")
Back to Journals » Cancer Management and Research » Volume 10
SALL4 activates TGF-β/SMAD signaling pathway to induce EMT and promote gastric cancer metastasis
Authors Zhang X, Zhang P, Shao M, Zang X, Zhang J, Mao F , Qian H, Xu W
Received 15 June 2018
Accepted for publication 31 August 2018
Published 10 October 2018 Volume 2018:10 Pages 4459—4470
DOI https://doi.org/10.2147/CMAR.S177373
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Xu Zhang,1,2,* Peng Zhang,1,* Meng Shao,1 Xueyan Zang,1 Jiayin Zhang,1 Fei Mao,1 Hui Qian,1,2 Wenrong Xu1,2
1Jiangsu Key Laboratory of Medical Science and Laboratory Medicine, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu 212013, China; 2Zhenjiang Key Laboratory of Gastrointestinal Cancer, Jiangsu University, Zhenjiang, Jiangsu 212013, China
*These authors contributed equally to this work
Background: Increasing evidence suggests that SALL4 plays oncogenic roles in cancer development and progression. We have previously shown that SALL4 is highly expressed in gastric cancer, and its upregulation is associated with lymph node metastasis and poor prognosis. The role of SALL4 in gastric cancer metastasis and the underlying mechanism remain unclear.
Materials and methods: The biological roles of SALL4 in gastric cancer cell mobility, migration, and invasion were investigated by wound healing, transwell migration assay, and Matrigel invasion assay. The effects of SALL4 on epithelial–mesenchymal transition (EMT) in gastric cancer cells were examined by quantitative real-time PCR and Western blot. The downstream target genes of SALL4 were identified by microarray. The regulation of TGF-β1 by SALL4 in gastric cancer cells was analyzed by luciferase reporter assay and chromatin immunoprecipitation assay.
Results: SALL4 knockdown inhibited, while SALL4 overexpression promoted the motility, migration, and invasion abilities of gastric cancer cells in vitro. SALL4 knockdown also suppressed the peritoneal metastasis of gastric cancer cells in nude mice. SALL4 knockdown suppressed, while SALL4 overexpression induced the activation of TGF-β/SMAD signaling pathway and triggered EMT in gastric cancer cells. TGF-β1 was identified as a direct target gene of SALL4. The results of chromatin immunoprecipitation study and luciferase reporter assay further confirmed that SALL4 bound to the promoter of TGF-β1 gene and activated its expression. TGF-β1 knockdown reversed SALL4-mediated promotion of gastric cancer cell motility, migration, and invasion, indicating that TGF-β1 acts as a downstream effector of SALL4. Furthermore, the expression of TGF-β1 was found to be closely associated with that of SALL4 in gastric cancer tissues.
Conclusion: SALL4 promotes the metastasis of gastric cancer, at least partly, by directly activating TGF-β1, suggesting that SALL4 may serve as a new target for gastric cancer therapy.
Keywords: gastric cancer, SALL4, TGF-β1, EMT, metastasis
Introduction
Gastric cancer is the fourth most common malignancy and the third leading cause of cancer-related deaths worldwide.1 Most patients are diagnosed at a late stage with lymph node or distant metastases or with relapse after prior curative surgical therapy, which makes the 5-year survival rate relatively low.2 Peritoneal metastasis is the most frequent metastatic and recurrent site, which has a very poor prognosis. The identification of molecules that play important roles in metastasis and clarification of their mechanisms will provide novel diagnostic markers and therapeutic targets for gastric cancer.
SALL4 is a zinc finger transcription factor that maintains the self-renewal and pluripotency of embryonic stem cells.3 SALL4 expression gradually decreases during development and is even absent in most adult tissues.4 However, the recent findings show that SALL4 is reexpressed in cancer. The increased expression of SALL4 has been reported in acute myeloid leukemia (AML),5,6 liver cancer,7,8 colon cancer,9 breast cancer,10 endometrial cancer,11 lung cancer,12,13 and glioma.14 Further studies reveal that SALL4 plays important roles in cancer development, progression, and metastasis. For instance, SALL4 is found to be constitutively expressed in AML, in which it inhibits cell apoptosis via interaction with β-catenin.6 In liver cancer, SALL4 inhibits PTEN expression by recruiting nucleosome remodeling and deacetylase complex and activates Akt signaling pathway to promote cell proliferation.15,16 In endometrial cancer, SALL4 promotes cell proliferation, migration, invasion, and drug resistance via the upregulation of c-Myc.11 The potent roles of SALL4 in multiple processes of carcinogenesis have made it a novel biomarker for cancer diagnosis and treatment.17,18
We have previously reported the elevated expression of SALL4 in human gastric cancer.19 SALL4 upregulation was associated with lymph node metastasis and predicted poor prognosis in gastric cancer patients. The functional roles of SALL4 in gastric cancer metastasis and the underlying molecular mechanisms remain unclear. In this study, we reported that SALL4 overexpression promoted, while SALL4 knockdown inhibited the metastatic potential of gastric cancer cells in vitro and in vivo. SALL4 induced epithelial–mesenchymal transition (EMT) in gastric cancer cells through the upregulation of TGF-β1 expression and the subsequent activation of TGF-β/SMAD signaling pathway. TGF-β1 knockdown abrogated the promoting role of SALL4 in gastric cancer metastasis. Our findings indicate that SALL4 promotes gastric cancer metastasis via direct regulation of TGF-β1, a strong EMT inducer, which represents a new mechanism for the oncogenic roles of SALL4 in cancer.
Materials and methods
Clinical specimens
A total of 19 paired gastric cancer and adjacent noncancerous tissues (5 cm away from the tumor edge) were obtained from the Department of General Surgery, the Affiliated People’s Hospital of Jiangsu University, between April 2015 and September 2016. Written informed consent was obtained from all the patients, and this study was approved by the Institutional Ethical Committee of Jiangsu University. All of the tissues were frozen in liquid nitrogen and then stored at −80°C for further use. The patients included in this study had not received any preoperative therapies.
Cell culture
Human gastric cancer cell lines MGC-803 and HGC-27 were purchased from the Institutes for Biological Sciences at the Chinese Academy of Sciences (Shanghai, China) and cultured in high-glucose Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum (FBS; Gibco®, Invitrogen Life Technologies, Carlsbad, CA, USA). All the cells were cultured in a humidified incubator with 5% CO2 at 37°C. For TGF-β1 inhibitor experiment, the cells were cultured in the presence or absence of Disitertide (HY-P0118, 50 µg/mL; MedChemExpress, Shanghai, China).
Gene transfection
The cells were seeded in 6-well plates at a density of 2×105/well and cultured in 37°C incubator overnight. The overexpressing plasmid and silencing siRNAs (GeneChem, Shanghai, China) were transfected into the cells by using LipoFiter™ transfection reagent (Hanbio, Shanghai, China) in a serum-free medium. The cells were changed to a complete medium at 6 hours after transfection and cultured for another 30 hours. The SALL4-targeting shRNA lentivirus was provided by GeneChem. The cells were transfected with lentivirus at a multiplicity of infection of 100 for 24 hours and then selected with puromycin (0.8 µg/mL) for 3 days. Table S1 lists the target sequences of shRNAs and siRNAs.
Luciferase reporter assay
In total, 293 T cells were cotransfected with SALL4-overexpressing plasmid and the luciferase reporter vector containing the promoter region of TGF-β1 as indicated. At 36 hours after transfection, the cells were collected and lysed. The luciferase activity was detected by using the dual luciferase assay kit (Promega, Madison, WI, USA).
Microarray analysis
RNA samples from control and SALL4-targeting shRNA-transfected MGC-803 cells (three samples/group) were collected and used for gene expression analysis by using Agilent Human lncRNA microarray v2.0 4 × 180K (OE Biotech, Shanghai, China).
