Back to Journals » Journal of Inflammation Research » Volume 18
SETD7 Dual Role in Disease and Opportunities for Therapeutic Intervention: Current Perspectives
Authors Baboni F, Tembo KM ![]() , Zhou X, Li Q, Dai C, Zhao Y, Batoko S, Lan P, Chen Z
, Zhou X, Li Q, Dai C, Zhao Y, Batoko S, Lan P, Chen Z
Received 16 April 2025
Accepted for publication 24 July 2025
Published 4 September 2025 Volume 2025:18 Pages 12191—12225
DOI https://doi.org/10.2147/JIR.S534623
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Subhasis Chattopadhyay
Farouk Baboni,1 Kingsley Miyanda Tembo,2,3 Xi Zhou,1 Qingwen Li,1 Chen Dai,1 Yuanyuan Zhao,1 Samiratou Batoko,4 Peixiang Lan,1 Zhishui Chen1
1Institute of Organ Transplantation, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology; Key Laboratory of Organ Transplantation, Ministry of Education; NHC Key Laboratory of Organ Transplantation; Key Laboratory of Organ Transplantation, Chinese Academy of Medical Sciences, Wuhan, People’s Republic of China; 2The Center for Biomedical Research, Department of Respiratory and Critical Care Medicine, Key Laboratory of Pulmonary Diseases of Health Ministry, Tongji Hospital, Tongji Medical College, Huazhong University of Sciences and Technology, Wuhan, People’s Republic of China; 3Healit Research International and TRACE Research and Innovation, Untold Global Healit Zambia, Lusaka, Zambia; 4School of Economics, Zhongnan University of Economics and Law, Wuhan, 430073, People’s Republic of China
Correspondence: Zhishui Chen, Email [email protected] Peixiang Lan, Email [email protected]
Abstract: SET domain-containing lysine methyltransferase 7 (SETD7) is a critical enzyme that methylates lysine residues on both histone and non-histone proteins, thereby regulating gene expression and protein function. This methyltransferase plays a versatile and context-dependent role in a wide range of physiological processes, including cell differentiation, reactive oxygen species (ROS) signaling, oxidative stress regulation, and energy metabolism. SETD7’s dual nature is highlighted by its paradoxical involvement in various diseases such as cancer, asthma, and Alzheimer’s disease, where it can either promote or suppress pathological progression depending on the cellular environment and molecular context. The multifaceted functions of SETD7 underscore its importance in maintaining cellular homeostasis but also present significant challenges for therapeutic targeting. Although selective inhibitors like Cyproheptadine and (R)-PFI-2 have recently been identified, the development of highly specific and effective therapies remains complex due to SETD7’s broad regulatory roles and the potential for unintended effects on normal physiological processes. These challenges necessitate nuanced therapeutic strategies, including the exploration of combination treatments and context-specific modulation to maximize efficacy while minimizing adverse outcomes. This review comprehensively explores SETD7’s structure, subcellular localization, and diverse biological functions in both normal and disease states. By elucidating the dual and context-dependent nature of SETD7, it aims to provide a framework for future research focused on unraveling its molecular mechanisms and advancing targeted therapeutic approaches that leverage its unique regulatory capabilities.
Keywords: lysine methyltransferase, gene expression, cancer, oxidative stress, inflammation, transcription factors
Introduction
SET domain-containing lysine methyltransferase 7 (SETD7) is a pivotal enzyme that catalyzes the monomethylation of lysine residues on both histone and non-histone proteins, with lysine being its primary amino acid target.1 Initially identified as a histone H3 lysine 4 (H3K4) methyltransferase, SETD7 primarily modifies lysine residues to regulate chromatin structure and gene expression.1,2 Beyond histones, SETD7 methylates various non-histone substrates including transcription factors (TF’s) and regulatory proteins, thereby modulating their stability, activity, and interactions. This lysine methylation occurs on substrates containing specific consensus motifs, facilitated by SETD7’s SET domain and N-terminal MORN repeats, enabling precise substrate recognition and catalytic efficiency.1,2
Functionally, SETD7 orchestrates a broad spectrum of cellular processes such as cell proliferation, differentiation, reactive oxygen species (ROS) signaling, oxidative stress response, and endoplasmic reticulum stress adaptation. Through these activities, it plays essential roles in maintaining cellular homeostasis and regulating physiological pathways including metabolism and immune responses.1–3 The role of SETD7 in disease pathogenesis is notably complex and highly context-dependent, varying with cell type, disease model, and molecular environment. SETD7 can act as either a promoter or suppressor of pathological processes depending on the specific cellular context and its target proteins.1,4 For example, in breast cancer, SETD7 inhibition is associated with a more undifferentiated luminal cell phenotype, mediated by its effects on the stability of proteins such as E2F1 and DNMT1.1,5 Conversely, SETD7 enhances stem cell properties by stabilizing factors like LIN28A and hypoxia-inducible factor 1α (HIF-1α), while also promoting differentiation through repression of SOX2, Yes-associated protein (YAP), and STAT3 activities, and by increasing serum response factor (SRF) activity.1
SETD7 also plays a significant role in oxidative stress regulation by modulating ROS signaling via mitochondria and the nuclear factor erythroid 2-related factor 2 (NFE2L2)/Antioxidant Response Element (ARE) pathway, thereby influencing energy metabolism and inflammatory responses.3 Furthermore, SETD7 has been shown to modulate the Hippo signaling pathway and hypoxia-inducible factor 1α (HIF-1α), suggesting its role in maintaining tissue homeostasis in chondrocyte differentiation.6
In disease contexts beyond cancer, SETD7 has been implicated in asthma, where it amplifies tumor necrosis factor-alpha (TNF-α)-induced proliferation and migration of airway smooth muscle cells through NF-κB/CD38 signaling.7 In Alzheimer’s disease, SETD7-mediated lysine methylation promotes enrichment of soluble Tau protein, suggesting a role in neurodegeneration.8 This dual and sometimes paradoxical nature of SETD7 highlights it as a versatile regulator whose effects are strongly influenced by cellular context, substrate availability, and signaling milieu. Such complexity poses significant challenges for therapeutic targeting. Although selective inhibitors like Cyproheptadine and (R)-PFI-2 have been developed, the broad and context-dependent functions of SETD7 require careful consideration to avoid unintended disruption of its physiological roles.2,9,10
This review aims to elucidate the multifaceted and context-dependent functions of SETD7 in disease occurrence and pathogenesis, providing an overview of current knowledge and identifying critical areas for future research. A deeper understanding of SETD7’s molecular mechanisms will be essential to harness its therapeutic potential across a diverse spectrum of pathological conditions.
SETD7 Localization, Structure and Function
Localization
SETD7, a 41-kDa lysine monomethyltransferase, exhibits both nuclear and cytoplasmic localization, which has been demonstrated to vary in diverse cell types and cellular conditions.1,2 For instance, in murine embryonic fibroblasts (MEFs), it is seen to retain the YAP in the cytoplasm,11 whereas in human monocytes, it co-localizes with NFκB-p65 within the nucleus and cytoplasm.12 This dual location is also found in the osteosarcoma-derived human cell line U2-OS, where SETD7 is present in both the nucleus and cytoplasm but accumulates in the nucleus upon DNA damage.13 Its localization is also regulated by interactions with other cellular factors such as nuclear factor-kappa-B (NF-κB), which has been shown to recruit SETD7 to the gene promoters that are dependent on NF-κB signaling, indicating a role in nuclear functions. Further investigation has shown that this recruitment suggests that its nuclear localization might be mediated through interactions with particular proteins rather than through conventional nuclear localization signals. Unlike the remainder of the SET domain-containing lysine methyltransferases (KMTs), SETD7 lacks a defined nuclear target and sends signals outside often resulting in a more pronounced cytoplasmic presence.1,12
Furthermore, the cellular distribution of SETD7 is recognized as being influenced by external factors such as transcriptional inhibitors and environmental conditions like high glucose levels, resulting in its concentration within the endothelial cell nucleus.14 The existence of membrane occupation and recognition nexus (MORN) motifs in SETD7 indicates potential engagement with the plasma membrane, although no membrane protein substrates have been identified yet.2,15–18 The complex localization pattern underscores the pleiotropic effects of SETD7 throughout the cell, emphasizing the necessity for additional research to fully understand the processes regulating its transit and function in different cellular compartments, as its impact reach many areas. Understanding SETD7’s varied localization within the cell provides a foundation for examining its structural components, which are critical to its function as a methyltransferase.
Structure
SETD7 domain is highly conserved within the methyltransferase family and has a crucial role in their enzymatic activity.19,20 The structure of SETD7 is split into two main domains: the N-terminal domain, comprising of a sequence of opposing β strands, and the C-terminal domain (also known as the SET domain), characterized by a compact composition comprised of β strands organized into three separate sheets encircling a pseudo-knot. The SET domain, present in almost all methyltransferases, is crucial for their catalytic role.1,19,20 Surrounding the SET domain are various regions, specifically pre-SET, i-SET, and post-SET, located either at the N-terminal end or outside the C-terminal terminal of the SET domain. The i-SET and post-SET (sometimes referred to as c-SET) regions, alongside the SET domain, facilitate the establishment of binding sites for substrates and cofactors, along with the formation of small hydrophobic channel where the substrate lysine and cofactor converge within the core enzyme.1
The pre-SET (or n-SET) region and N-terminal domain provide stability to the enzyme and the SET domain, while the pre-SET region also serve as a binding site for other proteins or DNA.1,21 This configuration is unique among methyltransferases, as the substrate and cofactor S-adenosylmethionine (SAM) binds to separate locations on opposite faces of the SET domain. This separation facilitates a sequential binding process where SAM binds first, followed by the substrate. This mechanism is essential for the precise methylation of the target lysine residue, highlighting the intricate design of SETD7 for its function.1 The structure of SETD7 possesses a preserved SET domain, which is important to its methyltransferase activity. This domain enables the relocation of the methyl groups from SAM to the lysine residue on its target proteins.22,23 The Human Protein Atlas offers a comprehensive structural prediction of SETD7, emphasizing regions of antigenicity and potential functional domains.24 This structural information is crucial for understanding how SETD7 interacts with its substrates and performs its enzymatic function. A visual representation of these structural elements is provided in Figure 1, which illustrates the key domains and motifs of SETD7.
 |
Figure 1 Conceptual illustration of SETD7 structure. SETD7 consist of two main domains, the N-terminal and the C-terminal. The N-terminal domain comprises a sequence of three opposing β strands, MORN motifs 1, 2 and 3 (Medium orchid). The C-terminal includes β strands organized into three separate sheets, consisting of the SET domains (Medium slate blue), surrounded by the c-SET (or post-SET) (Khaki) and n-SET (or pre-SET) domains (Light sky blue), and features an additional region known as i-SET (Medium purple). MORN: Membrane occupation and recognition nexus. |
Function
SETD7 exerts significant control over gene expression and chromatin architecture through its methyltransferase activity on both histone and non-histone substrates. Its canonical role involves catalyzing the monomethylation of lysine 4 on histone H3 (H3K4me1), a modification that is strongly associated with transcriptional activation.1,20 This epigenetic mark is typically enriched at enhancer regions and TSS of actively expressed genes, promoting a more open chromatin conformation and facilitating access for transcriptional machinery and regulatory proteins.1
Histone Methylation
SETD7 primarily functions as a histone methyltransferase, specifically monomethylating lysine 4 on histone H3 (H3K4me1). This modification is linked with transcriptional activation mainly due to its proximity to enhancer regions and transcription initiation sites of transcriptionally active genes and is vital for regulating gene expression. Its main function is the precise monomethylation of histone H3 at lysine 4 (H3K4me1), a modification intimately associated with transcriptional activation. This specific methylation event is commonly seen near enhancer regions and transcription start sites (TSSs) of actively expressed genes, thereby acting as a vital regulator of gene expression and impacting essential cellular functions.1,25–28
This histone-lysine N-methyltransferase affects target substrates through a series of well-orchestrated steps characterized by specificity and efficiency in lysine methylation. The enzyme possesses SET domain, essential for catalyzing the relocation of a methyl group from SAM to a lysine residue on the substrate.23,29–31 Initially, SETD7 identifies and binds to its target substrate through highly specific interactions facilitated by the enzyme’s structural domains, including the SET, i-SET, and pre-SET regions. The N-terminal of MORN repeats enhance substrate binding affinity via charge to charge interactions with positively charged sequences on the substrate proteins. This selective binding guarantees that only appropriate proteins are modified by SETD7.1
After binding to its substrate, SETD7 facilitates the relocation of a methyl group from SAM to the ε-amino group of the target lysine residue.22,23,29,32,33 The process begins with the attachment of SAM to the enzyme at a distinct site on the SET domain, distinct from the substrate binding site. This ordered mechanism allows for the formation of a triadic complex comprising the enzyme, SAM, and the substrate. A constricted hydrophobic channel guides the substrate lysine towards the cofactor SAM, shielding it from solvent interference and creating an optimal environment for catalysis.1,19,20,22,23
Upon successful methylation, the modified protein undergoes changes in activity, stability, and localization, significantly impacting many cellular processes encompassing transcriptional regulation, cell cycle enhancement, and responses to environmental signals. Importantly, the specific location of methylation whether in the nucleus or cytoplasm often depends on the nature of the target protein.1,21 The H3K4me1 mark serves as a signal, recruiting “reader” proteins that further enhance the transcription of particular genes, underscoring the sophisticated regulatory mechanisms regulated by SETD7.1,23,34
SETD7’s role in gene regulation is multifaceted as it interacts with numerous TF’s and chromatin remodeling complexes, like p300 and BRG1, to modulate the gene expression in cell differentiation and proliferation processes.3,18,25–28,35–38 For instance, SETD7-mediated H3K4me1 can maintain transcriptionally active chromatin by preventing the activity of repressive complexes such as NuRD and Suv39h1, thereby inhibiting transcriptional silencing and promoting subsequent acetylation of histones by p300, which further enhances gene activation.1,18,39
Non-Histone Protein Methylation
SETD7, traditionally recognized for its histone methyltransferase activity, exhibits a broader regulatory scope by methylating a diverse array of non-histone substrates. This expanded substrate specificity encompasses TF’s, chromatin remodelers, cell cycle regulators, and proteins integral to signaling pathways, thereby positioning SETD7 as a multifaceted enzyme with critical roles in cellular homeostasis and disease pathogenesis.
Among the non-histone proteins targeted by SETD7 are key transcription factors and transcriptional regulators such as E2F1, RUNX2, YAP, FOXO, TP53 (p53), NF-κB, TAF10, and β-catenin. These modifications can have diverse effects, either enhancing or repressing transcription depending on the specific substrate and cellular context.1,40,41
For example, SETD7 methylation of E2F1 at K185 can destabilize E2F1 by promoting its ubiquitination and proteasomal degradation, thereby inhibiting its transcriptional activity. However, in some contexts, this methylation is necessary for full E2F1-mediated transcriptional activation prior to its eventual degradation.1,39,42,43 In prostate cancer, SETD7 methylates FOXA1 at K270, repressing its chromatin binding and acting as a transcriptional repressor, which counterbalances the activity of the demethylase LSD1. Loss of SETD7 expression in CRPC leads to increased FOXA1 chromatin binding, reactivation of cryptic enhancers, and upregulation of oncogenic transcriptional programs, highlighting a tumor suppressor function for SETD7 in this context.41
SETD7 is recruited to specific genomic loci primarily through interactions with transcription factors and chromatin remodeling complexes, rather than through intrinsic DNA-binding specificity. Additionally, SETD7 binds methylated H3K36 within gene bodies to facilitate RNA polymerase II-dependent transcription, demonstrating its role in both the establishment and maintenance of active chromatin states.44 The activity of SETD7 is further modulated through crosstalk with other chromatin-modifying enzymes, such as histone acetyltransferases and demethylases, allowing the integration of multiple epigenetic signals to fine-tune chromatin accessibility and gene regulation.30,39,44,45
Epigenetic and chromatin regulators such as DNMT1 and other histone methyltransferases also fall within SETD7’s substrate repertoire. Methylation of these proteins affects their stability and function, thereby influencing the epigenetic landscape and gene expression profiles.1,5 Hormone receptors such as in estrogen receptor alpha (ERα) and androgen receptor (AR) are also substrates of SETD7. Methylation typically stabilizes these receptors, augmenting their transcriptional activity and impacting hormone-responsive pathways implicated in various diseases, including cancers.46,47
In addition to transcription factors and hormone receptors, SETD7 methylates proteins involved in key signaling pathways. These include the KEAP1-NRF2 complex, JAK2/STAT3, HIF1α, members of the SMYD family, and PD-L1. Through these modifications, SETD7 regulates cellular responses to oxidative stress, inflammation, hypoxia, and immune evasion, highlighting its central role in maintaining cellular homeostasis and modulating disease progression. This dynamic interplay is essential for orchestrating complex transcriptional responses during development, differentiation, and in response to environmental cues.41
This broader substrate specificity extends to proteins that regulate cellular homeostasis and those implicated in various disease processes. These proteins are crucial for various biological processes, such as cell cycle regulation, the maintenance of redox balance, and the mediation of signal transduction.1–4,48,49 The methylation of these proteins can modify their stability, subcellular localization, and interaction with other molecules, thereby influencing their activity and downstream effects. For example, methylation might stabilize a protein, leading to increased activity, or it may destabilize a protein, targeting it for degradation. Furthermore, methylation can alter the subcellular localization of a protein, affecting its ability to interact with other molecules and influencing its role in cellular processes.1–4,50–52 The consequences of SETD7-mediated methylation on non-histone proteins are often multifaceted and intricately linked to the specific proteins modified and cellular environments. For example, methylation might enhance the stability of a particular protein, leading to increased activity and downstream effects related to disease development. Conversely, methylation can destabilize a protein, leading to its degradation and subsequently impacting cellular pathways.1,2,53,54
This multifaceted functionality underscores the complexity of SETD7’s role in disease pathogenesis.1–3,50–52 The methylation of these proteins by SETD7 also significantly affects their stability, localization, and activity, with cascading effects on multiple cellular pathways.1,2,54 While some non-histone targets might be modified in the nucleus (eg, TF’s bound to DNA), others may be modified in the cytoplasm before translocation to the nucleus or while residing in the cytoplasm. The location of modification depends on the specific non-histone protein involved.1,2,54 This ability to modulate a wide array of downstream cellular pathways makes SETD7 a central player in maintaining cellular homeostasis and contributes to its significant influence in various disease contexts.
SETD7 therefore serves as a multifunctional enzyme important in the regulation of gene expression via methylation mechanisms activities on both histone and non-histone substrates. Its diverse roles in cellular homeostasis and disease highlight its potential as target for treatment of several disorders. Further research into the mechanisms governing SETD7’s actions may provide deeper insights into its contributions to both normal physiology and pathological states. Given SETD7’s broad functionality in histone and non-histone methylation, its role in specific cellular processes and diseases is of significant interest. Predicted interactions and function partners of SETD7, as well as SETD7’s functional consequences are shown in Figure 2 and Table 1, respectively.
