")
Back to Journals » Journal of Inflammation Research » Volume 14
Regulation of IFN-Is by MEF2D Promotes Inflammatory Homeostasis in Microglia
Authors Lu F, Wang R, Xia L, Nie T, Gao F, Yang S, Huang L, Shao K, Liu J, Yang Q
Received 20 February 2021
Accepted for publication 26 May 2021
Published 29 June 2021 Volume 2021:14 Pages 2851—2863
DOI https://doi.org/10.2147/JIR.S307624
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Fangfang Lu,1,2,* Ronglin Wang,3,* Li Xia,4,* Tiejian Nie,2 Fei Gao,4 Shaosong Yang,5 Lu Huang,4 Kaifeng Shao,2 Jiankang Liu,1 Qian Yang2
1Center for Mitochondrial Biology and Medicine, The Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi’an Jiaotong University, Xi’an, 710049, Shaanxi, People’s Republic of China; 2Department of Experimental Surgery, Tangdu Hospital, Airforce Medical University of PLA, Xi’an, Shaanxi, 710038, People’s Republic of China; 3Department of Oncology, Tangdu Hospital, Airforce Medical University of PLA, Xi’an, Shaanxi, 710038, People’s Republic of China; 4Department of Neurosurgery, Tangdu Hospital, Airforce Medical University of PLA, Xi’an, Shaanxi, 710038, People’s Republic of China; 5Department of Neurosurgery, The First Medical Center of Chinese PLA General Hospital, Beijing, 100853, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qian Yang; Jiankang Liu Tel/Fax +86-029-84777007
; +86-029-82665849
Email [email protected]; [email protected]
Background: Microglia play an essential role in the central nervous system immune response. The transcription factor myocyte enhancer factor-2 D (MEF2D) is known to participate in stress regulation in various cell types and is easily activated in microglia. MEF2D has been shown to transcriptionally regulate several cytokine genes in immune cells and directly regulates the inflammatory response, suggesting that MEF2D may act as a key stimulus response regulator of microglia and is involved in the regulation of brain microhomeostasis. To uncover the molecular mechanism of MEF2D in the inflammatory system, in the present study, we investigated the global effect of MEF2D in activated microglia and explored its potential regulatory network.
Methods: Experiments with a recombinant lentiviral vector containing either shRNA or overexpressing MEF2D were performed in the murine microglial BV2 cell line. Transcriptome sequencing and global gene expression patterns were analysed in lipopolysaccharide-stimulated shMEF2D BV2 cells. Pro- and anti-inflammatory factors were assessed by Western blot, qPCR or ELISA, and microglial activity was assessed by phagocytosis and morphologic analysis. The direct binding of MEF2D to the promoter region of interferon regulatory factor 7 (IRF7) was tested by ChIP-qPCR. The interferon-stimulated genes (ISGs) were tested by qPCR.
Results: MEF2D actively participated in the inflammatory response of BV2 microglial cells. Stably expressed RNAi-induced silencing of MEF2D disrupted the microglial immune balance in two ways: (1) the expression of proinflammatory factors, such as NLRP3, IL-1β, and iNOS was promoted; and (2) the type-I interferon signalling pathway was markedly inhibited by directly modulating IRF7 transcription. In contrast, overexpression of MEF2D significantly reduced the expression of NLRP3 and iNOS under LPS stimulation and alleviated the level of immune stress in microglia.
Conclusion: These findings demonstrate that MEF2D plays an important role in regulating inflammatory homeostasis partly through transcriptional regulation of the type-I interferon signalling pathway.
Keywords: microglia, MEF2D, IRF7, type-I interferons, inflammatory homeostasis
Introduction
Microglia, the resident immune cells of the central nervous system (CNS), are the primary mediators of the immune response and immune surveillance in the CNS.1 Microglia play an important role in brain development, maintenance of the neural environment in ageing,2 and in neurodegeneration.3–5 Modest microglial activation is necessary and beneficial for CNS homeostasis, but uncontrolled neuroinflammation induced by excessive or chronically activated microglia is a salient feature in most neurological diseases, such as Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis.6–8 Therefore, maintaining the hierarchical and balanced microglial immune response is the key to a healthy brain. Although considerable research has focused on microglial inflammatory homeostasis, the convergent regulatory mechanisms are not fully understood.
The myocyte enhancer factor-2 (MEF2) family, initially identified as transcription factors in the muscle lineage,9 has four mammalian isoforms, MEF2A, B, C and D. MEF2s are an important modulator of development, proliferation, differentiation and immunity in various cell types. In neurons, MEF2s function as converging factors to regulate neuronal proliferation, differentiation, survival, and synapse development.10,11 MEF2 transcriptional activity is tightly regulated by extracellular stimuli, such as neurotrophic stimulation and calcium influx.12,13 Dysregulation of MEF2s by toxic signals contributes to many neurodegenerative diseases.14–16 Our previous study demonstrated that MEF2D promotes the survival of dopamine neurons in the SNc of a Parkinson’s mouse model.11,14 MEF2 proteins are also expressed in cells of the immune system, such as T cells, B cells and microglia. In mammals, MEF2C is phosphorylated by p38 mitogen-activated protein kinases (p38 MAPK) in myeloid linage cells, resulting in increased transcription of c-jun,17 and MEF2D directly regulate the transcription of interleukin-2 (IL-2) and interleukin-10 (IL-10) genes in T cells and microglia.13,18 In Drosophila, MEF2s have been identified as a critical transcriptional switch between metabolism and immunity, playing an important role in the innate immune response.19 These pioneering studies on the immune regulation of MEF2s in various species and cell types suggest that MEF2s may also be involved in the microglial regulation of the immune response in the CNS.
In a previous study, we found that MEF2D was induced by lipopolysaccharide (LPS) in microglia18 and participated in the regulation of several cytokines. As an important stress response protein, the function of MEF2D in microglia needs to be further explored in detail. Here, we applied RNA interference (RNAi) technology, chromatin immunoprecipitation quantitative real-time PCR (ChIP-qPCR) and transcriptome sequencing to identify the regulatory role of MEF2D in activated microglia. Our data showed that compared to the negative control, RNAi-induced silencing of MEF2D promoted the expression of inducible nitric oxide synthase (iNOS), NAcht leucine-rich repeat protein 3 (NLRP3), and downstream mature IL-1β and significantly blocked the interferon signalling pathway. MEF2D could directly bind to MEF2 consensus sites in the promoter region of IRF7 to regulate its transcription. These results indicate that MEF2D is an integral sensor that regulates the innate immune response in activated microglia.