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation assay was performed in MGC-803 cells by using a commercial kit (Millipore, Darmstadt, Germany). After cross-linking with 1% formaldehyde at 37°C for 10 minutes, the cells were harvested in sodium dodecyl sulfate lysis buffer, and the DNA was shredded to fragments of 200 bp by sonication. The precleared chromatin was incubated with the antibodies against SALL4 or nonspecific IgG overnight. Protein G Agarose beads were added and incubated at 4°C for 1 hour. After reversing the cross-links, the DNA was isolated and used for PCR. Table S2 shows the specific primers for PCR detection of the responsive element in TGF-b1 gene promoter.
Cell scratch assay
The confluent cell monolayers were wounded by scratching with a 10-µL pipette tip and then cultured for 36 hours. Cell migration over the scraped area was photographed at 0 and 36 hours, respectively.
Transwell migration assay
The transfected cells were collected and seeded into the upper chamber (8 µm) at a density of 1×105 cells/well (Corning Inc., Corning, NY, USA). The lower chamber was filled with 500 µL culture medium supplemented with 10% FBS; 12 hours later, the cells on the upper surface of the membrane were removed with a cotton swab. Then, the lower cells were fixed with formaldehyde and stained with crystal violet for 30 minutes. The number of migrated cells was counted under a microscope.
Matrigel invasion assay
The diluted basement Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) was added into each chamber and let to polymerize at 37°C for 30 minutes. The transfected cells were seeded into the upper chamber at a density of 2×105 cells/well. The lower chamber was filled with 500 µL culture medium supplemented with 10% FBS. The cells were allowed to invade to the lower membrane for 24 hours. Subsequently, the cells on the upper surface of the membrane were removed with a cotton swab. The lower cells were then fixed with formaldehyde and stained with crystal violet for 30 minutes. The number of migrated cells was counted under a microscope.
Quantitative real-time (qRT) PCR
Total RNA was extracted using TRIzol reagent (Invitrogen Life Technologies) and reverse-transcribed into cDNA using miScript reverse transcription kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). The relative expression of target genes was detected on a Bio-Rad CFX96 qRT-PCR system with the SYBR Green method (β-actin served as an internal control). Table S2 lists the sequences of the primers.
Western blot analysis
The cells were washed twice with PBS and lysed with radioimmunoprecipitation assay buffer containing 1% protease inhibitors. Equal amounts of proteins were separated on 12% sodium dodecyl sulfate–polyacrylamide gels and transferred onto polyvinylidene fluoride membranes, followed by blocking with 5% nonfat milk for 1 hour. The membranes were incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: anti-E-cadherin (4695S; Cell Signaling Technology, Beverly, MA, USA), anti-N-cadherin (4370S; Cell Signaling Technology), anti-Slug (9585S; Cell Signaling Technology), anti-Vimentin (5741S; Cell Signaling Technology), anti-Twist (46702S; Cell Signaling Technology), anti-p-SMAD2 (3108S; Cell Signaling Technology), SMAD2 (5399S; Cell Signaling Technology), anti-p-SMAD3 (9520S; Cell Signaling Technology), SMAD3 (9523S; Cell Signaling Technology), and anti-GAPDH (MB001; Bioworld Technology, St. Louis Park, MN, USA). After incubation with the secondary antibodies (Bioworld Technology) at 37°C for 1 hour, the bands were visualized with a chemiluminescent detection system.
Animal study
BALB/c nude mice aged 4–6 weeks were purchased from the Shanghai Laboratory Animal Center (Shanghai, China) and maintained in accordance with the institutional policies. Control or sh-SALL4 MGC-803 cells were collected in PBS and intraperitoneally injected into the mice (2×106 cells/mice, n=5). At 6 weeks after injection, the mice were sacrificed, and the number of metastatic nodules was counted. The protocol was approved by the Animal Use and Care Committee of Jiangsu University.
Immunofluorescence
For immunofluorescent staining, the cells seeded on cover slips were fixed and incubated with primary monoclonal antibody against N-cadherin and p-SMAD3 (Cell Signaling Technology) followed by incubation with fluorescence-labeled secondary antibody for 30 minutes at room temperature. The cells were counterstained with Hoechst33342 for 30 seconds. Finally, the cells were photographed under a microscope (DeltaVision OMX SR; GE Healthcare BioSciences, Piscataway, NJ, USA).
Statistical analysis
All the results were expressed as mean ± SD. Statistical analyses were performed using Student’s t-test with GraphPad Prism Version 5.0 software (GraphPad Software, La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
SALL4 gene silencing inhibits, while gene overexpression promotes the migration and invasion of gastric cancer cells
To test the importance of SALL4 in gastric cancer metastasis, we knocked down SALL4 gene expression by lentivirus-mediated shRNA in MGC-803 cells (with a relatively high level of SALL4). The efficiency of gene silencing by SALL4 shRNA was verified by qRT-PCR and Western blot (Figure 1A). Cell scratch assay results showed that SALL4 knockdown reduced the motility of MGC-803 cells (Figure 1B). Transwell migration and Matrigel invasion assay results showed that MGC-803 cells in SALL4 knockdown group had lower migration rate and invasion abilities than that in the control group (Figure 1C). On the contrary, SALL4 overexpression by plasmid transfection promoted the migration and invasion abilities of HGC-27 cells (with a relatively low level of SALL4; Figure 1D–F). These data suggest that SALL4 knockdown inhibits, while overexpression promotes the migration and invasion of gastric cancer cells.
