:10.1007/s11427-017-9285-1
28. Tian L, Cao W, Yue R, et al. Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. J Pharmacol Sci. 2019;139(4):352–360. doi:10.1016/j.jphs.2019.02.008
29. Su X, Yao Z, Li S, Sun H. Synergism of Chinese herbal medicine: illustrated by Danshen compound. Evid Based Complement Altern Med. 2016;2016:1–10. doi:10.1155/2016/7279361
30. Wu TW, Zeng LH, Fung KP, et al. Effect of sodium tanshinone IIA sulfonate in the rabbit myocardium and on human cardiomyocytes and vascular endothelial cells. Biochem Pharmacol. 1993;46(12):2327–2332. doi:10.1016/0006-2952(93)90624-6
31. Xu H, Ji Chen K. Integrating traditional medicine with biomedicine towards a patient-centered healthcare system. Chin J Integr Med. 2011;17(2):83–84. doi:10.1007/s11655-011-0641-2
32. Shang Q, Xu H, Huang L. Tanshinone IIA: a promising natural cardioprotective agent. Evid Based Complement Altern Med. 2012;2012:1–7. doi:10.1155/2012/716459
33. Zhang D, Lu X, He S, et al. Sodium tanshinone IIA sulfonate protects against Aβ‐induced cell toxicity through regulating Aβ process. J Cell Mol Med. 2020;24(6):3328–3335. doi:10.1111/jcmm.15006
34. Chen L, He W, Peng B, et al. Sodium Tanshinone IIA sulfonate improves post-ischemic angiogenesis in hyperglycemia. Biochem Biophys Res Commun. 2019;520(3):580–585. doi:10.1016/j.bbrc.2019.09.106
35. Zhou X, Pan Y, Wang Y, et al. Tanshinones induce tumor cell apoptosis via directly targeting FHIT. Sci Rep. 2021;11(1):12217. doi:10.1038/s41598-021-91708-z
36. Cheng Y, An Q, Wang J, Wang Y, Dong J, Yin J. RasGRF1 participates in the protective effect of tanshinone IIA on depressive like behaviors of a chronic unpredictable mild stress induced mouse model. Gene. 2020;754:144817. doi:10.1016/j.gene.2020.144817
37. Yang N, Chen H, Gao Y, et al. Tanshinone IIA exerts therapeutic effects by acting on endogenous stem cells in rats with liver cirrhosis. Biomed Pharmacother. 2020;132:110815. doi:10.1016/j.biopha.2020.110815
38. Yang G, Jia L, Wu J, et al. Effect of tanshinone IIA on oxidative stress and apoptosis in a rat model of fatty liver. Exp Ther Med. 2017. doi:10.3892/etm.2017.5162
39. Wang N, Zhang X, Ma Z, et al. Combination of tanshinone IIA and astragaloside IV attenuate atherosclerotic plaque vulnerability in ApoE(-/-) mice by activating PI3K/AKT signaling and suppressing TRL4/NF-κB signaling. Biomed Pharmacother. 2020;123:109729. doi:10.1016/j.biopha.2019.109729
40. Zhu H, Chen Z, Ma Z, et al. Tanshinone IIA protects endothelial cells from H 2 O 2 -induced injuries via PXR activation. Biomol Ther. 2017;25(6):599–608. doi:10.4062/biomolther.2016.179
41. Zhu J, Xu Y, Ren G, et al. Tanshinone IIA Sodium sulfonate regulates antioxidant system, inflammation, and endothelial dysfunction in atherosclerosis by downregulation of CLIC1. Eur J Pharmacol. 2017;815:427–436. doi:10.1016/j.ejphar.2017.09.047
42. Qun Jia L, Lin Yang G, Ren L, et al. Tanshinone IIA reduces apoptosis induced by hydrogen peroxide in the human endothelium-derived EA.hy926 cells. J Ethnopharmacol. 2012;143(1):100–108. doi:10.1016/j.jep.2012.06.007
43. He L, Liu YY, Wang K, et al. Tanshinone IIA protects human coronary artery endothelial cells from ferroptosis by activating the NRF2 pathway. Biochem Biophys Res Commun. 2021;575:1–7. doi:10.1016/j.bbrc.2021.08.067
44. Chen HW, Yen CC, Kuo LL, et al. Benzyl isothiocyanate ameliorates high-fat/cholesterol/cholic acid diet-induced nonalcoholic steatohepatitis through inhibiting cholesterol crystal-activated NLRP3 inflammasome in Kupffer cells. Toxicol Appl Pharmacol. 2020;393:114941. doi:10.1016/j.taap.2020.114941
45. Rajamäki K, Lappalainen J, Öörni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5(7):e11765. doi:10.1371/journal.pone.0011765
46. Ma X, Lu D, Liu Y, et al. Multiplexed quantitative evaluation on mitochondrial toxicity of tris (2,3-dibromopropyl) phosphate in hepatocyte. Ecotoxicol Environ Saf. 2021;221:112425. doi:10.1016/j.ecoenv.2021.112425