Materials and Methods
Cell Culture
The BV2 cell line was provided by Dr. Mao Zixu (Department of Pharmacology and Neurology, Emory University School of Medicine, Atlanta, GA 30322, USA). Dr. Mao purchased this cell line from the ATCC cell bank. BV2 cells were cultured in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12) (HyClone, Logan City, Utah, USA) supplemented with 5% foetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and incubated with 5% CO2 at 37°C.
Lentivirus Infection
Recombinant lentivirus vectors to silence (MEF2D knockdown by shRNA (shMEF2D)) or enhance (overexpression of MEF2D (OE-MEF2D)) MEF2D gene expression were obtained commercially from Hanbio, Shanghai, China. The following RNAi sequence of MEF2D was used: GCTGGATACTTGGACATTAAA. BV2 cells were seeded in a 6-well plate and infected with lentivirus in 1 mL of medium for 12 h before replacement with fresh medium. Then, after 72 h of incubation, puromycin (5 μg/mL) was used to select cell lines stably expressing shMEF2D or OE-MEF2D for at least 3 generations.
Luciferase Assay
The MEF2-firefly reporter vector was obtained from the Mao laboratory (Department of Pharmacology and Neurology, Emory University School of Medicine, Atlanta, GA 30322, USA). The BV2 cells stably expressing shMEF2D and negative control (NC) were transfected with vectors of MEF2-firefly reporter and the reference Renilla reporter. Then, the cells were seeded in 48-well plates and stimulated with 500 ng/mL of LPS for 24 h. The luciferase assay was performed using a Dual-Luciferase® Reporter Assay Kit (Promega, Madison, WI, USA) following procedures provided by the manufacturer.
Immunoblotting
Nuclear and cytoplasmic proteins were extracted using a kit (Sango Biotech, Shanghai, China) according to the manufacturer’s instructions. Total protein was extracted with ice-cold lysis buffer containing protease inhibitors. The protein concentration was determined using a BCA protein assay kit (Thermo, Rockford, USA). Equal amounts of protein were separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred onto polyvinylidene fluoride (PVDF) membranes. After 2 h of blocking with 5% fat-extracted milk at room temperature, the membranes were incubated with primary antibodies against MEF2D (BD, #610774), NLRP3 (Cell Signalling, Danvers, USA (CST), #13158), Nrf2 (CST, #12721), iNOS (Abcam, Cambridge, UK, ab178945) and β-actin (Proteintech, Chicago, USA #66009-1-Ig) overnight at 4°C, washed with tris-buffered saline with Tween 20 (TBST) buffer three times, and then incubated with horseradish peroxidase (HRP) secondary antibody for 2 h at room temperature. Protein bands were visualized using electrochemiluminescence (ECL) and analysed using ImageJ software.
Quantitative Real-Time PCR (qPCR)
Total RNA was extracted using TRIzol reagent (Roche, Basel, Switzerland). cDNA was reverse transcribed using a First Strand cDNA Synthesis Kit (Roche). Quantification of mRNA was performed using qPCR SYBR Green Master Mix (Yeasen, Shanghai, China). All target mRNA levels were normalized to β-actin as a control standard. The primers are listed in Table S1.
ChIP-qPCR
The ChIP assay was performed with a ChIP Assay Kit (Millipore, Massachusetts, USA). The quantity of DNA was analysed by qPCR, and gene-specific primers of the promoter are listed in Table S1.
Enzyme-Linked Immunosorbent Assay (ELISA)
The quantity of IL-1β in the supernatant of BV2 cells stably expressing NC and shMEF2D was tested using mouse enzyme-linked immunosorbent assay kits (R&D Systems, Minnesota, USA) according to the manufacturer’s procedures.
3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) Assay
BV2 cells stably expressing NC and shMEF2D were seeded in 96-well plates (5 × 103 cells/well) and treated with LPS (500 ng/mL) for 24 and 48 h. Twenty microlitres of MTT (5 mg/mL) (Millipore) was added to each well and incubated for 4 hours. Then, after removing the supernatant, 150 μL of dimethyl sulfoxide (DMSO) (Millipore) was added to each well and mixed thoroughly for 10 min. The optical density was measured at 490 nm.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labelling (TUNEL) Staining
For terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) staining, the FragEL™ DNA Fragmentation Detection Kit (Millipore) was used according to the manufacturer’s instructions.
Transcriptome Sequencing (RNA-Seq)
BV2 cells stably expressing negative control and shMEF2D were stimulated with LPS (500 ng/mL) for 12 h, and whole lysates were collected in 1 mL of TRIzol reagent. Total RNA was extracted, and a TruSeq® RNA LT Sample Prep Kit v2 (Illumina, San Diego, USA) was used to construct cDNA libraries. The cDNA fragments were sequenced using an Illumina HiSeq2500. Biological triplicate RNA sequencing was performed on 12 independent RNA samples from BV2 cell lines.
Phagocytosis
Fluorescent beads (AAT Bioquest, Protonex Red 600 latex beads) were administered for 4 h to BV2 cells stably expressing NC, shMEF2D, or OE-MEF2D. After washing, uptake of fluorescent beads was analysed by laser scanning confocal microscopy (Nikon, Minato, Japan).
Statistical Analyses
Data are expressed as the mean ± standard error of the mean (SEM) from at least three independent experiments. Data were analysed by either one-way ANOVA or two-way ANOVA as appropriate. Statistical analyses were carried out using SPSS 19.0. P-values < 0.05 were considered statistically significant.
Results
MEF2D Participated in the Inflammatory Response of BV2 Cells
Previous studies have shown that MEF2D participates in MPTP-induced microglial activity. This prompted us to explore the transcriptional regulation network of MEF2D. We chose the LPS-induced BV2 microglial cell model. The murine BV2 cell line retains most of the immunological properties ascribed to active microglia and is widely used in neuroinflammation research.20 After LPS treatment, the level of MEF2D gradually increased and reached a significantly higher level at 24 h (Figure 1A left). Because MEF2D is a transcription factor, nuclear and cytoplasmic proteins were fractionated to confirm the nuclear distribution of MEF2D (Figure 1A right). After LPS treatment, MEF2D was only elevated in the nucleus, but the cytoplasmic level did not change, indicating that MEF2D may play a transcriptional role in inflammation.