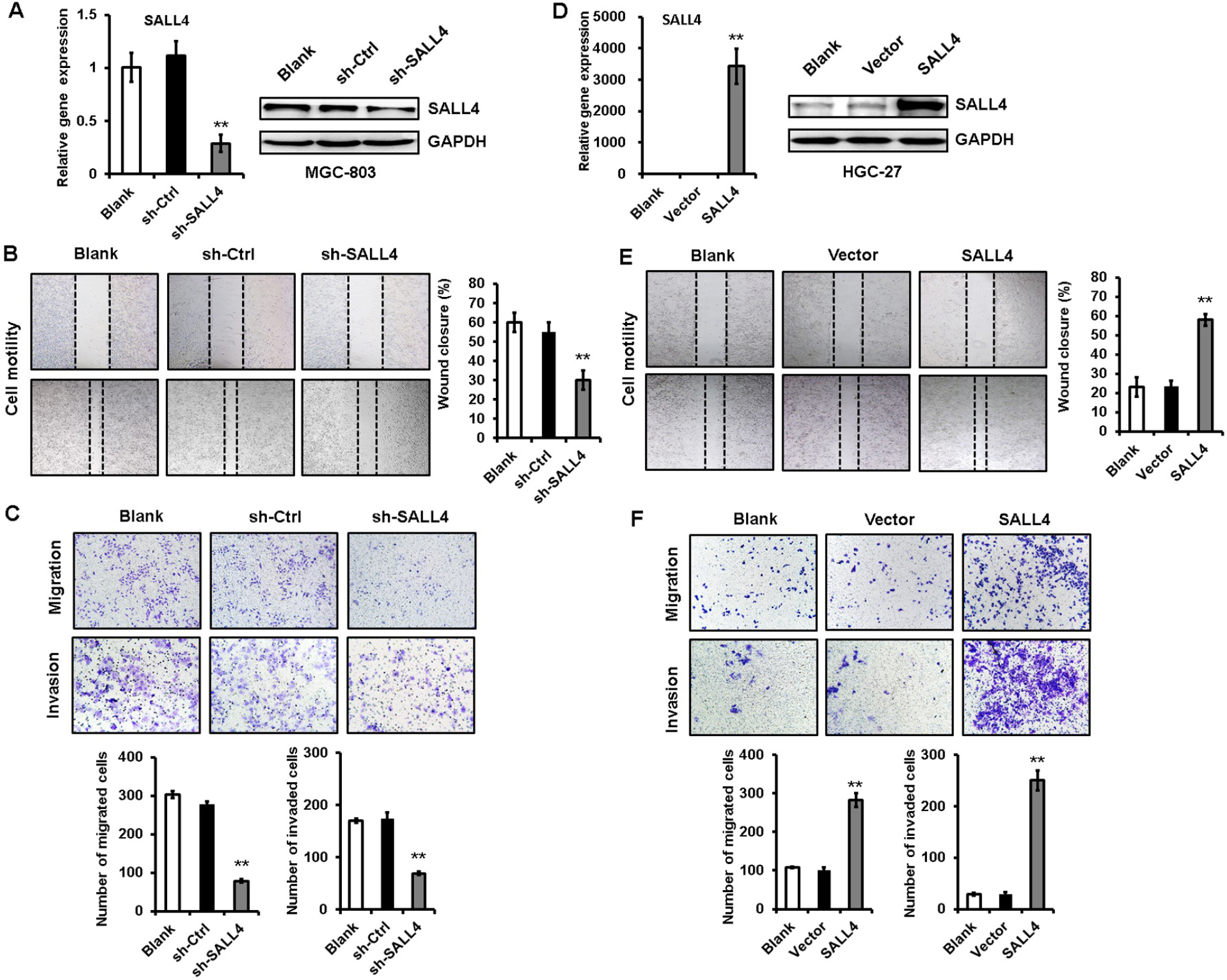 | Figure 1 SALL4 knockdown inhibits, while overexpression promotes the migration and invasion of gastric cancer cells. Notes: (A) The efficiency of SALL4 knockdown was verified by using qRT-PCR and Western blot. (B) Cell scratch assay for the motility of MGC-803 cells with SALL4 gene knockdown. (C) Transwell migration and Matrigel invasion assays for the migration and invasion abilities of MGC-803 cells with SALL4 gene knockdown. (D) The efficiency of SALL4 overexpression was verified by using qRT-PCR and Western blot. (E) Cell scratch assay for the motility of SALL4-overexpressing HGC-27 cells. (F) Transwell migration and Matrigel invasion assays for the migration and invasion abilities of SALL4-overexpressing HGC-27 cells. **P<0.01. Abbreviation: qRT-PCR, quantitative real-time PCR. |
SALL4 gene silencing suppresses, while gene overexpression promotes EMT in gastric cancer cells
EMT has been linked to enhanced metastatic potential in gastric cancer cells. To test whether SALL4 promotes gastric cancer cell migration and invasion by inducing EMT, we observed the morphology changes of SALL4-overexpressing and SALL4-silencing gastric cancer cells and detected the expression of EMT-related genes and proteins in these cells. As shown in Figure 2A, MGC-803 cells in the control group displayed a mesenchymal-like phenotype, while those in SALL4 knockdown group displayed an epithelial-like phenotype. On the contrary, HGC-27 cells in the control group displayed an epithelial-like phenotype, while those in SALL4 overexpression group displayed a mesenchymal-like phenotype. In addition, SALL4 knockdown increased the expression level of epithelial marker E-cadherin and decreased that of mesenchymal markers N-cadherin and Vimentin in MGC-803 cells (Figure 2B and C). SALL4 knockdown also reduced the expression levels of EMT transcription factors Slug and Twist. However, SALL4 overexpression led to the decreased level of E-cadherin and increased levels of N-cadherin, Vimentin, Slug, and Twist in HGC-27 cells (Figure 2D and E). The results of fluorescent staining further confirmed a decreased expression of N-cadherin in the SALL4 knockdown group while an increased expression of N-cadherin in the SALL4 overexpression group compared with the control group (Figure 2F). These findings indicate that SALL4 knockdown suppresses, while overexpression induces EMT in gastric cancer cells.
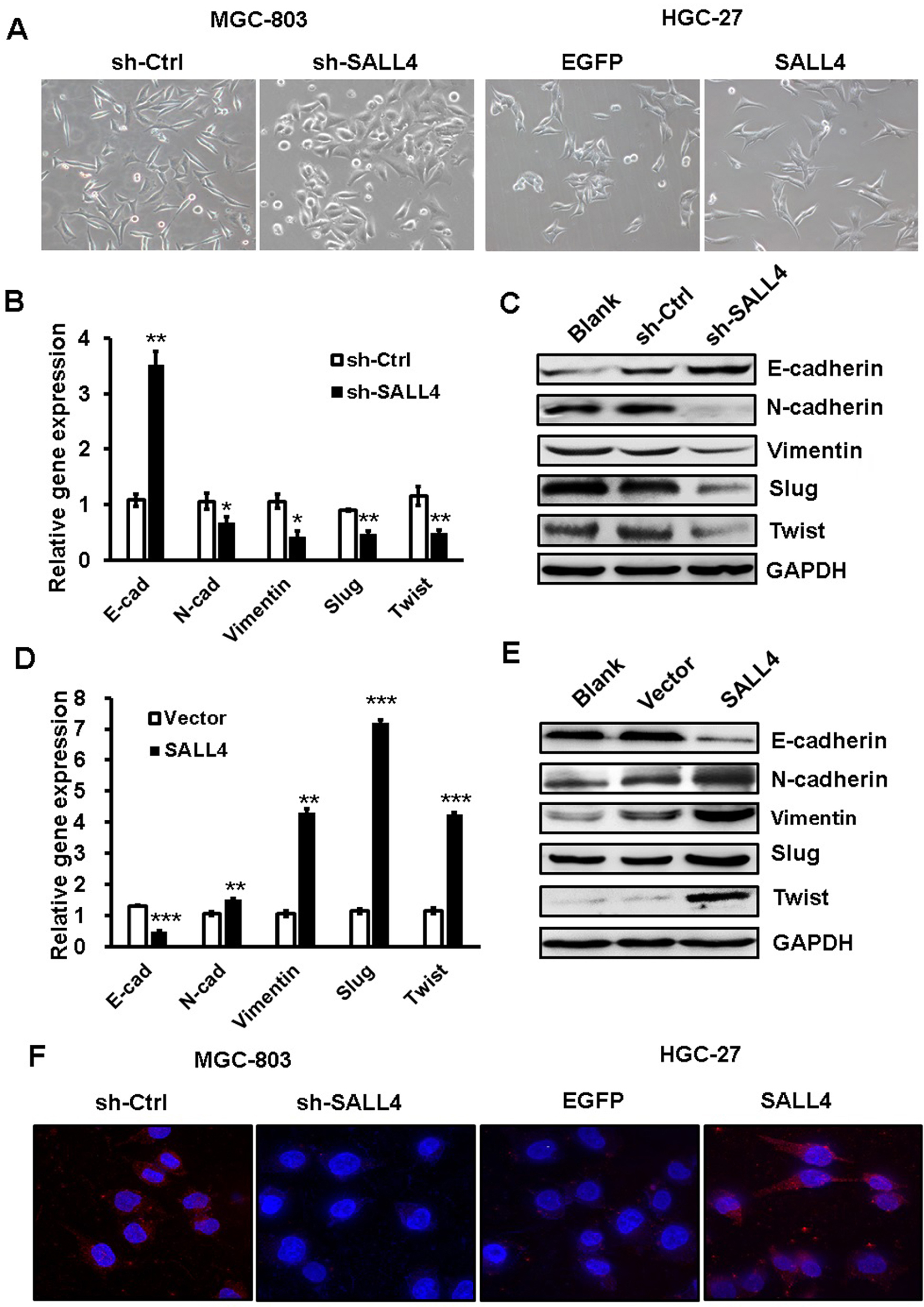 | Figure 2 SALL4 knockdown suppresses, while overexpression induces EMT in gastric cancer cells. Notes: (A) The morphology changes in SALL4-depleted MGC-803 cells and SALL4-overexpressing HGC-27 cells. (B, C) The expression of E-cadherin, N-cadherin, Vimentin, Slug, and Twist in SALL4-depleted MGC-803 cells was determined by using qRT-PCR (B) and Western blot (C). (D, E) qRT-PCR (D) and Western blot (E) assays for the expression of E-cadherin, N-cadherin, Vimentin, Slug, and Twist in SALL4-overexpressing HGC-27 cells. (F) Immunofluorescent staining of N-cadherin in SALL4-depleted MGC-803 cells and SALL4-overexpressing HGC-27 cells. *P<0.05, **P<0.01, ***P<0.001. Abbreviations: EMT, epithelial–mesenchymal transition; qRT-PCR, quantitative real-time PCR. |
TGF-β1 is identified as a downstream target of SALL4