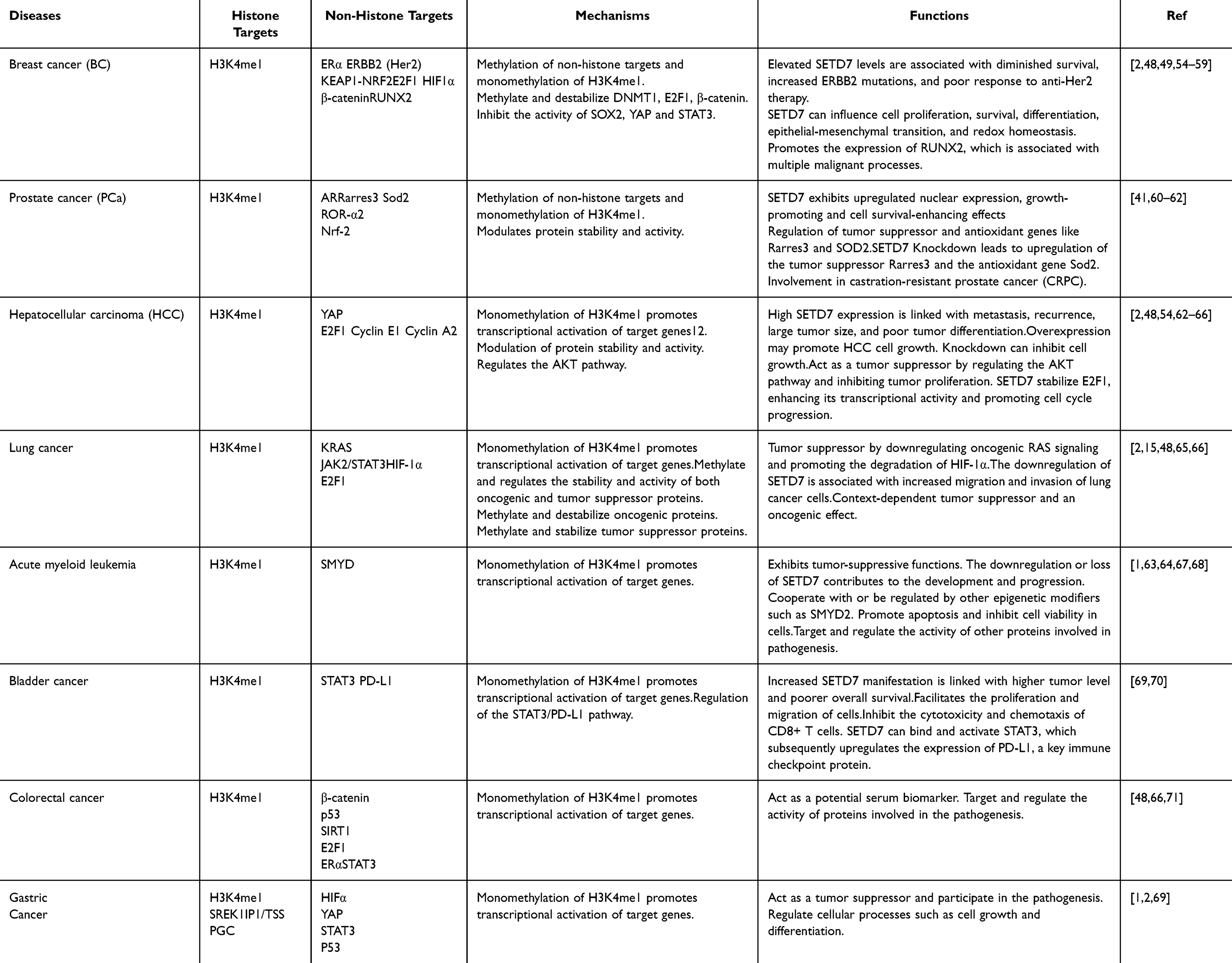 |
Table 1 Mechanisms and Function Consequences of SETD7 in Cancer |
 |
Figure 2 Predicted interactions and function partners of SETD7 as generated by STRINGs 10.0 software. Active predicted modes of action in the network display are shown in different colors. Possible interaction and functional association network of SETD7 with FOXO, TP53, DNMTI, and SIRT1. Confidence score sort 0.9. Known Interaction; Blue: From curated databases; Purple: Experimentally determined. Predicted interactions; Green: Gene neighborhood; Red: Gene fusions; Dark blue: Gene co-cocurence. Others; Lime Green: Textmining; Black: Co-expression; Light blue: Protein homology. |
SETD7 Role in Cell Cycle Regulation and Differentiation
SETD7 functions as a crucial epigenetic regulator involved in cell cycle regulation and differentiation.29 It primarily exerts its effects particularly through the monomethylation of H3K4, which is associated with activated transcription at enhancer regions and transcription start sites.1,29 This modification is essential for orchestrating gene expression programs fundamental for differentiation and is coupled with its activity on non-histone proteins, including pivotal regulators such as E2F1, retinoblastoma protein (pRb), and various transcription factors.1,25–29,37
In the context of cell cycle regulation, SETD7 is integral to the G1/S phase transition.54,71,72 It functions by methylating crucial cell cycle proteins like E2F1, which controls the expression of genes critical for cell cycle advancement, including cyclins and cyclin-dependent kinases (CDKs). Specifically, SETD7-mediated methylation of E2F1 at lysine 185 enhances its ubiquitination and subsequent degradation, thereby suppressing its transcriptional activity and promoting apoptosis.54,73 Additionally, SETD7’s methylation of pRb at lysines 873 and 810 is vital for maintaining its tumor suppressor functions, including facilitating cell cycle arrest and senescence in response to DNA damage.5,42 This established interplay positions SETD7 as a tumor suppressor that collaborates with pRb to ensure genomic stability.
Moreover, researchers have revealed that SETD7 is critical for several key cellular processes including development and differentiation. SETD7 expression, which is notably missing in pluripotent embryonic stem cells, is activated during the transition to a multipotent state, suggesting its involvement in promoting differentiation.5,44 Specifically, SETD7 is essential for mesendoderm specification and the differentiation of diverse cellular types including endothelial cells, cardiomyocytes, hepatocytes, and neuronal precursor cells.5,74 Interestingly, while SETD7’s methyltransferase activity is frequently associated with these differentiation processes, it is not always essential; this suggests that SETD7 may also regulate target genes through alternative mechanisms beyond direct methylation.5 Although there are indications that SETD7 may exhibit dimethyltransferase activity, it is generally accepted that its main function is as a monomethyltransferase, with its enzymatic activity potentially serving as a foundation for further methylation by other enzymes.1,3
SETD7 also plays a pivotal role in differentiation, particularly in stem cells.75 Its expression is upregulated during stem cell differentiation, where it is necessary for silencing pluripotency genes like OCT4 and NANOG while activating lineage-specific genes.75,76 Knockdown of SETD7 in human embryonic stem cells leads to impaired differentiation and persistence of pluripotency factors. Mechanistically, SETD7 exerts its influence by methylating linker histone H1 and non-histone proteins such as LIN28A, which affects their localization and function during differentiation.75–77 Furthermore, SETD7 modulates critical developmental signaling pathways, such as Wnt/β-catenin, facilitating mesoderm formation during hematopoietic differentiation.78
The dual roles of SETD7 in both promoting and repressing cell proliferation depend on the cellular context.1,72 Its involvement in signaling pathways, including the regulation of non-coding RNAs and the Hippo pathway, broadens its epigenetic influence on cell fate decisions.1,2,11 This multifaceted regulation highlights SETD7’s potential as a therapeutic target in cancer and regenerative medicine, particularly given its implications in diseases characterized by disrupted cell cycle control and differentiation processes. Therefore, SETD7 stands out as a key regulatory player that integrates signals for cell proliferation and differentiation, emphasizing its significance in maintaining cellular and developmental homeostasis.
SETD7 Influence on Stem Cell Plasticity
SETD7 influences stem cell plasticity through a dynamic process of lysine methylation on non-histone proteins, where it adds methyl groups that mark proteins for ubiquitin-dependent degradation. This methylation is reversible, as demethylases like LSD1 can remove these marks. The balance between methylation by SETD7 and demethylation by LSD1 regulates protein stability, controlling key processes such as embryonic development, stem cell pluripotency, self-renewal, and differentiation.79
SETD7 profoundly influences stem cell plasticity by orchestrating the complex balance between maintaining pluripotency and promoting differentiation through epigenetic and posttranslational modifications.1,37,75,76 Its expression markedly increases during the differentiation of human embryonic stem cells (hESCs), where it plays a pivotal role in silencing pluripotency genes such as OCT4 and NANOG. This silencing is mediated by SETD7 methylating linker histone H1, which induces conformational changes that reduce H1 recruitment to these key pluripotency loci, facilitating chromatin remodeling necessary for the transition from a stem-like state to a differentiated state.37,76 Knockdown of SETD7 disrupts this process, causing delayed silencing of pluripotency genes and impaired activation of differentiation programs, thereby hindering proper stem cell fate transitions.37,75,76
SETD7 negatively influences stem cell plasticity by promoting senescence during Mesenchymal stem cell (MSC) expansion. Inhibiting SETD7 delays senescence, preserves the regenerative capacity of human adipose-derived mesenchymal stem cells (hAD-MSCs), and enhances their effectiveness in tendon repair. Thus, SETD7 is a promising target for improving stem cell-based therapies by maintaining stem cell plasticity and function during culture.80
Beyond histone modification, SETD7 also methylates numerous non-histone proteins critical for stem cell function. It stabilizes stemness-associated factors like LIN28A and HIF1α, supporting self-renewal, while simultaneously inhibiting pluripotency regulators such as SOX2, YAP, and STAT3, which promotes differentiation. For instance, SETD7 methylates SOX2, leading to its degradation and reduced transcriptional activity, thereby facilitating exit from pluripotency. Additionally, SETD7 enhances the transcriptional activity of SRF, a regulator of muscle and neural lineage genes, further influencing lineage specification.1,37,75,79
SETD7’s regulatory reach extends to key signaling pathways that govern stem cell fate. It modulates the Wnt/β-catenin pathway, which is crucial for mesodermal lineage specification. During hematopoietic differentiation of hESCs, SETD7 enhances lateral plate mesoderm formation by facilitating β-catenin degradation, a function independent of its histone methylation activity.78 In adult skeletal muscle stem cells (MuSCs), SETD7 regulates the nuclear accumulation of β-catenin, promoting the transition from proliferative to differentiation-primed states. Inhibition of SETD7 in MuSCs enhances their expansion and regenerative capacity, highlighting its role in adult stem cell plasticity and potential therapeutic applications.63 Moreover, SETD7 influences the subcellular localization and activity of its targets, such as retaining YAP in the cytoplasm through methylation, which impacts the Hippo signaling pathway involved in cell proliferation and differentiation.2,10,11 This dynamic regulation of protein localization and function underscores SETD7’s versatility in controlling stem cell plasticity.
Collectively, SETD7 integrates epigenetic remodeling and direct methylation of TFs and signaling molecules to finely tune gene expression programs that govern stem cell maintenance, lineage commitment, and differentiation. Its ability to both stabilize stemness factors and repress pluripotency regulators enables stem cells to respond adaptively to developmental cues and environmental changes.1,2,10,11,37,63,75,76,78 This multifaceted role positions SETD7 as a key molecular switch in stem cell biology, with significant implications for developmental biology, regenerative medicine, and disease modeling.
SETD7 Involvement in Oxidative Stress and Metabolic Regulation
SETD7 is a key epigenetic regulator that influences oxidative stress responses and metabolic homeostasis through monomethylation of lysine residues on histone and non-histone proteins. This enzymatic activity modulates gene expression, protein stability, and cellular functions critical for maintaining redox balance and energy metabolism.3,60
In oxidative stress regulation, SETD7 impacts ROS signaling by methylating and modulating several transcription factors and proteins. It directly methylates and stabilizes NRF2, a master regulator of antioxidant response elements (AREs), enhancing the expression of antioxidant enzymes such as HMOX1 and Superoxide Dismutase 2 (SOD2).3,60 However, SETD7 also methylates NF-κB p65 (RELA), promoting pro-inflammatory signaling that can increase ROS production.3 Additionally, SETD7 influences mitochondrial function by regulating factors like PPARGC1A, which affects mitochondrial biogenesis and antioxidant capacity.3 Inhibition or knockdown of SETD7 reduces NF-κB-induced oxidative stress and pro-inflammatory cytokine production while enhancing mitochondrial antioxidant functions and NRF2 activity, leading to improved ROS clearance and cellular resistance to oxidative damage.3,13,30 SETD7 also methylates p53 at K372, modulating its pro-apoptotic activity under oxidative stress, thus influencing cell fate decisions in response to severe stress.3,13,30 In temporal lobe epilepsy with hippocampal sclerosis, elevated SETD7 and histone methyltransferases increase repressive methylation, suppressing antioxidant enzymes like Nrf2. This epigenetic repression impairs defence against oxidative stress, promoting seizure recurrence.81
The involvement of SETD7 in oxidative stress is further demonstrated by its role in mediating oxidative stress-induced protein modifications in atherosclerosis. Specifically, Inhibiting SETD7’s catalytic activity markedly decreased the formation of nitrotyrosine (NT) and 4-hydroxynonenal (4HNE)-protein adducts in the aorta of mice with atherosclerosis. These adducts are biomarkers of oxidative damage caused by ROS derived from NADPH oxidase, which contribute to the formation of peroxynitrite (ONOO-) and lipid peroxidation products like 4HNE.82
Regarding metabolic regulation, SETD7 methylates key metabolic regulators including HIF-1α, NRF2, NF-κB,PPARγ, and SIRT1, thereby affecting cellular responses to hypoxia, energy sensing, fatty acid oxidation, glucose uptake, adipogenesis, and insulin sensitivity.1,3,67,83 SETD7 contributes to metabolic regulation and cellular stress resistance by stabilizing NRF2, which enhances antioxidant defenses, suppressing NF-κB-driven inflammation, thereby reducing ROS that can inhibit AMPK activity. Through these mechanisms, SETD7 may indirectly supports AMPK function, promoting energy homeostasis and improving metabolic flexibility, as demonstrated in obesity models where inhibition of SETD7 leads to enhanced metabolic adaptability.3,13,30,72,83,84 Other studies have also implicated SETD7 activation in worsened oxidative stress injury, resulting in significant dysfunction in RAECs by promoting Glutathione Peroxidase 4 (GPX4)-mediated lipid peroxidation. Of which, SETD7 deficiency was shown to decrease p53 mono-methylation and suppress FBXO45 transcription, which in turn prevented GPX4 protein degradation, thereby reducing lipid peroxidation and oxidative stress.85 In addition, SETD7 also stabilizes proteins like LIN28A and HIF-1α that contribute to stem cell properties and metabolic adaptation under stress. Through these actions, SETD7 links epigenetic modifications to metabolic pathways, influencing cell proliferation, differentiation, and energy metabolism.1,3
The dual role of SETD7 in both promoting antioxidant defenses via NRF2 and enhancing pro-inflammatory ROS generation via NF-κB highlights its complex regulatory function in oxidative stress. Its modulation of mitochondrial function and metabolic regulators further integrates oxidative stress responses with cellular metabolism. This multifunctionality positions SETD7 as a promising therapeutic target in diseases characterized by oxidative stress and metabolic dysregulation, including cancer, metabolic disorders like diabetes and obesity, neurodegenerative diseases, and inflammatory conditions.3,49
Mechanisms and Functional Consequences of SETD7 in Disease Pathogenesis
SETD7 is essential in modulating gene expression through histone and non-histone substrate modification, influencing key cellular processes that are critical for health maintenance. SETD7 has been associated with a range of Non-communicable diseases (NCDs), Neurodegenerative diseases and inflammatory diseases, as well as aging-associated disorders. Emerging evidence suggests that it may significantly influence several disease processes, highlighting its potential effects on human health.1,2 A schematic illustration of potential SETD7 functional consequences and mechanisms is presented in Figures 2 and 3.
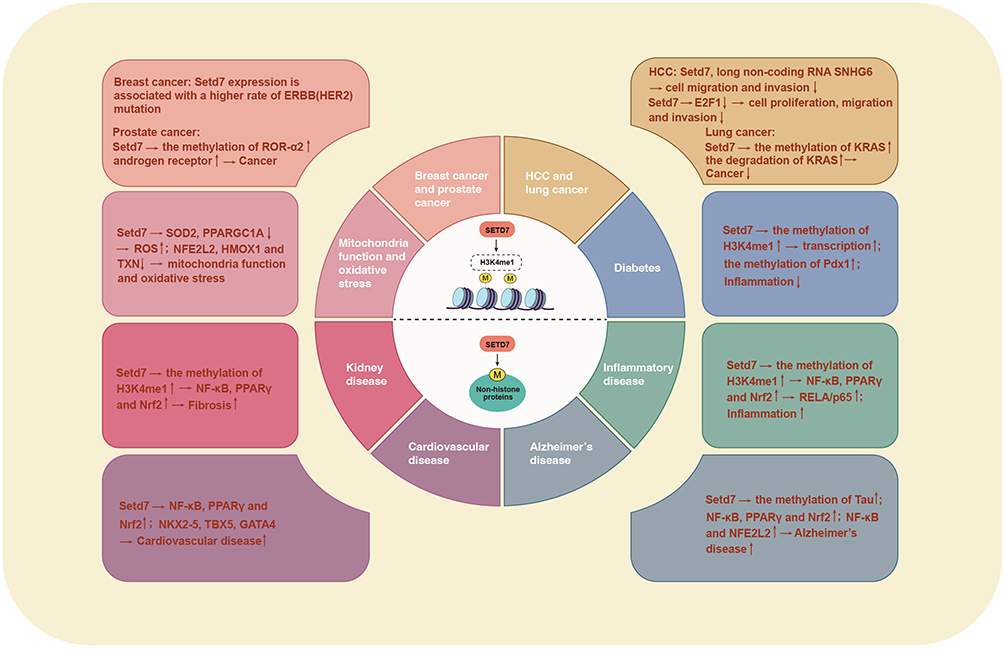 |
Figure 3 SETD7 mechanism and function in disease pathogenesis. This figure illustrates the dual role of SETD7 in various disease contexts, highlighting its function as both a promoter and suppressor of tumors. SETD7 is associated with disorders including cancer, cardiovascular disease, diabetes, inflammatory diseases, kidney disorders, and neurodegenerative disorders. Its regulatory impact extends to histone and non-histone proteins, influencing critical cellular activities including cell proliferation, movement, and immune responses. The figure emphasizes the complex interplay of SETD7 in disease mechanisms, underscoring its potential as a therapeutic target. |
SETD7 in Cancer
SETD7 has been recognized for its multifaceted role in cancer, acting as either a tumor promoter or a suppressor dependent on the cancer type and cellular microenvironment.54 The role of SETD7 in cancer is complicated, as it regulates a variety of cancer-related processes, such as inflammation-induced cell proliferation, differentiation, invasion, migration, metastasis, and EMT, as well as response to hypoxia and oxidative stress.48,54,86 The expression and associated function of SETD7 has been investigated in many cancers and are presented in Table 1.