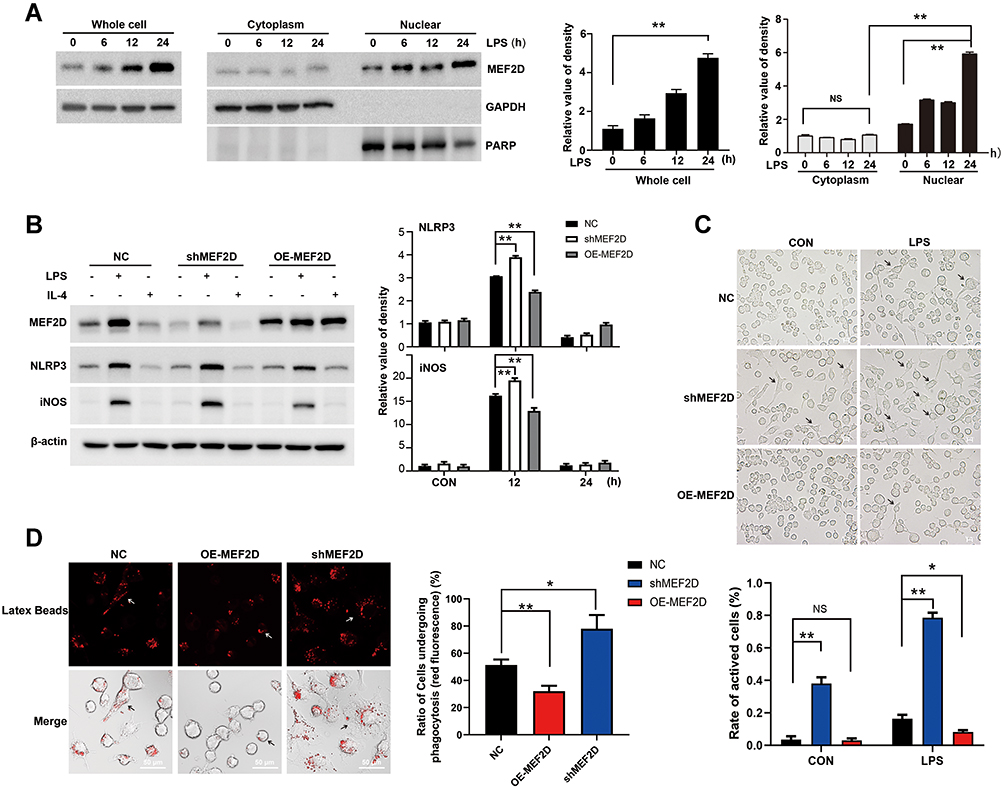 |
Figure 1 MEF2D participated in the inflammatory response of BV2 cells. (A) LPS induced an increase in the level of MEF2D, which was mainly distributed in the nucleus in BV2 cells. BV2 cells were exposed to 500 ng/mL of LPS for 6, 12 or 24 h, and nuclear and cytoplasmic proteins were fractionated to quantify MEF2D by immunoblotting. (A right graph) The relative quantification of MEF2D is shown. (B) BV2 cells were transfected with recombinant lentivirus expressing either control shRNA, shRNA-MEF2D (shMEF2D), or overexpression-MEF2D (OE-MEF2D), and stable cell lines were selected followed by treatment with LPS for 12 h or IL-4 for 18 h. The protein levels of the proinflammatory factors NLRP3 and iNOS were tested by immunoblotting. (B right graphs) The relative quantification data are shown. Data in A and B from three independent experiments are expressed as the mean ± SEM and were analysed by two-way ANOVA (**P ≤ 0.01). (C) The morphological effects of MEF2D on BV2 cells were determined. BV2 cells stably expressing NC, shMEF2D, or OE-MEF2D were treated with LPS for 12 h, and the morphological changes were analysed by optical microscopy. Arrows indicate activated microglia. The scale bars represent 50 μm. (C bottom graph) Active cells were defined as those with a ratio of long axis to short axis greater than 1.5. (D) The effect of MEF2D on phagocytosis was determined in BV2 cells. Fluorescent beads were added to the cells for 4 h. Immunofluorescence staining was performed to measure phagocytosis capacity. Arrows indicate microglia undergoing phagocytosis. Scale bars represent 50 μm. (D right chat) The ratio of red fluorescent positive cells reflects the phagocytic activity of microglia. The data shown in C and D are from at least 300 cells of 5 vision fields in three independent experiments, and analysed by two-way ANOVA (*P ≤ 0.05, **P ≤ 0.01). |
To verify the regulatory effect of MEF2D in inflammation, we used a lentiviral vector to generate BV2 cells stably expressing shMEF2D and OE-MEF2D. After LPS stimulation, the expression of the proinflammatory factors NLRP3 and iNOS was significantly increased by MEF2D knockdown and diminished by overexpression of MEF2D compared with NC (Figure 1B). Because IL-4 inhibits macrophage production of proinflammatory cytokines, we also administered IL-4 and found that the MEF2D level did not significantly change between treated and untreated groups of the different stable cell lines (Figure 1B). These data indicate that MEF2D is more inclined to participate in the proinflammatory reaction process.
Morphology is also an important indicator of the activation state of microglia. In the resting state, BV2 microglial cells were round with small cell bodies, while under LPS stimulation, the morphology changed significantly: the cell body became larger and the protrusions became increasingly thicker. Note that knockdown of MEF2D made BV2 cells turbulent at baseline. The rate of activated shMEF2D BV2 cells was significantly higher than that of NC cells at 12 h after LPS treatment. However, overexpression of MEF2D mitigated the activation state even following LPS treatment (Figure 1C). Immune phagocytosis is another main feature of activated microglia. The data consistently showed that knockdown of MEF2D enhanced the immune phagocytic function of BV2 cells, while overexpression of MEF2D partially alleviated the immunophagocytic activity (Figure 1D). These data suggest that MEF2D might act as an inhibitor of excessive inflammation to be tightly involved in regulating inflammatory homeostasis.
MEF2D Knockdown Promoted the Proinflammatory Response of BV2 Cells
Our above data showed that MEF2D may participate in the inflammatory response of microglia. To further test this hypothesis, we used BV2 cells stably expressing shMEF2D, in which the knockdown efficiency was approximately 80% of the background compared to that of NC cells. Even with LPS stimulation for 24 h, the expression of MEF2D in shMEF2D BV2 cells significantly deceased (Figure 2B and E). MTT and TUNEL assays were performed, and the data showed that knockdown of MEF2D did not affect the survival and proliferation of BV2 cells, even under LPS stress conditions (Figure S1A and B).