To demonstrate the mechanism for SALL4-mediated induction of EMT and the promotion of gastric cancer cell migration and invasion, we performed microarray to determine the differentially expressed genes between control and SALL4 knockdown MGC-803 cells. As shown in Figure 3A, 1,539 differentially expressed genes were identified, of which 1,001 genes were upregulated and 538 genes were downregulated. Gene ontology and KEGG pathway analysis results showed that SALL4 was associated with cell adhesion molecules, calcium signaling pathway, and glycolysis (data not shown). In the downregulated genes, we choose TGF-β1 as the potential target for further study as it is a well-known EMT inducer. In consistent with the results of microarray, qRT-PCR and Western blot results confirmed that TGF-β1 expression was decreased in SALL4 knockdown MGC-803 cells (Figure 3B). We confirmed the regulation of TGF-β1 by SALL4 using luciferase reporter and chromatin immunoprecipitation (ChIP) assays. Luciferase reporter assay results showed that SALL4 overexpression increased the luciferase activities of TGF-b1 gene promoter (Figure 3C). ChIP assay results showed that SALL4 could bind to the 540- to −301-bp region of the promoter of TGF-b1 gene (Figure 3D). Finally, we determined the expression of SALL4 and TGF-β1 genes in gastric cancer tissues and found that the expression levels of SALL4 and TGF-β1were closely associated (Figure 3E). Our results suggest that SALL4 may bind to TGF-b1 gene promoter and transactivate its expression.
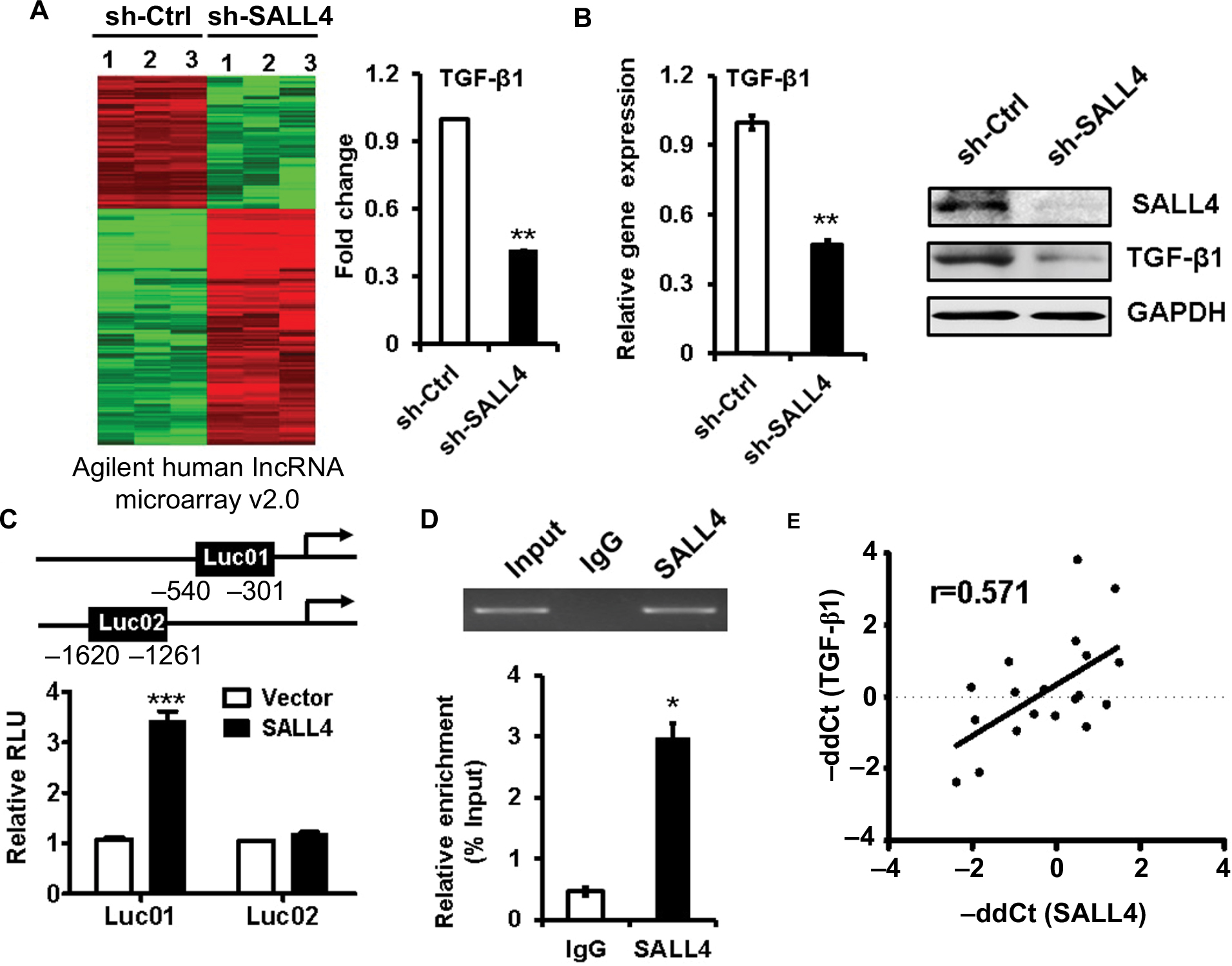 | Figure 3 TGF-β1 is identified as a downstream target of SALL4. Notes: (A) The differentially expressed genes between sh-Ctrl- and sh-SALL4-transfected MGC-803 cells were determined by using microarray analyses. The relative fluorescence intensity for TGF-β1 gene was shown. (B) qRT-PCR and Western blot assays for the expression of TGF-β1 gene in MGC-803 cells with SALL4 gene knockdown. (C) Luciferase reporter assays for the relative luciferase activity of TGF-β1 gene promoter. (D) ChIP assay for the binding of SALL4 to the promoter region of TGF-β1 gene. (E) Correlation analysis of SALL4 and TGF-β1 expression in 19 paired gastric cancer and adjacent noncancerous tissues. *P<0.05, **P<0.01, ***P<0.001. Abbreviations: ChIP, Cardiovascular Health Improvement Project; qRT-PCR, quantitative real-time PCR. |
SALL4 gene silencing suppresses, while gene overexpression promotes the activation of TGF-β/SMAD signaling pathway
TGF-β induces EMT mainly through SMAD signaling pathway. We next wanted to know the effects of SALL4 on SMAD signaling pathway. qRT-PCR and Western blot results showed that SALL4 knockdown inhibited the expression of TGF-β1 and the phosphorylation of SAMD2 and SMAD3 in MGC-803 cells (Figure 4A and B). By contrast, SALL4 overexpression enhanced the expression of TGF-β1 and the phosphorylation of SAMD2 and SMAD3 in HGC-27 cells (Figure 4C and D). The results of immunofluorescent staining further confirmed the decreased nuclear translocation of p-SMAD3 in SALL4-silencing MGC-803 cells while the increased nuclear translocation of p-SMAD3 in SALL4-overexpressing HGC-27 cells compared with control cells (Figure 4E). These results suggest that SALL4 could control the activation of TGF-β/SMAD signaling pathway.
