Back to Journals » Cancer Management and Research » Volume 12
Simultaneous Presentation of Leukemic Non-Nodal Mantle Cell Lymphoma and Gamma-Delta T-Large Granular Lymphocytic Leukemia in a Patient with Rheumatoid Arthritis
Authors Gorodetskiy VR ![]() , Probatova NA, Kupryshina NA, Palshina SG
, Probatova NA, Kupryshina NA, Palshina SG ![]() , Obukhova TN
, Obukhova TN ![]() , Sidorova YV
, Sidorova YV ![]() , Ryzhikova NV
, Ryzhikova NV ![]() , Sudarikov AB
, Sudarikov AB ![]()
Received 11 May 2020
Accepted for publication 3 August 2020
Published 30 September 2020 Volume 2020:12 Pages 9449—9457
DOI https://doi.org/10.2147/CMAR.S261910
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Vadim R Gorodetskiy,1 Natalya A Probatova,2 Natalia A Kupryshina,3 Svetlana G Palshina,1 Tatiana N Obukhova,4 Yulia V Sidorova,5 Natalya V Ryzhikova,5 Andrey B Sudarikov5
1Department of Intensive Methods of Therapy, V.A. Nasonova Research Institute of Rheumatology, Moscow, Russia; 2Department of Pathology, N.N. Blokhin Russian Cancer Research Center, Moscow, Russia; 3Hematopoiesis Immunology Laboratory, N.N. Blokhin Russian Cancer Research Center, Moscow, Russia; 4Cytogenetic Laboratory, National Research Center for Hematology, Moscow, Russia; 5Laboratory of Molecular Hematology, National Research Center for Hematology, Moscow, Russia
Correspondence: Vadim R Gorodetskiy
Department of Intensive Methods of Therapy, V.A. Nasonova Research Institute of Rheumatology, Kashirskoye Shosse 34А, Moscow 115522, Russia
Tel +7 916 517-81-92
Fax +7 499 6144468
Email [email protected]
Abstract: The peculiar features of T-cell large granular lymphocytic leukemia (T-LGLL) are its association with autoimmune disorders (particularly with rheumatoid arthritis (RA)) and a broad spectrum of B-cell lymphoproliferative disorders. However, association of T-LGLL with mantle cell lymphoma (MCL) is extremely rare. Here, we describe a case of an 80-year-old man admitted with suspected Felty’s syndrome. The blood count showed white blood cells at 2.2× 109/L, with 3% neutrophils, 88% lymphocytes, and at 0.66× 109/L LGLs. The spleen had been removed 43 months prior to the admission due to suspected B-cell splenic lymphoma. Re-examination of the spleen revealed cyclin D1+ and SOX11− lymphocytes in the inner part of the unexpanded mantle zones of the white pulp follicles, thus displaying a so-called in situ histologic pattern of MCL, and in small clusters in the red pulp. The splenic cords were moderately expanded by lymphocytes expressing CD3, TIA1, and granzyme B but not CD4 and CD8. Monoclonal rearrangements of the immunoglobulin heavy chain gene and the T-cell receptor (TCR) gamma and delta chain genes, polyclonal rearrangements of the TCR beta chain gene, mutation of the signal transducer and activator of transctiption 3 gene (c.1940A>T; p.N647I), and t(11;14)(q13;q32) translocation were identified in the spleen sample. Flow cytometry of bone marrow revealed a population of TCR γδ+, CD3+, CD4−, CD5−, CD7+, CD8−, CD16−, CD56−, and CD57− lymphocytes. Fragment analysis demonstrated identical TCR gene clonal rearrangement patterns in the spleen and bone marrow samples. In this study, we describe the first case of simultaneous presentation of γδ T-LGLL and leukemic non-nodal MCL (L-NN-MCL) in a patient with RA and present morphological findings of L-NN-MCL in the spleen.
Keywords: composite lymphoma, spleen, mantle cell lymphoma, in situ, large granular lymphocyte leukemia, rheumatoid arthritis
Introduction
T-cell large granular lymphocytic (T-LGL) leukemia is a rare chronic lymphoproliferative disorder typically characterized by monoclonal expansion of CD3+ cytotoxic large granular lymphocytes, cytopenia, and splenomegaly.1,2 Most cases of T-LGL leukemia express the alpha-beta (αβ) T-cell receptor (TCR), but a rare variant of gamma-delta (γδ) TCR-positive T-LGL leukemia has also been reported.3,4 The most common phenotype of γδ T-LGL leukemic cells is CD4–/CD8– or CD4–/CD8+.5,6 Activating somatic mutations in the signal transducer and activator of transcription 3 (STAT3) gene have been reported in approximately 40% of patients with T-LGL leukemia.7,8 A peculiar feature of T-LGL leukemia is its association with autoimmune disorders, particularly rheumatoid arthritis (RA), which is diagnosed in approximately 15% of patients with T-LGL leukemia.8,9 Another well-recognized feature of T-LGL leukemia is its association with a broad spectrum of B-cell lymphoproliferative disorders.10,11 Yabe et al found no statistically significant difference in the prevalence of second hematologic malignancies in αβ T-LGL leukemia or γδ T-LGL leukemia.6
Two main clinical variants of mantle cell lymphoma (MCL): classical MCL and leukemic non-nodal MCL (L-NN-MCL) are noted by 2016 World Health Organization classification.12 Patients with L-NN-MCL present with peripheral blood, bone marrow, and sometimes splenic involvement.12,13 L-NN-MCL is characterized by expression of CD19, CD20, and CD5 markers and cyclin D1 overexpression due to t(11;14)(q13;q32) translocation. Unlike classical MCL, L-NN-MCL follows an indolent course, is SOX11 negative and does not involve lymph nodes.12,14 The histopathological features of L-NN-MCL in the spleen have not been previously reported.