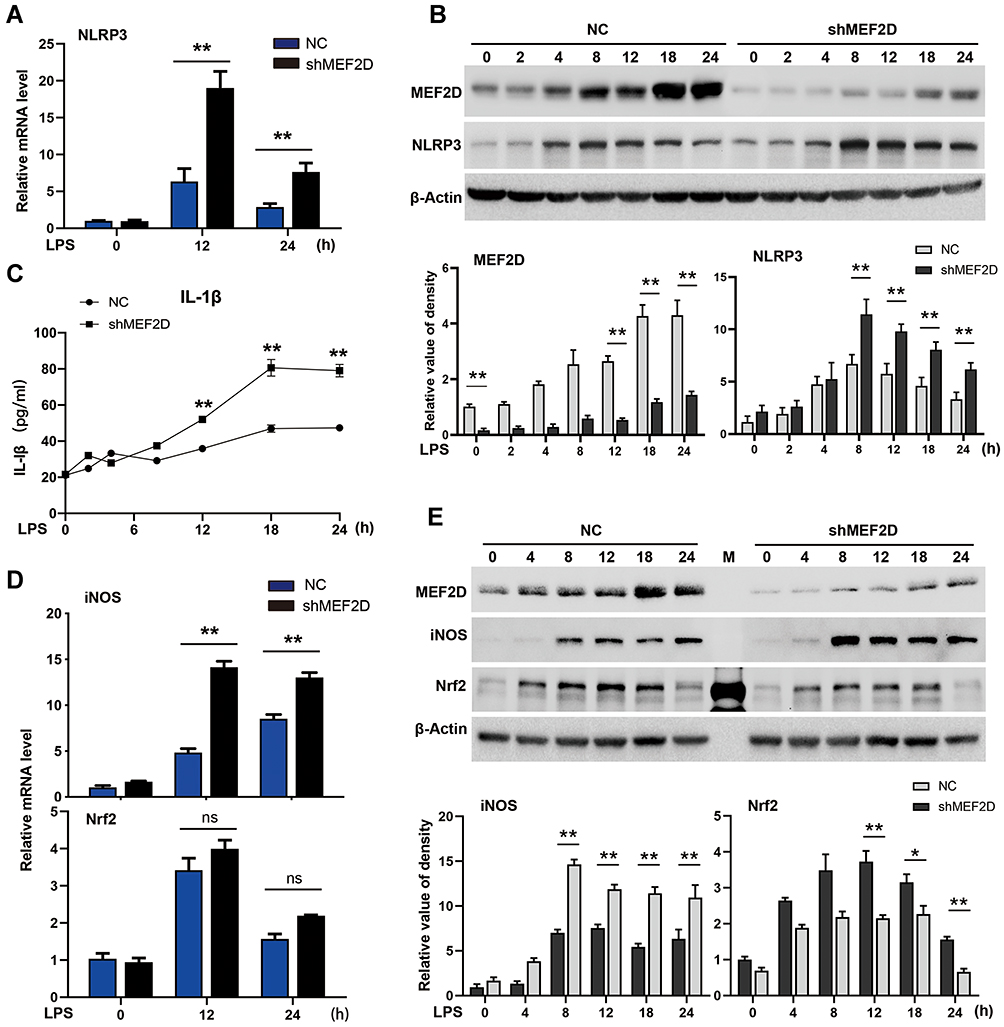 |
Figure 2 MEF2D knockdown hyperactivates BV2 cells. BV2 stable NC and shMEF2D cell lines were treated with LPS. (A and B) The mRNA and protein levels of the preinflammatory factor NLRP3 were tested by qPCR and Western blot. (B bottom graph) Relative quantification data are shown. (C) The effect of shMEF2D on the secretion of the preinflammatory cytokine IL-1β following LPS treatment was determined. The secreted levels of IL-1β were tested by ELISA. (D and E) The mRNA and protein levels of the preinflammatory factor iNOS and the oxidative stress regulator Nrf2 were tested by qPCR and immunoblotting, respectively. (E bottom graph) Relative quantification data are shown. The data from three independent experiments are expressed as the mean ± SEM and were analysed by two-way ANOVA (*P ≤ 0.05, **P ≤ 0.01). |
To determine the role of MEF2D in activated BV2 cells, we performed time course studies of several key inducible inflammatory factors in shMEF2D BV2 cells under LPS treatment. NLRP3 is an innate immune proinflammatory factor that mediates the cleavage-mature process of IL-1β. Its function is crucial for the regulation of neuroinflammation mediated by microglia. Knockdown of MEF2D significantly promoted the expression of NLRP3 after LPS treatment for 8 h, and the trend lasted for at least 24 h in BV2 cells (Figure 2A and B). The secretion level of the downstream IL-1β cytokine in shMEF2D cells was significantly higher after 12 h of LPS stimulation than that of NC cells (Figure 2C). The mRNA and protein levels of another proinflammatory factor, iNOS, were also increased markedly by knockdown of MEF2D compared with NC after 8 h of treatment with LPS (Figure 2D and E). In contrast, the protein but not the mRNA level of the anti-inflammatory factor nuclear factor-erythroid 2-p45 derived factor 2 (Nrf2) was decreased (Figure 2D and E). These results strongly confirm that knockdown of MEF2D markedly enhances the susceptibility and proinflammatory response of BV2 microglial cells.
