Back to Journals » Journal of Inflammation Research » Volume 15
Research Progress on the Mechanism of Sepsis Induced Myocardial Injury
Authors Bi CF, Liu J, Yang LS ![]() , Zhang JF
, Zhang JF ![]()
Received 10 May 2022
Accepted for publication 19 July 2022
Published 26 July 2022 Volume 2022:15 Pages 4275—4290
DOI https://doi.org/10.2147/JIR.S374117
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Cheng-Fei Bi,1– 3,* Jia Liu,2,4,* Li-Shan Yang,1 Jun-Fei Zhang1– 3
1Department of Emergency Medical, General Hospital of Ningxia Medical University, Yinchuan, People’s Republic of China; 2School of Clinical Medicine, Ningxia Medical University, Yinchuan, People’s Republic of China; 3Key Laboratory of Hui Ethnic Medicine Modernization, Ministry of Education, Ningxia Medical University, Yinchuan, People’s Republic of China; 4Medical Experimental Center, General Hospital of Ningxia Medical University, Yinchuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li-Shan Yang; Jun-Fei Zhang, Email [email protected]; [email protected]
Abstract: Sepsis is an abnormal condition with multiple organ dysfunctions caused by the uncontrolled infection response and one of the major diseases that seriously hang over global human health. Besides, sepsis is characterized by high morbidity and mortality, especially in intensive care unit (ICU). Among the numerous subsequent organ injuries of sepsis, myocardial injury is one of the most common complications and the main cause of death in septic patients. To better manage septic inpatients, it is necessary to understand the specific mechanisms of sepsis induced myocardial injury (SIMI). Therefore, this review will elucidate the pathophysiology of SIMI from the following certain mechanisms: apoptosis, mitochondrial damage, autophagy, excessive inflammatory response, oxidative stress and pyroptosis, and outline current therapeutic strategies and potential approaches in SIMI.
Keywords: sepsis induced myocardial injury, mechanism, signaling pathway
Introduction
Sepsis is an abnormal compound condition of life-threatening organ dysfunction caused by maladjustment of infection response and one of the major diseases that seriously hang over global human health. The third international consensus contends that septic shock is a subset of sepsis and its identification in adult adhere to clinical hypotension criteria, defines sepsis as an uncontrolled response of the host to infection.1 Some scholars confirmed the higher effectiveness of the third version consensus in predicting septic shock through analysis of the ICU database in England.2 Septic patients’ mortality in ICU decreased from 35.0% in 2000 to 18.4% in 2012.3 Although the in-hospital mortality of sepsis has decreased significantly worldwide, the survival rate after discharge is still low due to secondary organ damage.4 A meta-analysis of 13 international electronic databases showed that sepsis is characterized by high morbidity and mortality, and these data are dominated by high-income countries.5 However, current surveillance data on sepsis in low- and middle-income countries are still scarce.6 It can be seen that we still have an onerous task to study sepsis. In particular, certain groups of people (such as pregnant women, infants, the elderly, and those with immune deficiency) are at higher risk of sepsis because of their immune function is relatively weak.7,8 In recent years, despite significant advances in anti-infective therapy and organ function support technologies, infection still remains the leading cause of death worldwide.9 Sepsis is also a common cause of death in patients in ICU. Due to under-reporting of information, the real situation of high septic mortality is greatly inconsistent with the reported status quo.10 Then the diagnosis and treatment of sepsis are complicated due to its characteristics of multiple etiologies.11 So it is particularly important to study the mechanism of sepsis, which lays the solid foundation for the therapeutic prospect of sepsis in our country.
The early manifestation of sepsis is systemic inflammatory response syndrome (SIRS), which can be cured by supportive therapy.12 When severe sepsis develops, however, multiple organ damages occur and may still result in death after proper treatment.12 As the major organ regularly compromised by sepsis and always damaged by septic shock, the heart and its injury during sepsis have been studied in clinical and basic study over the years.13 Cardiac dysfunction, one of the common complications of sepsis, has attracted more and more attention in the field of ICU’s management in recent years.14 Sepsis induced multiple organ dysfunction is the main reason for the high incidence and fatality of sepsis in ICU.15 So, it can be seen that some septic patients may die in large part from the secondary cardiac dysfunction. The identification, diagnosis, treatment, and prognosis of infectious cardiomyopathy, a common complication of sepsis, remain the huge challenge for physicians in ICU.16 The significantly reduced cardiac function induced by sepsis is closely related to excessive inflammatory response, oxidative stress, apoptosis and limited autophagy.17 Obviously, the autophagy-oxidative stress-inflammation-cell death process provides a potential therapeutic strategy for cardio-protection in sepsis.18 Based on this, the summary of basic research progress on SIMI will be conducive to the improvement of its therapeutic strategy.
At present, accumulating studies on the mechanism of SIMI treatment have involved foregoing aspects.19 However, these research directions are relatively fragmented and scattered, and we need to sort out and summarize them so as to conduct further research on SIMI in the future. In addition, previous researches on the mechanism of SIMI are mainly based on a single omics technology, and there is no similar systematic review on the research progress of SIMI. In summary, this paper of the mechanism of SIMI will take up the following aspects: apoptosis, mitochondrial damage, autophagy, excessive inflammatory response, oxidative stress and pyroptosis (Figure 1). The objective of this present review is to sum up some mechanisms of SIMI, to summary the most commonly discussed and researched underlying signaling pathways of myocardial depression in sepsis, and to concisely generalize prevalent therapeutic strategies and potential targets.
 |
Figure 1 Mechanisms of myocardial injury induced by sepsis. |
Apoptosis of SIMI
In the pathophysiological process of sepsis, apoptosis is an inevitable result, which may induce cell death to some extent.20 Studies on the pathophysiology of sepsis suggest that apoptosis occurs in parenchymal cells and one of the adverse effects of parenchymal apoptosis is the multiple organ dysfunctions.21 However, the specific causes are still unclear and need to be explored. Apoptosis is a widely discussed molecular mechanism of myocardial depression in sepsis.22 This review will summarize the apoptosis of SIMI from the following three aspects.
Apoptosis-Related Proteins of SIMI
In recent years, there are more and more studies focus on apoptosis-related proteins (Figure 2), which involve Bcl-2 as well as Bax and caspase-3. The first is an anti-apoptotic protein, while the latter two belong to pro-apoptotic proteins.23 The present review divided the mechanisms of apoptosis-related proteins in SIMI into two parts: promoting and inhibiting of apoptosis. On the one hand, there are two cases regarding the promotion of apoptosis. Firstly, pro-apoptotic proteins are activated by some signals. For example, high expression of pro-apoptotic signaling pathway (TNF-α/Bax) induced by extracellular histone promotes myocardial apoptosis in sepsis.24 Then, the activation of caspase-3 plays a vital role in myocardial apoptosis during sepsis.25,26 It has been reported that the activation of Caspase-3 is the final result of endogenous and exogenous apoptotic pathways, which further promotes the formation of apoptotic bodies by regulating DNA27 and trigger cell death. Inflammatory cytokines which were produced during sepsis also induce myocardial apoptosis by activating caspase. Secondly, the inhibition of anti-apoptotic protein, such as miR-21 promotes apoptosis of SIMI by inhibiting Bcl-2 expression.28 Therefore, the above studies indicate that the onset of SIMI may be related to the activation of Bax and caspase-3 and the inhibition of Bcl-2, which induce the apoptosis of cardiomyocytes during sepsis.
 |
Figure 2 Apoptosis-related proteins of SIMI. |
On the other hand, the inhibition of apoptosis was just the opposite, examples are as follows. JNK/Bax, the pro-apoptotic signaling pathway, ameliorates SIMI when it is inhibited.29 JNK is one of the five characteristic signaling pathways of MAPK, and its phosphorylation promotes apoptosis. Studies have confirmed to be able to inhibit apoptosis of cardiomyocytes by increasing protein expression of Bcl-2 and decreasing the expression of Bid, t-Bid and caspase-9 in mice with SIMI, further confirmed that the pathogenesis of SIMI includes Bcl-2 mediated endogenous anti-apoptosis pathway and caspase-8/Bid/ t-bid/Caspase-9 exogenous pro-apoptosis pathway and the imbalance between the two ways.30 Bid, one of the pro-apoptotic proteins, is cleaved by activated Caspase-8 into t-bid (the active form of Bid), which promotes apoptosis after it enters the mitochondria. Therefore, Bcl-2 is expected to be a potential therapeutic target for SIMI. X-linked apoptosis inhibitor protein (XIAP) is the strongest apoptosis inhibitor in IAP family, which can directly inhibit Caspases and regulate cell apoptosis in multiple ways. MiR-181a-5p/XIAP signaling pathway was found to be involved in oxidative stress and inflammatory responses in LPS-induced H9c2 cells.31 It is conceivable that the regulation of apoptotic pathways also affects the progression of oxidation and inflammation during sepsis.
At present, the studies on apoptosis-related proteins in SIMI mainly focus on the above mentioned. There are other types of apoptosis-related proteins, such as other members of the Bcl-2 family (Bak, Bcl-XL, et al),32 but whether they are expressed in cardiomyocytes remains unknown. Therefore, we still need to pay a lot of efforts to explore the mechanism of apoptosis in SIMI.