In this study, we describe the first case of simultaneous presentation of L-NN-MCL and γδ T-LGL leukemia in a patient with RA and present morphological findings of L-NN-MCL in the spleen.
Materials and Methods
Immunohistochemical Studies
Immunohistochemical studies were carried out on formalin-fixed paraffin-embedded (FFPE) tissues. The following antibodies, at the dilutions suggested by the manufacturers, were used: CD2 (clone AB75, Dako, Glostrup, Denmark), CD3 (Polyclonal, Dako), CD4 (clone: 4B12, Dako), CD5 (clone 4C7, Dako), CD8 (clone: C8/144B, Dako), CD20 (clone L26, Dako), CD23 (clone DAK-CD23, Dako), CD43 (clone DF-T1, Dako), CD79a (clone JCB117, Dako), cyclin D1 (clone EP12, Dako), BCL2 (clone 124, Dako), BCL6 (clone PG-B6p, Dako), VS38 (clone VS38c, Dako), SOX11 (clone MRQ-58, Cell Marque, Rocklin, CA), granzyme B (clone GrB-7, Dako), TIA1 (clone 2G9, Immunotech, France), IgA (Polyclonal, Dako), IgG (Polyclonal, Dako), IgM (Polyclonal, Dako), IgD (Polyclonal, Dako), and Ki-67 (clone MIB-1, Dako). After dewaxing and heat-induced antigen retrieval, immunostaining was performed on an Autostainer Link 48 (Dako, Denmark) according to the manufacturer’s instructions. All immunostained samples were counterstained with hematoxylin.
Flow Cytometric Analysis
An eight-color flow cytometric analysis was performed on peripheral blood and bone marrow specimens. Lymphocytes were gated for analysis using CD45 and side scatter plots. Cells were stained with a panel of fluorescence-labeled monoclonal antibodies, including CD3, CD4, CD5, CD7, CD8, CD16, CD19, CD20, CD23, CD38, CD45, CD56, CD57, TCR αβ chain, TCR γδ chain, and immunoglobulin kappa (κ) and lambda (λ) light chains. Flow cytometry analysis was performed using a BD FACSCanto™ II (Becton Dickinson, San Jose, CA, USA) and FCS express Version 3 (De Novo Software, Los Angeles, CA) software.
Conventional Cytogenetics and Fluorescence in situ Hybridization Analysis
Chromosome G-banding analysis was performed on bone marrow aspirate specimen. Cells were cultured for 24 h without mitogens and 72 h with phorbol-12-myristate-13-acetate at 50 ng/mL plus lipopolysaccharide at 100 μg/mL as stimulators for metaphase induction. Colcemid (0.02 μg/mL) was added to the culture for the last hour of incubation. Chromosome preparation and staining were performed according to standard protocols. Thirty metaphases were analyzed. Chromosomes were classified according to the International System for Human Cytogenetic Nomenclature.15
Fluorescence in situ hybridization (FISH) analysis for t(11;14)(q13;q32) was performed on bone marrow smears and FFPE tissue sections of the spleen using an LSI IGH/CCND1 dual-color, dual-fusion translocation probe (Abbott Molecular, USA) according to the manufacturer’s instructions. For the FFPE spleen tissue, an LSI D7S522/CEP7 Dual Color probe and a CEP 8 Spectrum Orange probe (Abbott Molecular, USA) were used for detection of isochromosome 7q (i(7q)) and trisomy 8, respectively. A total of 200 interphase nuclei for each probe were analyzed. Image capture and acquisition were processed using Axio Scope, A1 and Axio Imager, a Z2 (Carl Zeiss, Germany) microscope and Ikaros and the Isis imaging system (MetaSystems, Germany).
Polymerase Chain Reaction Analysis
Evaluation of B- and T-Cell Clonality
Genomic DNA was extracted from blood, bone marrow and spleen tissue samples. Evaluation of B-cell clonality was used based on the immunoglobulin heavy chain (IGH) framework 1, 2, and 3 assays (VH-JH rearrangements). T-cell clonality was examined based on rearrangements of the TCRG (Vγ–Jγ), TCRB (Vβ–Jβ, Dβ–Jβ) and TCRD (Vδ-Dδ-Jδ) genes. Both IGH and TCR clonality assays were done according to BIOMED-2 standardized protocol.16 Polymerase chain reaction (PCR) was carried out using an automated DNA Engine thermocycler (BioRad, Hercules, USA), and fragments were detected using an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA); the data were analyzed with GeneMapper software version 4.0 (Applied Biosystems, Foster City, CA).
Evaluation of STAT3 Gene Mutations
Allele-specific (AS) TaqMan Real-Time PCR assays were employed to determine point somatic mutations of gene STAT3 (p.Y640F; p.N647I; p.D661V; p.D661Y; p.D661H; p.D661N). DNA (200–400 ng) was added to 25 μL of the reaction mixture containing 10 pmol of WT (wild type) specific or MT (mutated type) specific forward primer, 10 pmol of common reverse primer, and 7.5 pmol of the fluorescent probe. AS-PCR was performed in triplicate (3 WT + 3 MT) using a StepOne Real-Time PCR System (Applied Biosystems, USA). PCR conditions were preliminary denaturation at 95°C for 5 min, followed by 45 cycles at 95°C for 30 s, 62°C for 30 s, and 72°C for 30 s. A mixture of DNA from healthy donors was used as a negative control. Samples with mutations confirmed by Sanger sequencing were used as positive controls. The primer and probe sequences are shown in Supplement 1.