Differently Expressed Genes in Response to MEF2D Knockdown Enriched in the Immune System of BV2 Cells
To investigate the regulatory function of MEF2D in microglia, we used RNA-seq and analysed the global gene expression patterns in LPS-stimulated BV2 cells with or without stable MEF2D knockdown. To monitor the changes in inflammatory factors in the acute stage, transcriptome sequencing was performed 12 h after LPS treatment (Figure 3D). The knockdown efficiency and transcriptional activity were tested for quality control (Figure 3A and B). RNA-seq transcriptional analysis was performed using three biological replicates for each treatment: NC, NC + LPS stimulation, shMEF2D, and shMEF2D + LPS stimulation. Principal component analysis (PCA) showed good separation and a high level of consistency between biological replicates of the same population in BV2 cells (Figure 3C). Using a false discovery rate, P≤0.01, and fold change≥1.5 log2 as the cut-off values, we identified 310 differentially expressed genes in MEF2D knockdown cells relative to controls without LPS treatment (Figure 3E). The genes were enriched in the gene ontology (GO) annotations of “tissue development”, “cell adhesion”, and “cell differentiation” (Figure 3F) and were in the top Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway classes “infectious diseases”, “cancers”, “digestive system”, “neurodegenerative diseases” and “cellular community” (Table S2). Under LPS stimulation for 12 h, 242 genes were altered between shMEF2D and NC BV2 stable cell lines (Figure 3E) and were enriched in the GO annotations “response to stimulus”, “immune system process” and “innate immune response” (Figure 3F) and involved in the top KEGG pathway classes of “immune system”, “infectious diseases”, “cancers” and “signal transduction” (Table S2). These results strongly confirm that MEF2D could impact the immune response and that knockdown of MEF2D could change the immunoregulatory function of BV2 microglial cells.
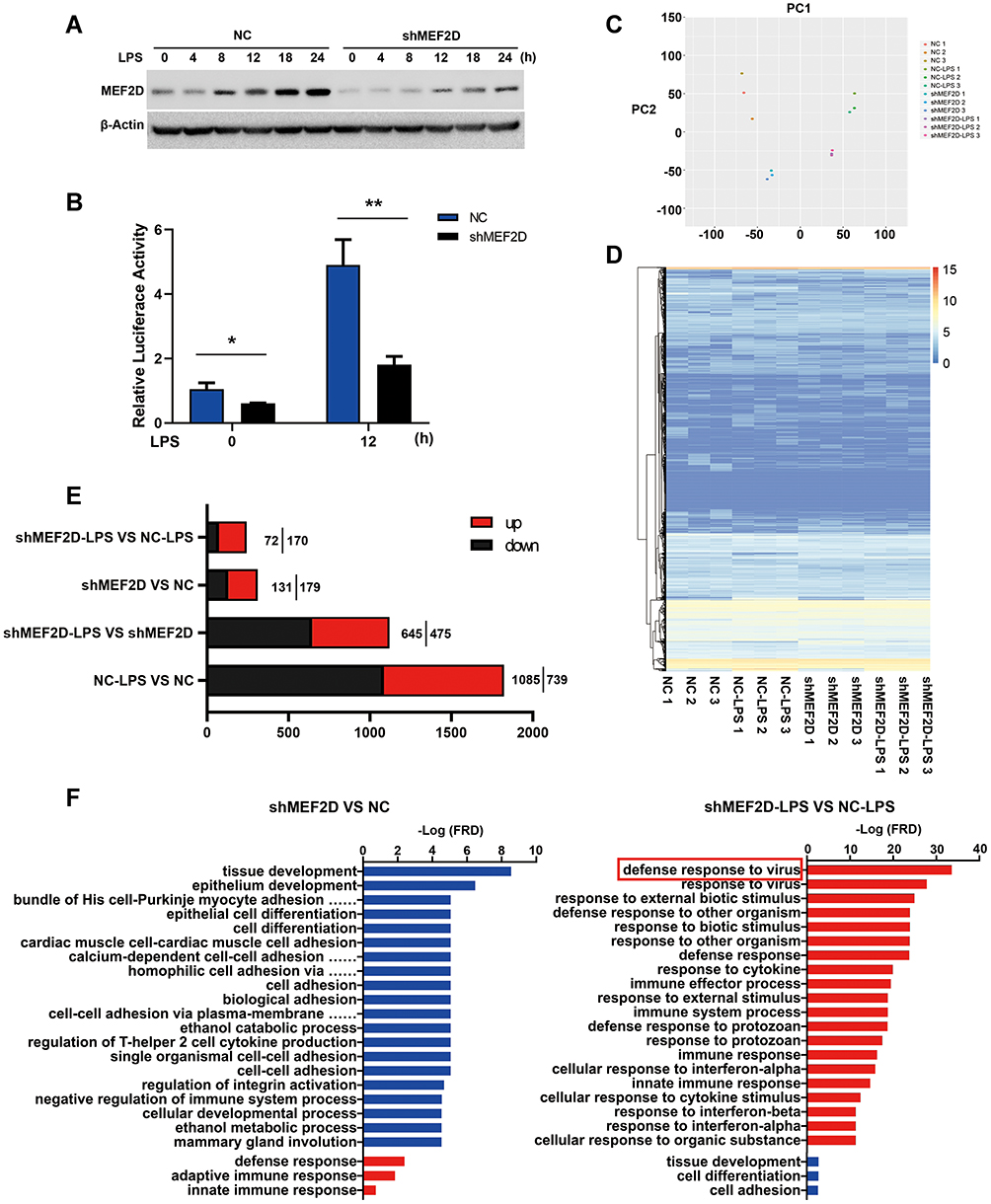 |
Figure 3 Functional annotation and canonical pathways of MEF2D knockdown in BV2 cells. (A and B) The knockdown efficiency of shRNA-MEF2D was tested by Western blot and luciferase reporter assays. The data from three independent experiments are expressed as the mean ± SEM and were analysed by two-way ANOVA (*P ≤ 0.05, **P ≤ 0.01). (C) The first principal component (PC1) separates the NC, NC + LPS, shMEF2D and LPS + shMEF2D samples, while the second principal component (PC2) separates the biological replicates of the same population in BV2 cells. (D) A heat map representing RNA-Seq shared differentially expressed genes between shMEF2D and NC cells stimulated with LPS is shown. Heat maps were generated with Multi Experiment Viewer (version 4.8) software. (E) The number of differentially expressed genes between different groups was determined. (P ≤ 0.01, and log2-fold change ≥1.5). (F) Gene ontology analysis of the functional annotations of differentially expressed genes of MEF2D knockdown compared to NC with (right) or without (left) LPS treatment in BV2 cells is shown. (P ≤ 0.01, and log2-fold change ≥1.5). |
Knockdown of MEF2D Significantly Blocked the Interferon Response System
The interferon signalling pathway is an essential part of the innate immune response in microglia by participating in the immune response to a variety of endogenous and exogenous stimuli, triggering phenotypical plasticity and the cascade of interferon-stimulated genes (ISGs). Interferons (IFNs) have been implicated in the dysregulation of the immune response in autoimmune diseases21 and neurodegeneration.22,23 Strikingly, we found that some of the downregulated genes in shMEF2D cells compared to NC cells after LPS treatment for 12 h clustered in the interferon signalling pathway, such as CXCL10, IFIT1, IL12B, IL-6, IRF7, ISG15, and MX2. (Figure 4A). Most of the suppressed ISGs were induced mainly by the IFN-Is pathway. These data strongly confirm that MEF2D knockdown obviously inhibits the interferon response system. Thus, MEF2D is an indispensable element for the full activation of the interferon signalling pathway.