Classical Signaling Pathway of Apoptosis in SIMI
Current studies on the mechanism of apoptosis following with SIMI mainly focus on MAPK and PI3K/AKT/mTOR signaling pathways and come up with those two inhibited ways can attenuate cardiac depression.33 Recently, gene expression profiles suggest the high-expression of angiotensin II type 1 receptor (AT1R) and initiation of the MAPK signaling pathway in SIMI models.34 Evidence suggests that miR-101-3p up-regulates and inhibits its specific target dual specificity phosphatase-1 (DUSP1) in LPS-induced myocardial injury mice, further activates MAPK and NF-κB signaling pathways to induce apoptosis.35 It is verified properly that activation of the MAPK pathway is demonstrated by its phosphorylation.36 Then, the TNF-α/P38-MAPK/caspase-3 signaling pathway was activated in SIMI to induce apoptosis of cardiomyocytes, which can stand the test of experiment.37 Going back 20 years, many studies had been conducted to validate the regulation of TNF-α against SIMI’s treatment and TNF-α has been confirmed to possess the ability of depressing myocardial contractility.38 In brief, the signaling pathways related to TNF-α and MAPK participate in the apoptosis during SIMI.
It is noteworthy that the PI3K/AKT/mTOR signaling pathway negatively regulates myocardial cell apoptosis in SIMI.39 As a pro-apoptotic protein, PTEN is the primary phosphatase which negatively regulates PI3K/Akt pathway.40 Therefore, the activation of PI3K/Akt pathway leads to the inhibition of apoptosis and mitigates SIMI.41,42
This review will also introduce other pathways about the apoptosis mechanism of SIMI from genomic and proteomics perspectives: p53 and NF-κB. In SIMI, splenic reservoir CD11b+Gr-1+ cells rapidly migrated into circulation and the heart, further inducing myocardial cell apoptosis via promoting the high expression of P53 mediated by the inhibition of mTOR.43 It has been found that myocardial apoptosis in sepsis caused by TRAF6-mediated phosphorylation of NF-κB44 and ubiquitination of Akt2.45 Increased glycolytic metabolism induced by sepsis produces large amounts of lactate, which leads to SIMI by activating the TLR4-mediated NF-kB signaling pathway relating to apoptosis.46 Similarly, some scholars researched the TLR2/NF-κB signaling pathway at the gene level and found that inhibition of this pathway can alleviate SIMI.47 Therefore, I believe that TLR2/NF-κB signaling pathway may be involved in myocardial apoptosis. In addition, miR-208-5p overexpression activates the NF-κB/HIF-1α signaling pathway during sepsis, resulting in increased HIF-1α expression in T cells and inducing myocardial apoptosis in SIMI.48 However, miR-146a mitigated SIMI by targeting regulation of ErbB4 to inhibit NF-κB signaling.49 Other scholars also studied the myocardial protection effects of some extracts in sepsis from the NF-kB signaling pathway and obtained beneficial results.50–52 The aforementioned studies suggested that activation of NF-kB signaling pathway involves sepsis-induced apoptosis of cardiomyocytes, and also provided a target for sepsis treatment.
The researches of classical signaling pathway of SIMI always attract our attention in the past, present and future. Therefore, based on this review, our research direction will be more closely related to clinical practice, which will provide potential strategies for the treatment of septic patients.
Other Aspects of Apoptosis in SIMI
Notch pathway is mainly composed of four parts: Notch receptor, Notch ligand, CSLDNA binding protein and downstream target gene. Notch receptor signaling between adjacent cells regulates cell differentiation, proliferation and apoptosis. Notch signaling pathway is involved in apoptosis of SIMI.53 In addition, the study of Li et al found that interferon gene stimulator (STING) led to SIMI by activating NLRP3 mediated inflammation and apoptosis, and proposed a hypothesis on the mechanism of STING-IRF3 promoting myocardial cell inflammation and apoptosis in SIMI.54 Silent information regulator 1 (SIRT1) regulates the expression of DNA as well as apoptosis and autophagy by deacetylation of substrate proteins, especially proteins of cardio-protection. Some articles have proposed a reliable theory that the activation of SIRT1 improves SIMI by promoting autophagy and inhibiting apoptosis.55 However, the absence of SIRT1 signaling exacerbated SIMI via weakening its inhibition of the NF-κB signaling pathway.56 This is the result of proteomics. From genomic perspective, the binding of LncRNA ZFAS1 and miR-34b-5p in sepsis contributes to the over-expression of SIRT1, further improving apoptosis of cardiomyocytes.57 Similarly, LncRNA CRNDE also attenuates SIMI through miR-29a/SIRT1 signaling pathway.58
More and more studies have shown that long non-coding RNAs regulate apoptosis pathways by binding to target genes and affect proliferation differentiation of cells, thus leading to disease. For example, LncRNA HOTAIR highly binds to PDCD4 mediated by Lin28 to trigger inflammatory storms and apoptosis in sepsis, thereby promoting to SIMI.59 In sepsis, miR-194 activates its downstream signaling pathway of Wnt/β-catenin, which mediates apoptosis and induces SIMI.60 Then, some studies have shown that miR-642a inhibits apoptosis of LPS-induced H9C2 cells by promoting cell viability and migration, which was the result of the absence of LncRNA LUCAT1.61 Last but not least, ROS-mediated endoplasmic reticulum (ER) stress can also lead to apoptosis of septic cardiomyocytes.62 So, whether oxidative stress also plays an irreplaceable role in the pathophysiology of SIMI? The answer is yes.
Mitochondria are the first barrier of apoptosis, and the specific mechanism of inducing apoptosis in SIMI has not been clarified. The details of ER, another pathway of apoptosis, are still obscure in triggering SIMI. In short, in the process of studying the clinical treatment and management of SIMI, we still have a huge difficulty in pathogenesis to overcome.
Mitochondrial Injury of SIMI
Mitochondrial injury is a key link in the process from cellular injury to the initiation of sepsis.63 The degree of mitochondrial damage is closely related to the severity of SIMI, and the relationship is directly proportional.64
Pathophysiology and Improvement of Damaged Mitochondria in SIMI
Some scholars observed mitochondrial damage in cardiomyocytes of septic patients through immunohistochemistry.65 Electron microscopic analysis also observed large numbers of swollen mitochondria in cardiomyocytes in rats exposed to LPS.66 Mitochondrial dysfunction is the primary prerequisite for the pathophysiological process of SIMI, which consist of ATP production, Ca2+ homeostasis, mitochondrial permeability transition pore (MPTP) and over-produced oxygen-free radicals.67 First, sepsis induces the accumulation of calpain-1 in mitochondria, which impedes ATP synthesis and leads to mitochondrial damage.68 Second, it is confirmed that the activation of the PINK1/PKA/NCLX signaling pathway maintain mitochondrial calcium homeostasis in cardiomyocytes during sepsis.69 Similarly, activation of the Akt/GSK-3β signaling pathway mediated by p2X7 maintains mitochondrial calcium homeostasis, which ameliorates myocardial depression induced by sepsis.70 Most notably, MPTP is significantly opening during sepsis and its closures improve SIMI.71 Some basic studies that can improve the over-opening of MPTP should be put on the agenda to provide a reference for the clinical treatment of SIMI.
Certain pathways are also involved in the mechanism of mitochondrial damage, although they cannot be explained by the phenomenon described above. TLR2, a member of the Toll-like receptors (TLRs) family, monitors and recognizes a variety of disease-associated molecular patterns (PAMP) and is the body’s first barrier against infectious diseases. TLR2-mediated mitochondrial dysfunction, one of the mechanisms of SIMI, was negatively correlated with the expression of miR-410-3p.72 In the process of studying the mechanism of mitochondrial damage in SIMI, some scholars proposed the concept of Mst1/Drp1/mitochondrial fission/F-actin signaling pathway and believed that this pathway is involved in mitochondrial dysfunction.73
Mitochondrial dysfunction has become a hot topic in the pathophysiology of SIMI,74 and its repairs will be a potential molecular mechanism of ameliorating apoptosis.75 Previous studies have observed swelling and destruction of mitochondrial structures in LPS-induced sepsis mice. When studying the protective mechanism of mitochondrial damage in SIMI, it was found that NaHS improves mitochondrial function by activating the PGC-1α/Nrf2 signaling pathway.76
Mitophagy in SIMI
Damaged mitochondria remove selectively broken proteins and organelles and maintain the stability of the intracellular environment via autophagy, which is known as mitochondrial autophagy. Mitochondrial damage is common in the development of sepsis. Therefore, mitophagy in SIMI has become a novel therapy. Mitochondrial dysfunction has been reported to play a fundamental role in SIMI. In sepsis, UPRmt activation precedes mitochondrial autophagy, with UPRmt repairing mitochondrial proteins and mitochondrial autophagy repairing mitochondrial numbers.77 In the absence of UPRmt, the myocardial protection of mitochondrial autophagy was blocked; therefore, it is hypothesized that UPRmt is suppressed in SIMI and thus interrupts the cardio-protective effect of mitochondrial autophagy. However, specific mechanisms have been slow to be discovered. The recovery of mitochondrial damage in sepsis is a reversible process, which is inseparable from the enhancement of PARK2/Parkin-mediated mitophagy.78 Then the enhanced mitophagy of SIMI is also regulated by the PINK1/Parkin,79,80 TFEB66 and Akt/mTOR81 signaling pathway. Uncoupling protein 2 (UCP2) protects mitochondria from sepsis by promoting AMPK-activated autophagy during SIMI.82
Autophagy of SIMI
Autophagy plays an important role in maintaining the homeostasis of cardiovascular cells.83 More and more studies concentrating on autophagy had shown that the activation of autophagy can improve cardiac dysfunction induced by sepsis.84,85 It is conceivable that the inhibition of autophagy pathway plays a crucial role in the induction of SIMI. But recent papers put forward an opposite view: inhibition of the autophagy pathway ameliorates sepsis-induced cardiac systolic dysfunction.86,87 Of course, the general trend in basic research still holds that defects in autophagy in healthy cells are a prerequisite for diseases.88,89 In addition, inhibition of autophagy is a prerequisite for apoptosis. In other words, treatment based on the activation of autophagy improved apoptosis in SIMI. Previous basic studies on the autophagy of SIMI are mainly focused on the regulation of pathways, and this review will elucidate it from the following specific pathways.