Breast Cancer
In BC, SETD7 has been shown to be a potential prognostic marker associated with increased expression in HER2-enriched molecular subtype of BC.48,49 High levels of SETD7 expression have been associated with a higher rate of ERBB2 (HER2) mutations in breast tumors, and poor survival outcomes.48 This high SETD7 expression has also been linked to poorer responsiveness to anti-Her2 therapy, along with higher stromal, lower immune scores, and elevated cancer-associated fibroblasts and endothelial cells.48,49,55,56 The predictive value of SETD7 as a prognostic marker differs across molecular subtypes, being most notable in Her2-enriched cancers and least significant in basal-like cancers. In basal-like cancers, higher SETD7 expression is associated with worse recurrence-free survival.48 Additionally, SETD7-related genes are predominantly associated with immune response mechanisms, and with the upregulation of SETD7 in breast cancer tissues can be associated with a poorer prognosis due to its complex roles in regulating cell proliferation and survival.48,57
Conversely, functional studies have shown that SETD7 may also possess tumor-suppressor functions which are tied to its ability to methylate and induce the degradation of oncogenic proteins such as Dnmt1, E2F-1, and HIF-1α. Additionally, the increased expression of KEAP1 and reduced expression of GATA1 and vascular endothelial growth factor (VEGF) A, along with the induction of RUNX2 have been associated with increased metastatic potential in breast cancer. SETD7 may therefore play a significant function in mitigating oxidative stress by downregulating KEAP1 and increasing the expression of GATA1 and VEGFA, while simultaneously regulating the expression of genes associated with antioxidant responses.54
This anti-oncogenic regulatory mechanism of SETD7 is further qualified by Kaplan-Meier data, which clearly demonstrates that breast cancer patients with elevated SETD7 expression tend to have a slightly longer overall survival, with a median of 40.44 months, compared to 36 months for those with lower expression levels as shown in Figure 4A.87 Other studies have reported similar findings, in which the median survival duration for patients with low SETD7 expression was approximately half the duration for those with high SETD7 expression, specifically 9.5 years compared to 18.1 years,49 while others have shown a correlation between high SETD7 mRNA levels and improved overall survival.40,48 These findings imply that SETD7 may have potential as a prognostic marker for breast cancer.
SETD7 exhibits a complex role in breast cancer, serving both as a potential prognostic marker and a regulator of oncogenic processes. Its high expression is associated with the HER2-enriched subtype and poorer outcomes in certain contexts, yet it also displays tumor-suppressive functions by regulating key oncogenic proteins. The dual nature of SETD7 is further underscored by its involvement in mitigating oxidative stress and influencing metastatic potential. Notably, elevated SETD7 expression has been linked to improved overall survival in some studies, suggesting its potential utility as a prognostic marker. This multifaceted role of SETD7 highlights the need for further investigation to fully elucidate its mechanisms and to explore its therapeutic and prognostic implications in breast cancer.
Prostate Cancer
SETD7 has been associated with the emergence and advancement of prostate cancer (PCa), indicating its significant role in tumor advancement and viability. Researchers has shown that the knockdown of SETD7 ameliorates LNCaP cell proliferation and enhances apoptosis by inducing caspase-3 expression.41,54 The downregulation of SETD7 has also been demonstrated to inhibit the colony-forming capacity of PCa cells in soft agar, while simultaneously increasing the formation of ROS and promoting DNA damage in cancer cells. The suppression of SETD7 expression heightened H2O2-induced DNA damage while diminishing H3K4me1 accumulation at Nrf2 and Gstt2 promoter regions. These findings indicate that SETD7 downregulation correlates with decreased expression of downstream Nrf2 targets, further linking it to colony formation efficiency, and suggests that SETD7 functions as a transcription activator in PCa cells.60
Furthermore, SETD7 methylates histone demethylases Jumonji C domain-containing 2A, 2B and 2D (JMJD2B, JMJD2A, and JMJD2D) on multiple lysine residues, regulating their oncogenic activities. SETD7 enhances JMJD2B’s coactivation of the transcription factor Ju-nana (JUN) and selectively modulates interactions with others like E26 transformation-variant 1 (ETV1).88,89 Methylation of JMJD2A promotes tumor growth and invasion via genes such as Matrix Metalloproteinase-1 (MMP1) and Nucleophosmin 3 (NPM3). JMJD2D methylation stimulates cancer cell proliferation and invasion through pathways involving Casitas B-lineage lymphoma C (CBLC) and Pleiomorphic Adenoma Gene-Like 1 (PLAGL1). The positive correlation of JMJD2 family members, SETD7, and transcription factors in tumors highlights their cooperative role in prostate cancer progression. This methylation axis offers a promising therapeutic target.88,89
Additionally, SETD7 knockdown has also be shown to induce the upregulation of the tumor suppressor Rarres3 (retinoic acid receptor responder 3) and the antioxidant gene SOD2, suggesting a potential tumor-suppressive function. In castration-resistant PCa (CRPC), it has been associated with reading frame alterations which may affect chromatin accessibility and RNA processing during transcription.41,61 Therefore, reducing SETD7 levels disrupts the cancerous properties of prostate cells, leading to increased cell death and impaired tumor growth. The elevation of ROS and subsequent DNA damage may enhance the therapeutic potential of targeting SETD7, as this could sensitize PCa cells to conventional treatments and improve overall treatment efficacy. Thus, SETD7 presents itself as a promising candidate for therapeutic inhibition based strategies in PCa, underscoring the need for further exploration of its mechanisms and its potential as a disease biomarker.
Hepatocellular Carcinoma
In hepatocellular carcinoma (HCC), SETD7 interacts with the long non-coding RNA SNHG6, which destabilizes SETD7 mRNA, leading to increased protein levels and reduced cell migration and invasion.54,90 Research has shown that SETD7 expression is substantially elevated in HCC tumor tissues than in adjacent non-cancerous liver tissues, with significant increases observed in both mRNA and protein concentrations across specific HCC cell lines such as Bel-7404, QGY-7703, and SMMC-7721.62,64 This elevated expression of SETD7 has been strongly associated with aggressive characteristics of tumors, as well as metastasis, larger tumor size, poor differentiation, recurrence, and worse overall survival outcomes.62,64,65
Yet paradoxically, high SETD7 expression has also been linked to improved overall survival in large-scale genomic analyses. This dichotomy may arise from SETD7’s ability to modulate E2F-1 activity, where cytoplasmic interactions downregulate cell cycle proteins like cyclin E1 and cyclin A2, counteracting proliferation, migration, and invasion.2,54,62 Kaplan-Meier analysis on the prognostic significance of SETD7 expression in liver cancer demonstrated that patients with high SETD7 expression had a higher median survival time of 71 months, compared to 45.7 months for those with lower expression levels as shown in Figure 4B.87
The contrasting clinical outcomes, poor prognosis in tumor microenvironments versus survival benefits in broader genomic datasets, highlights context-dependent mechanisms, potentially influenced by tissue-specific signaling networks or competing epigenetic modifiers. Further research is needed to resolve these disparities and clarify SETD7’s therapeutic potential, particularly its interplay with non-coding RNAs and cell cycle regulators in HCC progression.
Lung Cancer
In lung cancer, SETD7 has been identified as a potential tumor suppressor, with its expression significantly downregulated in lung cancer tissues and cell lines compared to non-cancerous lung tissues and bronchial epithelial cells. Studies have shown that both protein and mRNA levels of SETD7 are lower in lung tumor specimens than in adjacent non-malignant tissues.54,65 For instance, SETD7 expression is reduced by 46.1%, 58.1%, and 35.3% in the H661, H1299, and A549 lung cancer cell lines, respectively, compared to normal BEAS-2B cells. This downregulation suggests that SETD7 acts as an anti-oncogene in lung cancer, where its loss is associated with increased cellular invasion and migration without affecting cell viability.65
SETD7’s tumor suppressive role in lung cancer is further supported by its impact on metastatic potential under non-stress conditions. It regulates the JAK2/STAT3 signaling pathway, which is crucial for cell migration and invasion.65,91 Additionally, SETD7 promotes KRAS degradation through methylation in non-small cell lung cancer (NSCLC), contributing to its tumor suppressive effects.4,15 Additionally, SETD7 can inhibit tumor progression by suppressing proliferation, angiogenesis, and migration through pathways involving MMP2, TWIST1, VEGFA, and the JAK/STAT pathway.54 Kaplan-Meier analysis indicates that higher SETD7 expression in lung cancer patients is associated with improved overall survival, with a median survival of 96.2 months for the high expression group versus 60 months for the low expression group as shown in Figure 4C.87
However, other studies have reported conflicting reports suggesting that high SETD7 mRNA levels may be linked to shorter survival in some NSCLC cases in stress conditions. SETD7 has therefore been shown to act as a tumor promoter by reducing the sensitivity of lung cancer cells to chemotherapy. This is supported by studies showing that downregulating SETD7 or inhibiting its activity increases chemotherapy sensitivity, potentially through the regulation of E2F-1 and independently of p53 status.54,92
SETD7’s role in lung cancer is therefore complex and context-dependent. The dual nature of SETD7 highlights the need for further research to fully understand its role in lung cancer in different contexts, and to explore its potential as a therapeutic target. Despite these complexities, higher SETD7 expression is generally associated with improved survival in lung cancer patients, suggesting that its tumor suppressive functions may predominate in many clinical scenarios.
Other Cancers
Furthermore, SETD7 has also been implicated in others cancers, such as acute myeloid leukemia, gastric cancer, colorectal cancer, glioma, and renal cell carcinoma. Studies have shown the correlation between increased SETD7 expression with cancer progression and metastasis, highlighting its significance as a prognostic marker in these malignancies.2,69,93–95 In gastric cancer, researchers have demonstrated that SETD7 knockdown increases tumor cell proliferation, migration, and invasion. While the high SETD7 expression is reported to induce tumor suppressor functions.96 In contrast, additional analysis of genomic data repositories further linked high SETD7 expression in gastric cancer with decreased overall survival, with a median survival of 40.2 months compared to 63.9 months for low expression cohorts as shown in Figure 4D.87
In acute myeloid leukaemia (AML), the relationship between SETD7 expression and survival is nuanced. Kaplan-Meier analysis reveals a marginal difference in median survival between high and low SETD7 expression groups, with 17.3 months for high expression versus 18.3 months for low expression (Figure 4E).87 However, studies show that SETD7 knockdown reduces apoptosis, while overexpression increases it, highlighting its complex role in controlling cell death and survival. The variability in SETD7’s impact across different AML subtypes and the influence of compensatory pathways underscore the need for more detailed research.54,64,68
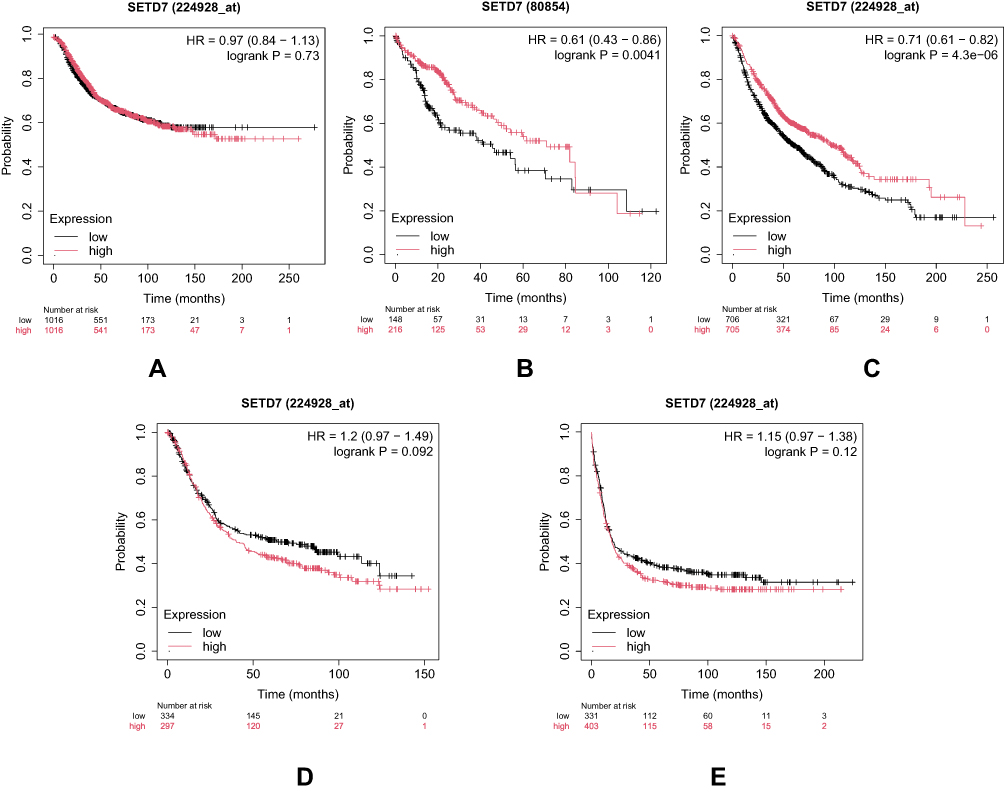 |
Figure 4 SETD7 predicts the prognosis of cancer patients using Kaplan-Meier survival plot analysis of data obtained from GEO, EGA, TCGA, Metabric, Impact, and PubMed repositories. (A) Breast cancer (B) Liver cancer (C) Lung (D) Gastric cancer (E) AML. |
SETD7’s involvement in bladder, renal cell carcinoma and epithelial tumors, further illustrates its dual potential. In epithelial tumors, SETD7 expression loss or downregulation has been negatively correlated with the degree of cancer advancement, suggesting that SETD7 may function as a potential oncogene in this context.5,49,70 Additionally, in bladder cancer, SETD7 promotes immune evasion and correlates with poor prognosis,70 while in renal cell carcinoma, SETD7 stabilizes oncoproteins, contributing to tumor progression.72 The diverse roles of SETD7 across these malignancies highlight its potential as both a biomarker for prognosis and a target for novel treatments. However, discrepancies in its prognostic significance and functional mechanisms necessitate further investigation to clarify its roles and therapeutic potential.
The dysregulation of SETD7-mediated methylation can lead to cellular abnormalities and have an impact in disease progression of multiple cancer types.97 The expression and function of SETD7 varies across these cancers, with potential roles as both an oncogene and tumor suppressor as shown in Tables 1 and 2.68–70 However, the underlying mechanisms remain unclear; it is uncertain whether SETD7 is actively deactivated during the process of tumorigenesis or if cells lacking SETD7 are indicative of adult stem or progenitor cells proliferating within tumorous tissues.5 This complex interplay of SETD7’s expression and function across different malignancies underscores its dual potential as a biomarker for prognosis and as a target for developing novel treatments. Additional studies are essential to understand the specific pathways by which SETD7 influences tumor biology and to clarify its roles as both an oncogene and a tumor suppressor. Table 2 summarizes the roles and prognostic values of SETD7 across various cancer types as presented in Table 1, highlighting its potential in varies context.
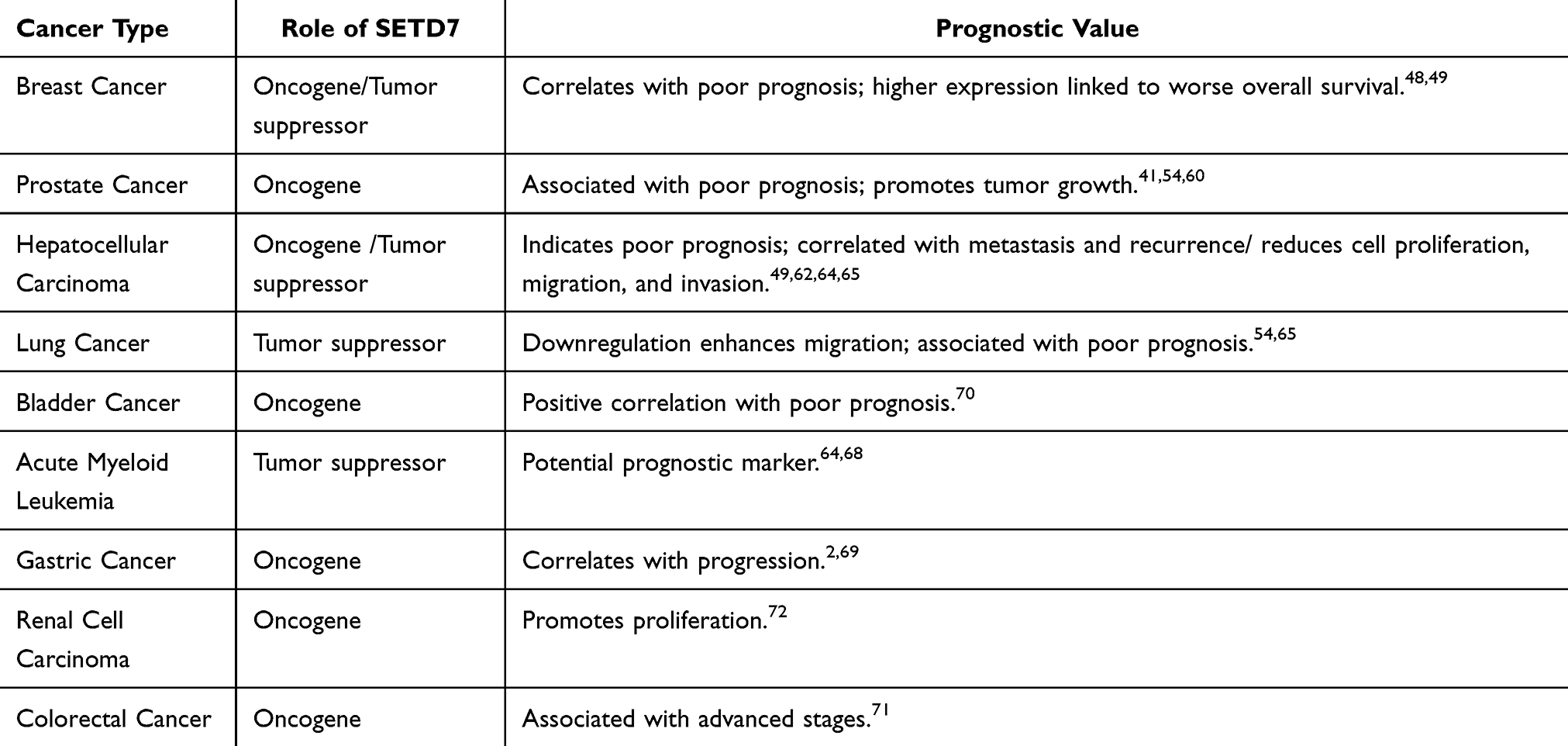 |
Table 2 Roles and Prognostic Values of SETD7 Across Various Cancer Types |
SETD7 in Diabetes
SETD7 has emerged as a viable target for intervention in metabolic disorders, particularly due to its role in transcriptional regulation and glucose homeostasis.1,67 Evidence suggests that in diabetes, SETD7 catalyzes the monomethylation of histone H3 at lysine 4 (H3K4me1), which is correlated with transcriptional activation. Additionally, SETD7 interacts with non-histone proteins critical for glucose regulation, including the Pancreatic and Duodenal Homeobox Protein 1 (Pdx1).1,67,98,99 This protein is essential for pancreatic growth and functionality, and its dysfunction is linked to diabetes. Studies indicate that SETD7 enhances the transcriptional activity of Pdx1, potentially through methylation of specific lysine residues Lys123 and Lys131. This connection between SETD7 and Pdx1 is vital, as Pdx1 regulates genes necessary for insulin secretion and glucose sensing.1,98–100
Adult Pancreas Side Population (SP) cells represent a rare subset approximately 1% of pancreatic cells, characterized by their ability to efflux Hoechst dye and exhibit stem cell-like properties. These cells express the key transcription factor Pdx1 but initially do not produce insulin. Importantly, under conditions of β-cell injury or diabetic stress, SP cells expand and can differentiate into functional insulin-producing β cells, thereby contributing to pancreatic regeneration and glucose homeostasis.99 SETD7 plays a pivotal role in regulating these SP cells, primarily through its epigenetic and transcriptional control mechanisms. One critical interaction is between SETD7 and Pdx1, a master regulator of pancreatic development and β-cell function. SETD7 enhances the transcriptional activity of Pdx1, potentially via methylation at specific lysine residues (Lys123 and Lys131), which is essential for the activation of genes involved in insulin secretion and glucose sensing.99,101 This relationship underscores SETD7’s importance in promoting the differentiation and functional maturation of SP cells into insulin-producing β cells, a process vital for maintaining glucose homeostasis and adapting to metabolic stress.