47. Le Y, Shen H, Yang Z, Lu D, Wang C. Comprehensive analysis of organophosphorus flame retardant-induced mitochondrial abnormalities: potential role in lipid accumulation. Environ Pollut. 2021;274:116541. doi:10.1016/j.envpol.2021.116541
48. Liu X, Xu Y, Cheng S, et al. Geniposide combined with notoginsenoside R1 attenuates inflammation and apoptosis in atherosclerosis via the AMPK/mTOR/Nrf2 signaling pathway. Front Pharmacol. 2021;12. doi:10.3389/fphar.2021.687394
49. Xie T, Wang C, Jin Y, et al. CoenzymeQ10-induced activation of AMPK-YAP-OPA1 pathway alleviates atherosclerosis by improving mitochondrial function, inhibiting oxidative stress and promoting energy metabolism. Front Pharmacol. 2020;11. doi:10.3389/fphar.2020.01034
50. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi:10.1172/JCI13505
51. Ming Li Z, Wen Xu S, Qing Liu P. Salvia miltiorrhizaBurge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol Sin. 2018;39(5):802–824. doi:10.1038/aps.2017.193
52. Amaya-Montoya M, Pinzón-Cortés JA, Silva-Bermúdez LS, et al. ApoE and apoC-III-defined HDL subtypes: a descriptive study of their lecithin cholesterol acyl transferase and cholesteryl ester transfer protein content and activity. Lipids Health Dis. 2020;19(1):106. doi:10.1186/s12944-020-01291-x
53. Li YH, Tseng WK, Yin WH, et al. Prognostic effect of high-density lipoprotein cholesterol level in patients with atherosclerotic cardiovascular disease under statin treatment. Sci Rep. 2020;10(1):21835. doi:10.1038/s41598-020-78828-8
54. Khera AV, Demler OV, Adelman SJ, et al. Cholesterol efflux capacity, high-density lipoprotein particle number, and incident cardiovascular events. Circulation. 2017;135(25):2494–2504. doi:10.1161/CIRCULATIONAHA.116.025678
55. Qian Z, Zhao Y, Wan C, et al. Pyroptosis in the initiation and progression of atherosclerosis. Front Pharmacol. 2021;12. doi:10.3389/fphar.2021.652963
56. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
57. Zhao Y, Shi J, Shao F. Inflammatory caspases: activation and cleavage of gasdermin-D in vitro and during pyroptosis. Methods Mol Biol. 2018;131–148. doi:10.1007/978-1-4939-7519-8_9
58. Yang M, Lv H, Liu Q, et al. Colchicine alleviates cholesterol crystal-induced endothelial cell pyroptosis through activating AMPK/SIRT1 pathway. Oxid Med Cell Longev. 2020;2020:1–18. doi:10.1155/2020/9173530
59. Abdul-Muneer PM, Alikunju S, Mishra V, et al. Activation of NLRP3 inflammasome by cholesterol crystals in alcohol consumption induces atherosclerotic lesions. Brain Behav Immun. 2017;62:291–305. doi:10.1016/j.bbi.2017.02.014
60. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8
61. Qi D, Young LH. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends Endocrinol Metab. 2015;26(8):422–429. doi:10.1016/j.tem.2015.05.010
62. Wu S, Zou MH. AMPK, mitochondrial function, and cardiovascular disease. Int J Mol Sci. 2020;21(14):4987. doi:10.3390/ijms21144987
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Electroacupuncture Ameliorates Depression-Like Behaviors in Post-Stroke Rats via Activating AMPK-Mediated Mitochondrial Function
Ding Z, Gao J, Feng Y, Wang M, Zhao H, Wu R, Zheng X, Feng X, Lai M
Neuropsychiatric Disease and Treatment 2023, 19:2657-2671
Published Date: 4 December 2023
FSCN1 is a Potential Therapeutic Target for Atherosclerosis Revealed by Single-Cell and Bulk RNA Sequencing
Zhang L, Jiang H, Li L, Sun Z, Qian Y, Wang Z
Journal of Inflammation Research 2024, 17:9683-9696
Published Date: 25 November 2024
TMAO Induces Vascular Endothelial Cells Pyroptosis Through TET2-CYTB-ROS Pathway
Xia L, Wang Z, Chen X
Journal of Inflammation Research 2025, 18:8719-8733
Published Date: 2 July 2025
Knowledge Mapping of CircRNAs in AS Research from 2010 to 2025: Spotlight on Lipid Metabolism, Pyroptosis and Exosomes
Wu K, He S, Shen Y, Li J, Li B, Chen G, Lei Y, Chen G, Li Q, Li M
Journal of Inflammation Research 2026, 19:580929
Published Date: 4 February 2026