There are negative regulation pathways PI3K/Akt, mTOR and positive regulation pathway AMPK relating to autophagy (Figure 3).90 For example, some scholars hold that cell damage is associated with inhibition of autophagy by activation of the PI3K/Akt/Bcl-2 signaling pathway during sepsis.91 However, other scholars believe that the activation of autophagy mediated by the activated PI3K/Akt signaling pathway can attenuate SIMI.92 Then, mTOR (Target of rapamycin) is a serine/threonine kinase. The mTOR signaling pathway promotes substance metabolism, participates in the regulation of apoptosis and autophagy. Suppressing of mTOR signaling pathway can also enhance autophagy in sepsis and thus improve cardiac dysfunction.93 Platelet-derived exosomes promote neutrophil extracellular trap (NETs) production in sepsis by activating autophagy-related Akt/mTOR signaling pathway, which precipitate myocardial depression induced by sepsis.94 NETs are the defense mechanism of neutrophils against infectious diseases and play an important role in the process of tissue damage induced by sepsis.95 Clearly, there is a positive correlation between the number of NETs and the severity of disease. When sepsis-induced cardiomyopathy occurs, the cardiac autophagy pathway is inhibited.96 Of note, the mechanism of inhibition of autophagy in SIMI is related to restricted phosphorylation of AMPK.82 The present studies show that the attenuation of SIMI is achieved through autophagy, which is activated by phosphorylated AMPK.97,98 As an autophagy-initiating kinase, the activating of ULk1 by AMPK through phosphorylating Ser 317 and Ser 777 may ensure that autophagy is initiated. Of course, due to the negative feedback loop formed between ULk1 and AMPK, ULk1 is involved in not only the activation but also the termination of autophagy.99 It had been confirmed that the mechanism of autophagy reaction in SIMI may be related to the phosphorylation of ULk1 based on the activation of AMPK and the suppression of the mTOR.100,101
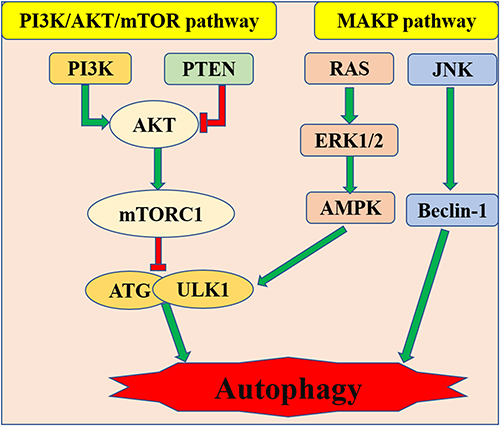 |
Figure 3 PI3K/Ak/mTOR and AMPK pathway relating to autophagy in SIMI. |
In addition, several momentous but uncommon targets for autophagy have been identified and contributed to the clinical treatment of SIMI. Studies have demonstrated protection of sepsis-induced myocardial damage by regulating JNK signaling pathway-dependent autophagy.102 Sirtuin6 (SIRT6), a member of the Sirtuin family of NAD+-dependent enzymes,103 has a specific role of improving SIMI via activating autophagy.104 However, we still have a lot of room for improvement in the treatment of SIMI regarding the regulation of autophagy, especially in epigenetics.
Over-Inflammation in SIMI
Moderate inflammation is good for the body, while its excessive response is bad. The real causes of cardiac dysfunction induced by sepsis are closely related to excessive inflammatory response.22
The Role of Pro-Inflammatory Cytokines in Over-Inflammation
Some pro-inflammatory cytokines have previously been reported to inhibit myocardial function,105 and this involves mechanisms of increased apoptosis and activated inflammation.106 In addition, inflammatory cytokines can also inhibit the contractility of cardiac myocytes.107 When sepsis occurs, the body can produce a large number of pro-inflammation factors (such as TNF-α, interleukin-6 (IL-6), IL-1β, etc.) and angiotensin II and lead to the damage of macrophages,108 clearly, the activation of inflammatory storms is a key to SIMI. IL-18 plays a decisive role in the mechanism of sepsis induced cardiac inflammation, which is positively correlated with the inhibition of PLN and Akt phosphorylation mediated by increased PP2A activity.109 Activation of phosphorylated Akt also provides a promising research basis for the treatment of SIMI.110 There is a study demonstrated that the NLRP3 inflammasomes were activated in SIMI, and TXNIP111 as well as GSDMD112 mediated the activation of NLRP3 inflammasomes. However, the activation of NLRP3 inflammasomes depends on the production of reactive oxygen species (ROS).112 Proteomic studies have also shown that inflammatory responses in sepsis are induced by activation of the TLR4/NLRP3 signaling pathway.113 Therefore, therapeutic strategies based on the inhibition of NLRP3 inflammasome will be the key to our study of SIMI.114,115
The Role of Macrophages in Over-Inflammation
The explosive inflammatory response during sepsis is also driven by macrophage infiltration.116 In SIMI, up-regulation of 5-LOx expression stimulates LTB4 overexpression and activates the BLT1/IL-12P35 signaling pathway, leading to polarization of M1 macrophages and inducing excessive inflammatory responses.117
The Role of Signaling Pathways in Over-Inflammation
On the one hand, activation of pro-inflammatory signaling pathways is the key to the pathophysiology of SIMI, such as JAK2/STAT3,118 IGF-1/PI3K/Akt/GSK-3β119 and Fas/FasL120 signaling pathway.
On the other hand, the mechanisms of RAS during sepsis consist of MAPK, NF-κB and TLR4 signaling pathways, which all mediate inflammation response through the connection of the three of them.121 Excessive inflammatory responses mediated by NF-κBp65 signaling have been reported extensively.122,123 It is also confirmed that TLR4/NF-κB is a key inflammatory storm-induced signaling pathway124,125 and subsequently triggers TNF-α and IL-18 expression126 in SIMI, but its negative regulation by miR-146a improves cardiac function.127 Activation of TLR4 during sepsis promotes nuclear NF-κB translocation, which accelerates cytokine secretion by cardiomyocytes. In addition, TLR4/JNK signaling pathway plays a critical role in regulating myocardial dysfunction during sepsis.128 Previous studies have shown that inhibition of MAPK/NF-κBp65 signaling reduces inflammatory storms and ameliorates myocardial injury in SIMI.129,130 In fact, activation of the MAPK/NF-κB pathway will eventually induce large amounts of TNF, which is produced by cardiomyocytes.131 MiR-23b132 and miR-29a133 mitigated inflammatory responses in SIMI by inhibiting the NF-κB signaling pathway. Similarly, miR-23a suppressed sepsis-induced inflammatory responses by inhibiting the SIRT1/NF-κB signaling pathway as well.134 The inflammatory storm of cardiomyocytes in sepsis may be also caused by inhibition of phosphorylation of AMPK signaling pathway.135
The Role of Genomics in Over-Inflammation
In the past five years, a large number of experiments have explored novel treatment strategies for SIMI from the perspectives of genomics and epigenetics and provided theoretical basis for the clinical management of patients with SIMI. Upregulation of miR-21 stimulated the production of a large number of pro-inflammatory factors in SIMI.136 Study has shown that miR-29a was overexpressed in the cell and animal models of sepsis.137 Of note, overexpression of miR-29a increased LPS-induced inflammation and injury in H9c2 cells. LncRNA PVT1 induces polarization of M1 macrophages and deteriorates myocardial injury in SIMI by activating miR-29a/HMGB1 signaling pathway.138 Other pathways that PVT1 leading to SIMI are mediated by PVT1/miR-24/KLF6139 and MAPK/NF-κB,140 which activate inflammation. LncRNA PTENP1 triggers inflammatory storms in sepsis by regulating the expression of miR-106b-5p and causes myocardial injury.141 Similarly, LncRNA H19 with low expression plays a pro-inflammatory role in sepsis by promoting the expression of miR-874.142
However, different microRNAs have different regulatory effects on inflammation in sepsis. For example, miR-181b mitigated the inflammatory response on SIMI via down-regulating the targeted gene HMGB1.143 MiR-494-3p, a potential therapy of SIMI, can inhibit PTEN-mediated inflammatory response.144 In addition, miR-215-5p,145 miR-141146 and so on play the anti-inflammatory role in the treatment of SIMI by acting with their respective target genes. Wharton’s jelly-derived mesenchymal stem cells (WJ-MSCs) ameliorate myocardial depression during sepsis through activating the cholinergic anti-inflammatory pathway (CAP) mediated by α7-nicotinic acetylcholine receptors (α7nAChRs).147 The MSC-mediated anti-inflammatory effect of SIMI is largely dependent on the regulation of exosomal miR-223.148
Oxidative Stress in SIMI
Septic patients are often in a state of oxidative stress.149 Oxidative damage of cardiomyocytes with dysfunction has been documented in abundant literatures on the pathophysiology of sepsis.150 Then, the oxidative stress that occurs in sepsis is inseparable from mitochondrial dysfunction.151 Treatment with mitochondrial antioxidants has been reported to alleviate SIMI.152 Some studies assessed oxidative stress in SIMI by measuring the expression levels of superoxide dismutase (SOD) and malondialdehyde (MDA) through proteomics.153,154 The former reflects the body’s antioxidant potential, while the latter reflects the body’s oxidative stress ability. During researching the pathogenesis of SIMI, nuclear factor erythroid-2 related factor 2 (Nrf2) signaling pathway has been confirmed to be involved in the regulation of oxidative stress.155 Nrf2, an anti-oxidative stress signaling pathway factor, is at the level of low expression during SIMI156 and inhibits sepsis-induced ROS generation.157 Oxidative stress, the result of an imbalance between ROS production and antioxidant defense systems, is characterized by an overproduction of ROS.