In diabetes, elevated glucose levels trigger SETD7-dependent epigenetic modifications in hematopoietic stem and progenitor cells. These changes promote the development of a senescence-associated secretory phenotype and increase the expression of NF-κB p65. As a result, there is sustained inflammation and an increase in proinflammatory monocyte production, establishing a link between SETD7 activity, chronic immune dysfunction, and the heightened risk of atherosclerosis observed in type 2 diabetes.101 However, within the pancreas, SETD7’s activity supports the adaptive response of SP cells, aiding in the regeneration and maintenance of β-cell mass.99,101
Further supporting this connection, research by Zhong et al, demonstrated that both global knockout and endothelial cell-specific deletion of SETD7 may led to partial restoration of vascular function and a reduction in high glucose-induced inflammatory responses in STZ-induced and db/db mouse models. Their findings indicate that SETD7 exacerbates diabetic endothelial dysfunction by facilitating FBXO45-mediated ubiquitylation and subsequent degradation of GPX4, thereby contributing to vascular inflammation and injury in diabetes.85
In addition, the interaction between SETD7 and PPARγ (Peroxisome Proliferator-Activated Receptor Gamma) has been recognized as a crucial regulatory mechanism in maintaining β-cell mass and function. PPARγ directly binds with the SETD7 promoter, promoting its expression under glycemic adaptation. Improved glucose homeostasis and β-cell performance could indirectly have anti-inflammatory effects by reducing metabolic stress and inflammation associated with diabetes.67 SETD7’s role in the adaptive response of pancreatic β-cells in maintenance of glucose homeostasis and its depletion may result in impaired glucose handling and decreased expression of essential β-cell genes in islets, highlighting its necessity for the normal function and survival of pancreatic β-cells.67
Furthermore, targeting SETD7 and its associated pathways shows promise as a strategy for managing diabetes, particularly in the context of diabetes-related kidney disease. This approach indicates the potential to attenuate the progression of diabetes-related kidney disease and other kidney diseases by modulating SETD7 activity or expression. While the specific non-histone targets are not clearly identified, SETD7’s regulation of gene expression through H3K4 monomethylation facilitates transcriptional stimulation of genes that play a key role in glucose homeostasis and β-cell function.1,67 Therefore, understanding how SETD7 influences diabetes related pathways could offer fresh perspectives on disease mechanisms and potential treatment approaches. The functional consequences of SETD7 in diabetes are shown in Table 3.
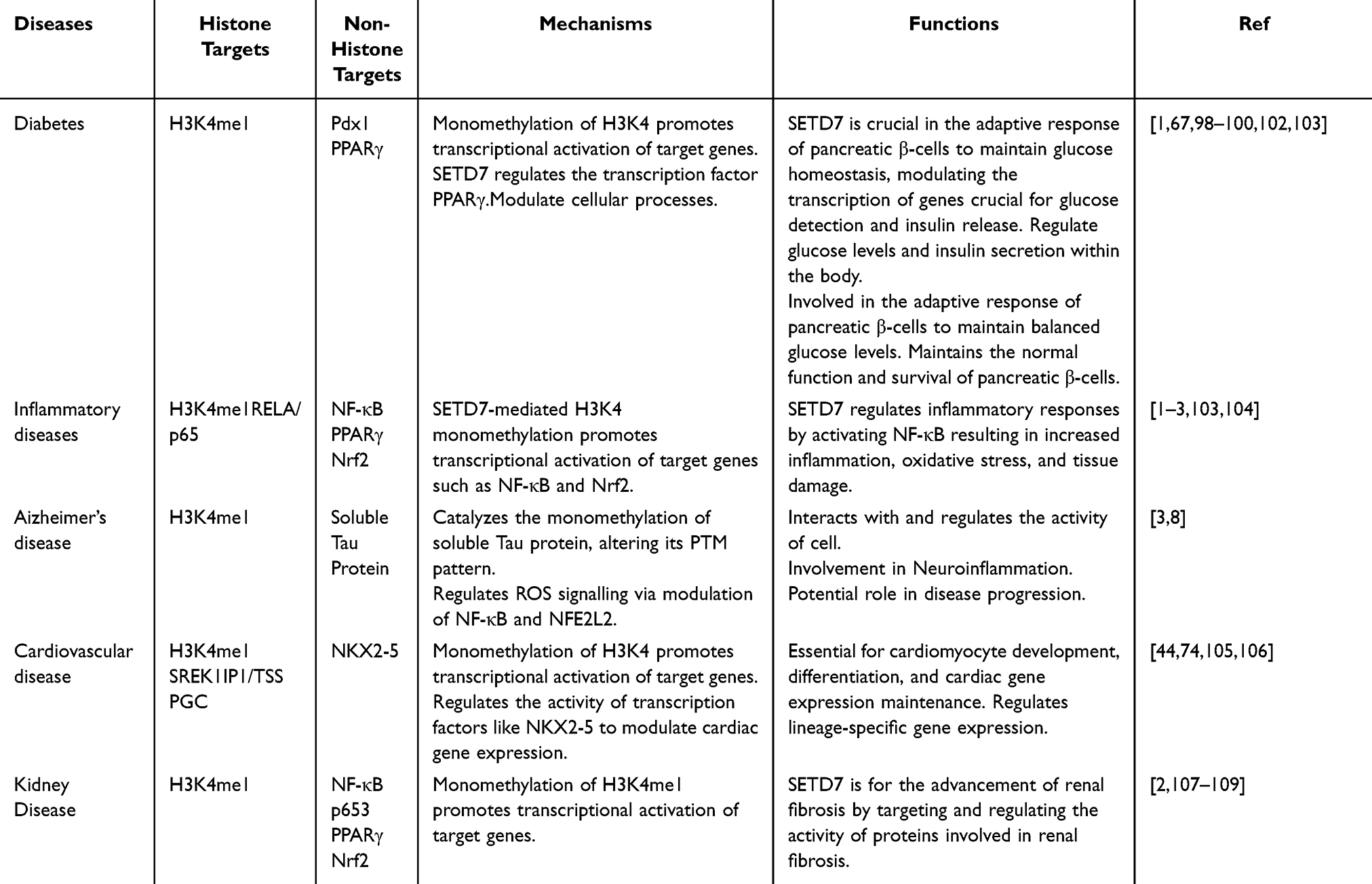 |
Table 3 Mechanisms and Function Consequences of SETD7 in Other Diseases |
Cardiovascular Diseases
In cardiovascular disease, SETD7’s vital role has been proven in regulating gene expression and promoting cardiac differentiation through epigenetic mechanisms. During cardiomyocytes differentiation, SETD7 enhances the transcription of cell-type-specific genes, with its expression progressively increasing throughout the differentiation process.74 Utilizing cardiomyocytes generated from both human embryonic stem cells and human induced pluripotent stem cells (iPSC) in vitro, researchers have proven that SETD7 is pivotal in directing the commitment of cardiac lineage, as well as regulating the manifestation of lineage-specific genes and essential co-factors during different stages of differentiation.44 Also, as hepatocytes and neuroprogenitor cells (NPCs) differentiate, SETD7 undergoes continuous upregulation, thereby modulating the manifestation of genes specific to a certain lineage and their interaction with different regulatory co-factors throughout cardiomyocytes differentiation.44
Furthermore, SETD7 is also important for sustaining the cardiac-specific gene expression in adult cardiomyocytes.44,74 In adult heart diseases, monocytes from patients with type 2 diabetes exhibit SETD7-dependent epigenetic alterations, which are linked to processes such as endothelial dysfunction and oxidative stress. Inhibiting SETD7 has therefore been shown to alleviate the burden of CVD in diabetes patients, indicating its promise as a therapeutic target.110 In myocardial ischemia-reperfusion injury, SETD7 is believed to play a key role in preserving cardiac-specific gene expression in the adult heart, and its inhibition has been shown to prevent myocardial ischemic injury.111 Additionally, SETD7 is essential in the regulation of target genes implicated in inflammation and immune responses, by attenuating inflammation and the epigenetic regulation of cardiac genes during cardiac differentiation, thereby modulating cellular processes. SETD7 is also important for calcium functions in fully developed cardiomyocytes, where calcium signaling is crucial for muscle contraction and, consequently, for regulating the heartbeat.44
This multifunctional enzyme is essential for cardiomyocytes differentiation, cardiac function, and protecting the heart during myocardial ischemic injury, while also being involved in various cardiovascular diseases and myocardial inflammation in obesity-related heart failure where the heart’s ability to eject blood is preserved.44,110–113 These diverse functions highlight SETD7 as a potential therapeutic target for conditions involving cardiac differentiation, myocardial injury, and cardiovascular diseases. Therefore, SETD7’s role in CVD is multifaceted and its inhibition has shown promise as a treatment target in terms of myocardial ischemia-reperfusion injury, especially in patients with diabetes. Targeting SETD7 could therefore offer novel therapeutic strategies for managing cardiovascular diseases, emphasizing the importance of ongoing research to better understand its mechanisms and potential applications. The functional consequence of SETD7 in Cardiovascular diseases is shown in Table 3.
Kidney Disease
In kidney disease, SETD7 exerts its influence through various mechanisms and functions across a range of targets which encompasses both histone and non-histone protein targets. It not only catalyzes the monomethylation of H3K4, which is linked to transcriptional activation, but it also targets and regulates proteins associated with renal inflammatory responses in fibrosis, such as NF-κB p65, as well as other TF’s like NF-κB, PPARγ, and Nrf2.2,114 SETD7 has a significant impact on the advancement of renal fibrosis, a prominent feature of chronic kidney disease. Its levels are increased in the kidneys of individuals with chronic kidney disease and other fibrotic conditions.115,116 It also functions as a crucial epigenetic regulator, promoting the fibrotic phenotype in the kidney.115 Increased recruitment of SETD7 and the resulting H3K4me1 markers have been associated with the upregulation of profibrotic genes, such as those encoding extracellular matrix proteins and inflammatory mediators.116,117 Additionally, the use of specific inhibitor’s such as PFI-2 and Sinefungin to block SETD7 pharmacologically, has been shown to decrease renal fibrosis in experimental models of kidney damage and illness.115,116,118 This demonstrates that attenuation of renal fibrosis and modulating its activity or expression could potentially mitigate the advancement of chronic kidney disease and the shift from acute kidney injury to chronic kidney disease.115,116,119
The role of SETD7 therefore extends to the promotion of renal fibrosis and the modulation of gene expression and cellular processes specifically related to kidney disease. These include transcriptional activation of genes involved in renal fibrosis, suppression of myofibroblast accumulation, and attenuation of inflammation by reducing pro-inflammatory cytokine and chemokine production in the kidneys, as well as oxidative stress.2,3,115,116 This multifaceted enzyme therefore has a considerable role in the advancement of renal fibrosis, marking it a possible treatment target in chronic kidney disease and related fibrotic conditions. The functional consequence of SETD7 in Kidney diseases is shown in Table 3.
SETD7 in Neurodegenerative Diseases
SETD7 has also emerged as a significant player in neurodegenerative conditions, especially in relation to Parkinson’s disease (PD) and AD, and alcohol-induced neurodegenerative conditions. Its primary function involves the methylation of histones and non-histone proteins, which regulates gene manifestation critical for neuronal health and survival.
Alzheimer’s Disease
Increased SETD7 manifestation has been detected in the parahippocampal cortex of people with dementia compared to those with mild cognitive decline or non-dementia donors.8 This has also been observed in specific brain regions of patients with advanced Braak stages of AD, indicating a relationship between SETD7 levels and disease progression.8,120 SETD7 has been linked with AD through the regulation of the PTM pattern of soluble Tau, a protein linked to the pathology of AD. The abundance of SETD7-induced monomethylation on soluble Tau in AD has been reported by Bichmann et al.8
SETD7 is also believed to have a significant role in mature brain cells. Specifically, SETD7 methylation of tau protein associated with AD at site K132, leads to subsequent methylation at K130, with many of the methylation domains located in the microtubule binding domain of Tau.8,37,121 This methylation of tau is linked to its movement from nerve endings to the cell body and nuclei of neurons, increased phosphorylation, build-up of abnormal tau structures, and the progression of AD.8,121 Studies have shown the interaction of SETD7 with neuroinflammatory pathways, where it upregulates pro-inflammatory cytokines through the stimulation of TF’s like NF-κB, thereby exacerbating neurodegenerative processes.34 Similarly, additional studies have shown that SETD7 enhances NF-κB activity and promotes the synthesis of pro-inflammatory cytokines in response to oxidative stress, linking its function to inflammation that can worsen neuronal damage.3 SETD7’s ability to modify non-histone proteins impacts their stability and degradation, influencing various cellular signaling pathways vital for neuronal function.1,15
Furthermore, targeting H3K4 methylation using small molecule inhibitors has shown promise in restoring synaptic functionality and memory in AD murine models, indicating it could be a viable therapeutic strategy for treating AD.37,122,123 The inhibition of SETD7 has also been shown to alleviate cognitive dysfunction via the suppression of NOD-like receptor protein 3 (NLRP3) inflammasome activation in isoflurane-induced aged mice.105 While SETD7 expression is only modestly increased in AD, its role in Tau methylation and aggregation could be significant and highlights its prospective use in developing AD treatments.8,121 Based on reported evidence, the exact role of SETD7 in AD necessitates further studies to elucidate its impact on the onset and advancement of AD. Research into SETD7-specific inhibition could advance precision treatments for AD. The functional consequence of SETD7 in Alzheimer’s disease is shown in Table 3.
Parkinson’s Disease
PD is a significant neurodegenerative disorder primarily affecting the motor system, characterized by tremors, rigidity, and coordination issues. Unlike AD, PD is not strictly age-dependent, as it has the potential to impact people of all ages, regardless of their age group, however it is more frequently identified in elderly persons. A defining characteristic of PD is the gradual loss of dopamine-producing neurons in the brain, resulting in the distinctive motor symptoms.124 SETD7 could be involved in the development of PD via its functions in histone methylation and regulation of key proteins associated with neurodegeneration. Its activity influences gene expression through methylation, which is crucial in neurodegenerative processes in PD.1,125,126 SETD7 is also thought to interact with pathways that mediate neuroinflammation, a significant aspect of PD pathology.3
Increased expression of SETD7 has been detected in certain brain regions of individuals with neurodegenerative disorders, indicating its potential as a biomarker and potential therapeutic target.8,123 Chronic neuroinflammation can exacerbate neurodegenerative processes and SETD7’s ability to modulate inflammatory responses may be crucial in the progression of the disease.105,127 SETD7 has been shown to modulate oxidative stress via upregulation of pro-inflammatory cytokines resulting from the stimulation of TF’ssuch as NF-κB, thus linking its activity to inflammatory responses that can worsen neuronal damage.3 The ability of SETD7 to modify non-histone proteins also implies that it may impact the stability and degradation of proteins critical to neuronal function, including those involved in cellular signaling pathways.1,2,15 When exposed to inflammatory cytokines such as TNF-α, the JAK-STAT signaling pathway enhances the upregulation of SETD7, leading to increased expression and activity of this enzyme.
This leads to increased neuroinflammation and apoptosis of dopaminergic neurons, further contributing to the neurodegenerative process. Researchers have indicated that targeting SETD7 may enhance cellular resilience against oxidative stress and damage caused by energy deprivation mitigating the neurodegenerative effects of PD, thus highlighting its potential as a therapeutic target.2,128–130 Although specific studies on SETD7 in PD are limited, the parallels in neurodegenerative processes raise important questions about its involvement in PD and its potential role in developing novel therapeutic strategies.
Alcohol Induced Neurodegeneration
In addition to being implicated in various neurodegenerative processes, SETD7 has also been associated with alcohol-induced neurodegeneration. It has been shown that Alcohol use disorder can lead to significant neurotoxicity and is linked to cognitive decline and neurodegenerative conditions such as cerebellar degeneration, alcohol addiction, fetal alcohol syndrome/fetal alcohol spectrum disorders, and alcoholic dementia.126 Although extensive studies have been conducted, the underlying molecular mechanisms of neuronal diseases caused by alcohol consumption are still not well elucidated. Extensive research involving human participants and animal models of alcohol exposure over the last few decades has identified significant epigenetic changes, particularly in histone acetylation, histone methylation, and DNA modifications, which are crucial for regulating gene expression in alcohol use disorders.126
Understanding the role of SETD7 as a methyltransferase in this context is therefore crucial, as it may influence the molecular mechanisms underlying alcohol-related brain damage. By modulating the methylation of specific histones and non-histone proteins,126 SETD7 could influence the manifestation of genes implicated in neuroinflammation and neuronal death, implying a significant role for SETD7 in the advancement of alcohol-induced neurodegeneration.131 Studies has demonstrated that alcohol exposure during early development can cause neuronal damage at various phases of cerebral development, resulting in neuronal dysfunction in adulthood. Alongside modifications in histone acetylation and DNA methylation, alterations in histone methylation have also been observed in numerous investigations on the effects of alcohol exposure on development126 For instance, GD7 acute ethanol exposure has been demonstrated to increase H3K9me2, decrease H3K27me3 and alter Kdm4c,G9a, Kdm1a, Setdb1, Uhrf1 and Ezh2 mRNA levels.131 It also leads to decreased expression of the Pomc gene, while lower concentrations of β-endorphin protein are observed in the arcuate nucleus of the hypothalamus in male offspring. This is associated with diminished staining of the histone modifications H3K4me2 and H3K4me3, as well as increased H3K9me2 in this brain region. In addition to the changes in histone modifications, altered expression of three genes encoding histone methyltransferase (HMT) family enzymes (Setd7, Setdb1, and G9a) has also been observed in the offspring prenatally exposed to alcohol.107 These epigenetic changes have ultimately been linked with impaired hypothalamic function,such as dysregulation of the stress response axis.107,108
Given its involvement in the molecular pathways affected by alcohol, SETD7 shows great potential as a target for therapeutic intervention for mitigating the adverse health impacts of alcohol abuse globally. The dysregulation of SETD7 in response to alcohol exposure highlights its potential role in the epigenetic modifications that contribute to neurodegeneration. By influencing histone methylation and other critical cellular processes, SETD7 may help regulate gene expression associated with neuroinflammatory responses and neuronal survival. Therefore, targeting SETD7 could offer new strategies for preventing or reversing alcohol-induced neurodegeneration, potentially enhancing cognitive abilities and promoting general brain well-being in individuals with alcohol use disorders. Furthermore, as research continues to unravel the complexities of SETD7’s role in neurobiology, it may pave a path for novel therapeutic approaches that not only address the neurological consequences of alcohol abuse, but also address the broader implications for mental health and addiction recovery. Thus, SETD7 represents a significant focal point in the quest to combat this global health crisis posed by alcohol-related neurodegenerative diseases.