Case Report and Results
In December 2018, an 80-year-old man was admitted to the V. A. Nasonova Research Institute of Rheumatology with suspected Felty’s syndrome.
According to medical records, in December 2014, he sought medical help because of progressive weakness, shortness of breath under minor physical exertion, and swelling of the lower extremities. The complete blood count at that time showed a hemoglobin level of 10.1 g/dL, platelets at 191×109/L, and white blood cells at 2.7×109/L, with 23% neutrophils (absolute neutrophil count 0.621×109/L), 67% lymphocytes, and 10% monocytes. Blood chemistry markers of liver and kidney function were within the normal ranges. The serum lactate dehydrogenase level was 426 IU/L (normal range, 208–378 IU/L), and the β2-microglobulin level was 14.8 mg/L (normal range, 1.16–2.52). An examination was performed, including upper gastrointestinal endoscopy and chest and abdomen computed tomography (CT), and splenomegaly (measuring 6.0×12.0×18.0 cm) was detected. Flow cytometry of the patient’s blood revealed a small population of lymphocytes (0.42×109/L) with a CD19+, CD5+, CD23−, CD38+ phenotype and monotypic surface lambda light chain expression (Figure 1A and B). Hematoxylin and eosin stained sections of a bone marrow core biopsy revealed hypercellular, relative to the age norm, bone marrow with trilineage hematopoiesis and moderate interstitial lymphocytic infiltration. Lymphocytes expressed CD3 and were negative for CD20, cyclin D1, and SOX11. Splenectomy was performed in May 2015 due to suspicion of splenic lymphoma, and B-cell spleen lymphoma was diagnosed. The patient was in good condition with no therapy until spring 2017, when he developed episodes of joint pain and fever.
 |
Figure 1 Flow cytometry of mantle cell lymphoma in the peripheral blood (2015) shows CD19/CD5 coexpression (depicted in dark blue) (A) and λ light-chain restriction (B). |
On admission, a complete blood count showed a hemoglobin level of 10.1 g/dL, platelets at 188×109/L, and white blood cells at 2.2×109/L, with 3% neutrophils (absolute neutrophil count – 0.066×109/L), 88% lymphocytes, 1% monocytes, 3% eosinophils, and 5% basophils. The absolute large granular lymphocyte count was 0.66×109/L. His rheumatoid factor level was 116 IU/mL (normal range, 0–15 IU/mL), anti-cyclic citrullinated peptide antibody level was above 300 U/mL (normal range, 0–5 U/mL), and C-reactive protein level was 40.9 mg/L (normal range, 0–5 mg/L). Magnetic resonance imaging of the right hand revealed erosive arthritis of the wrist joint and metacarpophalangeal joint of the second finger. A CT scan of the chest and abdominal ultrasound did not indicate enlargement of the lymph nodes.
Repeated examination of the spleen revealed that the white pulp was histologically unremarkable and represented by lymphoid follicles, some with germinal centers. In the red pulp, small clusters of lymphoid cells and moderate expansion of the splenic cords by infiltration of medium-sized slightly pleomorphic lymphoid cells were observed (Figure 2A). In the centers of the follicles, a network of CD23-positive follicular-dendritic cells was well presented. The cells of the lymphoid follicles and small clusters in red pulp expressed CD20 and CD79a. The mantle zones of the lymphoid follicles were not expanded. In addition, the cells of the mantle zones strongly expressed BCL2, IgM, IgD and weakly expressed CD5 (Figure 2B). The expression of cyclin D1 was observed in cell nuclei in the inner layers of the mantle zones (Figure 2C). Of note, these cyclin D1-positive cells did not co-express SOX11. The immunophenotype of the cells in the small clusters in the red pulp was similar to that of the cells in the inner mantle zones. Namely, these cells expressed cyclin D1 (Figure 2D) and BCL2 (Figure 2E) and weakly expressed CD5 (Figure 2F). Lymphocytes infiltrating the splenic cords expressed CD3 (Figure 2G), TIA1, and granzyme B (Figure 2H) but not CD4 and CD8. CD8 expression was observed in some perisinusoidal lymphoid cells and in the vascular endothelium. Cells with t(11;14)(q13;q32) translocation and monoclonal rearrangements of the IGH gene were identified in the spleen tissue. In addition, monoclonal rearrangements of the TCR gamma and delta chain genes, but not the TCR beta chain gene, were identified. Mutation of the STAT3 gene (c.1940A>T; p.N647I) was revealed. Trisomy 8 and i(7q), essential for differential diagnosis of γδ T-LGL leukemia and hepatosplenic T-cell lymphoma (γδ HSTCL), were not detected.