In previous studies, the mechanism of oxidative stress in SIMI has been elucidated from epigenetic and specific signaling pathways. For example, miR-124-3p has been tested to alleviate SIMI by inhibiting oxidative stress,116 the activation of PKCβ2 signal triggers sepsis-induced oxidative damage in cardiomyocytes, which is the result of activating autophagy,158 and the inhibition of NF-κB signaling pathway also ameliorates sepsis induced myocardial oxidative damage.159 Obviously, research on antioxidant therapy for sepsis is still a potential prospect at present.
Pyroptosis in SIMI
Pyroptosis is affected by oxidative stress, apoptosis and inflammation, and its controlled condition influences the progression of sepsis. Some cytokines, inflammasomes, caspase family, interleukin and GSDMD, are involved in the molecular mechanism of pyroptosis.160 Interference with these aforementioned molecules has been reported to improve the development of cardiovascular disease.161 Studies on the pathophysiology of apoptosis in sepsis have largely concentrated on exploring signaling pathways mediated by GSDMD and NLRP3.162
Accordingly, the mechanism of pyroptosis on SIMI is mediated by ER/SIRT1/NLRP3/GSDMD signaling pathway, which is the most complete pathway discovered so far.163 Of note, GSDMD plays an integral role in apoptosis,164 and some certain activated Caspases (e.g, caspase-4, 5, and caspase-11) cleave it and further trigger pyroptosis. The same is true in sepsis. In addition, pyroptosis is a kind of cell death containing an inflammatory response, which is mediated by GSDMD.165 So, NLRP3 inflammasome is an important signal molecule of pyroptosis in SIMI.166 Some scholars have confirmed that activation of NLRP3 inflammasome induces cardiomyocyte’s pyroptosis in sepsis by studying the cardio-protective effect of emodin, and the indirect regulation of pyroptosis by NLRP3 inflammasome is mediated by caspase-1,167 ROS and NF-κB signaling pathway.161 Then, C1q/tumor necrosis factor-related protein 1 (CTRP1) is a promoter that inhibits pyroptosis of cell, its inhibition effect of pyroptosis is triggered by binding to overexpressed Nrf2. Nrf2, an important regulator of cellular defense against stress response, can protect against cytotoxic injury by regulating the expression of many cellular protective genes. When sepsis occurs, low expression of Nrf2 leads to non-activation of CTRP1 binding sites, thus inducing pyroptosis of cardiomyocytes.168
Previous studies have researched the mechanism of pyroptosis from genomics and laid a foundation for the treatment of SIMI. LncRNA x–inactive specific transcript (XIST) contributes to the treatment of SIMI by affecting pyroptosis mediated by miR-150-5p/c-Fos axis.169 LncRNA ZFAS1/miR-138-5p/SESN2 signaling pathway also improved SIMI by inhibiting pyroptosis.170 Exosomes in the blood of septic patients promote myocardial pyroptosis by activating NF-κB-dependent miR-885-5p/HMBOX1 signaling pathway.171 Therefore, some LncRNAs serve as the competing endogenous RNA (ceRNA) of its specific target genes (microRNA) to up-regulate the expression of cell-stress-related proteins, thereby alleviating sepsis-induced myocardial pyroptosis.
Recently, other emerging pathways of pyroptosis have being studied during sepsis, and whether they work in SIMI is still not clear.
Prospects for Therapeutic Strategies in SIMI
Current therapeutic strategies of SIMI are based on the above six pathophysiological mechanisms. For example, on the one hand, Melatonin et al98 take the role of improving SIMI through activating AMPK-mediated autophagy; on the other hand, Helium126 and Berberine124 et al exert their effect on SIMI by inhibiting inflammation after acting on the TLR4/NF-κB signaling pathway (see Table 1 for details). These basic literatures listed in Table 1 also indicates that the treatment of SIMI under the same mechanism includes both Traditional Chinese medicines and Western medicines, and one drug can ameliorate SIMI by acting on different mechanisms. Based on this, this review proposes whether the combined treatment of Chinese and Western drugs can enhance the treatment effect of SIMI. Then, the molecular mechanisms involved in SIMI are not independent of each other, as the signaling pathways that mediate each mechanism are interlinked. Therefore, the research on multi-targeted monotherapy of SIMI may become the next hot topic.
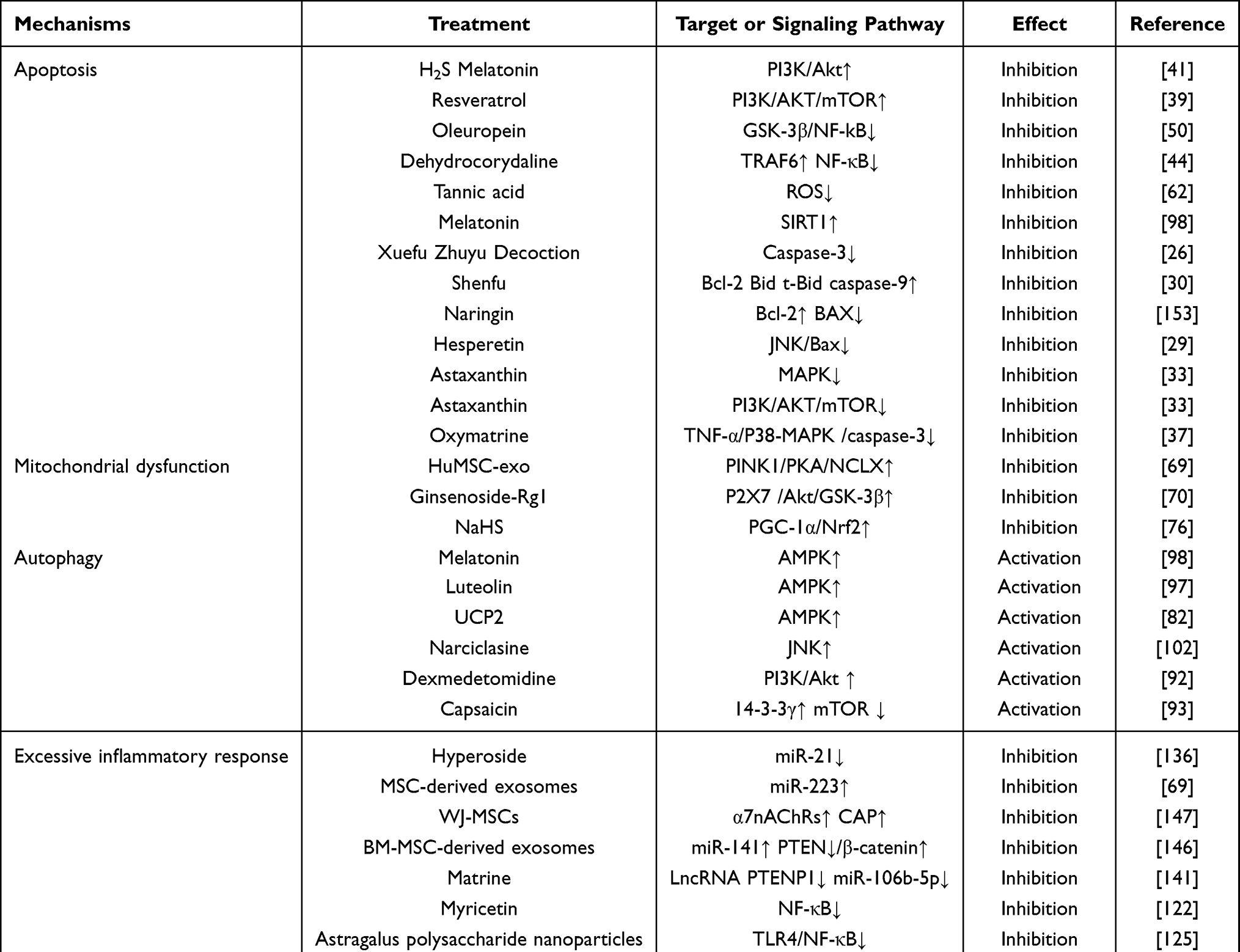 |
Table 1 Confirmed Therapeutic Strategies Based on 6 Types of Mechanisms in SIMI |
Conclusion
There are some signaling pathways that induce both apoptosis and autophagy: p53, Death-associated protein kinase (DAPK), Bcl-2 family and JNK.172 In most of the cell stress stages, however, autophagy is activated at the early stage and apoptosis is activated at the late stage both by the above four signals, so there is a mutually negative regulatory relationship between autophagy and apoptosis.173 Some researchers have studied the interaction mechanism between autophagy and apoptosis in cancer and summarized some LncRNAs, ROS and Beclin-1 also participated in it.174 To sum up, whether the interaction between autophagy and apoptosis also exists in SIMI and which regulatory factors are involved will be the focus of our subsequent study.
In the same way, there are the common signaling pathways between apoptosis and inflammatory response, such as the TLR4/NF‑κB.175 It has been confirmed that apoptosis is the trigger of inflammation and this cascade reaction can be inhibited by RIPK1.176 Activation of some autophagy-related signaling pathways can protect cells from inflammation response,177 therefore, autophagy is the key to anti-inflammation. Autophagy is activated by inflammatory responses found in the early stages of myocardial stress,178 which may exacerbate myocardial tissue damage. Therefore, whether early application of anti-inflammatory therapy in clinical patients of SIMI is reasonable deserves further investigation.
Oxidative stress is often thought to be the cause of chronic inflammatory disease.179 In addition, there is a mutual promotion between inflammation and oxidative stress,180 both mediated by the common signaling pathway: NF‑κB.181 Mitochondrial dysfunction is closely related to oxidative stress because of the excessive production of ROS in mitochondria.182 Collectively, mitochondrial dysfunction appears to be the mediation between inflammation and oxidative stress. In fact, the essence of pyroptosis is inflammation.183 Then, caspases184 and NLRP354 trigger the activation of pyroptosis, inflammation and apoptosis. In view of this, we aim to find the monotherapy strategy of multitarget for patients with SIMI that may improve the prognosis of them by reducing the side effects of multidrug use.