Inflammatory and Immune-Related Conditions
Inflammatory Conditions
SETD7-mediated H3K4 monomethylation facilitates transcriptional stimulation of target genes associated with inflammation and immune responses. It may also target and regulate other proteins involved in inflammatory pathways, potentially worsening tissue damage caused by oxidative stress in different inflammatory diseases.1–3 SETD7 interacts with and regulates the TF’s including NF-κB, PPARγ, and Nrf2, which are key regulators of inflammatory responses, as shown in Table 3.2,3 It also targets the RELA/p65 promoter region modulating NF-κB expression through monomethylation of H3K4 which regulate the production of pro-inflammatory cytokines and chemokines.3
Therefore, by suppressing the production of pro-inflammatory signaling molecules, SETD7 inhibition can effectively attenuate inflammatory responses.2,3,102 Additionally, this inhibition can then suppress oxidative stress and promote antioxidant responses through the regulation of Nrf2 and its target genes.3,102 SETD7’s involvement in inflammatory diseases highlights its broader impact on immune responses and inflammatory pathways. By modulating nonhistone proteins through lysine methylation, SETD7 may contribute to the regulation of inflammatory signaling pathways, immune cell function, and the pathogenesis of inflammatory conditions. More investigation is crucial to define the specific pathways through which SETD7 influences inflammation and to explore its therapeutic potential in managing inflammatory diseases.
Rheumatoid Arthritis
SETD7 significantly contributes to the onset and progression of rheumatoid arthritis (RA) by affecting various pathways that influence inflammatory processes and joint destruction. SETD7 is involved in the modulation of pro-inflammatory cytokines, which are crucial in the pathophysiology of RA. By methylating non-histone proteins, SETD7 can alter the stability and activity of TF’s, such as NF-κB, which is pivotal for the manifestation of cytokines like IL-6 and TNF-α, thus contributing to the chronic inflammation characteristics of RA.132–134 SETD7 has been shown to influence immune responses through modulation of TF’s involved in T cell signaling, activation and differentiation, which potentially exacerbates joint inflammation.
In addition, SETD7 through its methylation activity has been shown to interact with various signaling pathways implicated in RA, including the JAK/STAT pathway, which affects cellular responses to cytokines and growth factors that drive inflammation and tissue remodeling in the joints. As such, SETD7 has been implicated in processes such as angiogenesis and chondrocyte apoptosis, both critical for the progression of RA.2,84,135 Its expression in chondrocytes is also upregulated by inflammatory cytokines like TNF-α, facilitated through JAK-STAT signaling pathway. This upregulation enhances the synthesis of chemokines and VEGF, thereby facilitating angiogenesis. Additionally, it inhibits the nuclear localization of HIF-1α, a crucial regulator of hypoxic responses, which mediates chondrocyte apoptosis and contributes to the inflammatory environment in RA.136 Silencing SETD7 in the chondrogenic cell line ATDC5 suppresses the Hippo signaling pathway, leading to reduced YAP phosphorylation and elevated nuclear accumulation of YAP and HIF-1α, which promoted the expression of genes involved in chondrogenic differentiation.6 Dysregulation of genes involved in immune tolerance can lead to inappropriate immune responses against self-antigens, further perpetuating the cycle of inflammation and joint destruction.136–138
The involvement of SETD7 in RA has significant implications for understanding disease progression and developing therapeutic strategies. SETD7’s role in modulating pro-inflammatory cytokines and influencing immune responses suggests that targeting this enzyme could reduce chronic inflammation and joint destruction associated with RA. Its interaction with key signaling pathways, such as JAK/STAT, highlights the potential for SETD7 inhibition to mitigate angiogenesis and chondrocyte apoptosis, which are critical for disease progression. Furthermore, SETD7’s impact on immune tolerance and its response to inflammatory cytokines underscores the need for further research into its role in perpetuating autoimmune responses in RA. Overall, SETD7 emerges as a promising therapeutic target for managing RA by addressing both inflammatory and immune dysregulation aspects of the disease.
Asthma
SETD7 has been implicated in the pathogenesis of asthma through several functional mechanisms that influence inflammatory responses and contribute to airway remodeling. Scientist’s has demonstrated that SETD7 regulates gene expression related to airway smooth muscle cell proliferation and contraction.7 SETD7 methylation activity on specific TF’s, such as TNF-α, promote the expression of contractile proteins and growth factors that exacerbate airway remodeling, resulting in increased airway resistance due to hyperplasia and hypertrophy of ASM cells.7,104
At a cell level, SETD7 affects various immune cell types involved in asthma, including eosinophils, mast cells, and T-helper cells. By modifying key TF’s in these cells, SETD7 influences their activation, survival, and cytokine production. As a result, SETD7 regulates the levels of pro-inflammatory cytokines such as IL-4, IL-5, and IL-13, which are central to the pathophysiology of asthma.7,104,139,140 By methylating TF’s involved in Th2 cell differentiation and activation, SETD7 may also boost the production of these cytokines, thereby increasing inflammation and airway hyper-responsiveness.7,104,141 SETD7 also plays a crucial role in regulating inflammation by enhancing inflammatory signaling pathways, notably through the stimulation of NF-κB and the upregulation of CD38 expression.3,7,104,113
Similarly, SETD7’s role in oxidative stress responses in asthma has also been reported, with SETD7 being shown to interact with pathways that regulate ROS and oxidative stress responses. The inhibition of SETD7 has been revealed to counteract NF-κB-induced oxidative stress and the generation of pro-inflammatory cytokines in macrophages and bronchial epithelial cells.3 By modulating the manifestation of antioxidant genes through histone methylation, SETD7 may impact the oxidative environment in asthmatic airways, influencing inflammation and tissue damage.3,142
In contrast, SETD7 has also been demonstrated to regulate epithelial cell function in the airways, where it affects the expression of genes essential for sustaining epithelial integrity and barrier effectiveness. Therefore, dysregulation of SETD7 may lead to impaired epithelial barrier function, facilitating allergen penetration and promotion of an asthmatic response.3,7,106
SETD7’s involvement in asthma pathogenesis therefore has significant implications for understanding disease mechanisms and developing therapeutic strategies. By modulating inflammatory responses and contributing to airway remodeling, SETD7 plays a crucial role in asthma progression. Targeting SETD7 could offer a novel approach to managing asthma by reducing inflammation, airway remodeling, and oxidative stress, highlighting its potential as a therapeutic target for mitigating asthma severity and improving patient outcomes.
SETD7 Inhibitors and Their Therapeutic Potential
The therapeutic potential of SETD7 is multifaceted and diverse, with significant implications for treating various diseases and disorders. As a key epigenetic regulator, SETD7 influences gene expression and chromatin architecture, extending its impact beyond histone modification to the methylation of non-histone proteins, which can profoundly affect cellular processes, underscoring the importance of SETD7 in disease pathophysiology.1,2,143 To harness this potential, researchers have focused on developing potent and specific SETD7 inhibitors. These efforts aim to provide effective treatments for conditions where SETD7 upregulation plays a critical role, offering new avenues for managing diseases where SETD7 dysregulation is implicated.10 Despite these efforts, the current repertoire of SETD7 specific compounds remains limited, with the majority demonstrating weak inhibitory activity.144
Early SETD7 Inhibitors
Early SETD7 inhibitor development focused on structural analogs of the cofactor S-adenosylmethionine (SAM). Researchers designed derivatives by replacing sulfur with nitrogen and adding alkylamino chains to mimic lysine side chains, leading to compounds like DAAM-3, which showed about 50% inhibition of SETD7 at 10 μM.144,145 Structural studies revealed that DAAM-3 binds the cofactor site and extends its n-hexyl group into the lysine-binding pocket.38,144
(R)-PFI-2
Subsequent advances yielded more potent inhibitors, such as (R)-PFI-2, a first-in-class inhibitor. Unlike earlier SAM-based designs, (R)-PFI-2 occupies the substrate-binding groove and interacts directly with SAM’s methyl group, enabling substrate-competitive inhibition.10,146 This mechanistic distinction highlights the evolution of SETD7-targeted drug design. (R)-PFI-2 is the leading and most commonly employed SETD7 inhibitor to date and analogues as substrates and inhibitors. This compound was identified through a high-throughput screening of approximately 150,000 molecules from Pfizer’s chemical library.9,10 This compound has demonstrated remarkable efficacy with an IC50 of 2.0nM, and demonstrates over 1000-fold selectivity compared to a set of 18 other methyltransferases and DNMT1.144 The molecular architecture of SETD7’s catalytic region is complex, and (R)-PFI-2 has revealed an unusual mode of action, characterized by a substrate-competitive and SAM-dependent binding model. Molecular dynamics modeling has elucidated the differing effectiveness of (R)-PFI-2 and its enantiomer by revealing distinct bioactive activation thresholds, thereby providing valuable insights for future drug development that consider structural nuances and the impact of chirality on SETD7 inhibition.144,147
The cellular activity of (R)-PFI-2 has underscored the impact of SETD7-mediated methylation on YAP’s cellular distribution and transcriptional activity.10 Exposure of densely cultured MCF7 cells to increasing doses of (R)-PFI-2 led to a commensurate increase in YAP accumulation within the nucleus. This nuclear enrichment of YAP coincided with elevated expression of its downstream targets, including AREG and CYR61. These results suggest that continuous SETD7 functioning is crucial for maintaining proper YAP distribution within this particular cell line. This compound is an extremely specific and powerful inhibitor of SETD7, possessing an inhibition constant (Ki) of 0.33nM.10,144 It has been used extensively in research to explore the biological roles of SETD7 in cellular processes and disease mechanisms. While primarily a research tool, its high specificity makes it a potential candidate for therapeutic applications, pending further clinical studies.
SETD7 also suppresses osteoclastogenesis by enhancing the activation and nuclear localization of Transcriptional co-activator with PDZ-binding motif (TAZ), a transcriptional coactivator that inhibits NF-κB signaling. This inhibition reduces osteoclast formation in vitro and prevents bone loss in vivo, highlighting (R)-PFI-2’s potential as a therapeutic agent for osteoporosis.148 Similarly, the (S)-PFI-2 enantiomer has been demonstrated to inhibit SETD7, albeit with a significantly reduced potency, exhibiting an IC50 of approximately 1.0 μM. Notably, (R)-PFI-2 is substantially more potent than its (S)-enantiomer, which is about 500-fold less active. This stark difference in inhibitory efficacy underscores the importance of chirality in SETD7 inhibition, as (R)-PFI-2’s superior potency is attributed to its unique interaction with the SETD7 active site, involving direct contact with the cofactor S-adenosylmethionine and a substrate-competitive mechanism.10,144,147,149
Amustaline
Beyond the (R)-PFI-2 hydrochloride-based inhibitors, Amustaline, a small molecule drug, is currently under evaluation in Phase 3 trials for its potential in preventing graft-versus-host disease (GVHD) in recipients of hematopoietic stem cell transplants. This compound selectively targets activated T cells, which are the primary drivers of GVHD, thereby showcasing its therapeutic promise. Amustaline’s ability to efficiently target and interact with intracellular components makes it an attractive candidate for managing GVHD, a condition characterized by severe immune responses where donor cells attack the recipient’s tissues, leading to significant morbidity and mortality in transplant patients.150,151
Cyproheptadine as a SETD7 Inhibitor
Cyproheptadine (R=-OH), a known antihistamine, has emerged as a SETD7 inhibitor (IC50 = 1 μM) through high-throughput screening.144,152–154 Additional screening utilizing a fluorogenic substrate-based histone methyltransferase assay also identified Cyproheptadine, as a strong inhibitor of SETD7.152,153 Crystallography revealed its binding to the substrate pocket, inducing conformational changes in SETD7’s C-terminal region and lysine-access pathway.144,152
While less potent than (R)-PFI-2, Cyproheptadine competitively blocks substrate methylation and demonstrates therapeutic potential in ERα-positive BC models.152 It promotes ERα degradation via proteasomal pathways,144,152 suppresses estrogen-driven proliferation in MCF7 cells,152,153,155 and reduces blood urea nitrogen (BUN) levels in diabetic models.156,157 These findings also accentuate SETD7’s pivotal role in orchestrating active transcription through H3K4 demethylation in the regulation of NF-kB-mediated inflammatory response,2,3,12,105,158 and positions SETD7 as a promising therapeutic target against ischemia-reperfusion injury (IRI) in both diabetic and non-diabetic contexts.156,157 The pharmacological properties of Cyproheptadine therefore extend beyond its antihistamine action, providing a strong basis for the enhancement and creation of effective SETD7 inhibitors based on its structure.153,155
Cyproheptadine also exhibits cytotoxic effects on lung cancer cells and reduces their invasion and migration. In vivo, SETD7 knockout mice show fewer lung metastatic nodules, and cyproheptadine treatment further suppresses tumor growth. Metabolomic analysis indicates that both SETD7 deletion and cyproheptadine treatment alter tumor metabolism, particularly reducing amino acids like arginine, serine, and glycine. These findings suggest that SETD7 is a promising target for preventing lung metastasis and that cyproheptadine may act, at least partly, through pathways involving SETD7 to inhibit NSCLC progression.154
DC21
The identification of DC21 as a SETD7 inhibitor, with an IC50 of 15.93 μM, was facilitated through a combination of virtual screening and biochemical analysis of an in-house library derived from the Specs ligand database.159,160 Notably, DC21 inhibits the proliferation of MCF7 breast cancer cells with an IC50 of 25.84 μM at the cellular level, highlighting its potential as a guiding compound for developing more potent SETD7-targeting therapeutic agents in various diseases, including cancer.29,159 As a chemical probe, DC21 plays a crucial role in elucidating the physiological functions of SETD7, providing valuable insights into the design of tumor-targeting therapies.29 Additionally, DC21 exhibits favorable selectivity towards several other epigenetic targets, such as G9a, NSD1, SUV39H1, MOF, and DOT1L, underscoring its utility as a molecular tool for investigating SETD7’s biological roles and enhancing the efficacy of SETD7 inhibitors.29,159
5′-Deoxy-5′-Methylthioadenosine
5′-Deoxy-5′-methylthioadenosine (MTA) has been identified as a SETD7 inhibitor with the capacity to modulate inflammatory pathways, offering potential therapeutic benefits in both viral infections and cancer. Notably, MTA inhibits Hepatitis C virus (HCV) replication by targeting SETD7, which is implicated in promoting viral replication through the suppression of interferon signaling pathways.64,70,161 In the context of cancer, MTA has been shown to mitigate tumor growth and invasiveness in hepatocellular carcinoma (HCC) and BC by inhibiting SETD7, leading to reduced methylation of key proteins involved in cell proliferation and metastasis.62,64,70 Additionally, MTA enhances the efficacy of cancer therapies, such as chemotherapy, by increasing cancer cells’ sensitivity to DNA-damaging agents.64,161 As a SETD7 inhibitor, MTA highlights its therapeutic potential across multiple diseases, particularly in cancer and viral infections, where its ability to modulate epigenetic regulation and immune responses could provide novel treatment strategies.
Sinefungin
Sinefungin, a fungicidal agent, has been identified as one of the first potent and broadly effective inhibitors of SETD7, competing with S-adenosylmethionine (SAM) and exhibiting an IC50 of 2.5 μM.45,162 Notably, sinefungin has been shown to reduce the expression of mesenchymal markers and extracellular matrix proteins by inhibiting H3K4me1 in the kidneys of mice with unilateral ureteral obstruction (UUO), a model of renal fibrosis.45 Furthermore, sinefungin suppresses TGF-β1-induced smooth muscle actin expression and H3K4me1 in both NRK-52E and NRK-49F cell lines, thereby decreasing renal fibrosis.45,118 The modification of sinefungin by adding an n-propyl group resulted in N-propyl sinefungin, which demonstrated enhanced inhibitory activity with an IC50 of 2.2 μM.45,163 This structural optimization highlights the potential for improving sinefungin’s efficacy as a SETD7 inhibitor, offering insights into the development of therapeutic agents targeting fibrotic diseases.
Other Promising SETD7 Inhibitors
Efforts to optimize Cyproheptadine’s structure by introducing chemical substituents on its dibenzocycloheptene component led to the discovery of a derivative with a 2-hydroxy group which exhibited enhanced potency with an IC50 of 0.41 μM.103,139,153 Another promising compound, DC-S100, was identified using virtual screening based on pharmacophore and docking techniques. Following further refinement, modified N-phenyl benzamide compounds, DC-S238 and DC-S239, demonstrated strong and specific suppression of SETD7, with IC50 values of 4.88 μM and 4.59 μM, respectively,153,160,164 with both compounds also displaying selectivity for DNMT1, DOT1L, EZH2, NSD1, SETD8, and G9a.160 Notably, DC-S239 showed proliferation-suppressing effects on BC MCF7 and leukemia cell lines (HL60, MV4-11), while having minimal impact on human colon cancer (HCT116) and lymphoma (DHL4) cells, indicating its stronger activity against BC and leukemia.144 Further exploration using scaffold changes and 2D fingerprint comparisons led to the discovery of DC-S303 (IC50 = 1.1 μM), while Morin (IC50 = 6.02 μM) and scutellarein (IC50 = 1.96 μM) were identified as potential SETD7 inhibitors through virtual screening of ligand databases.45,165
Additionally, leveraging structural insights from SAM, researchers designed SETin-1 using a pharmacophore-based strategy that incorporates “privileged scaffolds”. This approach yielded a competitive SETD7 inhibitor with moderate potency (IC50 = 10 μM), as validated by ELISA-based enzymatic assays.45,163,165 While less potent than state-of-the-art inhibitors like (R)-PFI-2 (IC50 = 0.33 μM), SETin-1 provides a valuable proof-of-concept for SAM-mimetic design and highlights the potential of scaffold-based optimization to refine selectivity and efficacy. Its discovery underscores the importance of integrating cofactor-binding patterns and privileged pharmacophores in targeting epigenetic enzymes like SETD7, which are implicated in diseases ranging from cancer to inflammatory disorders.
Despite the limited number of selective compounds, extensive research has focused on developing potent SETD7 inhibitors. Notable advancements include Cyproheptadine, which shows promise in treating ERα-associated diseases like breast cancer, and (R)-PFI-2, an extremely specific and powerful inhibitor. Moreover, Amustaline, currently in phase 3 trials, holds potential for preventing graft-versus-host disease. These findings highlight the need for continued exploration and optimization of SETD7 inhibitors for therapeutic applications in diseases such as cancer and transplantation medicine, offering opportunities for impactful interventions in the future. List of some of the important SETD7 Inhibitors with therapeutic potential is shown in Table 4
 |
Table 4 SETD7 Inhibitors and Their Therapeutic Potential |
Challenges in Developing Specific and Potent SETD7 Inhibitors
The development of highly specific and potent inhibitors for SETD7 faces several significant challenges that stem from both the structural characteristics of the enzyme and its complex biological functions. SETD7 targets a range of substrates, including histone and non-histone proteins, through a highly specialized catalytic pocket designed to accommodate natural lysine residues and the cofactor SAM.1,9,10 Designing inhibitors that effectively compete with these natural substrates requires molecules that fit precisely into this catalytic site. Even minor structural changes in potential inhibitors can cause steric hindrance or reduce binding affinity, making it difficult to develop compounds that are both effective and selective.9 Currently, only a limited number of small molecules, such as (R)-PFI-2, have been identified as potent and selective SETD7 inhibitors.2,9,10 While (R)-PFI-2 demonstrates strong binding affinity and specificity by targeting the histone lysine binding site in a SAM-dependent manner, creating analogs that maintain or improve upon these properties while enhancing drug-like characteristics remains a complex task.10 Achieving a balance between specificity and potency is particularly challenging because SETD7 shares conserved structural features with other methyltransferases, increasing the risk that inhibitors might affect related enzymes and cause off-target effects. Furthermore, SETD7’s diverse and context-dependent roles in cellular pathways, acting as either a tumor suppressor or promoter depending on the biological environment adds another layer of complexity.2,54 This makes it difficult to predict and control the therapeutic outcomes of SETD7 inhibition without unintended consequences. The limited pharmacological data and lack of clinical applications for existing inhibitors highlight the need for more comprehensive research focused on detailed structural analyses and high-throughput screening of novel compounds. Such efforts are essential to overcome these obstacles and pave the way for the development of highly specific, potent, and safe SETD7 inhibitors that can be effectively used in therapeutic settings.