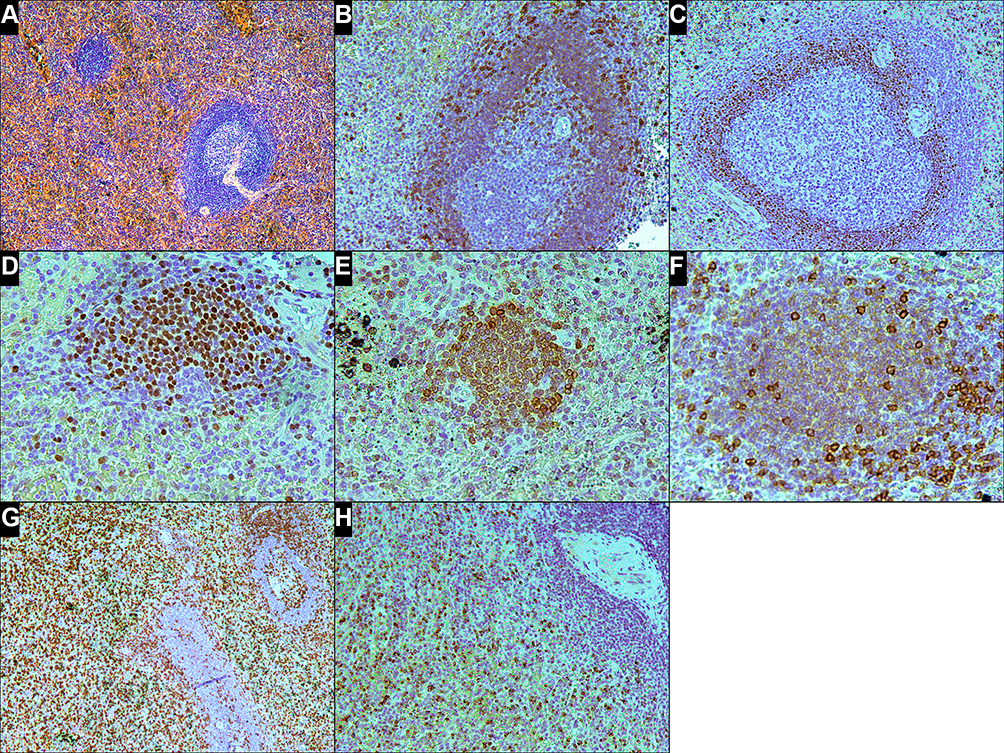 |
Figure 2 Histologic and immunophenotypic findings in the spleen involved leukemic non-nodal mantle cell lymphoma and T-cell large granular lymphocytic leukemia. (A) Follicle with a germinal center and unexpanded mantle zone, a cluster of small lymphoid cells, and moderate expansion of splenic cords by small lymphoid cells (H&E, x100). (B) CD5 immunohistochemical staining shows a brightly positive signal in reactive T-lymphocytes and weakly colored mantle zone cells (CD5 stain, x200). (C) Cyclin D1 expression in the nuclei of cells, mostly in the inner mantle zone (cyclin D1 stain, x200). (D) Small cluster of cyclin D1-positive lymphocytes in the red pulp (cyclin D1 stain, x400). (E and F) Lymphocytes in the small cluster express BCL2 (E, BCL2 stain, x400) and weakly express CD5 (brightly colored cells are reactive T-lymphocytes) (F, CD5 stain, x400). (G and H) Neoplastic T cells in the splenic cords express CD3 (G, CD3 stain, x100) and granzyme B (H, granzyme B stain, x200). |
Bone marrow aspirate showed hypercellular bone marrow with fewer than 5% blasts, and lymphocytes at 13.4%. The neutrophilic series showed approximately 22% giant hypersegmented hypergranular cells (Figure 3A). The number of segmented neutrophils was slightly reduced. Approximately 10% of erythroid precursors had megaloblastoid features and nuclear fragmentation (Figure 3B). Megakaryocytes also exhibited abnormal morphology, such as multiple separated nuclei (Figure 3C). G-band analysis did not reveal clonal chromosomal aberrations. Histological examination of trephine biopsy revealed an increase in bone marrow cellularity (Figure 4A). Based on immunohistochemical CD3 staining, both small clusters and interstitial infiltration by small T-cells were found (Figure 4B). A population of TCR γδ+, CD3+, CD4−, CD5−, CD7+, CD8−, CD16−, CD56−, and CD57− lymphocytes was detected by flow cytometry (Figure 5A–C). Fragment analysis showed identical TCR gene clonal rearrangement patterns in the bone marrow and spleen samples, but no mutations of STAT3 gene were detected in the bone marrow. In addition, monoclonal rearrangements of the IGH gene and cells with t(11;14)(q13;q32) translocation were also not revealed in the bone marrow. The data are presented in Table 1.