In the past 20 years, the mechanisms of SIMI have been studied in a wide range of fields, including proteomics and genomics. In addition, the purposes of research based on SIMI mechanism is also different, some are to explore the treatment of SIMI, while others are just to explore the specific pathway leading to SIMI. However, the ultimate goal of all basic research is better clinical treatment and management. Although studies on the mechanism of SIMI have become mature, none of the six mechanisms mentioned above, including apoptosis, mitochondrial damage, autophagy, excessive inflammatory response, oxidative stress and pyroptosis, has a specific and complete signaling pathway that can explain how SIMI occurs. There are still many signaling molecules lacking in the studied pathway, and also many signaling pathways related to sepsis are not reflected in the studies of SIMI; so it is imperative that we continue to explore and refine the existing mechanisms of SIMI.
Funding
This study was supported by the Natural Science Fund of Ningxia (2022AAC03593) and Ningxia Medical University (XZ2021015).
Disclosure
The authors declare that there is no conflicts of interest in this work.
References
1. Shankar-Hari M, Phillips GS, Levy ML, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):775–787. doi:10.1001/jama.2016.0289
2. Shankar-Hari M, Harrison DA, Rubenfeld GD, Rowan K. Epidemiology of sepsis and septic shock in critical care units: comparison between sepsis-2 and sepsis-3 populations using a national critical care database. Br J Anaesth. 2017;119(4):626–636.
3. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA. 2014;311(13):1308–1316.
4. Prescott HC, Angus DC. Enhancing recovery from sepsis: a review. JAMA. 2018;319(1):62–75.
5. Fleischmann-Struzek C, Mellhammar L, Rose N, et al. Incidence and mortality of hospital- and ICU-treated sepsis: results from an updated and expanded systematic review and meta-analysis. Intensive Care Med. 2020;46(8):1552–1562.
6. Fleischmann C, Scherag A, Adhikari NK, et al. Assessment of global incidence and mortality of hospital-treated sepsis. current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–272.
7. Lin GL, McGinley JP, Drysdale SB, Pollard AJ. Epidemiology and immune pathogenesis of viral sepsis. Front Immunol. 2018;9:2147.
8. Fleischmann C, Reichert F. Global incidence and mortality of neonatal sepsis: a systematic review and meta-analysis. Arch Dis Child. 2021;106(8):745–752.
9. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211.
10. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. 2018;392(10141):75–87. doi:10.1016/S0140-6736(18)30696-2
11. Gauer R, Forbes D, Boyer N. Sepsis: diagnosis and management. Am Fam Physician. 2020;101(7):409–418.
12. Faix JD. Biomarkers of sepsis. Crit Rev Clin Lab Sci. 2013;50(1):23–36. doi:10.3109/10408363.2013.764490
13. Merx MW, Weber C. Sepsis and the heart. Circulation. 2007;116(7):793–802. doi:10.1161/CIRCULATIONAHA.106.678359
14. Genga KR, Russell JA. Update of sepsis in the intensive care unit. J Innate Immun. 2017;9(5):441–455. doi:10.1159/000477419
15. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
16. Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. 2021;18(6):424–434.
17. Lv X, Wang H. Pathophysiology of sepsis-induced myocardial dysfunction. Mil Med Res. 2016;3:30.
18. Li F, Lang F, Zhang H, Xu L, Wang Y, Hao E. Role of TFEB mediated autophagy, oxidative stress, inflammation, and cell death in endotoxin induced myocardial toxicity of young and aged mice. Oxid Med Cell Longev. 2016;2016:5380319.
19. Yang F, Zhao LN, Sun Y, Chen Z. Levosimendan as a new force in the treatment of sepsis-induced cardiomyopathy: mechanism and clinical application. J Int Med Res. 2019;47(5):1817–1828.
20. Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6(11):813–822.
21. Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med. 2000;28(4 Suppl):N105–113.
22. Fernandes CJ
23. Yang D, Jiang Y, Qian H, Liu X, Mi L. Silencing Cardiac Troponin I-interacting kinase reduces lipopolysaccharide-induced sepsis-induced myocardial dysfunction in rat by regulating apoptosis-related proteins. Biomed Res Int. 2021;2021:5520051.
24. Wang L, Wang Z, Liu X, et al. Effects of extracellular histones on left ventricular diastolic function and potential mechanisms in mice with sepsis. Am J Transl Res. 2022;14(1):150–165.
25. Favory R, Neviere R. Significance and interpretation of elevated troponin in septic patients. Crit Care. 2006;10(4):224.
26. Meng F, Lai H, Luo Z, et al. Effect of xuefu zhuyu decoction pretreatment on myocardium in sepsis rats. Evid-Based Compl Alt Med. 2018;2018:2939307.
27. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516.
28. Li Y, Sun G, Wang L. MiR-21 participates in LPS-induced myocardial injury by targeting Bcl-2 and CDK6. Inflamm res. 2022;71(2):205–214.
29. Yang Z, Liu Y, Deng W, et al. Hesperetin attenuates mitochondria-dependent apoptosis in lipopolysaccharide-induced H9C2 cardiomyocytes. Mol Med Rep. 2014;9(5):1941–1946.
30. Xu P, Zhang WQ, Xie J, Wen YS, Zhang GX, Lu SQ. Shenfu injection prevents sepsis-induced myocardial injury by inhibiting mitochondrial apoptosis. J Ethnopharmacol. 2020;261:113068.
31. Luo S, Huang X, Liu S, Zhang L, Cai X, Chen B. Long non-coding RNA small nucleolar RNA host gene 1 alleviates sepsis-associated myocardial injury by modulating the miR-181a-5p/XIAP axis in vitro. Ann Clin Lab Sci. 2021;51(2):231–240.
32. Reed JC. Apoptosis-regulating proteins as targets for drug discovery. Trends Mol Med. 2001;7(7):314–319.
33. Xie WJ, Hou G, Wang L, Wang SS, Xiong XX. Astaxanthin suppresses lipopolysaccharide‑induced myocardial injury by regulating MAPK and PI3K/AKT/mTOR/GSK3β signaling. Mol Med Rep. 2020;22(4):3338–3346.
34. Zhang T, Yin YC, Ji X, et al. AT1R knockdown confers cardioprotection against sepsis-induced myocardial injury by inhibiting the MAPK signaling pathway in rats. J Cell Biochem. 2020;121(1):25–42.
35. Xin Y, Tang L, Chen J, Chen D, Wen W, Han F. Inhibition of miR‑101‑3p protects against sepsis‑induced myocardial injury by inhibiting MAPK and NF‑κB pathway activation via the upregulation of DUSP1. Int J Mol Med. 2021;47(3):45.
36. Martin EL, Ranieri VM. Phosphorylation mechanisms in intensive care medicine. Intensive Care Med. 2011;37(1):7–18.
37. Zhang M, Wang X, Bai B, Zhang R, Li Y, Wang Y. Oxymatrine protects against sepsis-induced myocardial injury via inhibition of the TNF-α/p38-MAPK/caspase-3 signaling pathway. Mol Med Rep. 2016;14(1):551–559.
38. Meng X, Harken AH. The interaction between Hsp70 and TNF-alpha expression: a novel mechanism for protection of the myocardium against post-injury depression. Shock. 2002;17(5):345–353.
39. Shang X, Lin K, Yu R, et al. Resveratrol protects the myocardium in sepsis by activating the phosphatidylinositol 3-Kinases (PI3K)/AKT/Mammalian target of rapamycin (mTOR) pathway and inhibiting the nuclear factor-κB (NF-κB) signaling pathway. Med Sci Monitor. 2019;25:9290–9298.
40. Ge C, Liu J, Dong S. miRNA-214 protects sepsis-induced myocardial injury. Shock. 2018;50(1):112–118.
41. Liu J, Li J, Tian P, et al. H2S attenuates sepsis-induced cardiac dysfunction via a PI3K/Akt-dependent mechanism. Exp Ther Med. 2019;17(5):4064–4072.
42. An R, Zhao L, Xi C, et al. Melatonin attenuates sepsis-induced cardiac dysfunction via a PI3K/Akt-dependent mechanism. Basic Res Cardiol. 2016;111(1):8.
43. Fu C, Xu Q, Tang S, et al. The mobilization of splenic reservoir myeloid-derived suppressor cells in sepsis-induced myocardial injury. Am J Transl Res. 2020;12(11):7114–7126.
44. Li Y, Zhang L, Zhang P, Hao Z. Dehydrocorydaline protects against sepsis-induced myocardial injury through modulating the TRAF6/NF-κB pathway. Front Pharmacol. 2021;12:709604.
45. Zhang Y, Xu X, Ceylan-Isik AF, et al. Ablation of Akt2 protects against lipopolysaccharide-induced cardiac dysfunction: role of Akt ubiquitination E3 ligase TRAF6. J Mol Cell Cardiol. 2014;74:76–87.
46. Zheng Z, Ma H, Zhang X, et al. Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis. 2017;215(9):1396–1406.
47. Wang SM, Liu GQ, Xian HB, Si JL, Qi SX, Yu YP. LncRNA NEAT1 alleviates sepsis-induced myocardial injury by regulating the TLR2/NF-κB signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(11):4898–4907.
48. Ouyang H, Tan Y, Li Q, et al. MicroRNA-208-5p regulates myocardial injury of sepsis mice via targeting SOCS2-mediated NF-κB/HIF-1α pathway. Int Immunopharmacol. 2020;81:106204.