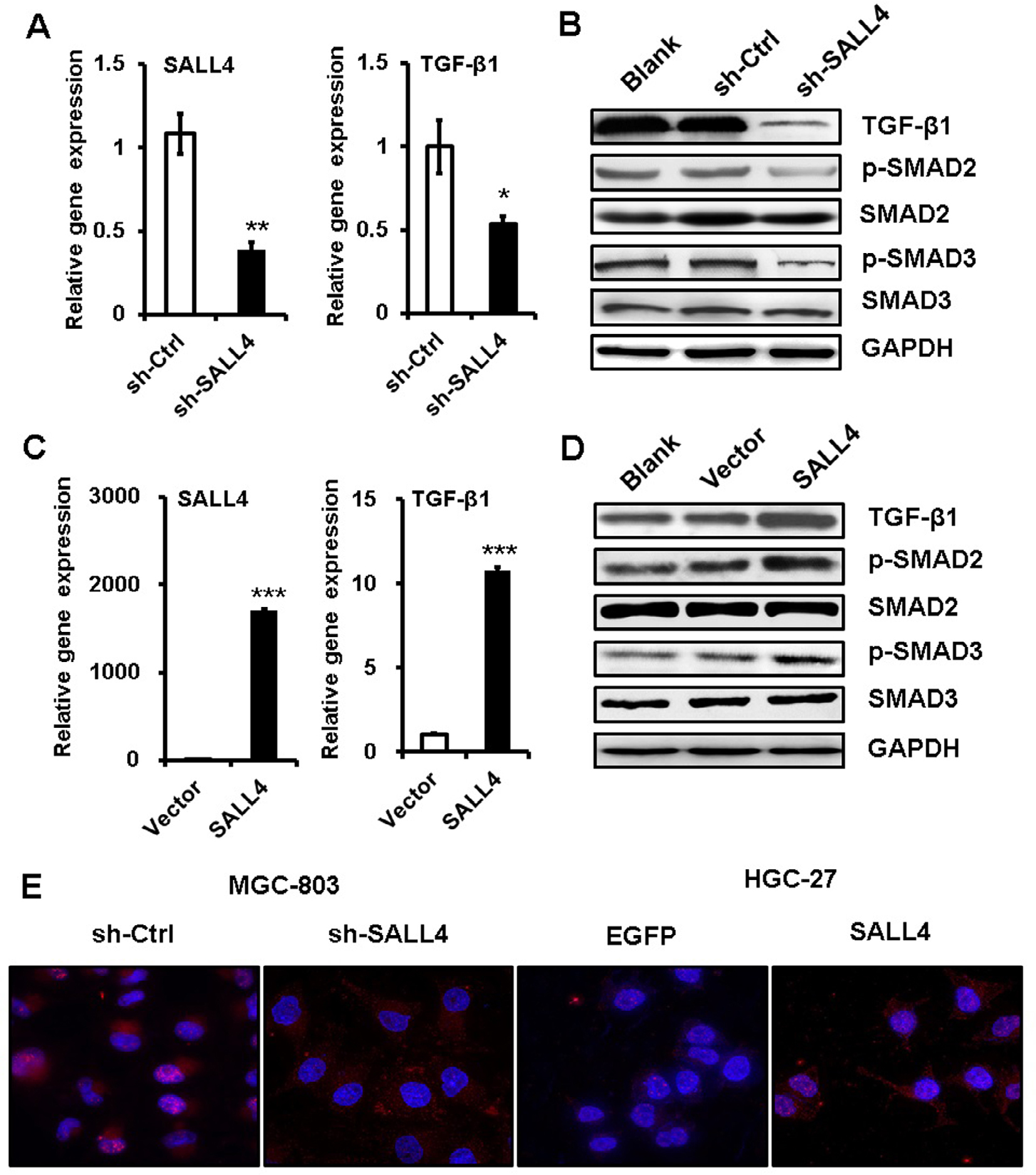 | Figure 4 SALL4 regulates the activation of TGF-β/SMAD signaling pathway. Notes: (A) qRT-PCR analyses of SALL4 and TGF-β1 expression in SALL4-depleted MGC-803 cells. (B) Western blot assays for the expression of phosphorylated SMA2/3 in SALL4-depleted MGC-803 cells. (C) qRT-PCR analyses of SALL4 and TGF-β1 expression in SALL4-overexpressing HGC-27 cells. (D) Western blot assays for the expression of phosphorylated SMA2/3 in SALL4-overexpressing HGC-27 cells. (E) Immunofluorescent staining of p-SMAD3 in SALL4-depleted MGC-803 cells and SALL4-overexpressing HGC-27 cells. *P<0.05, **P<0.01, ***P<0.001. Abbreviation: qRT-PCR, quantitative real-time PCR. |
TGF-β1 inhibition abrogates the promotion of gastric cancer cell migration and invasion by SALL4 overexpression
To demonstrate the importance of TGF-β1 in the oncogenic roles of SALL4 in gastric cancer, we depleted TGF-β1 expression in SALL4-overexpressing HGC-27 cells by using siRNA. The efficiency of TGF-β1 gene knockdown was shown in Figure 5A and B. Compared with SALL4-overexpressing group, TGF-β1 gene knockdown group displayed reduced abilities of cell motility, migration, and invasion (Figure 5C and E). In addition, we used a TGF-β1 inhibitor (HY-P0118) to block the function of TGF-β1 in SALL4-overexpressing HGC-27 cells. The results of wound healing, transwell migration, and Matrigel invasion assays showed that HY-P0118 treatment remarkably reversed the cell motility, migration, and invasion abilities of SALL4-overexpressing HGC-27 cells (Figure 5D and F). We also found that the induced EMT phenotype and activation of TGF-β/SMAD signaling pathway in gastric cancer cells by SALL4 overexpression were reversed by TGF-β1 gene knockdown (Figure 5G and H), indicating that SALL4 promotes gastric cancer cell migration and invasion and induces EMT through the upregulation of TGF-β1.