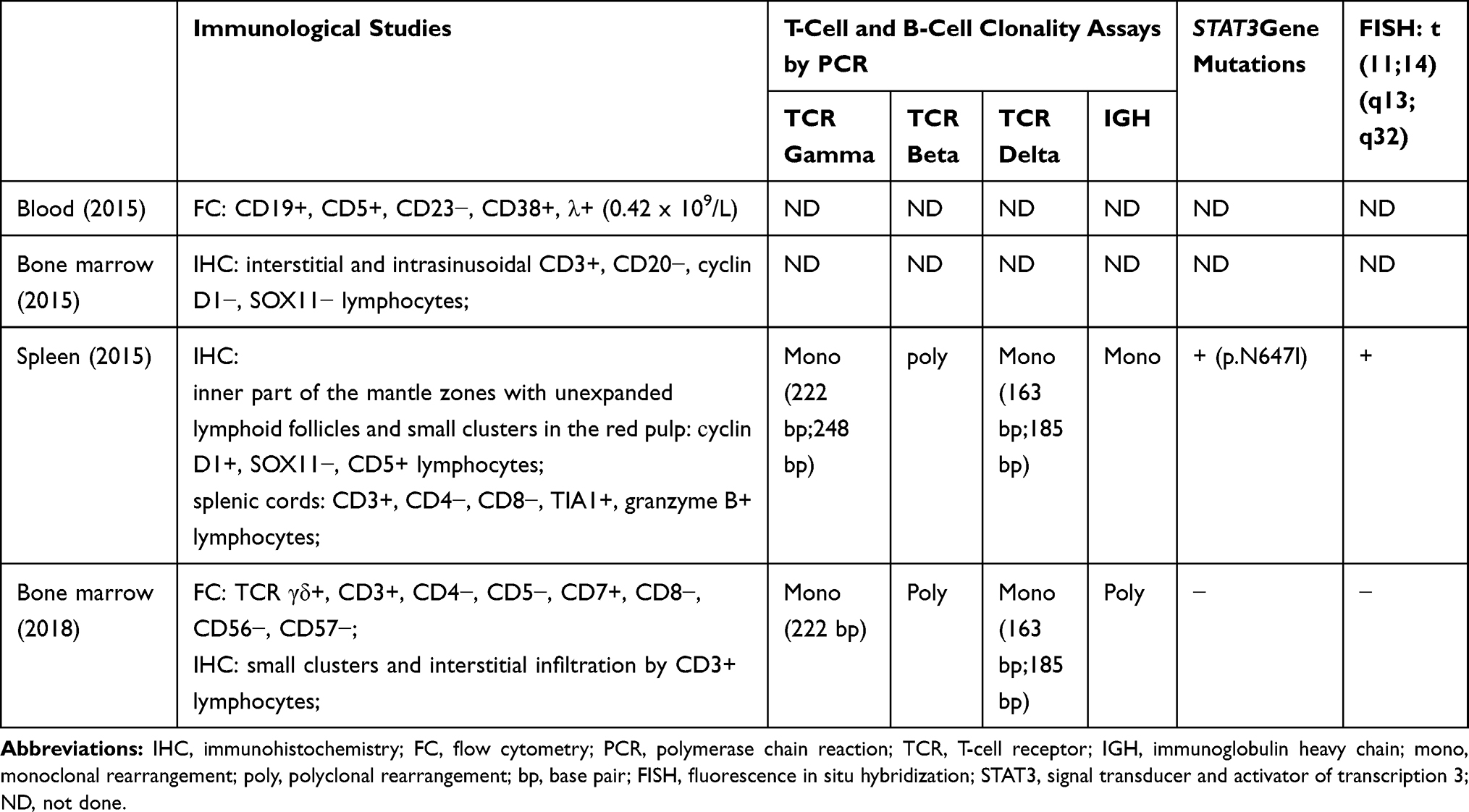 |
Table 1 Findings in Spleen, Blood and Bone Marrow Biopsy Specimens |
 |
Figure 3 Bone marrow aspirate smear (2018). (A) Giant hypersegmented hypergranular neutrophilic cells (arrows). (B) Nuclear fragmentation of an erythroid precursor (arrow). (C) Abnormal megakaryocytes displaying multiple separated nuclei. Romanowsky, x1000. |
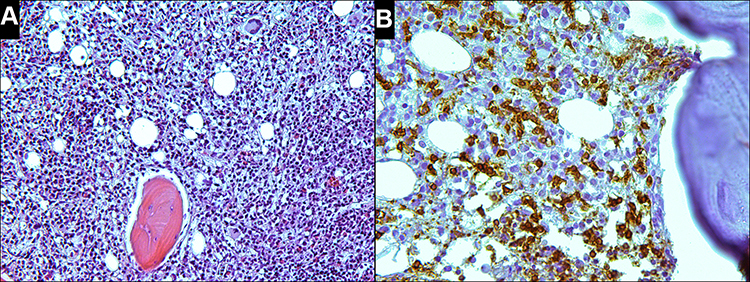 |
Figure 4 (A) Biopsy of the bone marrow (2018) shows hypercellular, relative to the age norm, bone marrow. The bone marrow contains approximately 20% adipose tissue (H&E, x200). (B) CD3 staining shows infiltration by small T-cells with irregular nuclei (x400). |
 |
Figure 5 Flow cytometry of T-cell large granular lymphocytic leukemia in bone marrow (2018) shows CD3+/TCR γδ+ (depicted in green) (A), CD5−/TCR γδ+ (B), and CD7+/TCR γδ+ (C) expression. |
In summary, all findings suggested that a patient with RA had spleen involvement of both γδ T-LGL leukemia and L-NN-MCL. Although the patient was lost to follow-up, we were informed that he died in July 2019 without progression of lymphoma. An autopsy was not performed.
Discussion
L-NN-MCL is considered a variant of MCL with distinct clinical and biological features. Overall, the histopathological features of L-NN-MCL have not been sufficiently characterized. Espinet et al studied upper and lower gastrointestinal tract tissue samples from asymptomatic individuals with monoclonal expansion of cyclin D1-positive mature B-cells in the peripheral blood in the absence of nodal enlargement and splenomegaly.17 The immunohistochemical analysis in all 5 studied cases identified cyclin D1-positive, SOX11-negative small lymphoid cells in lymphoid aggregates lacking germinal centers and in the mantle zones of secondary follicles, with no significant expansion of these areas. To our knowledge, the histopathological features of L-NN-MCL in the spleen have not been previously reported. The clinical picture and phenotype of MCL in our case corresponded to L-NN-MCL, though the number of tumor lymphocytes in the peripheral blood was low. In the spleen, cyclin D1-positive, SOX11-negative lymphocytes were accidentally found in the inner part of the unexpanded mantle zones of the follicles of the white pulp (indicative of the so-called in situ histologic pattern of MCL) and as small clusters in the red pulp.