49. An R, Feng J, Xi C, Xu J, Sun L. miR-146a attenuates sepsis-induced myocardial dysfunction by suppressing IRAK1 and TRAF6 via targeting ErbB4 expression. Oxid Med Cell Longev. 2018;2018:7163057.
50. Xing C, Xu L, Yao Y. Beneficial role of oleuropein in sepsis-induced myocardial injury. Possible involvement of GSK-3beta/NF-kB pathway. Acta cirurgica brasileira. 2021;36(1):e360107.
51. Li X, Cheng Q, Li J, He Y, Tian P, Xu C. Significance of hydrogen sulfide in sepsis-induced myocardial injury in rats. Exp Ther Med. 2017;14(3):2153–2161.
52. Wang X, Zingarelli B, O’Connor M, et al. Overexpression of Hsp20 prevents endotoxin-induced myocardial dysfunction and apoptosis via inhibition of NF-kappaB activation. J Mol Cell Cardiol. 2009;47(3):382–390.
53. Chen DD, Wang HW, Cai XJ. Transcription factor Sp1 ameliorates sepsis-induced myocardial injury via ZFAS1/Notch signaling in H9C2 cells. Cytokine. 2021;140:155426.
54. Li N, Zhou H, Wu H, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215.
55. Zhang WX, He BM, Wu Y, Qiao JF, Peng ZY. Melatonin protects against sepsis-induced cardiac dysfunction by regulating apoptosis and autophagy via activation of SIRT1 in mice. Life Sci. 2019;217:8–15.
56. Han D, Li X, Li S, et al. Reduced silent information regulator 1 signaling exacerbates sepsis-induced myocardial injury and mitigates the protective effect of a liver X receptor agonist. Free Radic Biol Med. 2017;113:291–303.
57. Chen DD, Wang HW, Cai XJ. Long non-coding RNA ZFAS1 alleviates sepsis-induced myocardial injury via target miR-34b-5p/SIRT1. Innate Immun. 2021;27(5):377–387.
58. Zhu Y, Sun A, Meng T, Li H. Protective role of long noncoding RNA CRNDE in myocardial tissues from injury caused by sepsis through the microRNA-29a/SIRT1 axis. Life Sci. 2020;255:117849.
59. Ni SY, Xu WT, Liao GY, Wang YL, Li J. LncRNA HOTAIR promotes LPS-induced inflammation and apoptosis of cardiomyocytes via Lin28-Mediated PDCD4 stability. Inflammation. 2021;44(4):1452–1463.
60. Zhai Y, Ding N. MicroRNA-194 participates in endotoxemia induced myocardial injury via promoting apoptosis. Eur Rev Med Pharmacol Sci. 2018;22(7):2077–2083.
61. Wang J, Xin S, Yang R, Jiang J, Qiao Y. Knockdown of lncRNA LUCAT1 attenuates sepsis‑induced myocardial cell injury by sponging miR-642a. Mamm Genome. 2021;32(6):457–465.
62. Yang YP, Zhao JQ, Gao HB, et al. Tannic acid alleviates lipopolysaccharide‑induced H9C2 cell apoptosis by suppressing reactive oxygen species‑mediated endoplasmic reticulum stress. Mol Med Rep. 2021;24(1):e34.
63. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107.
64. Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22.
65. Takasu O, Gaut JP, Watanabe E, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187(5):509–517.
66. Unuma K, Aki T, Funakoshi T, Yoshida K, Uemura K. Cobalt protoporphyrin accelerates TFEB activation and lysosome reformation during LPS-induced septic insults in the rat heart. PLoS One. 2013;8(2):e56526.
67. Larche J, Lancel S, Hassoun SM, et al. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol. 2006;48(2):377–385.
68. Ni R, Zheng D, Wang Q, et al. Deletion of capn4 protects the heart against endotoxemic injury by preventing ATP synthase disruption and inhibiting mitochondrial superoxide generation. Circ Heart Fail. 2015;8(5):988–996.
69. Zhou Q, Xie M, Zhu J, et al. PINK1 contained in huMSC-derived exosomes prevents cardiomyocyte mitochondrial calcium overload in sepsis via recovery of mitochondrial Ca(2+) efflux. Stem Cell Res Ther. 2021;12(1):269.
70. Liu Z, Pan H, Zhang Y, et al. Ginsenoside-Rg1 attenuates sepsis-induced cardiac dysfunction by modulating mitochondrial damage via the P2X7 receptor-mediated Akt/GSK-3β signaling pathway. J Biochem Mol Toxicol. 2022;36(1):e22885.
71. Hu Y, Yan JB, Zheng MZ, et al. Mitochondrial aldehyde dehydrogenase activity protects against lipopolysaccharide‑induced cardiac dysfunction in rats. Mol Med Rep. 2015;11(2):1509–1515.
72. Zuo T, Tang Q, Zhang X, Shang F. MicroRNA-410-3p binds to TLR2 and alleviates myocardial mitochondrial dysfunction and chemokine production in LPS-Induced sepsis. Mol Ther Nucleic Acids. 2020;22:273–284.
73. Shang X, Li J, Yu R, et al. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/F-actin signaling pathway. J Mol Histol. 2019;50(2):91–103.
74. Fang X, Wang J. [Role of mitochondrial dysfunction in the pathogenesis of septic cardiomyopathy]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2018;30(2):189–192. Chinese.
75. De Kock I, Van Daele C, Poelaert J. Sepsis and septic shock: pathophysiological and cardiovascular background as basis for therapy. Acta Clin Belg. 2010;65(5):323–329.
76. Liang D, Huang A, Jin Y, et al. Protective effects of exogenous NaHS against sepsis-induced myocardial mitochondrial injury by enhancing the PGC-1α/NRF2 pathway and mitochondrial biosynthesis in mice. Am J Transl Res. 2018;10(5):1422–1430.
77. Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021;45:102049.
78. Piquereau J, Godin R, Deschenes S, et al. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy. 2013;9(11):1837–1851.
79. Li J, Shi W, Zhang J, Ren L. To explore the protective mechanism of PTEN-Induced Kinase 1 (PINK1)/Parkin mitophagy-mediated extract of Periplaneta americana on lipopolysaccharide-induced cardiomyocyte injury. Med Sci Monitor. 2019;25:1383–1391.
80. Shi J, Chen Y, Zhi H, An H, Hu Z. Levosimendan protects from sepsis-inducing cardiac dysfunction by suppressing inflammation, oxidative stress and regulating cardiac mitophagy via the PINK-1-Parkin pathway in mice. Ann Transl Med. 2022;10(4):212.
81. Zhang E, Zhao X, Zhang L, et al. Minocycline promotes cardiomyocyte mitochondrial autophagy and cardiomyocyte autophagy to prevent sepsis-induced cardiac dysfunction by Akt/mTOR signaling. Apoptosis. 2019;24(3–4):369–381.
82. Mao JY, Su LX, Li DK, Zhang HM, Wang XT, Liu DW. The effects of UCP2 on autophagy through the AMPK signaling pathway in septic cardiomyopathy and the underlying mechanism. Ann Transl Med. 2021;9(3):259.
83. Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120(11):1812–1824.
84. Zhao P, Kuai J, Gao J, Sun L, Wang Y, Yao L. Delta opioid receptor agonist attenuates lipopolysaccharide-induced myocardial injury by regulating autophagy. Biochem Biophys Res Commun. 2017;492(1):140–146.
85. Jia J, Gong X, Zhao Y, et al. Autophagy enhancing contributes to the organ protective effect of alpha-lipoic acid in septic rats. Front Immunol. 2019;10:1491.
86. Luo Y, Fan C, Yang M, et al. CD74 knockout protects against LPS-induced myocardial contractile dysfunction through AMPK-Skp2-SUV39H1-mediated demethylation of BCLB. Br J Pharmacol. 2020;177(8):1881–1897.
87. Turdi S, Han X, Huff AF, et al. Cardiac-specific overexpression of catalase attenuates lipopolysaccharide-induced myocardial contractile dysfunction: role of autophagy. Free Radic Biol Med. 2012;53(6):1327–1338.
88. Klionsky DJ, Petroni G, Amaravadi RK, et al. Autophagy in major human diseases. EMBO J. 2021;40(19):e108863.
89. Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–1576.
90. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662.
91. Yuan MJ, Wang T. The new mechanism of Ghrelin/GHSR-1a on autophagy regulation. Peptides. 2020;126:170264.
92. Yu T, Liu D, Gao M, et al. Dexmedetomidine prevents septic myocardial dysfunction in rats via activation of α7nAChR and PI3K/Akt- mediated autophagy. Biomed Pharmacother. 2019;120:109231.
93. Qiao Y, Wang L, Hu T, Yin D, He H, He M. Capsaicin protects cardiomyocytes against lipopolysaccharide-induced damage via 14-3-3γ-mediated autophagy augmentation. Front Pharmacol. 2021;12:659015.
94. Jiao Y, Li W, Wang W, et al. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit Care. 2020;24(1):380.
95. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189(6):2689–2695.
96. Chen P, An Q, Huang Y, Zhang M, Mao S. Prevention of endotoxin-induced cardiomyopathy using sodium tanshinone IIA sulfonate: involvement of augmented autophagy and NLRP3 inflammasome suppression. Eur J Pharmacol. 2021;909:174438.
97. Wu B, Song H, Fan M, et al. Luteolin attenuates sepsis‑induced myocardial injury by enhancing autophagy in mice. Int J Mol Med. 2020;45(5):1477–1487.
98. Di S, Wang Z, Hu W, et al. The protective effects of melatonin against LPS-induced septic myocardial injury: a potential role of AMPK-mediated autophagy. Front Endocrinol (Lausanne). 2020;11:162.