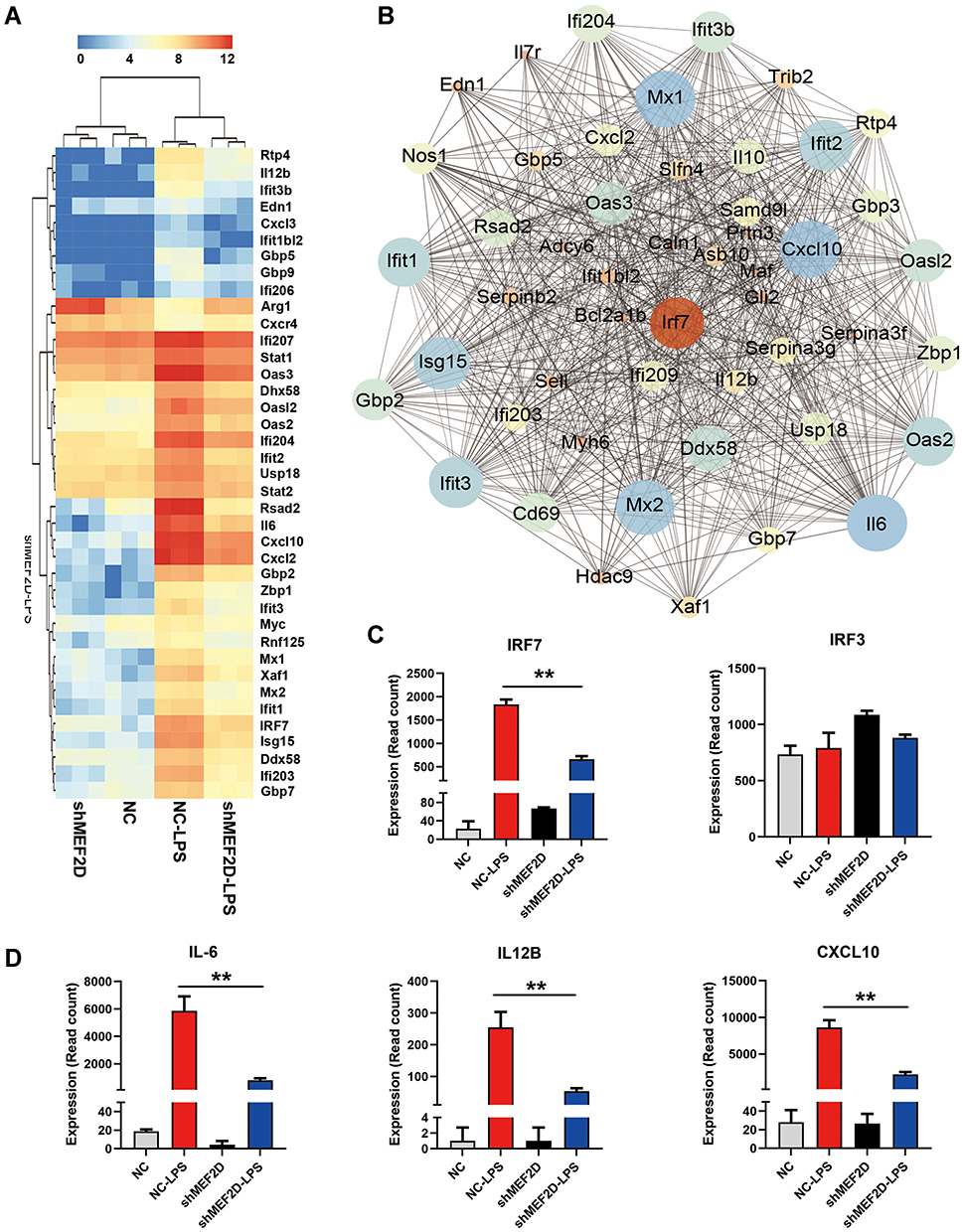 |
Figure 4 MEF2D knockdown significantly inhibited the interferon signalling pathway. (A) A heat map representation of the interferon-stimulated genes differentially expressed between shMEF2D and NC BV2 cells stimulated with LPS is shown. Data are from three independent experiments (P ≤ 0.01, and log2-fold change ≥ 1.5). Heat maps were generated with Multi Experiment Viewer (version 4.8) software. (B) The network of significantly differentially expressed genes was evaluated by RNA-seq in BV2 shMEF2D and NC stable cell lines with LPS treatment (P ≤ 0.01, and log2-fold change ≥ 1.5). (C and D) Transcript abundance (in read count) was evaluated using RNA-seq in the key regulators (IRF7 and IRF3) (C) and effectors (IL-6, IL12B and CXCL10) (D) of the type I interferon signalling pathway in different LPS-treated BV2 stable cell lines. The data from three independent experiments are expressed as the mean ± SEM and were analysed by two-way ANOVA (**P ≤ 0.01). |
IFN-Is are transcriptionally regulated24 and induced following recognition of pathogen components during infection by various host pattern recognition receptors. IRF7 and IRF3 are the most important transcription factors in the initiation of IFN-Is.25 The gene network indicated that IRF7 is the key transcription factor of the significantly and differentially expressed genes in BV2 shMEF2D and NC stable cell lines treated with LPS (Figure 4B). The transcript abundance (in read count) of IRF7 and IRF3 was evaluated, and the data showed that the IRF7 mRNA level was suppressed, while IRF3 was not affected (Figure 4C). Additionally, the transcript abundances of IFN-Is, such as IL-6, IL12B and CXCL10, were significantly suppressed by shMEF2D under LPS stimulation (Figure 4D). Because IRF7 is a master regulator in the initiation stage and in the second wave of the cascade,25,26 the RNA-seq data led us to speculate that IRF7 may be the key factor by which MEF2D regulates IFN-Is.