SETD7’s Potential in Combination Therapies
SETD7 serves as a central node in the regulation of diverse processes with critical implications for cell fate decisions, tissue development, and disease progression. Its dual capacity to act as both a tumor suppressor and promoter, depending on context and substrate, has sparked interest in its therapeutic targeting especially as part of combination therapies. Its broad influence on proliferation, differentiation, apoptosis, and immune responses makes it a compelling target for combination therapies aimed at enhancing treatment efficacy and overcoming resistance.1,39,41,44,100
Preclinical Evidence for Synergy
SETD7’s multifaceted roles in cancer biology position it as a promising target for combination therapies that exploit its regulatory influence on cell cycle progression, hormone receptor stability, oxidative stress response, and multiple signaling pathways.3,50,92,168,169 Recent studies demonstrate a synergistic anti-cancer effect when SETD7 inhibition is combined with CDK4 inhibitors, particularly in osteosarcoma. Osteosarcoma, a malignant bone tumor with limited therapeutic options, shows overexpression of CDK4, which correlates with poor prognosis. SETD7 promotes osteosarcoma cell proliferation by regulating the CDK4-cyclin D1 complex, essential for cell cycle progression. Inhibition or genetic ablation of SETD7 disrupts this complex, causing G1 cell cycle arrest and decreased phosphorylation of retinoblastoma protein (pRb), thereby suppressing tumor growth.4,54,170,171
The combination of SETD7 and CDK4 inhibitors not only enhances tumor suppression but also allows dose reductions of both agents, minimizing toxicity while maintaining efficacy. This approach offers a novel strategy to overcome resistance and improve treatment outcomes in osteosarcoma. In vivo studies confirmed that combined SETD7 and CDK4 inhibition significantly reduced tumor growth without notable adverse effects, highlighting the therapeutic potential of this combination.170
In hormone-driven cancers, SETD7 plays a pivotal role in modulating hormone receptor function in hormone-driven cancers such as BCa and PCa. In BCa, SETD7 methylates ERα, stabilizing the receptor and boosting its transcriptional activity, thereby contributing to tumor progression. This methylation also affects ERα’s stability, subcellular localization, and interactions with other proteins mechanisms that may underlie resistance to endocrine therapies.48,152
Tamoxifen is the most commonly used endocrine therapy for advanced or metastatic ER-positive BCa, particularly in premenopausal women, where it inhibits tumor growth by targeting ERα. However, many patients eventually develop resistance to tamoxifen. Despite this, ERα continues to drive tumor progression in tamoxifen-resistant BCa, confirming it remains a critical therapeutic target. Given SETD7’s role in stabilizing ERα through methylation, targeting SETD7-mediated modifications may offer a novel strategy to overcome tamoxifen resistance and enhance treatment efficacy.46,47
In PCa, SETD7 expression is elevated in androgen-dependent cells but decreases in castration-resistant prostate cancer (CRPC). Loss of SETD7 promotes tumor growth, metastasis, and reduces sensitivity to enzalutamide, a key anti-androgen therapy. Silencing SETD7 in FOXA1-positive PCa cell lines increases proliferation and migration under hormone-depleted conditions mimicking androgen deprivation therapy and desensitizes cells to enzalutamide. These findings suggest that SETD7 supports responsiveness to enzalutamide, and its loss contributes to treatment resistance.41 Therefore combining SETD7 inhibitors with anti-estrogen therapies or anti-androgen therapies may more effectively disrupt hormone receptor stability than hormone therapy alone, potentially overcoming endocrine resistance and improving clinical outcomes.
SETD7 plays a critical role in maintaining cellular redox homeostasis by modulating the KEAP1-NRF2 antioxidant pathway. NRF2 drives the expression of antioxidant enzymes that protect cells from oxidative stress, and SETD7 acts as a positive regulator by promoting transcription of these genes, thereby helping cancer cells manage ROS and survive under oxidative conditions.49 In breast cancer models, SETD7 knockdown reduces antioxidant enzyme expression, disrupts the glutathione redox balance (GSH/GSSG), and elevates ROS levels, leading to increased apoptosis.49 Similarly, downregulation of SETD7 enhances the sensitivity of HCC to Sorafenib, a ROS-inducing kinase inhibitor.172–174 Additionally, SETD7 regulates PARP1 expression and influences the response to cisplatin in breast cancer cells. Notably, the PARP1 inhibitor niraparib can overcome cisplatin resistance in triple-negative breast cancer (TNBC) cells following SETD7 knockdown.175
Furthermore, combining SETD7 inhibitors with therapies that induce oxidative stress, such as chemotherapy, radiotherapy, or targeted agents that elevate ROS, may sensitize tumors by overwhelming their antioxidant defenses. This approach exploits cancer cells’ dependence on SETD7-mediated antioxidant responses and holds promise for improving therapeutic outcomes.3,49,54
Therapeutic Implications Beyond Cancer
While much of the research on SETD7 has focused on its roles in cancer biology, emerging evidence highlights its significant potential as a therapeutic target in a range of non-cancer diseases, particularly metabolic, inflammatory, and cardiovascular disorders.
Metabolic Disorders and Diabetes
SETD7 is implicated in the regulation of glucose homeostasis and pancreatic β-cell function. In diabetes, SETD7-mediated methylation influences key transcription factors such as Pdx1, which is essential for insulin secretion and pancreatic development. Pharmacological inhibition of SETD7 has shown promise in restoring vascular function and reducing inflammation in diabetic models, suggesting a role in mitigating diabetic complications such as endothelial dysfunction and atherosclerosis. Moreover, SETD7 inhibitors like (R)-PFI-2 have demonstrated efficacy in promoting neovascularization and improving tissue perfusion in diabetic mice, indicating potential for therapeutic use in diabetic vascular disease.1,98–100
Inflammatory and Immune-Related Conditions
SETD7 modulates inflammatory signaling pathways, including NF-κB and STAT3, which are central to chronic inflammation and immune dysfunction. By regulating these pathways, SETD7 contributes to persistent inflammation seen in metabolic syndrome and cardiovascular diseases. Targeting SETD7 may therefore help alleviate chronic inflammatory states and improve immune homeostasis.3
Cardiovascular Disease
In heart failure, particularly heart failure with preserved ejection fraction (HFpEF), SETD7 expression is elevated in immune cells and closely associated with increased pro-inflammatory gene expression. This suggests that SETD7 contributes to cardiac remodeling and inflammation, positioning it as a potential biomarker and therapeutic target in cardiovascular disease management.176 SETD7 also plays a critical role in ischemic stroke by epigenetically repressing Sirt5, a protective deacetylase. It does so by facilitating the recruitment of chromobox homolog 1 (Cbx1) to repressive histone marks (H3K9me2/3) at the Sirt5 promoter, leading to reduced Sirt5 expression. This repression results in elevated glutaminase activity, glutamate accumulation, and excitotoxic neuronal damage. Inhibition of SETD7 restores Sirt5 levels, decreases neurotoxic metabolites, and alleviates ischemic brain injury.177
Therefore, combining SETD7 inhibitors with agents that protect neuronal function or modulate excitotoxic pathways holds promise for synergistic neuroprotection. Such combination therapies could simultaneously reverse epigenetic repression and reduce glutamate-induced excitotoxicity, ultimately improving outcomes for patients suffering from ischemic stroke.
Neurodegenerative and Ischemic Conditions
SETD7 contributes to neuronal damage and inflammation in cerebral ischemia-reperfusion injury and ischemic stroke by epigenetically repressing protective genes and promoting excitotoxicity. Strategies that downregulate SETD7, such as delivery of microRNA miR-330-5p via mesenchymal stem cell-derived exosomes, have shown neuroprotective effects in preclinical models, highlighting its therapeutic potential in neurodegenerative diseases and stroke.178
Other Disease Contexts
SETD7 is also involved in osteoarthritis, were it is overexpressed in osteoarthritic cartilage, promoting chondrocyte apoptosis, inflammation, oxidative stress, and autophagy suppression. Upregulated via the JAK-STAT5 pathway and stabilized by YTHDF2, SETD7 inhibits chondrocyte differentiation and glycolysis through Hippo signaling. Its inhibition restores autophagy, reduces damage, and improves cell viability, making SETD7 and the YTHDF2/SETD7 axis promising targets for combined osteoarthritis therapies.179
SETD7 also contributes to noise-induced hearing loss (NIHL) by promoting cochlear inflammation through activation of the NF-κB pathway and NLRP3 inflammasome. The selective SETD7 inhibitor (R)-PFI-2 hydrochloride effectively suppresses NF-κB activation, reduces proinflammatory factor release, and prevents inflammasome assembly, thereby protecting cochlear hair cells from acoustic trauma.180
Inhibitors of SETD7 can reduce tissue damage and inflammation in these contexts, suggesting broader applications in degenerative and inflammatory diseases. Continued research and development of selective SETD7 inhibitors may provide novel treatment avenues across these varied disease states.
Key Considerations for Clinical Translation
The promising preclinical data on SETD7 inhibition across various disease models highlight several important factors to consider for successful clinical translation:
Selective and Potent Inhibitors
Developing highly selective small-molecule inhibitors of SETD7, such as (R)-PFI-2, is critical to minimize off-target effects and maximize therapeutic benefit. These inhibitors have demonstrated efficacy in vitro and in vivo by modulating SETD7’s methyltransferase activity, impacting cellular proliferation, oxidative stress, and inflammation.10,146 Continued optimization of pharmacokinetics and specificity is essential for clinical application.
Disease Context and Target Validation
SETD7 exhibits context-dependent roles, acting as either an oncogene or tumor suppressor depending on tissue type and disease state. For example, it promotes proliferation in clear cell renal carcinoma but suppresses pathological cardiac hypertrophy1,4,87 Thorough validation of SETD7’s role in specific diseases and patient populations is necessary to identify where inhibition will be beneficial versus detrimental.
Biomarker Development
Reliable biomarkers for SETD7 expression and activity will aid patient stratification and monitoring of therapeutic response. Nuclear localization of SETD7 and its expression levels correlate with disease progression in cancers such as prostate and colorectal cancer, suggesting potential as diagnostic or prognostic markers.88,89
Combination Therapy Potential
Given SETD7’s involvement in multiple pathways, cell cycle regulation, hormone receptor stability, oxidative stress response, and inflammation, combining SETD7 inhibitors with existing therapies (eg, CDK4/6 inhibitors, hormone therapies, chemotherapy) may enhance efficacy and overcome resistance. Preclinical studies outlined support synergistic effects and reduced toxicity with combination regimens.1,3,98–100
Safety and off-Target Effects
SETD7 methylates both histone and numerous non-histone proteins involved in essential cellular functions, raising concerns about unintended consequences of systemic inhibition, as it could disrupt multiple pathways involved in both normal cellular functions and disease progression.1,2,91 Careful dose optimization and monitoring for adverse effects on normal tissue homeostasis, stem cell function, and immune responses are required.
Translational Models and Clinical Trials
Robust preclinical models, including genetically engineered mice and patient-derived xenografts, have demonstrated therapeutic potential but also underscore complexity in SETD7 biology.174,181 Early-phase clinical trials will need to carefully evaluate safety, pharmacodynamics, and efficacy, with adaptive designs to address heterogeneity in patient responses.
Expanding Therapeutic Indications
Beyond cancer, SETD7 inhibitors show promise in metabolic diseases, cardiovascular disorders, and inflammatory conditions.182 Translational efforts should explore these indications, leveraging SETD7’s role in regulating oxidative stress, inflammation, and tissue remodeling.
Future Directions and Perspectives
SETD7 holds substantial potential as both a diagnostic marker and therapeutic target across a range of diseases, given its involvement in diverse cellular processes such as cell cycle regulation and gene expression. Its utility as a diagnostic tool is particularly evident in conditions like colorectal cancer, where it has demonstrated high sensitivity and specificity as a serum biomarker.71 Further research is essential to validate SETD7 expression levels as reliable biomarkers, especially in exploring its potential as a subtype-specific marker in cancers such as basal-like breast cancer, where high expression correlates with poor outcomes.48,49 Additionally, investigating SETD7 mutations or alterations in its target proteins, as well as developing epigenetic profiling techniques to assess SETD7-mediated methylation patterns, could enhance disease diagnosis and monitoring.1,2
Its role in cancer progression and immune escape, notably in bladder cancer, underscores its value as a therapeutic target. Therefore, a key focus should be on developing specific small molecule inhibitors targeting SETD7’s methyltransferase activity, which could be applied in conditions where SETD7 is overexpressed, such as in various cancers where it promotes tumor growth.62 Evaluating the potential of combining SETD7 inhibitors with other cancer treatments, like chemotherapy or targeted therapies, may enhance treatment efficacy, particularly in therapy-resistant cancers. Investigating subtype-specific therapies, such as SETD7 inhibition in basal-like breast cancer, is also warranted given the association between elevated SETD7 levels and unfavorable outcomes.48 Beyond cancer, exploring SETD7 modulation as a therapeutic strategy in diseases like diabetes, inflammatory disorders, and viral infections may reveal new treatment opportunities. Developing targeted delivery systems for SETD7 inhibitors or modulators and conducting mechanistic studies to understand SETD7’s context-dependent roles in various cellular processes and disease states will be crucial for informing the development of more effective and targeted therapies.1,2
Future research directions may involve exploring SETD7’s broader substrate specificity beyond histones, including receptors, signaling molecules, and TF’s.1,2,143 Investigating how SETD7 regulates non-histone proteins can provide valuable insights into its diverse functions in cellular pathways and disease processes. Furthermore, studying the molecular interactions of SETD7 with specific substrates could elucidate novel therapeutic targets for diseases such as lung fibrosis, cancers, diabetes, inflammatory conditions, and aging-associated disorders.
Conclusion
SETD7 is a multifaceted and essential regulator of various cellular activities, including cell growth, differentiation, and stress responses. Its enzymatic activity, characterized by the monomethylation of lysine residues on both histone and non-histone proteins, significantly impacts diverse biological processes such as metabolism, immunity, and cancer progression. The complex and context-dependent nature of SETD7’s function is evident in its ability to either stimulate or inhibit cell proliferation based on the specific protein target and cellular environment. For instance, in BC, inhibiting SETD7 is associated with a less differentiated phenotype, while in other contexts; it promotes stem cell phenotypes or differentiation. In AD, SETD7-mediated monomethylation of soluble Tau suggests its potential role in disease progression by influencing Tau localization and aggregation.8 Additionally, in asthma, SETD7 enhances NF-κB/CD38 signaling, promoting airway smooth muscle cell proliferation, which exacerbates airway inflammation.
Furthermore, SETD7’s involvement in ROS signaling and oxidative stress regulation underscores its role in maintaining cellular homeostasis and responding to environmental stressors. SETD7’s therapeutic potential arises from its involvement in modulating cellular mechanisms linked to regeneration, inflammation, oxidative stress, and cancer progression. Given the complexity and context-dependency of SETD7’s role in disease pathogenesis, future research efforts focused on understanding the intricate mechanisms by which SETD7 influences disease pathogenesis and exploring its diverse substrate interactions hold promise for developing innovative therapeutic strategies across a spectrum of diseases, including cancer, neurodegenerative disorders, and inflammatory conditions. Understanding the diverse functions of SETD7 will provide significant insights into its involvement in health and disease, laying the groundwork for advancements in targeted therapies across various conditions.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is supported by National Natural Science Foundation of China (82271807, 82071771 to PL), Hubei Province Science Fund for Distinguished Young Scholars (2022CFA057 to PL), the Non-profit Central Research Institute Fund of Chinese (2023-PT320-07 to PL), National Natural Science Foundation of China (82371794 to ZC) and Hubei Provincial Department of Science and Technology, Hubei Provincial Key Research and Development Program (2022BCA015 to ZC).
Disclosure
The authors declare no conflicts of interest.
References
1. Batista I de AA, Helguero LA. Biological processes and signal transduction pathways regulated by the protein methyltransferase SETD7 and their significance in cancer. Signal Transduct Target Ther. 2018;3(1):19. doi:10.1038/s41392-018-0017-6
2. Chiang C, Yang H, Zhu L, et al. The epigenetic regulation of nonhistone proteins by SETD7: new targets in cancer. Front Genet. 2022:13. doi:10.3389/fgene.2022.918509.
3. He S, Owen DR, Jelinsky SA, Lin LL. Lysine methyltransferase SETD7 (SET7/9) regulates ROS signaling through mitochondria and NFE2L2/ARE pathway. Sci Rep. 2015;5. doi:10.1038/srep14368
4. Zhang BW, Huang T, Yang YF, Li MY, Shao GB. Lysine methyltransferase SETD7 in cancer: functions, molecular mechanisms and therapeutic implications. Mol Biol Rep. 2025;52(1). doi:10.1007/s11033-025-10494-3
5. Soshnikova N. Functions of SETD7 during development, homeostasis and cancer. Stem Cell Investig. 2019;6:26. doi:10.21037/sci.2019.06.10
6. Li M, Ning J, Wang J, Yan Q, Zhao K, Jia X. SETD7 regulates chondrocyte differentiation and glycolysis via the Hippo signaling pathway and HIF-1α. Int J Mol Med. 2021;48(6). doi:10.3892/ijmm.2021.5043
7. Wu Y, Zou F, Lu Y, et al. SETD7 promotes TNF-α-induced proliferation and migration of airway smooth muscle cells in vitro through enhancing NF-κB/CD38 signaling. Int Immunopharmacol. 2019;72:459–466. doi:10.1016/j.intimp.2019.04.043
8. Bichmann M, Prat Oriol N, Ercan-Herbst E, et al. SETD7-mediated monomethylation is enriched on soluble Tau in Alzheimer’s disease. Mol Neurodegener. 2021;16(1). doi:10.1186/s13024-021-00468-x
9. Porzberg MRB, Lenstra DC, Damen E, Blaauw RH. (R) -PFI-2 Analogues as Substrates and Inhibitors of Histone Lysine Methyltransferase SETD7. 2023:202300457. doi:10.1002/cmdc.202300457
10. Barsyte-Lovejoy D, Li F, Oudhoff MJ, et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc Natl Acad Sci U S A. 2014;111(35):12853–12858. doi:10.1073/pnas.1407358111
11. Oudhoff MJ, Braam MJS, Freeman SA, et al. SETD7 controls intestinal regeneration and tumorigenesis by regulating wnt/β-catenin and hippo/yap signaling. Dev Cell. 2016;37(1):47–57. doi:10.1016/j.devcel.2016.03.002
12. Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-κB-dependent inflammatory genes: relevance to diabetes and inflammation. J Biol Chem. 2008;283(39):26771–26781. doi:10.1074/jbc.M802800200
13. Ivanov GS, Ivanova T, Kurash J, et al. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007;27(19):6756–6769. doi:10.1128/mcb.00460-07
14. Okabe J, Orlowski C, Balcerczyk A, et al. Distinguishing hyperglycemic changes by set7 in vascular endothelial cells. Circ Res. 2012;110(8):1067–1076. doi:10.1161/CIRCRESAHA.112.266171
15. Chiang CY, Fan S, Zheng H, et al. Methylation of KRAS by SETD7 promotes KRAS degradation in non-small cell lung cancer. Cell Rep. 2023;42(9):113003. doi:10.1016/j.celrep.2023.113003
16. Chiang C, Xiao T, Fan S, et al. Methylation of KRAS at lysine 182 and 184 by SETD7 promotes KRAS degradation. bioRxiv. doi:10.1101/2021.02.10.429309
17. Garbino A, Van Oort RJ, Dixit SS, et al. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175–186. doi:10.1152/physiolgenomics.00017.2009.-Junctophilins
18. Wang H, Cao R, Xia L, et al. Purification and functional characterization of a histone h3-lysine 4-specific methyltransferase. 2001;Vol 8.