99. Loffler AS, Alers S, Dieterle AM, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7(7):696–706.
100. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
101. Ren J, Xu X, Wang Q, Ren SY, Dong M, Zhang Y. Permissive role of AMPK and autophagy in adiponectin deficiency-accentuated myocardial injury and inflammation in endotoxemia. J Mol Cell Cardiol. 2016;93:18–31. doi:10.1016/j.yjmcc.2016.02.002
102. Tang R, Jia L, Li Y, Zheng J, Qi P. Narciclasine attenuates sepsis-induced myocardial injury by modulating autophagy. Aging. 2021;13(11):15151–15163. doi:10.18632/aging.203078
103. Tasselli L, Zheng W, Chua KF. SIRT6: novel mechanisms and links to aging and disease. Trends Endocrinol Metab. 2017;28(3):168–185. doi:10.1016/j.tem.2016.10.002
104. Yuan X, Chen G, Guo D, Xu L, Gu Y. Polydatin alleviates septic myocardial injury by promoting SIRT6-Mediated autophagy. Inflammation. 2020;43(3):785–795. doi:10.1007/s10753-019-01153-4
105. Cain BS, Meldrum DR, Dinarello CA, et al. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med. 1999;27(7):1309–1318. doi:10.1097/00003246-199907000-00018
106. Wang M, Markel TA, Meldrum DR. Interleukin 18 in the heart. Shock. 2008;30(1):3–10. doi:10.1097/SHK.0b013e318160f215
107. Chen X, Peng Y, Ge S, Li Y. Inflammation and cardiac dysfunction during sepsis, muscular dystrophy, and myocarditis. Burns Trauma. 2013;1(3):109–121. doi:10.4103/2321-3868.123072
108. Zhou Q, Pan X, Wang L, Wang X, Xiong D. The protective role of neuregulin-1: a potential therapy for sepsis-induced cardiomyopathy. Eur J Pharmacol. 2016;788:234–240. doi:10.1016/j.ejphar.2016.06.042
109. Okuhara Y, Yokoe S, Iwasaku T, et al. Interleukin-18 gene deletion protects against sepsis-induced cardiac dysfunction by inhibiting PP2A activity. Int J Cardiol. 2017;243:396–403. doi:10.1016/j.ijcard.2017.04.082
110. Zhao P, Wang Y, Zeng S, Lu J, Jiang TM, Li YM. Protective effect of astragaloside IV on lipopolysaccharide-induced cardiac dysfunction via downregulation of inflammatory signaling in mice. Immunopharmacol Immunotoxicol. 2015;37(5):428–433. doi:10.3109/08923973.2015.1080266
111. Yang C, Xia W, Liu X, Lin J, Wu A. Role of TXNIP/NLRP3 in sepsis-induced myocardial dysfunction. Int J Mol Med. 2019;44(2):417–426. doi:10.3892/ijmm.2019.4232
112. Dai S, Ye B, Zhong L, et al. GSDMD mediates LPS-Induced septic myocardial dysfunction by regulating ROS-dependent NLRP3 inflammasome activation. Front Cell Develop Biol. 2021;9:779432. doi:10.3389/fcell.2021.779432
113. Yang L, Zhang H, Chen P. Sulfur dioxide attenuates sepsis-induced cardiac dysfunction via inhibition of NLRP3 inflammasome activation in rats. Nitric Oxide. 2018;81:11–20. doi:10.1016/j.niox.2018.09.005
114. Guo T, Jiang ZB, Tong ZY, Zhou Y, Chai XP, Xiao XZ. Shikonin ameliorates LPS-Induced cardiac dysfunction by SIRT1-dependent inhibition of NLRP3 inflammasome. Front Physiol. 2020;11:570441. doi:10.3389/fphys.2020.570441
115. Wei S, Xiao Z, Huang J, Peng Z, Zhang B, Li W. Disulfiram inhibits oxidative stress and NLRP3 inflammasome activation to prevent LPS-induced cardiac injury. Int Immunopharmacol. 2022;105:108545. doi:10.1016/j.intimp.2022.108545
116. Wu M, Huang Z, Huang W, et al. microRNA-124-3p attenuates myocardial injury in sepsis via modulating SP1/HDAC4/HIF-1α axis. Cell Death Discov. 2022;8(1):40. doi:10.1016/j.phrs.2021.105781
117. Xie S, Qi X, Wu Q, et al. Inhibition of 5-lipoxygenase is associated with downregulation of the leukotriene B4 receptor 1/ Interleukin-12p35 pathway and ameliorates sepsis-induced myocardial injury. Free Radic Biol Med. 2021;166:348–357. doi:10.1016/j.freeradbiomed.2021.02.034
118. Zhang M, Wang X, Wang X, et al. Oxymatrine protects against myocardial injury via inhibition of JAK2/STAT3 signaling in rat septic shock. Mol Med Rep. 2013;7(4):1293–1299. doi:10.3892/mmr.2013.1315
119. He H, Chang X, Gao J, Zhu L, Miao M, Yan T. Salidroside mitigates sepsis-induced myocarditis in rats by regulating IGF-1/PI3K/Akt/GSK-3β signaling. Inflammation. 2015;38(6):2178–2184. doi:10.1007/s10753-015-0200-7
120. Niu J, Azfer A, Kolattukudy PE. Protection against lipopolysaccharide-induced myocardial dysfunction in mice by cardiac-specific expression of soluble Fas. J Mol Cell Cardiol. 2008;44(1):160–169. doi:10.1016/j.yjmcc.2007.09.016
121. Ning L, Rong J, Zhang Z, Xu Y. Therapeutic approaches targeting renin-angiotensin system in sepsis and its complications. Pharmacol Res. 2021;167:105409. doi:10.1016/j.phrs.2020.105409
122. Chen S, Fan B. Myricetin protects cardiomyocytes from LPS-induced injury. Herz. 2018;43(3):265–274. doi:10.1007/s00059-017-4556-3
123. Coldewey SM, Rogazzo M, Collino M, Patel NS, Thiemermann C. Inhibition of IκB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. Dis Model Mech. 2013;6(4):1031–1042.
124. Chen H, Liu Q, Liu X, Jin J. Berberine attenuates septic cardiomyopathy by inhibiting TLR4/NF-κB signalling in rats. Pharm Biol. 2021;59(1):121–128. doi:10.1080/13880209.2021.1877736
125. Xu X, Rui S, Chen C, et al. Protective effects of astragalus polysaccharide nanoparticles on septic cardiac dysfunction through inhibition of TLR4/NF-κB signaling pathway. Int J Biol Macromol. 2020;153:977–985. doi:10.1016/j.ijbiomac.2019.10.227
126. Zhang Y, Zhang J, Xu K, et al. Helium protects against lipopolysaccharide-induced cardiac dysfunction in mice via suppressing toll-like receptor 4-nuclear factor κB-tumor necrosis factor-Alpha/ Interleukin-18 signaling. Chin J Physiol. 2020;63(6):276–285. doi:10.4103/CJP.CJP_66_20
127. Xie J, Zhang L, Fan X, Dong X, Zhang Z, Fan W. MicroRNA-146a improves sepsis-induced cardiomyopathy by regulating the TLR-4/NF-κB signaling pathway. Exp Ther Med. 2019;18(1):779–785. doi:10.3892/etm.2019.7657
128. Chang C, Hu L, Sun S, et al. Regulatory role of the TLR4/JNK signaling pathway in sepsis‑induced myocardial dysfunction. Mol Med Rep. 2021;23(5). doi:10.3892/mmr.2021.11973
129. Shyni GL, Renjitha J, Somappa S, Raghu,KG. Zerumin A attenuates the inflammatory responses in LPS-stimulated H9c2 cardiomyoblasts. J Biochem Mol Toxicol. 2021;35(6):1–11. doi:10.1002/jbt.22777
130. Zhang J, Wang M, Ye J, et al. The anti-inflammatory mediator resolvin E1 protects mice against lipopolysaccharide-induced heart injury. Front Pharmacol. 2020;11:203. doi:10.3389/fphar.2020.00203
131. Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274(3):R577–595. doi:10.1152/ajpregu.1998.274.3.R577
132. Cao C, Zhang Y, Chai Y, et al. Attenuation of sepsis-induced cardiomyopathy by regulation of MicroRNA-23b is mediated through targeting of MyD88-Mediated NF-κB activation. Inflammation. 2019;42(3):973–986. doi:10.1007/s10753-019-00958-7
133. Song YX, Ou YM, Zhou JY. Gracillin inhibits apoptosis and inflammation induced by lipopolysaccharide (LPS) to alleviate cardiac injury in mice via improving miR-29a. Biochem Biophys Res Commun. 2020;523(3):580–587. doi:10.1016/j.bbrc.2019.11.129
134. Shi XJ, Jin Y, Xu WM, Shen Q, Li J, Chen K. MicroRNA-23a reduces lipopolysaccharide-induced cellular apoptosis and inflammatory cytokine production through Rho-associated kinase 1/sirtuin-1/nuclear factor-kappa B crosstalk. Chin Med J. 2021;134(7):829–839. doi:10.1097/CM9.0000000000001369
135. Wang Z, Bu L, Yang P, Feng S, Xu F. Alleviation of sepsis‑induced cardiac dysfunction by overexpression of Sestrin2 is associated with inhibition of p‑S6K and activation of the p‑AMPK pathway. Mol Med Rep. 2019;20(3):2511–2518. doi:10.3892/mmr.2019.10520
136. Zhang J, Liu Y, Liu L. Hyperoside prevents sepsis-associated cardiac dysfunction through regulating cardiomyocyte viability and inflammation via inhibiting miR-21. Biomed Pharmacother. 2021;138:111524. doi:10.1016/j.biopha.2021.111524
137. Pei X, Wu Y, Yu H, et al. Protective role of lncRNA TTN-AS1 in sepsis-induced myocardial injury via miR-29a/E2F2 axis. Cardiovasc Drugs Ther. 2021;36:399–412. doi:10.1007/s10557-021-07244-5
138. Luo YY, Yang ZQ, Lin XF, et al. Knockdown of lncRNA PVT1 attenuated macrophage M1 polarization and relieved sepsis induced myocardial injury via miR-29a/HMGB1 axis. Cytokine. 2021;143:155509. doi:10.1016/j.cyto.2021.155509