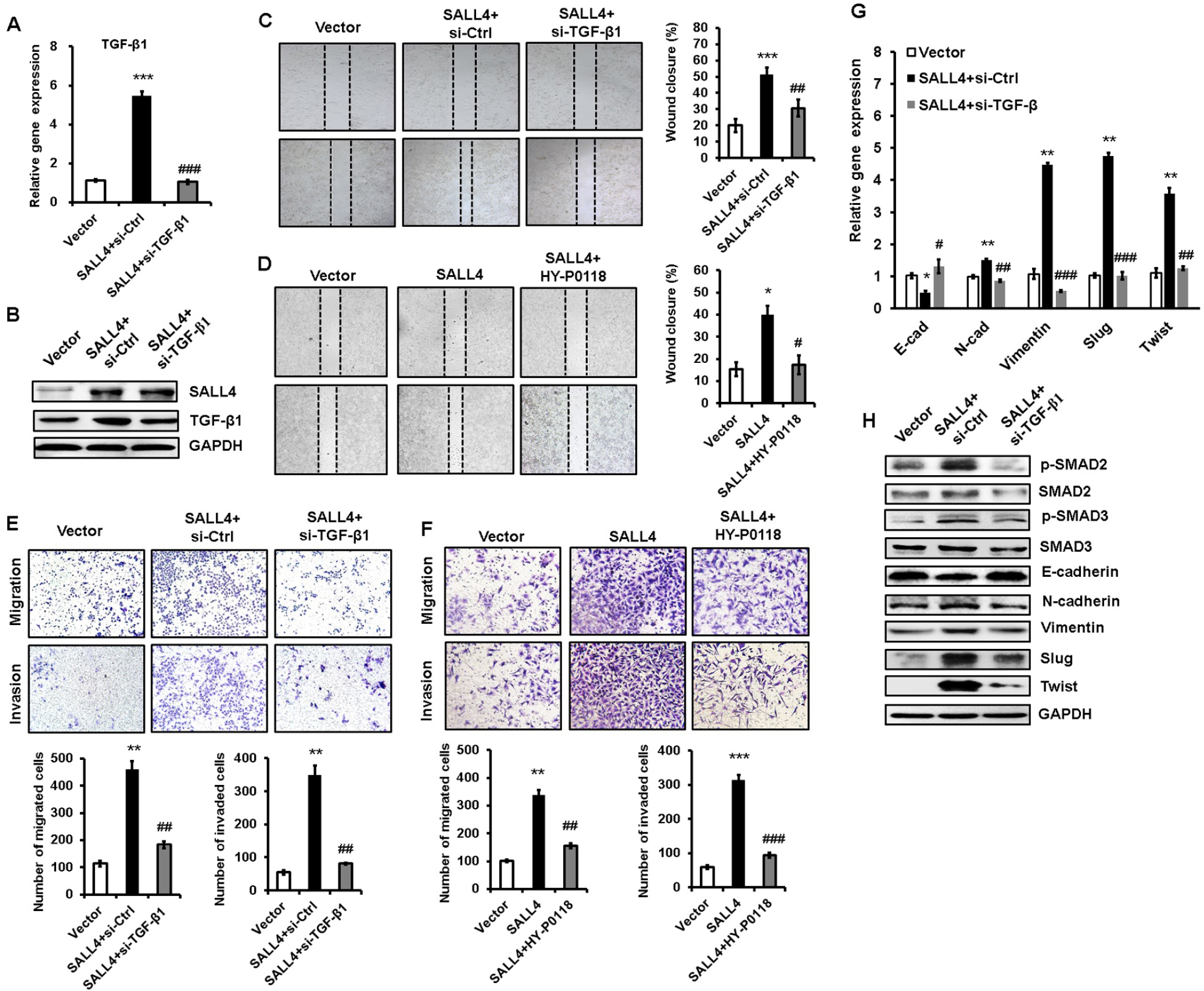 | Figure 5 TGF-β1 mediates the promoting role of SALL4 in gastric cancer cell migration and invasion. Notes: (A, B) qRT-PCR (A) and Western blot (B) analyses of TGF-β1 expression in SALL4-overexpressing HGC-27 cells with or without TGF-β1 knockdown. (C, D) Cell scratch assay for the motility of SALL4-overexpressing HGC-27 cells with or without TGF-β1 knockdown (C) or HY-P0118 treatment (D). (E, F) Transwell migration and Matrigel invasion assays for the migration and invasion abilities of SALL4-overexpressing HGC-27 cells with or without TGF-β1 knockdown (E) or HY-P0118 treatment (F). (G, H) qRT-PCR (G) and Western blot (H) analyses of EMT markers and SMAD2/3 in SALL4-overexpressing HGC-27 cells with or without TGF-β1 knockdown. *P<0.05, **P<0.01, ***P<0.001, compared to vector group; #P<0.05, ##P<0.01, ###P<0.001, compared to SALL4 group. Abbreviations: EMT, epithelial–mesenchymal transition; qRT-PCR, quantitative real-time PCR. |
SALL4 knockdown inhibits the peritoneal metastasis of gastric cancer in vivo
To further demonstrate the role of SALL4 in gastric cancer metastasis, we established a peritoneal metastasis model in nude mice. MGC-803 cells with or without SALL4 gene knockdown were injected into the abdominal cavity of nude mice. At 6 weeks after injection, the mice were sacrificed, and the metastatic tumor nodes in the abdomen were carefully examined. As shown in Figure 6A, SALL4 knockdown reduced the number of metastatic tumor nodes in mice. Hematoxylin and eosin staining results showed that there were more hepatic metastases in mice from the control group than that from the SALL4 knockdown group (Figure 6B). qRT-PCR and Western blot results showed that the expression of TGF-β1, p-SMAD2/3, N-cadherin, Vimentin, Slug, and Twist was decreased, while that of E-cadherin was increased in the SALL4 knockdown group (Figure 6C and D). These data suggest that SALL4 promotes gastric cancer metastasis via the activation of TGF-β/SMAD signaling pathway and induction of EMT.
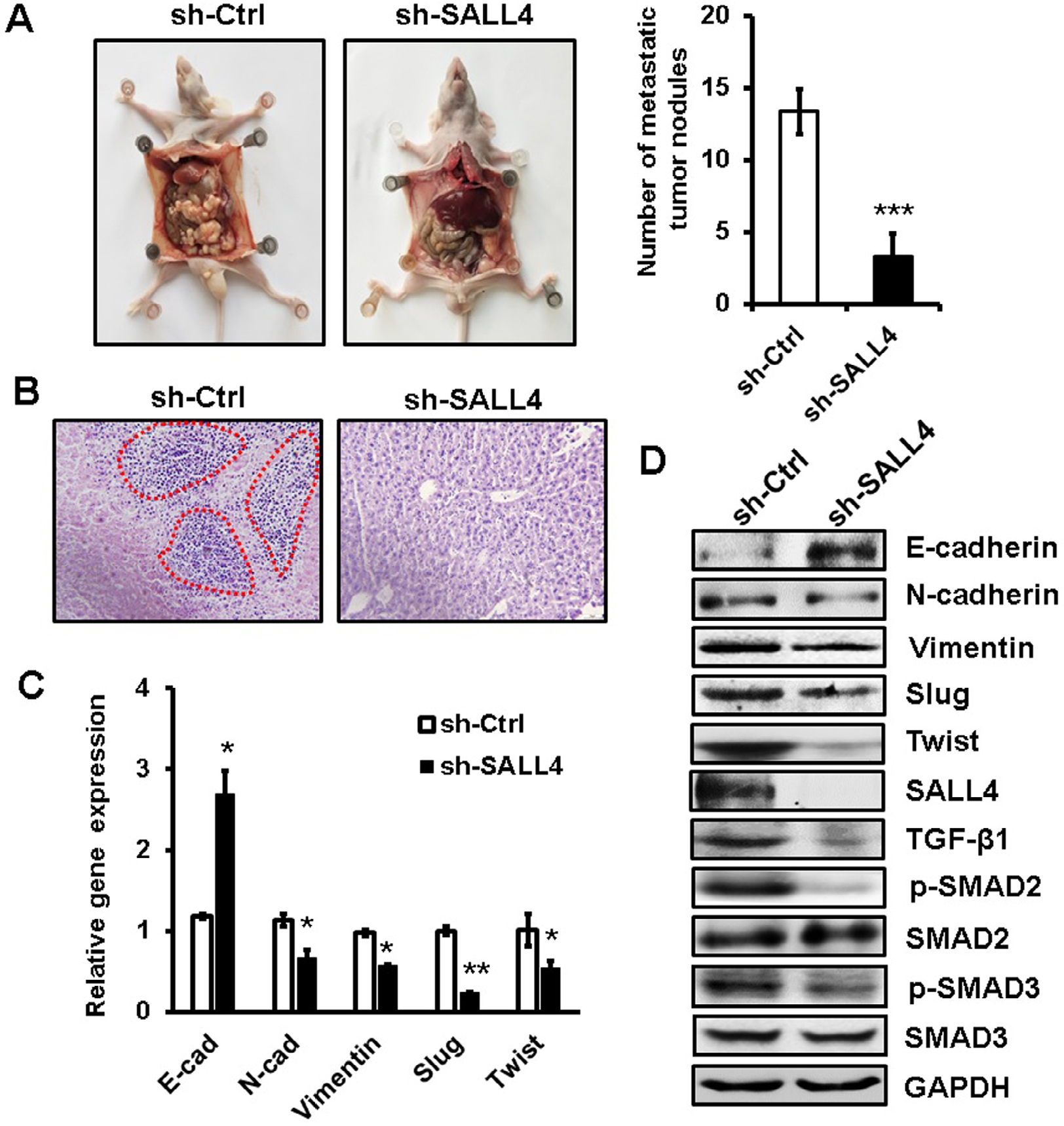 | Figure 6 SALL4 knockdown inhibits the peritoneal metastasis of gastric cancer in vivo. Notes: (A) The number of metastatic tumor nodes in the abdomen of mice from the control and SALL4 knockdown groups. (B) H&E staining of liver tissues in mice from the control and SALL4 knockdown groups. (C) qRT-PCR analyses of EMT markers in mouse metastatic tumor nodules from the control and SALL4 knockdown groups. (D) Western blot assays for EMT marker and SMAD2/3 expression in mouse metastatic tumor nodules from the control and SALL4 knockdown groups. *P<0.05, **P<0.01, ***P<0.001. Abbreviations: EMT, epithelial–mesenchymal transition; H&E, hematoxylin and eosin; qRT-PCR, quantitative real-time PCR. |
Discussion
In this study, we demonstrate that SALL4 plays a critical role in gastric cancer metastasis. SALL4 overexpression promoted, while SALL4 knockdown inhibited the metastatic potential of gastric cancer cells in vitro and in vivo. Mechanistically, SALL4 bound to the promoter region of TGF-b1 gene and activated its expression, resulting in the activation of TGF-β/SMAD signaling pathway and the induction of EMT. We further provided evidence that TGF-β1 knockdown reversed SALL4 overexpression-mediated promotion of gastric cancer cell migration and invasion, suggesting that TGF-β1 acts as a downstream mediator of SALL4’s function in gastric cancer metastasis. Although the involvement of SALL4 in regulating TGF-β pathway has been implicated in AML,6 to the best of our knowledge, this is the first report to suggest TGF-β1 as a direct target of SALL4 and to reveal the functional importance of TGF-β1 regulation to the oncogenic roles of SALL4 in cancer.