Detection of B-cells expressing cyclin D1 in samples histologically not satisfying the diagnosis of MCL has led to the assumption that such cases are pre-lymphoma lesions or the earliest involvement of MCL. For these cases, the term “in situ mantle cell neoplasia” (ISMCN) was introduced in the 2016 WHO classification of lymphoid neoplasms.12 Morphologically, ISMCN most commonly shows a monotonous population of cyclin D1-positive B-cells restricted to the inner layer of the non-expanded mantle zones of reactive follicles, with only scattered cells in the interfollicular areas.18 To our knowledge, only one report has described ISMCN in the spleen: Roullet et al described a case of composite spleen lymphoma with cyclin D1-positive lymphocytes detected around the neoplastic follicles of follicular lymphoma (FL) and in the mantle zone of nonneoplastic follicles.19
The relationship between ISMCN and overt MCL (classic MCL and L-NN-MCL) and the potential value of SOX11 expression in ISMCN for predicting evolution is not known. Indeed, a systematic literature search identified only 18 ISMCN cases in which SOX11 status was available.18,20,21 None of the 10 SOX11-negative ISMCN cases showed progression to overt MCL; however, in 3 of 8 SOX11-positive ISMCN cases, overt MCL was diagnosed several years before or several years after the diagnosis of ISMCN. These findings support the speculation that ISMCN can be a morphological manifestation of L-NN-MCL (in SOX11-negative cases) or the earliest manifestation (“pre mantle zone pattern” stage) of classical MCL (in SOX11-positive cases). Staging of our patient, performed at 43 months after splenectomy, did not reveal MCL, which is consistent with the assumption that SOX11-negative ISMCN appears to be a tissue counterpart of L-NN-MCL rather than the initial manifestation of classical MCL.
Coexistence of a B-cell lymphoproliferative disorder has been demonstrated in up to 27% of patients with T-LGL leukemia.10 The spectrum of the most common B-cell lymphoproliferative disorders includes monoclonal gammopathy of unknown significance (MGUS), multiple myeloma, chronic lymphocytic lymphoma, diffuse large B-cell lymphoma, and hairy cell leukemia.10,11,22-24 However, the association of T-LGL leukemia with MCL is extremely rare. To our knowledge, only 6 cases of the association of T-LGL leukemia with MCL have been described to date (Table 2).10,11,25-28 There are several models concerning the pathogenesis of the association of T-LGL leukemia with B-cell lymphoproliferative disorders, as discussed by Viny et al.10 Given that for all cases described in the literature, MCL preceded or was diagnosed simultaneously with T-LGL leukemia, it can be assumed that the evolution of T-LGL leukemia may be a consequence of uncontrollable clonal expansion in the polyclonal anti-MCL response. An additional argument to support this theory is the case of transient monoclonal T-LGL proliferation in the patient with MCL described by Stalika et al.28 A counterargument for the theory that the evolution of T-LGL leukemia has a reactive background is that 3 of 5 patients were in complete remission of MCL at the time of T-LGL leukemia diagnosis. Conversely, as some patients develop T-LGL leukemia after hematopoietic stem cell transplantation and/or rituximab therapy, it is possible that this therapy for MCL induces T-LGL leukemia. Composite T-cell and B-cell lymphomas are believed to derive from separate B- and T- precursor clones.29 However, it cannot be ruled out that some genetic events on the level of common T- and B-cell precursors may lead to the development of clonally related T- and B-cell lymphoid neoplasms.