139. Dai Q, Hong Y, Li J. PVT1 knockdown inhibited the biological behavior of LPS-induced cardiac fibroblasts by regulating miR-24. Genes Genomics. 2021;43(9):1003–1009. doi:10.1007/s13258-021-01104-0
140. Feng F, Qi Y, Dong C, Yang C. PVT1 regulates inflammation and cardiac function via the MAPK/NF-κB pathway in a sepsis model. Exp Ther Med. 2018;16(6):4471–4478. doi:10.3892/etm.2018.6814
141. Liu Y, Liu L, Zhang J. Protective role of matrine in sepsis-associated cardiac dysfunction through regulating the lncRNA PTENP1/miR-106b-5p axis. Biomed Pharmacother. 2021;134:111112. doi:10.1016/j.biopha.2020.111112
142. Fang Y, Hu J, Wang Z, et al. LncRNA H19 functions as an Aquaporin 1 competitive endogenous RNA to regulate microRNA-874 expression in LPS sepsis. Biomed Pharmacother. 2018;105:1183–1191. doi:10.1016/j.biopha.2018.06.007
143. Ling L, Zhi L, Wang H, Deng Y, Gu C. MicroRNA-181b inhibits inflammatory response and reduces myocardial injury in sepsis by downregulating HMGB1. Inflammation. 2021;44(4):1263–1273. doi:10.1007/s10753-020-01411-w
144. Wu P, Kong L, Li J. MicroRNA-494-3p protects rat cardiomyocytes against septic shock via PTEN. Exp Ther Med. 2019;17(3):1706–1716. doi:10.3892/etm.2018.7116
145. Yao Y, Xu K, Sun Y, et al. MiR-215-5p inhibits the inflammation injury in septic H9c2 by regulating ILF3 and LRRFIP1. Int Immunopharmacol. 2020;78:106000. doi:10.1016/j.intimp.2019.106000
146. Pei Y, Xie S, Li J, Jia B. Bone marrow-mesenchymal stem cell-derived exosomal microRNA-141 targets PTEN and activates β-catenin to alleviate myocardial injury in septic mice. Immunopharmacol Immunotoxicol. 2021;43(5):584–593. doi:10.1080/08923973.2021.1955920
147. Capcha JMC, Rodrigues CE, Moreira RS, et al. Wharton’s jelly-derived mesenchymal stem cells attenuate sepsis-induced organ injury partially via cholinergic anti-inflammatory pathway activation. Am J Physiol Regul Integr Comp Physiol. 2020;318(1):R135–R147. doi:10.1152/ajpregu.00098.2018
148. Wang X, Gu H, Qin D, et al. Exosomal miR-223 contributes to mesenchymal stem cell-elicited cardioprotection in polymicrobial sepsis. Sci Rep. 2015;5:13721. doi:10.1038/srep13721
149. Mantzarlis K, Tsolaki V, Zakynthinos E. Role of oxidative stress and mitochondrial dysfunction in sepsis and potential therapies. Oxid Med Cell Longev. 2017;2017:5985209. doi:10.1155/2017/5985209
150. Neri M, Riezzo I, Pomara C, Schiavone S, Turillazzi E. Oxidative-nitrosative stress and myocardial dysfunctions in sepsis: evidence from the literature and postmortem observations. Mediators Inflamm. 2016;2016:3423450. doi:10.1155/2016/3423450
151. Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107(1):57–64. doi:10.1093/bja/aer093
152. Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45(11):1559–1565. doi:10.1016/j.freeradbiomed.2008.09.003
153. Xianchu L, Lan PZ, Qiufang L, et al. Naringin protects against lipopolysaccharide-induced cardiac injury in mice. Environ Toxicol Pharmacol. 2016;48:1–6. doi:10.1016/j.etap.2016.09.005
154. Wang F, Jin Z, Shen K, et al. Butyrate pretreatment attenuates heart depression in a mice model of endotoxin-induced sepsis via anti-inflammation and anti-oxidation. Am J Emerg Med. 2017;35(3):402–409. doi:10.1016/j.ajem.2016.11.022
155. Zhang J, Yang Z, Liang Z, et al. Anti-Interleukin-16 neutralizing antibody treatment alleviates sepsis-induced cardiac injury and dysfunction via the nuclear factor Erythroid-2 related factor 2 pathway in mice. Oxid Med Cell Longev. 2021;2021:6616422. doi:10.1155/2021/6616422
156. Li D, Wang M, Ye J, et al. Maresin 1 alleviates the inflammatory response, reduces oxidative stress and protects against cardiac injury in LPS-induced mice. Life Sci. 2021;277:119467. doi:10.1016/j.lfs.2021.119467
157. Hao E, Lang F, Chen Y, et al. Resveratrol alleviates endotoxin-induced myocardial toxicity via the Nrf2 transcription factor. PLoS One. 2013;8(7):e69452. doi:10.1371/journal.pone.0069452
158. Lei S, Zhang Y, Su W, Zhou L, Xu J, Xia ZY. Remifentanil attenuates lipopolysaccharide-induced oxidative injury by downregulating PKCβ2 activation and inhibiting autophagy in H9C2 cardiomyocytes. Life Sci. 2018;213:109–115.
159. Li F, Lang F, Zhang H, et al. Apigenin alleviates endotoxin-induced myocardial toxicity by modulating inflammation, oxidative stress, and autophagy. Oxid Med Cell Longev. 2017;2017:2302896.
160. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128.
161. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019;52(2):e12563.
162. Gao YL, Zhai JH, Chai YF. Recent advances in the molecular mechanisms underlying pyroptosis in sepsis. Mediators Inflamm. 2018;2018:5823823.
163. Wei A, Liu J, Li D, et al. Syringaresinol attenuates sepsis-induced cardiac dysfunction by inhibiting inflammation and pyroptosis in mice. Eur J Pharmacol. 2021;913:174644.
164. Vande Walle L, Lamkanfi M. Pyroptosis. Current Biol. 2016;26(13):R568–R572.
165. Wang Q, Wu J, Zeng Y, et al. Pyroptosis: a pro-inflammatory type of cell death in cardiovascular disease. Clin Chim Acta. 2020;510:62–72.
166. Li Q, Zhang M, Zhao Y, Dong M. Irisin protects against LPS-Stressed cardiac damage through inhibiting inflammation, apoptosis, and pyroptosis. Shock. 2021;56(6):1009–1018.
167. Dai S, Ye B, Chen L, Hong G, Zhao G, Lu Z. Emodin alleviates LPS-induced myocardial injury through inhibition of NLRP3 inflammasome activation. Phytother Res. 2021;35(9):5203–5213.
168. Teng Y, Li N, Wang Y, et al. NRF2 inhibits cardiomyocyte pyroptosis via regulating CTRP1 in sepsis-induced myocardial injury. Shock. 2022;57(4):590–599.
169. Wang X, Li XL, Qin LJ. The lncRNA XIST/miR-150-5p/c-Fos axis regulates sepsis-induced myocardial injury via TXNIP-modulated pyroptosis. Lab Invest. 2021;101(9):1118–1129.
170. An L, Yang T, Zhong Y, Yin Y, Li W, Gao H. Molecular pathways in sepsis-induced cardiomyocyte pyroptosis: novel finding on long non-coding RNA ZFAS1/miR-138-5p/SESN2 axis. Immunol Lett. 2021;238:47–56.
171. Tu GW, Ma JF, Li JK, et al. Exosome-derived from sepsis patients’ blood promoted pyroptosis of cardiomyocytes by regulating miR-885-5p/HMBOX1. Front Cardiovasc med. 2022;9:774193.
172. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94.
173. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752.
174. Xie Q, Liu Y, Li X. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl Oncol. 2020;13(12):100871.
175. Ding Y, Wang L, Zhao Q, Wu Z, Kong L. MicroRNA‑93 inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting the TLR4/NF‑κB signaling pathway. Int J Mol Med. 2019;43(2):779–790.
176. Dannappel M, Vlantis K, Kumari S, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90–94.
177. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. 2021;54(3):437–453.
178. Wang X, Guo Z, Ding Z, Mehta JL. Inflammation, autophagy, and apoptosis after myocardial infarction. J Am Heart Assoc. 2018;7(9):43.
179. Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, Rahu N. Oxidative stress and inflammation: what polyphenols can do for us? Oxid Med Cell Longev. 2016;2016:7432797.
180. Siti HN, Kamisah Y, Kamsiah J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul Pharmacol. 2015;71:40–56.
181. Sul OJ, Ra SW. Quercetin prevents LPS-induced oxidative stress and inflammation by modulating NOX2/ROS/NF-kB in lung epithelial cells. Molecules. 2021;26(22):43.
182. Wiegman CH, Michaeloudes C, Haji G, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136(3):769–780.
183. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
184. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol. 2020;38:567–595.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Short Sleep Duration and Erectile Dysfunction: A Review of the Literature
Zhang F, Xiong Y, Qin F, Yuan J
Nature and Science of Sleep 2022, 14:1945-1961
Published Date: 27 October 2022
A Bibliometric and Knowledge Map Analysis of Osteoarthritis Signaling Pathways from 2012 to 2022
Li B, Zheng J
Journal of Pain Research 2022, 15:3833-3846
Published Date: 6 December 2022