EMT is a biological process in which cancer cells lose their epithelial features and gain mesenchymal markers, which enables tumor cells to become more migratory and invasive.20 The hallmark of EMT is the decrease in E-cadherin expression, accompanied by an increase in N-cadherin or Vimentin expression. The process of EMT is controlled by transcription factors such as Slug, Snail, ZEB1, and Twist, as well as certain signaling pathways, including TGF-β/SMAD and Wnt/β-catenin. Therefore, a further exploration of the regulatory mechanism for EMT in cancer metastasis has a great significance. He et al demonstrated that the inhibition of SALL4 reduces tumorigenicity involving EMT via Wnt/β-catenin pathway in esophageal squamous cell carcinoma.21 Liu et al suggested that SALL4 acts as an EMT and drug resistance inducer through the regulation of c-Myc in endometrial cancer.22 Itou et al showed that SALL4 suppresses the expression of E-cadherin while it increases the expression of ZEB1, maintaining cell dispersion in basal-like breast cancer.23 They further demonstrated that SALL4 upregulates integrin α6β1 expression to promote cell migration via the activation of focal adhesion dynamics in basal-like breast cancer cells.24 In this study, we showed that SALL4 overexpression increased TGF-β1 expression and activated TGF-β/SMAD signaling, suggesting that SALL4 acts as a potent EMT inducer via the regulation of distinct gene expression and signaling pathway activation.
SALL4 is involved in tumorigenesis, tumor growth, and tumor metastasis and drug resistance through the regulation of various downstream genes. In AML, Bmi-1 is identified as a target gene of SALL4. SALL4 overexpression induces increased levels of histones H3-K4 and H3-K79 methylation in the Bmi-1 gene promoter, which finally activates Bmi-1 expression.25 There is a strong correlation between SALL4 and Bmi-1 expression in human AML samples. SALL4 has been shown to interact with mixed-lineage leukemia (MLL) and co-occupies the HOXA9 promoter region with MLL in AML cells, leading to enhanced histone activation markers at the HOXA9 promoter region as well as increased HOXA9 expression.26 Li et al suggested that c-Myc is a key target gene of SALL4 in endometrial cancer, which could partially mediate the roles of SALL4 in metastasis and drug resistance.11 Kim et al demonstrated that SALL4 recruits ubiquitin E3 ligase CUL4B to destabilize heterochromatin protein 1α, promoting Glut1 expression and glycolysis and inducing drug resistance by enhancing DNA repair.27 We recently reported that the expression of CD44 and DANCR was controlled by SALL4 in gastric cancer cells to regulate their proliferation, migration, and invasion.28,29 We found in this study that the expression of SALL4 and TGF-β1 was positively correlated in gastric cancer tissues. The elevated expression of TGF-β1 has been linked with a worse overall survival in gastric cancer patients.30 The outcomes of gastric cancer patients with simultaneously high levels of SALL4 and TGF-β1 warrant a further study. Moreover, whether other cofactors are involved in the regulation of TGF-β1 by SALL4 and the epigenetic changes in SALL4 binding site in the TGF-b1 gene promoter need to be investigated in future studies.