19. Wilson JR, Jing C, Walker PA, et al. Crystal structure and functional analysisof the histone methyltransferase SET7/9. Cell. 2002;111:105–115. doi:10.1016/S0092-8674(02)00964-9
20. Qian C, Zhou MM. SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell Mol Life Sci. 2006;63(23):2755–2763. doi:10.1007/s00018-006-6274-5
21. Schapira M. Structural chemistry of human SET domain protein methyltransferases. 2011;Vol 5.
22. Daks A, Vasileva E, Fedorova O, Shuvalov O, Barlev NA. the role of lysine methyltransferase SET7/9 in proliferation and cell stress response. Life. 2022;12(3). doi:10.3390/life12030362
23. Gu B, Lee MG. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013;3(1). doi:10.1186/2045-3701-3-39
24. The human protein atlas. SETD7. https://www.proteinatlas.org/ENSG00000145391-SETD7/structure.
25. Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21(8):1273–1283. doi:10.1101/gr.122382.111
26. Cheng J, Blum R, Bowman C, et al. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell. 2014;53(6):979–992. doi:10.1016/j.molcel.2014.02.032
27. Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49(5):825–837. doi:10.1016/j.molcel.2013.01.038
28. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–285. doi:10.1038/nature09692
29. Yang S, Wang X, Bai J, Duan B. The role of SET domain containing lysine methyltransferase 7 in tumorigenesis and development. Cell Cycle. 2023;22(3):269–275. doi:10.1080/15384101.2022.2122257
30. Pradhan S, Chin HG, Estève PO, Jacobsen SE. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics. 2009;4(6):383–387. doi:10.4161/epi.4.6.9450
31. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi:10.1038/cr.2011.22
32. Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi:10.1038/nature01378
33. Ibáñez G, Mcbean JL, Astudillo YM, Luo M. An enzyme-coupled ultrasensitive luminescence assay for protein methyltransferases. Anal Biochem. 2010;401(2):203–210. doi:10.1016/j.ab.2010.03.010
34. Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011;21(4):564–578. doi:10.1038/cr.2011.42
35. Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339(2):240–249. doi:10.1016/j.ydbio.2009.08.017
36. Guenther MG, Jenner RG, Chevalier B, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci. 2005;102(24):8603–8608. doi:10.1073/pnas.0503072102
37. Daks A, Parfenyev S, Shuvalov O, et al. Lysine-specific methyltransferase Set7/9 in stemness, differentiation, and development. Biol Direct. 2024;19(1). doi:10.1186/s13062-024-00484-z
38. Niwa H, Handa N, Tomabechi Y, et al. Structures of histone methyltransferase SET7/9 in complexes with adenosylmethionine derivatives. Acta Crystallogr Sect D Biol Crystallogr. 2013;69(4):595–602. doi:10.1107/S0907444912052092
39. Keating S, El-Osta A. Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics. 2013;8(4):361–372. doi:10.4161/epi.24234
40. Lezina L, Aksenova V, Fedorova O, et al. KMT Set7 / 9 affects genotoxic stress response via the Mdm2 axis. Oncotarget. 2015;6(28):25843–25855. doi:10.18632/oncotarget.4584
41. Wang Z, Petricca J, Liu M, et al. SETD7 functions as a transcription repressor in prostate cancer via methylating FOXA1. Proc Natl Acad Sci U S A. 2023;120(33). doi:10.1073/pnas.2220472120
42. Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010;39(1):152–160. doi:10.1016/j.molcel.2010.06.006
43. Stark GR, Wang Y, Lu T. Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 2011;21(3):375–380. doi:10.1038/cr.2010.174
44. Lee J, Shao NY, Paik DT, et al. SETD7 drives cardiac lineage commitment through stage-specific transcriptional activation. Cell Stem Cell. 2018;22(3):428–444.e5. doi:10.1016/j.stem.2018.02.005
45. Dwivedi S, Chavan A, Paul AT. SET7, a lysine-specific methyl transferase: an intriguing epigenetic target to combat diabetic nephropathy. Drug Discov Today. 2023;28(10):103754. doi:10.1016/j.drudis.2023.103754
46. Zhu LEI, Xxia L, Shi L, Wu J, Jiayi Q, Xia TS. Rapamycin enhances the sensitivity of ER ‑ positive breast cancer cells to tamoxifen by upregulating p73 expression. Oncol Rep. 2019;41:455–464. doi:10.3892/or.2018.6842
47. Feng Q, Zhang Z, Shea MJ, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Nat Publ Gr. 2014;24(7):809–819. doi:10.1038/cr.2014.71
48. Monteiro FL, Stepanauskaite L, Williams C, Helguero LA. SETD7 expression is associated with breast cancer survival outcomes for SpeciFIc molecular subtypes: a systematic analysis of publicly available datasets. Cancers. 2022;14(24):6029. doi:10.3390/cancers14246029
49. Huang R, Li X, Yu Y, et al. SETD7 is a prognosis predicting factor of breast cancer and regulates redox homeostasis. Oncotarget. 2017;8(55):94080–94090.
50. Subramanian K, Jia D, Kapoor-Vazirani P, et al. Regulation of estrogen receptor alpha by the SET7 Lysine Methyltransferase. Mol Cell. 2008;30(3):336–347. doi:10.1016/j.molcel.2008.03.022
51. Chuikov S, Kurash JK, Wilson JR, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432(7015):353–360.
52. Kouskouti A, Scheer E, Staub A, Szlò Tora L, Talianidis I. Gene-Specific Modulation of TAF10 Function by SET9-Mediated Methylation Methylation of H3 at Lysine 4 and 36 by the SET1 and SET2 Family of Enzymes Has Been Demonstrated to Cor-Relate with Active Transcription (Kouzarides, 2002). 2004. Available from: http://www.molecule.org/.
53. Montenegro MF, Sánchez-Del-Campo L, González-Guerrero R, et al. Tumor suppressor SET9 guides the epigenetic plasticity of breast cancer cells and serves as an early-stage biomarker for predicting metastasis. Oncogene. 2016;35:6143–6152. doi:10.1038/onc.2016.154
54. Monteiro FL, Williams C, Helguero LA. A systematic review to define the multi-faceted role of lysine methyltransferase SETD7 in cancer. Cancers. 2022;14(6):1414. doi:10.3390/cancers14061414
55. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):1–14. doi:10.1186/s13059-017-1349-1
56. Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013:4. doi:10.1038/ncomms3612.
57. Si W, Zhou J, Zhao Y, Zheng J, Cui L. SET7/9 promotes multiple malignant processes in breast cancer development via RUNX2 activation and is negatively regulated by TRIM21. Cell Death Dis. 2020;11(2). doi:10.1038/s41419-020-2350-2
58. Gao L, Zhang J, Long Q, et al. SETD7 promotes metastasis of triple-negative breast cancer by YY1 lysine methylation. Biochim Biophys Acta - Mol Basis Dis. 2023;1869(7):166780. doi:10.1016/j.bbadis.2023.166780
59. Song H, Chu JW, Park SC, et al. Isoform‐specific lysine methylation of RORα2 by SETD7 is required for association of the TIP60 coactivator complex in prostate cancer progression. Int J Mol Sci. 2020;21(5). doi:10.3390/ijms21051622
60. AM BC. Histone methyltransferase Setd7 regulates Nrf2 signaling pathway by phenethyl isothiocyanate and ursolic acid in human prostate cancer cells. Physiol Behav. 2019;176(5):139–148. doi:10.1002/mnfr.201700840.Histone
61. Wang C, Sargsyan D, Zhang C, Wu R, Yang Y, Kong AN. Transcriptomic analysis of histone methyltransferase Setd7 knockdown and phenethyl isothiocyanate in human prostate cancer cells. Anticancer Res. 2018;38(11):6069–6083. doi:10.21873/anticanres.12957
62. Chen Y, Yang S, Hu J, Yu C, He M, Cai Z. Increased expression of SETD7 promotes cell proliferation by regulating cell cycle and indicates poor prognosis in hepatocellular carcinoma. PLoS One. 2016;11(5). doi:10.1371/journal.pone.0154939
63. Judson RN, Quarta M, Oudhoff MJ, et al. Inhibition of methyltransferase setd7 allows the in vitro expansion of myogenic stem cells with improved therapeutic potential. Cell Stem Cell. 2018;22(2):177–190.e7. doi:10.1016/j.stem.2017.12.010
64. Gu Y, Wang X, Liu H, Li G, Yu W, Ma Q. SET7/9 promotes hepatocellular carcinoma progression through regulation of E2F1. Oncol Rep. 2018;40(4):1863–1874. doi:10.3892/or.2018.6621
65. Cao L, Ren Y, Guo X, et al. Downregulation of SETD7 promotes migration and invasion of lung cancer cells via JAK2/STAT3 pathway. Int J Mol Med. 2020;45(5):1616–1626. doi:10.3892/ijmm.2020.4523
66. Gu Y, Zhang X, Yu W, Dong W. Oncogene or tumor suppressor: the coordinative role of lysine methyltransferase SET7/9 in cancer development and the related mechanisms. J Cancer. 2022;13(2):623–640. doi:10.7150/jca.57663
67. Jetton TL, Flores-Bringas P, Leahy JL, Gupta D. SetD7 (Set7/9) is a novel target of PPARγ that promotes the adaptive pancreatic β-cell glycemic response. J Biol Chem. 2021;297(5):101250. doi:10.1016/j.jbc.2021.101250
68. Gu Y, Wang Y, Wang X, Gao L, Yu W, Dong WF. Opposite effects of SET7/9 on apoptosis of human acute myeloid leukemia cells and lung cancer cells. J Cancer. 2017;8(11):2069–2078. doi:10.7150/jca.19143
69. Akiyama Y, Koda Y, Byeon SJ, et al. Reduced expression of SET7/9, a histone mono-methyltransferase, is associated with gastric cancer progression. Oncotarget. 2015;7(4):3966.
70. Lv J, Wu Q, Li K, et al. Lysine N-methyltransferase SETD7 promotes bladder cancer progression and immune escape via STAT3/PD-L1 cascade. Int J Biol Sci. 2023;19(12):3744–3761. doi:10.7150/ijbs.87182
71. Duan B, Bai J, Qiu J, et al. Histone-lysine N-methyltransferase SETD7 is a potential serum biomarker for colorectal cancer patients. eBioMedicine. 2018;37:134–143. doi:10.1016/j.ebiom.2018.10.036
72. Zhang J, Duan B, Li F, et al. SETD7 promotes cell proliferation and migration via methylation-mediated TAF7 in clear cell renal cell carcinoma. Int J Biol Sci. 2024;20(8):3008–3027. doi:10.7150/ijbs.93201
73. Munro S, Khaire N, Inche A, Carr S, La thangue NB. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010;29(16):2357–2367. doi:10.1038/onc.2009.511
74. Basuroy T, de la Serna IL. SETD7 in cardiomyocyte differentiation and cardiac function. Stem Cell Investig. 2019;6:6. doi:10.21037/sci.2019.08.01
75. Karimnia N, Rafehi H, Tuano NK, et al. Current perspectives in Set7 mediated stem cell differentiation. Non-Coding RNA. 2016;2(4):2–9. doi:10.3390/ncrna2040014
76. Castaño J, Morera C, Sesé B, et al. SETD7 regulates the differentiation of human embryonic stem cells. PLoS One. 2016;11(2):1–21. doi:10.1371/journal.pone.0149502
77. Kim SK, Lee H, Han K, et al. SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell Stem Cell. 2014;15(6):735–749. doi:10.1016/j.stem.2014.10.016
78. Wang D, Li Y, Xu C, et al. SETD7 promotes lateral plate mesoderm formation by modulating the Wnt/β-catenin signaling pathway. iScience. 2023;26(6). doi:10.1016/j.isci.2023.106917
79. Sun H, Zhang H. Lysine methylation-dependent proteolysis by the Malignant brain tumor (MBT) domain proteins. Int J Mol Sci. 2024;25:2248. doi:10.3390/ijms25042248
80. Wang J, Jian K, Yang Q, Gu C, Sheng J, Zhou Y. Retarding human adipose-derived MSCs senescence and promoting tendon repair using cell sheet engineering with a histone methyltransferase inhibitor. 1–15.
81. Kishore M, Pradeep M, Narne P, Jayalakshmi S. Regulation of Keap1 ‑ Nrf2 axis in temporal lobe epilepsy — hippocampal sclerosis patients may limit the seizure outcomes. Neurol Sci. 2023;44(12):4441–4450. doi:10.1007/s10072-023-06936-0
82. Adriana MS, Loredana VM, Gela LA, Muresian H, Simionescu M, Manea A. SET7 lysine methyltransferase mediates the up-regulation of NADPH oxidase expression, oxidative stress, and NLRP3 inflammasome priming in atherosclerosis. 2025.
83. Wang Z, Liu H. Roles of lysine methylation in glucose and lipid metabolism: functions, regulatory mechanisms, and therapeutic implications. Biomolecules. 2024;14(7):862. doi:10.3390/biom14070862
84. Daks A, Shuvalov O, Fedorova O, Parfenyev S, Simon HU, Barlev NA. Methyltransferase Set7/9 as a Multifaceted Regulator of ROS Response. Int J Biol Sci. 2023;19(8):2304–2318. doi:10.7150/ijbs.83158
85. Zhong W, Chen R, Zhao J, et al. SETD7 drives diabetic endothelial dysfunction through FBXO45-mediated GPX4 ubiquitylation. Cardiovasc Diabetol. 2025;24(1). doi:10.1186/s12933-025-02740-6
86. Dang Y, Ma X, Li Y, et al. Inhibition of SETD7 protects cardiomyocytes against hypoxia/reoxygenation-induced injury through regulating Keap1/Nrf2 signaling. Biomed Pharmacother. 2018;106:842–849. doi:10.1016/j.biopha.2018.07.007
87. Kaplan-Meier plotter. Available from: https://kmplot.com/analysis/.
88. dong KT, Gu R, Janknecht R. Methylation of the JMJD2B epigenetic regulator differentially affects its ability to coactivate the ETV1 and JUN transcription factors. 2023;14(6):101–115.
89. Gu R, dong KT, Song H, et al. SET7 / 9-mediated methylation affects oncogenic functions of histone demethylase JMJD2A. 2023;8(20):1–15.
90. Wang H, Pei M, Liu P, Guo D, Zhisu Liu ZZ, Zhang Z. lncRNA SNHG6 promotes hepatocellular carcinoma progression by interacting with HNRNPL/PTBP1 to facilitate SETD7/LZTFL1 mRNA destabilization. Cancer Lett. 2021;520:121–131. doi:10.1016/j.canlet.2021.07.009
91. Aziz N, Hong YH, Kim HG, Kim JH, Cho JY. Tumor-suppressive functions of protein lysine methyltransferases. Exp Mol Med. 2023;55(12):2475–2497. doi:10.1038/s12276-023-01117-7
92. Daks A, Mamontova V, Fedorova O, et al. Set7/9 controls proliferation and genotoxic drug resistance of NSCLC cells. Biochem Biophys Res Commun. 2021;572:41–48. doi:10.1016/j.bbrc.2021.07.086
93. Zhang M, Cai F, Guo J, et al. ACAT2 suppresses the ubiquitination of YAP1 to enhance the proliferation and metastasis ability of gastric cancer via the upregulation of SETD7. Cell Death Dis. 2024;15(4):1–18. doi:10.1038/s41419-024-06666-x
94. Li C, Feng SY, Chen L. SET7/9 promotes H3K4me3 at lncRNA DRAIC promoter to modulate growth and metastasis of glioma. Eur Rev Med Pharmacol Sci. 2020;24(23):12241–12250. doi:10.26355/eurrev_202012_24016
95. Zhang SL, Du X, Tan LN, et al. SET7 interacts with HDAC6 and suppresses the development of colon cancer through inactivation of HDAC6. Am J Transl Res. 2020;12(2):602–611.
96. van Heesbeen HJ, Smidt MP, Kadastik-Eerme L. Entanglement of genetics and epigenetics in parkinson’s disease. Front Neurosci. 2019;13:13. doi:10.3389/fnins.2019.00277
97. Cao L, Wang M, Xu K. Research progress of role and mechanism of SETD7 in tumor occurrence and progression. Chinese J Lung Cancer. 2023;26(1).
98. Li WC, Rukstalis JM, Nishimura W, et al. Activation of pancreatic-duct-derived progenitor cells during pancreas regeneration in adult rats. J Cell Sci. 2010;123(16):2792–2802. doi:10.1242/jcs.065268
99. Banakh I, Gonez LJ, Sutherland RM, Naselli G, Harrison LC. Adult pancreas side population cells expand after β cell injury and are a source of insulin-secreting cells. PLoS One. 2012;7(11):e48977. doi:10.1371/journal.pone.0048977
100. Kofent J, Zhang J, Spagnoli FM. The histone methyltransferase Setd7 promotes pancreatic progenitor identity. Dev. 2016;143(19):3573–3581. doi:10.1242/dev.136226
101. Vinci MC, Costantino S, Damiano G, et al. Persistent epigenetic signals propel a senescence ‑ associated secretory phenotype and trained innate immunity in - hematopoietic stem cells from diabetic patients. Cardiovasc Diabetol;.1–15. doi:10.1186/s12933-024-02195-1
102. Pan X, Fan J, Peng F, Xiao L, Yang Z. SET domain containing 7 promotes oxygen-glucose deprivation/reoxygenation-induced PC12 cell inflammation and oxidative stress by regulating Keap1/Nrf2/ARE and NF-κB pathways. Bioengineered. 2022;13(3):7253–7261. doi:10.1080/21655979.2022.2045830
103. Mohammed SA, Gorica E, Albiero M, et al. Targeting SETD7 rescues diabetes-induced impairment of angiogenic response by transcriptional repression of semaphorin-3G. Diabetes. 2025;74(6):969–982. doi:10.2337/db24-0997
104. Chetty A, Nielsen HC. Targeting airway smooth muscle hypertrophy in asthma: an approach whose time has come. J Asthma Allergy. 2021;14:539–556. doi:10.2147/JAA.S280247
105. Ma C, Yu X, Li D, et al. Inhibition of SET domain–containing (lysine methyltransferase) 7 alleviates cognitive impairment through suppressing the activation of NOD-like receptor protein 3 inflammasome in isoflurane-induced aged mice. Hum Exp Toxicol. 2022:41. doi:10.1177/09603271211061497.