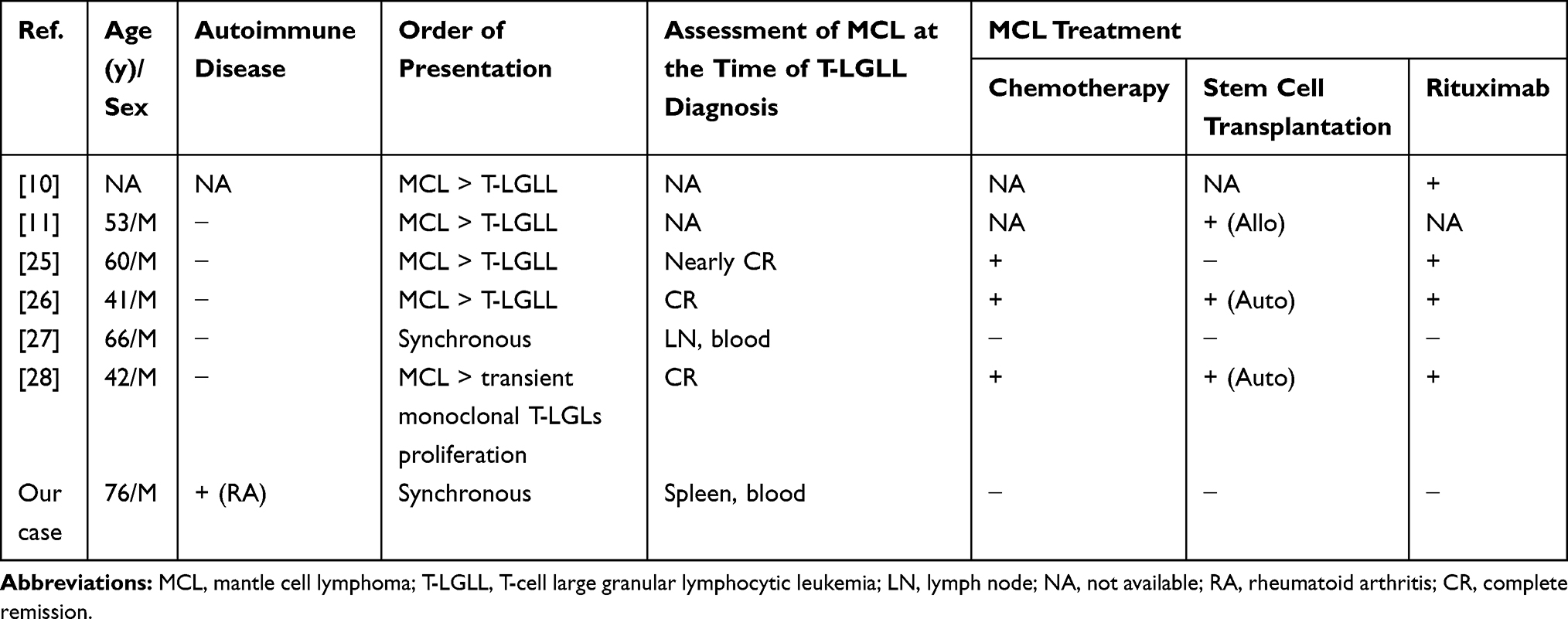 |
Table 2 Coexistence of MCL with T-LGL Leukemia (Literature Review) |
γδ T-LGL leukemia requires differential diagnosis with γδ HSTCL.30,31 Clinical features (older age and RA), granzyme B positivity of lymphocytes infiltrating the splenic cords and cytogenetic features (absence of i(7q) and trisomy 8) support the diagnosis of γδ T-LGL leukemia in our case. The diagnostic value of STAT3 gene mutations in differential diagnosis with γδ HSTCL remains unclear. STAT3 gene mutations have been found in approximately one-third of T-LGL leukemias and in 9% of HSTCLs.4,32
In our patient, the observed bone marrow hyperplasia and dysplastic changes may have indicated myelodysplastic syndrome and/or nutritional (vitamin B12/folic acid) deficiency. Because the patient was lost to follow-up, studies to exclude nutritional deficiency were not possible. The cytopenia in our patient was apparently due to both ineffective hematopoiesis and the pathogenetic mechanisms of T-LGL leukemia.
Conclusion
In summary, this is the first reported case of simultaneous of L-NN-MCL and γδ T-LGL leukemia presentation. In both neoplasms, the main tumor burden was localized in the spleen, whereas the peripheral blood and bone marrow were minimally (if at all) involved. Morphological findings of L-NN-MCL in the spleen manifested in the form cyclin D1-positive, SOX11-negative lymphocytes in the inner part of the unexpanded mantle zones of the follicles of the white pulp (which corresponded to ISMCN) and as small clusters in the red pulp.
Acknowledgment
The authors thank A. Firyulin for expert help in preparing the figures.
Disclosure
The authors declare that they have no conflicts of interest regarding this work.
References
1. Moignet A, Lamy T. Latest advances in the diagnosis and treatment of large granular lymphocytic leukemia. Am Soc Clin Oncol Educ Book. 2018;38:616–625. doi:10.1200/EDBK_200689
2. Cheon H, Dziewulska KH, Moosic KB, et al. Advances in the diagnosis and treatment of large granular lymphocytic leukemia. Curr Hematol Malig Rep. 2020;15(2):103–112. doi:10.1007/s11899-020-00565-6
3. Sandberg Y, Almeida J, Gonzalez M, et al. TCR gamma delta+ large granular lymphocyte leukemias reflect the spectrum of normal antigen-selected TCR gamma delta+ T-cells. Leukemia. 2006;20:505–513. doi:10.1038/sj.leu.2404112
4. Chan WC, Foucar K, Morice WG, et al. T-cell large granular lymphocytic leukemia. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.
5. Chen YH, Chadburn A, Evens AM, et al. Clinical, morphologic, immunophenotypic, and molecular cytogenetic assessment of CD4-/CD8- γδ T-cell large granular lymphocytic leukemia. Am J Clin Pathol. 2011;136:289–299. doi:10.1309/AJCPTFFQ18JMYKDF
6. Yabe M, Medeiros LJ, Wang SA, et al. Clinicopathologic, immunophenotypic, cytogenetic, and molecular features of γδ T-cell large granular lymphocytic leukemia: an analysis of 14 patients suggests biologic differences with αβ T-cell large granular lymphocytic leukemia. Am J Clin Pathol. 2015;144:607–619. doi:10.1309/AJCPJSA1E1YWSZEY
7. Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–1913. doi:10.1056/NEJMoa1114885
8. Sanikommu SR, Clemente MJ, Chomczynski P, et al. Clinical features and treatment outcomes in large granular lymphocytic leukemia (LGLL). Leuk Lymphoma. 2018;59(2):416–422. doi:10.1080/10428194.2017.1339880
9. Bareau B, Rey J, Hamidou M, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica. 2010;95(9):1534–1541. doi:10.3324/haematol.2009.018481
10. Viny AD, Lichtin A, Pohlman B, Loughran T, Maciejewski J. Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma. 2008;49(5):932–938. doi:10.1080/10428190801932635
11. Goyal T, Thakral B, Wang SA, et al. T-cell large granular lymphocytic leukemia and coexisting B-cell lymphomas: a study from the bone marrow pathology group. Am J Clin Pathol. 2018;149(2):164–171. doi:10.1093/ajcp/aqx146