Conclusion
In conclusion, our findings show that SALL4 promotes gastric cancer metastasis via the upregulation of TGF-β1 and the activation of TGF-β/SMAD signaling pathway, inducing EMT and enhancing migration and invasion abilities in gastric cancer cells. Our study not only reveals a new mechanism for the oncogenic roles of SALL4 in cancer, but also provides evidence for the potential of SALL4 as a diagnostic and therapeutic target for gastric cancer.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (grant numbers: 81672416 and 81572075), the Key Research and Development Project of Zhenjiang (grant number: SH2015034), the Major Research and Development Project of Jiangsu Province (grant number: BE2015667), the Starting Foundation for Senior Talents of Jiangsu University (grant number: 13JDG086), the 333 Project of Jiangsu Province, and the Foundation for Young Academic Leader of Jiangsu University.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654–2664. | ||
Zhang J, Tam WL, Tong GQ, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol. 2006;8(10):1114–1123. | ||
Oikawa T, Kamiya A, Kakinuma S, et al. Sall4 regulates cell fate decision in fetal hepatic stem/progenitor cells. Gastroenterology. 2009;136(3):1000–1011. | ||
Ma Y, Cui W, Yang J, et al. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood. 2006;108(8):2726–2735. | ||
Yang J, Chai L, Gao C, et al. SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood. 2008;112(3):805–813. | ||
Oikawa T, Kamiya A, Zeniya M, et al. Sal-like protein 4 (SALL4), a stem cell biomarker in liver cancers. Hepatology. 2013;57(4):1469–1483. | ||
Zeng SS, Yamashita T, Kondo M, et al. The transcription factor SALL4 regulates stemness of EpCAM-positive hepatocellular carcinoma. J Hepatol. 2014;60(1):127–134. | ||
Cheng J, Deng R, Wu C, et al. Inhibition of SALL4 suppresses carcinogenesis of colorectal cancer via regulating Gli1 expression. Int J Clin Exp Pathol. 2015;8(9):10092–10101. | ||
Kobayashi D, Kuribayshi K, Tanaka M, Watanabe N. SALL4 is essential for cancer cell proliferation and is overexpressed at early clinical stages in breast cancer. Int J Oncol. 2011;38(4):933–939. | ||
Li A, Jiao Y, Yong KJ, et al. SALL4 is a new target in endometrial cancer. Oncogene. 2015;34(1):63–72. | ||
Kobayashi D, Kuribayashi K, Tanaka M, Watanabe N. Overexpression of SALL4 in lung cancer and its importance in cell proliferation. Oncol Rep. 2011;26(4):965–970. | ||
Yanagihara N, Kobayashi D, Kuribayashi K, Tanaka M, Hasegawa T, Watanabe N. Significance of SALL4 as a drugresistant factor in lung cancer. Int J Oncol. 2015;46(4):1527–1534. | ||
Zhang L, Yan Y, Jiang Y, et al. The expression of SALL4 in patients with gliomas: high level of SALL4 expression is correlated with poor outcome. J Neurooncol. 2015;121(2):261–268. | ||
Yong KJ, Gao C, Lim JS, et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med. 2013;368(24):2266–2276. | ||
Lu J, Jeong HW, Jeong H, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One. 2009;4(5):e5577. | ||
Zhang X, Yuan X, Zhu W, Qian H, Xu W. SALL4: an emerging cancer biomarker and target. Cancer Lett. 2015;357(1):55–62. | ||
Gao C, Dimitrov T, Yong KJ, et al. Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood. 2013;121(8):1413–1421. | ||
Zhang L, Xu Z, Xu X, et al. SALL4, a novel marker for human gastric carcinogenesis and metastasis. Oncogene. 2014;33(48):5491–5500. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
He J, Zhou M, Chen X, et al. Inhibition of SALL4 reduces tumorigenicity involving epithelial-mesenchymal transition via Wnt/β-catenin pathway in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2016;35(1):98. | ||
Liu L, Zhang J, Yang X, Fang C, Xu H, Xi X. SALL4 as an epithelial-mesenchymal transition and drug resistance inducer through the regulation of c-Myc in endometrial cancer. PLoS One. 2015;10(9):e0138515. | ||
Itou J, Matsumoto Y, Yoshikawa K, Toi M. Sal-like 4 (SALL4) suppresses CDH1 expression and maintains cell dispersion in basal-like breast cancer. FEBS Lett. 2013;587(18):3115–3121. | ||
Itou J, Tanaka S, Li W, et al. The Sal-like 4 – integrin α6β1 network promotes cell migration for metastasis via activation of focal adhesion dynamics in basal-like breast cancer cells. Biochim Biophys Acta. 2017;1864(1):76–88. | ||
Yang J, Chai L, Liu F, et al. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci U S A. 2007;104(25):10494–10499. | ||
Li A, Yang Y, Gao C, et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J Clin Invest. 2013;123(10):4195–4207. | ||
Kim J, Xu S, Xiong L, Yu L, Fu X, Xu Y. SALL4 promotes glycolysis and chromatin remodeling via modulating HP1α-Glut1 pathway. Oncogene. 2017;36(46):6472–6479. | ||
Yuan X, Zhang X, Zhang W, et al. SALL4 promotes gastric cancer progression through activating CD44 expression. Oncogenesis. 2016;5(11):e268. | ||
Pan L, Liang W, Gu J, et al. Long noncoding RNA DANCR is activated by SALL4 and promotes the proliferation and invasion of gastric cancer cells. Oncotarget. 2018;9(2):1915–1930. | ||
Hawinkels LJ, Verspaget HW, van Duijn W, et al. Tissue level, activation and cellular localisation of TGF-beta1 and association with survival in gastric cancer patients. Br J Cancer. 2007;97(3):398–404. |
Supplementary materials
 | Table S1 Sequences of shRNA and siRNA |
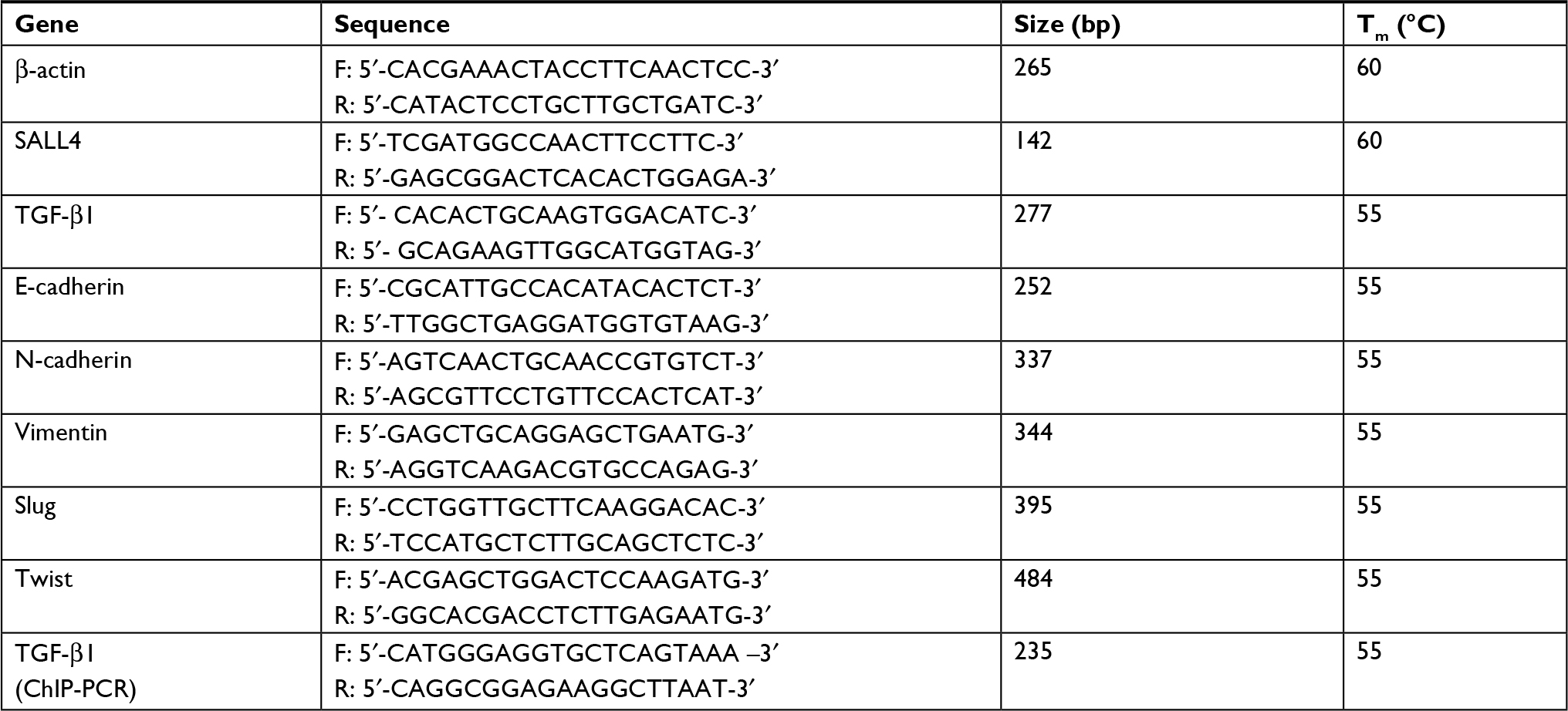 | Table S2 Sequences of PCR primers for target gene detection Abbreviation: ChIP, Chromatin immunoprecipitation. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.