106. Brook PO, Perry MM, Adcock IM, Durham AL. Epigenome-modifying tools in asthma. Epigenomics. 2015;7(6):1017–1032. doi:10.2217/epi.15.53
107. Bekdash RA, Zhang C, Sarkar DK. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in β-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res. 2013;37(7):1133–1142. doi:10.1111/acer.12082
108. Govorko D, Bekdash RA, Zhang C, Sarkar DK. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiatry. 2012;72(5):378–388. doi:10.1016/j.biopsych.2012.04.006
109. Ghafouri-Fard S, Gholipour M, Abak A, Mazdeh M, Taheri M, Sayad A. Expression analysis of NF-κB-Related lncRNAs in parkinson’s disease. Front Immunol. 2021;12:1–8. doi:10.3389/fimmu.2021.755246
110. Yang Y, Yuan RX, Luan Y. Histone methylation related therapeutic challenge in cardiovascular diseases. Front Cardiovasc Med. 2021;8. doi:10.3389/fcvm.2021.710053
111. Ambrosini S, Montecucco F, Kolijn D, et al. Targeting the methyltransferase SETD7 prevents myocardial ischemic injury: a translational study. Basic Sci Card Dis Ischaemia, Infarction, Cardioprot.
112. Ambrosini S, Montecucco F, Akhmedov A, et al. The methyltransferase SETD7 promotes myocardial ischemic injury by activating Hippo signalling. Eur Heart J. 2019:40. https://academic.oup.com/eurheartj/article/40/Supplement_1/ehz746.0339/5597912.
113. Costantino S, Ambrosini S, Mohammed SA, et al. A chromatin mark by SETD7 regulates myocardial inflammation in obesity-related heart failure with preserved ejection fraction. Eur Heart J. 2022:43. https://academic.oup.com/eurheartj/article/43/Supplement_2/ehac544.2883/6745715.
114. Zhao YB, Wei W, Lin XX, Chai YF, Jin H. The Role of Histone H3 Methylation in Acute Kidney Injury. Drug Des Devel Ther. 2022;16:2453–2461. doi:10.2147/DDDT.S376673
115. Liu B, Nie J, Liang H, et al. Pharmacological inhibition of SETD7 by PFI-2 attenuates renal fibrosis following folic acid and obstruction injury. Eur J Pharmacol. 2021:901. doi:10.1016/j.ejphar.2021.174097.
116. Yu C, Zhuang S, Peng Y, Ding J, Zhou W. Histone methyltransferases as therapeutic targets for kidney diseases. Front Pharmacol. 2019;10:10. doi:10.3389/fphar.2019.01393
117. Chen J, Guo Y, Zeng W, et al. ER stress triggers MCP-1 expression through SET7/9-induced histone methylation in the kidneys of db/db mice. Am J Physiol - Ren Physiol. 2014;306(8):20–26. doi:10.1152/ajprenal.00697.2012
118. Sasaki K, Doi S, Nakashima A, et al. Inhibition of SET domain–containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J Am Soc Nephrol. 2016;27(1):203–215. doi:10.1681/ASN.2014090850
119. Tanemoto F, Mimura I. Therapies targeting epigenetic alterations in acute kidney injury-to-chronic kidney disease transition. Pharmaceuticals. 2022;15(2):123. doi:10.3390/ph15020123
120. Allen M, Carrasquillo MM, Funk C, et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data. 2016;3:1–10. doi:10.1038/sdata.2016.89
121. Balmik AA, Chinnathambi S. Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun Signal. 2021;19(1):1–13. doi:10.1186/s12964-021-00732-z
122. Cao Q, Wang W, Williams JB, Yang F, Wang ZJ, Yan Z. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci Adv. 2020;6(50). doi:10.1126/sciadv.abc8096
123. Prasanth MI, Sivamaruthi BS, Cheong CSY, et al. Role of epigenetic modulation in neurodegenerative diseases: implications of phytochemical interventions. Antioxidants. 2024;13(5):606. doi:10.3390/antiox13050606
124. Ramesh S, ASPM A. Depletion of dopamine in Parkinson’s disease and relevant therapeutic options: a review of the literature. AIMS Neurosci. 2023;10(3):200–231. doi:10.3934/NEUROSCIENCE.2023017
125. Song H, Chen J, Huang J, et al. Epigenetic modification in Parkinson’s disease. Front Cell Dev Biol. 2023:1–10. doi:10.3389/fcell.2023.1123621.
126. Basavarajappa BS, Subbanna S. Histone methylation regulation in neurodegenerative disorders. Int J Mol Sci. 2021;22(9):4654. doi:10.3390/ijms22094654
127. Lind-Holm Mogensen F, Scafidi A, Poli A, Michelucci A. PARK7/DJ-1 in microglia: implications in Parkinson’s disease and relevance as a therapeutic target. J Neuroinflammation. 2023;20(1). doi:10.1186/s12974-023-02776-z
128. Kouli A, Torsney KM, Kuan W-L. Parkinson’s disease: etiology, neuropathology, and pathogenesis. In: Parkinson’s Disease: Pathogenesis and Clinical Aspects. 2018:3–26.
129. Shen J, Chen XC, Li WJ, et al. Identification of Parkinson’s disease-related pathways and potential risk factors. J Int Med Res. 2020;48(10). doi:10.1177/0300060520957197
130. Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers. 2019;11(12):2002. doi:10.3390/cancers11122002
131. Veazey KJ, Parnell SE, Miranda RC, Golding MC. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics and Chromatin. 2015;8(1). doi:10.1186/s13072-015-0031-7
132. Stylianou E. Epigenetics of chronic inflammatory diseases. J Inflamm Res. 2019;12:1–14. doi:10.2147/JIR.S129027
133. Rojano Rada J, Fernández Mestre M, Ramírez Morales C. Effect of epigenetics on rheumatoid arthritis. Medwave. 2023;23(03):e2619–e2619. doi:10.5867/medwave.2023.03.2619
134. Sánchez-Pernaute O. Epigenetic therapies, a step beyond biologics for rheumatoid arthritis. Reumatol Clin.
135. Yang C, Li D, Teng D, et al. Epigenetic regulation in the pathogenesis of rheumatoid arthritis. Front Immunol. 2022:13. doi:10.3389/fimmu.2022.859400.
136. Immunopaedia IADVANCEDIMMUNOLOGY. 2024. Available from: https://www.immunopaedia.org.za/immunology/advanced/1-transplantation/.
137. Yan Z, Gibson SA, Buckley JA, Qin H, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol. 2018;189:4–13. doi:10.1016/j.clim.2016.09.014
138. Todi H. The role of immune dysregulation in autoimmune disorders. Immunol Disord Immunother. 2023;8(2).
139. Kidd CDA, Thompson PJ, Barrett L, Baltic S. Histone modifications and asthma. the interface of the epigenetic and genetic landscapes. Am J Respir Cell Mol Biol. 2016;54(1):3–12. doi:10.1165/rcmb.2015-0050TR
140. Gubernatorova EO, Namakanova OA, Gorshkova EA, Medvedovskaya AD, Nedospasov SA, Drutskaya MS. Novel anti-cytokine strategies for prevention and treatment of respiratory allergic diseases. Front Immunol. 2021;12:601842. doi:10.3389/fimmu.2021.601842
141. Amrani Y, Chen H, Panettieri RA. Activation of tumor necrosis factor receptor 1 in airway smooth muscle: a potential pathway that modulates bronchial hyper-responsiveness in asthma ? Yassine Amrani, Hang Chen and Reynold A Panettieri Jr. Respir Res. 2000;1:49–53. doi:10.1186/rr12
142. Sheikhpour M, Ebrahimi Vargoorani M, Amiri V, Amiri V. A review of epigenetic changes in asthma: methylation and acetylation. Clin Epigenetics. 2021;13(1). doi:10.1186/s13148-021-01049-x
143. Gao L, Yu W, Song P, Li Q. Non-histone Methylation of SET7/9 and its Biological Functions. Recent Pat Anticancer Drug Discov. 2022;17(3):231–243. doi:10.2174/1574892816666211202160041
144. Feoli A, Viviano M, Cipriano A, Milite C, Castellano S, Sbardella G. Lysine methyltransferase inhibitors: where we are now. RSC Chem Biol. 2022;3(4):359–406. doi:10.1039/d1cb00196e
145. Mori S, Iwase K, Iwanami N, Tanaka Y, Kagechika H, Hirano T. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorganic Med Chem. 2010;18(23):8158–8166. doi:10.1016/j.bmc.2010.10.022
146. Lenstra DC, Damen E, Leenders RGG, et al. Structure–activity relationship studies on (R)-PFI-2 analogues as inhibitors of histone lysine methyltransferase SETD7. ChemMedChem. 2018;13(14):1405–1413. doi:10.1002/cmdc.201800242
147. Tang B, Li B, Li B, et al. Insights into the stereoselectivity of human SETD7 methyltransferase. RSC Adv. 2019;9(16):9218–9227. doi:10.1039/c9ra00190e
148. Xu R, Liu X, Zhang Y, et al. Activating transcriptional coactivator with PDZ-binding motif by (R) -PFI-2 attenuates osteoclastogenesis and prevents ovariectomized-induced osteoporosis. Biochem Pharmacol. 2024;219(54):115964. doi:10.1016/j.bcp.2023.115964
149. Niu Y, Shi D, Li L, Guo J, Liu H, Yao X. Revealing inhibition difference between PFI-2 enantiomers against SETD7 by molecular dynamics simulations, binding free energy calculations and unbinding pathway analysis. Sci Rep. 2017;7:7. doi:10.1038/srep46547
150. PATSNAP SB. Amustaline. 2024. Available from: https://synapse.patsnap.com/drug/2a79ee275c9746fc93d7cb0df5a06b71?csrfToken.
151. Brixner V, Kiessling AH, Madlener K, et al. Red blood cells treated with the amustaline (S-303) pathogen reduction system: a transfusion study in cardiac surgery. Transfusion. 2018;58(4):905–916. doi:10.1111/trf.14528
152. Takemoto Y, Ito A, Niwa H, et al. Identification of cyproheptadine as an Inhibitor of set domain containing lysine methyltransferase 7/9 (Set7/9) that regulates estrogen-dependent transcription. J Med Chem. 2016;59(8):3650–3660. doi:10.1021/acs.jmedchem.5b01732
153. Hirano T, Fujiwara T, Niwa H, et al. Development of novel inhibitors for histone methyltransferase set7/9 based on cyproheptadine. ChemMedChem. 2018;13(15):1530–1540. doi:10.1002/cmdc.201800233
154. Shannar A, Sarwar S, Dushyant P, Chou PJ, Peter RM. Cyproheptadine inhibits in vitro and in vivo lung metastasis and drives metabolic rewiring. Mol Biol Rep. 2024;51(1):1–12. doi:10.1007/s11033-024-10033-6
155. Fujiwara T, Ohira K, Urushibara K, et al. Steric structure–activity relationship of cyproheptadine derivatives as inhibitors of histone methyltransferase Set7/9. Bioorganic Med Chem. 2016;24(18):4318–4323. doi:10.1016/j.bmc.2016.07.024
156. AKVSABG HS. Cyproheptadine, a SET7/9 inhibitor, reduces hyperglycaemia-induced ER stress alleviating inflammation and fibrosis in renal tubular epithelial cells. Arch Physiol Biochem. doi:10.1080/13813455.2022.2105365
157. Sharma N, Sankrityayan H, Kale A, Gaikwad AB. Role of SET7/9 in the progression of ischemic renal injury in diabetic and non-diabetic rats. Biochem Biophys Res Commun. 2020;528(1):14–20. doi:10.1016/j.bbrc.2020.05.075
158. Fujimaki K, Ogihara T, Morris DL, et al. SET7/9 enzyme regulates cytokine-induced expression of inducible nitric-oxide synthase through methylation of Lysine 4 at histone 3 in the islet β cell. J Biol Chem. 2015;290(27):16607–16618. doi:10.1074/jbc.M115.661777
159. Hou Z, Min W, Zhang R, et al. Lead discovery, chemical optimization, and biological evaluation studies of novel histone methyltransferase SET7 small-molecule inhibitors. Bioorganic Med Chem Lett. 2020;30(9):127061. doi:10.1016/j.bmcl.2020.127061
160. Meng F, Cheng S, Ding H, et al. Discovery and optimization of novel, selective histone methyltransferase SET7 inhibitors by pharmacophore- and docking-based virtual screening. J Med Chem. 2015;58(20):8166–8181. doi:10.1021/acs.jmedchem.5b01154
161. Ea CK, Baltimore D. Regulation of NF-KB Activity through Lysine Monomethylation of P65. 2009;10.
162. Dowden J, Hong W, Parry RV, Pike RA, Ward SG. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases. Bioorganic Med Chem Lett. 2010;20(7):2103–2105. doi:10.1016/j.bmcl.2010.02.069
163. Kashyap S, Sandler J, Peters U, Martinez EJ, Kapoor TM. Using ‘biased-privileged’ scaffolds to identify lysine methyltransferase inhibitors. Bioorganic Med Chem. 2014;22(7):2253–2260. doi:10.1016/j.bmc.2014.02.024
164. Ding H, Lu WC, Hu JC, et al. Identification and characterizations of novel, selective histone methyltransferase SET7 inhibitors by scaffold hopping- and 2D-molecular fingerprint-based similarity search. Molecules. 2018;23(3):567. doi:10.3390/molecules23030567
165. Min W, Hou Z, Zhang F, et al. Computational discovery and biological evaluation of novel inhibitors targeting histone-lysine N-methyltransferase SET7. Bioorganic Med Chem. 2020;28(7):115372. doi:10.1016/j.bmc.2020.115372
166. Sowalsky AG, Xia Z, Wang L, et al. Whole transcriptome sequencing reveals extensive unspliced mRNA in metastatic castration-resistant prostate cancer. Mol Cancer Res. 2015;13(1):98–106. doi:10.1158/1541-7786.MCR-14-0273
167. Logan I, Shuttleworth V, Gaughan L, Sheerin N. Epigenetic regulators, including SETD7, as new targets for the treatment of chronic kidney disease. Lancet. 2016;387:S66. doi:10.1016/s0140-6736(16)00453-0
168. Ko S, Ahn J, Song CS, Kim S, Knapczyk-Stwora K, Chatterjee B. Lysine methylation and functional modulation of androgen receptor by set9 methyltransferase. Mol Endocrinol. 2011;25(3):433–444. doi:10.1210/me.2010-0482
169. Shen C, Wang D, Liu X, et al. SET7/9 regulates cancer cell proliferation by influencing β-catenin stability. FASEB J. 2015;29(10):4313–4323. doi:10.1096/fj.15-273540
170. Shi Y, Wang Z, Shao Y, et al. Combined SET7/9 and CDK4 inhibition act synergistically against osteosarcoma. Biochem Biophys Res Commun. 2024;708:149808. doi:10.1016/j.bbrc.2024.149808
171. Xie Q, Bai Y, Mei P, Yuan Z. Methylation-mediated regulation of E2F1 in DNA damage- induced cell death. J Recept Signal Transduct. 2011;2014.doi:10.3109/10799893.2011.552914.
172. Babaei Z, Panjehpour M, Parsian H, Aghaei M. SAR131675 receptor tyrosine kinase inhibitor induces apoptosis through Bcl- 2/Bax/Cyto c mitochondrial pathway in human umbilical vein endothelial cells. Anti-Cancer Agents Med Chem (Formerly Curr Med Chem - Anti-Cancer Agents). 2022;22(5):943–950. doi:10.2174/1871520621666210708102619
173. Tamura R, Carpenter CI, Thomas CM, et al. ROS-activatable prodrug of doxazolidine as novel cancer therapy paradigm. Adv Ther. 2024;2400340:1–9. doi:10.1002/adtp.202400340
174. Liang Y, Chen J, Yu Q, et al. Phosphorylated ERK is a potential prognostic biomarker for Sorafenib response in hepatocellular carcinoma. Cancer Med. 2017;6(12):2787–2795. doi:10.1002/cam4.1228
175. Myadelets D, Parfenyev S, Vasileva J, et al. Methyltransferase Set7/9 controls PARP1 expression and regulates cisplatin response of breast cancer cells. Biochem Biophys Res Commun. 2024;691:149328. doi:10.1016/j.bbrc.2023.149328
176. Benincasa G, Pepin ME, Russo V, et al. High ‑ resolution DNA methylation changes reveal biomarkers of heart failure with preserved ejection fraction versus reduced ejection fraction. Basic Res Cardiol. 2025;120(2):347–361. doi:10.1007/s00395-024-01093-7
177. Wang J, Tan S, Zhang Y, et al. Set7 / 9 aggravates ischemic brain injury via enhancing glutamine metabolism in a blocking Sirt5 manner. Cell Death &. 2024;31:511–523. doi:10.1038/s41418-024-01264-y
178. Liu W, Shen Y, Pan R, Qi X. mir-330-5p from mesenchymal stem cell-derived exosomes targets SETD7 to reduce inflammation in rats with cerebral ischemia-reperfusion injury. J Mol Histol. 2025;56(1):63. doi:10.1007/s10735-024-10347-6
179. Li L, Zhu J, Chen Y, et al. Interaction between YTH domain‐containing family protein 2 and SET domain‐containing lysine methyltransferase 7 suppresses autophagy in osteoarthritis chondrocytes, exacerbating cartilage damage. J Gene Med. 2025;27(1):1–13. doi:10.1002/jgm.70005
180. Ren D, Chen X, Liu H, Li M, Zheng L, Yong P. Exploring the efficacy of (R) -PFI-2 hydrochloride in mitigating noise-induced hearing loss by targeting NLRP3 inflammasome and NF- κ B pathway to reduce inner ear inflammation. J Otol. 2024;19(4):200–206. doi:10.1016/j.joto.2024.07.008
181. Guo T, Wen XZ, yu LZ, et al. ISL1 predicts poor outcomes for patients with gastric cancer and drives tumor progression through binding to the ZEB1 promoter together with SETD7. Cell Death Dis. 2019;10(2). doi:10.1038/s41419-018-1278-2
182. What are SETD7 inhibitors and how do they work? synapse by patsnap.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Prognostic Roles of Inflammation- and Nutrition-Based Indicators for Female Patients with Cancer
Yang M, Zhang Q, Ge Y, Tang M, Hu C, Wang Z, Zhang X, Song M, Ruan G, Zhang X, Liu T, Xie H, Zhang H, Zhang K, Li Q, Li X, Liu X, Lin S, Shi H
Journal of Inflammation Research 2022, 15:3573-3586
Published Date: 17 June 2022
The Effects and Pathogenesis of PM2.5 and Its Components on Chronic Obstructive Pulmonary Disease

Wang Q, Liu S
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:493-506
Published Date: 6 April 2023
Quercetin: A Flavonoid with Potential for Treating Acute Lung Injury

Huang M, Liu X, Ren Y, Huang Q, Shi Y, Yuan P, Chen M
Drug Design, Development and Therapy 2024, 18:5709-5728
Published Date: 6 December 2024
Inflammatory and Redox Blood Gene Expression Fingerprint of Severe Obstructive Sleep Apnoea in Patients With Mild Alzheimer’s Disease
Romero-ElKhayat L, Dakterzada F, Huerto R, Carnes-Vendrell A, Mínguez O, Pujol Sabaté M, Targa A, Barbé F, Milanesi E, Dobre M, Manda G, Cuadrado A, Piñol-Ripoll G
Journal of Inflammation Research 2025, 18:1609-1621
Published Date: 4 February 2025
Exercise Prescription Training in Chronic Obstructive Pulmonary Disease: Benefits and Mechanisms
Liu S, Yang A, Yu Y, Xu B, Yu G, Wang H
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1071-1082
Published Date: 15 April 2025