Transcriptional Regulation of IRF7 by MEF2D
To determine the key regulatory purpose of MEF2D, the downregulated genes in shMEF2D cells were screened with a putative MEF2 binding motif in the promoter sequences. Several genes, including IRF7, were predicted to possess potential sites (Figure 5A). To assess the direct binding of MEF2D, we carried out a ChIP-PCR assay. The PCR data revealed that MEF2D bound specifically to the regions of the IRF7 promoter that contained putative sites. Significant increases in binding occurred in response to LPS treatment in BV2 cells (Figure 5B and C). We also performed qPCR to confirm the subsequent IFN-Is. The data showed that LPS stimulation significantly activated the IFN-I signalling pathway, and knockdown of MEF2D markedly inhibited the elevation of IRF7, IFNA1, MX2, IFIT1, IFI27, ISG15, RSAD2, ZBP1 and DDX58 (Figure 5D). Together, these findings demonstrated that MEF2D directly regulates the transcriptional activity of IRF7, the key regulatory factor of IFN-Is.27,28 These results strongly support the conclusion that MEF2D is a powerful regulator of the immune system, mainly through the IFN-I signalling pathway in BV2 microglial cells.
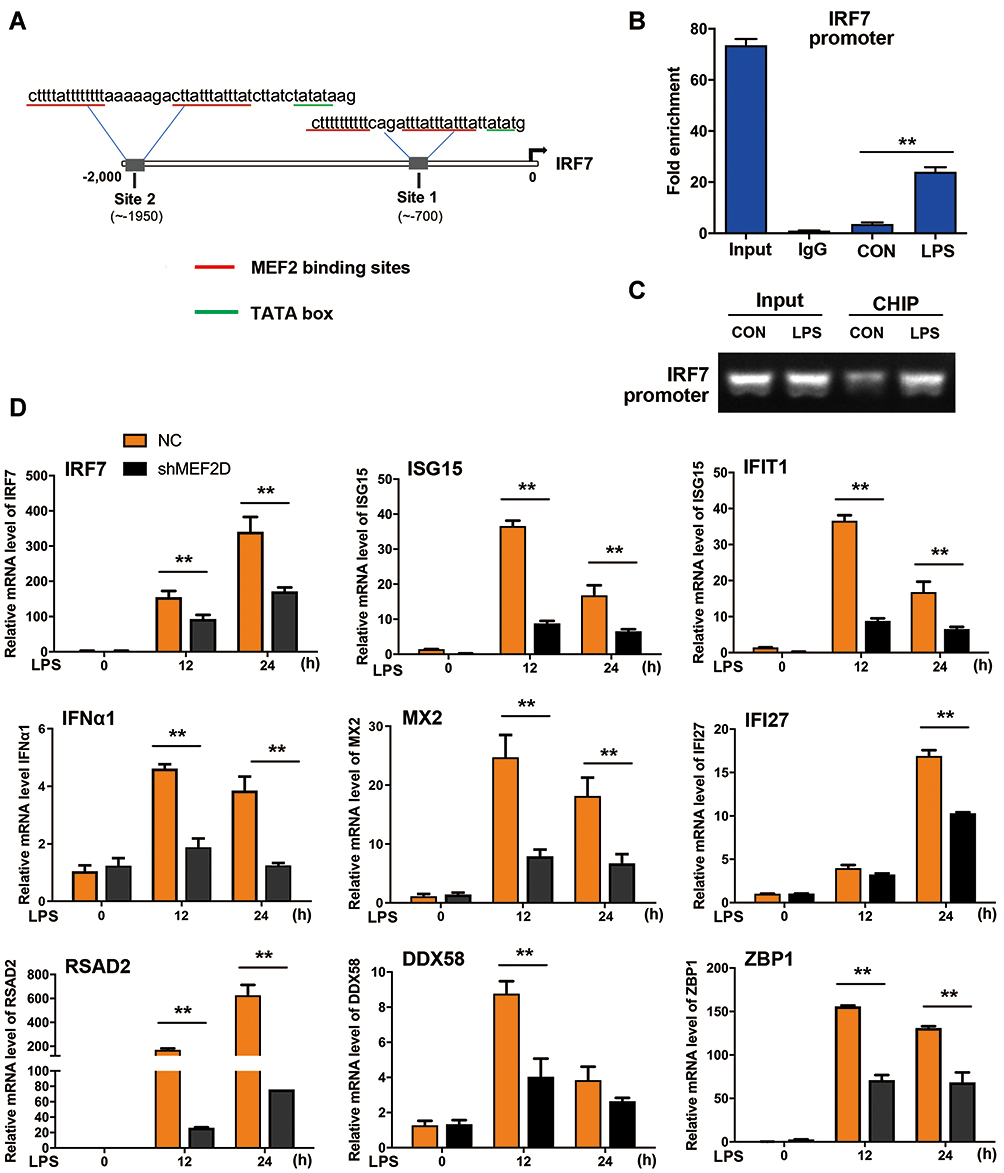 |
Figure 5 Regulation of the interferon signalling pathway by MEF2D. (A) MEF2 binding sites in the IRF7 promoter were identified. The underlined sequences in red indicate the MEF2 binding site, and those in blue indicate the auxiliary sequence TATA box. (B and C) Binding of MEF2D to the IRF7 promoter was assessed in BV2 cells. BV2 cells treated with LPS for 12 h were analysed by ChIP-qPCR and PCR. (D) MEF2D knockdown significantly reduced the key ISG levels. BV2 NC and shMEF2D stable cell lines were stimulated by LPS, and the mRNA levels of IRF7 and several ISGs, the key inducible factors of the interferon signalling pathway, were quantified by qPCR. The data from three independent experiments are expressed as the mean ± SEM and were analysed by two-way ANOVA (**P ≤ 0.01). |
Discussion
Microglia are the native macrophages in the brain. As highly plastic cells, microglia adopt diverse phenotypes and play seemingly paradoxical roles, ranging from neurotoxic to neuroprotective effects, to maintain micro homeostasis of the brain under pathological conditions. Obviously, the high plasticity of microglia requires a set of precise regulatory systems to control a wide range of functional activities under various conditions. Extensive studies have demonstrated that transcriptional regulation and epigenetic mechanisms, including microRNA regulation,29 histone modification and DNA methylation,30 are an important part of these regulatory systems. Recently, it was reported that microRNA-129-5p targets the high mobility group protein box1 to regulate inflammation in LPS-activated spinal microglial cells.31 Our study revealed that MEF2D, which sensitively responds to diverse stressors, was strongly induced in microglia under LPS stimulation and was associated with changes in microglial phenotypes and functions. Although MEF2D has been well studied in multiple model systems and promotes neuronal survival by mediating several survival and death signals,32,33 its role in the immune system needs to be further clarified. To uncover the specific regulatory function of MEF2D, we constructed stable MEF2D knockdown and MEF2D-overexpressing BV2 microglial cell lines. We found that compared to NC, knockdown of MEF2D did not affect BV2 cell proliferation or apoptosis but significantly increased the expression of proinflammatory factors, such as NLRP3, IL-1β and iNOS, following 12 h of LPS treatment. This result is different from our previous study in DA neurons.11 This may be due to the much lower level of endogenous MEF2D in microglia than in DA neurons. This inducible characteristic indicates that MEF2D is more likely to act as a stress regulator involved in the inflammatory response in microglia rather than a nutritional factor. Inhibiting the expression of MEF2D could disrupt the balance of inflammatory cytokines, leading to hyperactivation of proinflammation. Our data also indicate that knockdown of MEF2D markedly and specifically inhibits the IFN-I signalling pathway. IFN-I signals are tightly involved in the microglial inflammatory process. IFNs are a group of related antimicrobial cytokines critical for defending against viruses and other pathogens.34 Because of the elevated levels of the proinflammatory factors NLRP3, IL-1β, and iNOS, we can reasonably speculate that the inhibition of IFN-I signalling by MEF2D knockdown contributes to inadequate immune responses, which compensatively leads to overactivation of other proinflammatory signalling pathways. However, the exact mechanism by which MEF2D is involved in the crosstalk between classical proinflammatory signalling pathways, such as p38 MAPK, and the IFN signalling pathway needs further investigation.