12. Swerdlow SH, Campo E, Seto M, et al. Mantle cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.
13. Jain AG, Chang -C-C, Ahmad S, et al. Leukemic non-nodal mantle cell lymphoma: diagnosis and treatment. Curr Treat Options Oncol. 2019;20(12):85. doi:10.1007/s11864-019-0684-8
14. Fernàndez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010;70(4):1408–1418. doi:10.1158/0008-5472.CAN-09-3419
15. McGowan-Jordan J, Simons A, Basel SM, Editors. An International System for Human Cytogenomic Nomenclature. New York: Karger; 2016.
16. van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317.
17. Espinet B, Ferrer A, Bellosillo B, et al. Distinction between asymptomatic monoclonal B-cell lymphocytosis with cyclin D1 overexpression and mantle cell lymphoma: from molecular profiling to flow cytometry. Clin Cancer Res. 2014;20(4):1007–1019. doi:10.1158/1078-0432.CCR-13-1077
18. Carvajal-Cuenca A, Sua LF, Silva NM, et al. In situ mantle cell lymphoma: clinical implications of an incidental finding with indolent clinical behavior. Haematologica. 2012;97(2):270–278. doi:10.3324/haematol.2011.052621
19. Roullet MR, Martinez D, Ma L, et al. Coexisting follicular and mantle cell lymphoma with each having an in situ component: a novel, curious, and complex consultation case of coincidental, composite, colonizing lymphoma. Am J Clin Pathol. 2010;133(4):584–591. doi:10.1309/AJCP5RT4MRSDGKSX
20. Koletsa T, Markou K, Ouzounidou S, et al. In situ mantle cell lymphoma in the nasopharynx. Head Neck. 2013;35(11):E333–E337. doi:10.1002/hed.23206
21. Sabater-Marco V, Santonja-López N, Ortíz-Zuluaga S, et al. Orbital soft tissue composite lymphoma presenting as recurrence of a nodal lymphoma with mantle and follicular cell components: a case report, literature review and guideline for the treatment of patients. Rev Esp Patol. 2020;53:48–54.
22. Sidiqi MH, Aljama MA, Viswanatha DS, Dingli D. T-cell large granular lymphocytic leukemia and plasma cell disorders. Haematologica. 2019;104(3):e108–e110. doi:10.3324/haematol.2018.204099
23. Cheng J, Talamo G, Malysz J, Ochmann M, Lamy T, Loughran TP
24. Xie XY, Sorbara L, Kreitman RJ, Fukushima PI, Kingma DW, Stetler-Stevenson M. Development of lymphoproliferative disorder of granular lymphocytes in association with hairy cell leukemia. Leuk Lymphoma. 2000;37(1–2):97–104. doi:10.3109/10428190009057632
25. Lin CL, Hsieh YC, Chang ST, Chuang SS. Clonal lymphoproliferation of T cell large granular lymphocytes with pleomorphic nuclei following mantle cell lymphoma. Int J Hematol. 2013;97(2):294–296. doi:10.1007/s12185-013-1271-6
26. Papadaki T, Stamatopoulos K, Stavroyianni N, Paterakis G, Phisphis M, Stefanoudaki-Sofianatou K. Evidence for T-large granular lymphocyte-mediated neutropenia in Rituximab-treated lymphoma patients: report of two cases. Leuk Res. 2002;26(6):597–600. doi:10.1016/S0145-2126(01)00183-7
27. Xiao Z, Ni Y, Yin G, Wu H, Li J, Miao K. Mantle cell lymphoma concurrent with T-large granular lymphocytic leukemia: report of a case and review of literature. Int J Clin Exp Pathol. 2015;8(3):3365–3369.
28. Stalika E, Papalexandri A, Kannelis G, et al. Transient monoclonal CD3+ T large granular lymphocyte proliferation in a case of mantle cell lymphoma with rituximab-associated late onset neutropenia. Hematol Oncol. 2011;29(3):144–146. doi:10.1002/hon.963
29. Küppers R, Dührsen U, Hansmann ML. Pathogenesis, diagnosis, and treatment of composite lymphomas. Lancet Oncol. 2014;15(10):e435–e446. doi:10.1016/S1470-2045(14)70153-6
30. Chen YH, Peterson L. Differential diagnosis of CD4-/CD8- γδ T-cell large granular lymphocytic leukemia and hepatosplenic T-cell lymphoma. Am J Clin Pathol. 2012;137:496–497. doi:10.1309/AJCPCOPRE1HGD0FC
31. Yabe M, Medeiros LJ, Wang SA, et al. Distinguishing between hepatosplenic T-cell lymphoma and γδ T-cell large granular lymphocytic leukemia: a clinicopathologic, immunophenotypic, and molecular analysis. Am J Surg Pathol. 2017;41(1):82–93. doi:10.1097/PAS.0000000000000743
32. McKinney M, Moffitt AB, Gaulard P, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7(4):369–379. doi:10.1158/2159-8290.CD-16-0330
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.