IFNs can be divided into three classes based on sequence homology: types I, II, and III.35 Well-known IFN-Is are essential in initiating and regulating innate and adaptive immunity.34 In vitro studies proved that microglia are the main cell population in the CNS mediating the IFN-I response.36 The chronic production of IFN-Is partly contributes to the dysfunction of microglia and is implicated in the development of age-related neuropathological diseases and chronic neurodegeneration, such as age-associated cognitive decline,37 prion disease,38 and Alzheimer’s disease.39 IFN-Is exert antiviral and immunomodulatory activities by inducing ISGs. IRF7 is the master transcriptional regulator of the IFN-I-dependent immune response.27,40 The regulatory mechanism of IRF7 in activated microglia is unclear. Using targeted resequencing of 215 candidate genes involved in autoimmunity in Swedish cohorts, a rare regulatory variant rs200395694: G>T located in intron 4 of the MEF2D gene was identified to be associated with systemic lupus erythaematosus, a chronic inflammatory autoimmune disease with excessive production of IFN-Is and autoantibodies against nucleic acids as hallmarks.41 The region has properties of an active cell-specific enhancer.42 In the current study, we proved that as the key regulator of the IFN-I response, IRF7 is directly transcribed by MEF2D, explaining how IRF7 works in stress-induced microglia.
Conclusions
Imbalances in IFN-Is and the inflammatory response are often synchronized and are involved in ageing and neurodegenerative diseases. A properly controlled inflammatory response is the key to brain homeostasis. IFN-Is are pleiotropic cytokines with a critical role in the initiation, amplification and termination of the proinflammatory response in the CNS.43,44 Our data indicate that MEF2D, as an endogenous inducible factor, participates in the regulation of inflammatory homeostasis mainly by regulating the IFN-I signalling pathway. Our findings reveal that MEF2D, as a stress-sensing and regulatory molecule, implicitly and powerfully regulates IFN-I signalling pathways and is involved in the regulation of inflammatory homeostasis. It appears that MEF2D is inactive at the initial stage of inflammation but plays an inhibitory role in the amplification of the proinflammatory response. Thus, we speculate that MEF2D may be an important regulator mediating the balance between effective immune responses and excessive inflammation.
Abbreviations
ChIP-qPCR, Chromatin Immunoprecipitation quantitative real-time PCR; CNS, central nervous system; CXCL10, C-X-C motif chemokine 10; DDX58, DExD/H-Box Helicase 58; DMEM/F12, Dulbecco’s modified Eagle’s medium/F12; DMSO, dimethyl sulfoxide; ECL, electrochemiluminescence; ELISA, enzyme-linked immunosorbent assay; FBS, fetal bovine serum; GO, gene ontology; HEPES, N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid; HRP, horseradish peroxidase; IFI27, Interferon Alpha Inducible Protein 27; IFIT1, Interferon Induced Protein With Tetratricopeptide Repeats 1; IFNA1, Interferon Alpha 1; IFN-Is, type-I interferons; IL-10, Interleukin-10; IL-1β, Interleukin-1β; IL-2, Interleukin-2; IL-4, Interleukin-4; IL-6, Interleukin-6; IL12B, Interleukin 12B; iNOS, inducible nitric oxide synthase; IRF3, interferon regulatory factor 3; IRF7, interferon regulatory factor 7; ISGs, interferon-stimulated genes; ISG15, interferon-stimulated gene 15; KEGG, kyoto encyclopaedia of genes and genomes; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; MEF2, Myocyte enhancer factor-2; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MX2, MX Dynamin Like GTPase 2; NC, negative control; NLRP3, NAcht Leucine-rich repeat Protein 3; Nrf2, nuclear factor-erythroid 2-p45 derived factor 2; OE-MEF2D, overexpression MEF2D; PAGE, polyacrylamide gel electrophoresis; PCA, principal components analysis; PVDF, polyvinylidene fluoride; qPCR, quantitative real-time PCR; RNAi, RNA interference; RNA-seq, Transcriptome sequencing; RSAD2, Radical S-Adenosyl Methionine Domain Containing 2; shMEF2D, MEF2D knockdown by shRNA; TBST, tris buffered saline tween 20; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; ZBP1, Z-DNA-binding protein 1.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank Zixu Mao, Department of Pharmacology and Neurology, Emory University, USA, for providing BV2 cells and plasmids. Fangfang Lu, Ronglin Wang and Li Xia are co-first authors.
Funding
This work was supported by the National Natural Science Foundation of China, Grant No. 31930048 and 81720108016 (QY); 31770917, 31570777 and 91649106 (JL).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–318. doi:10.1016/0166-2236(96)10049-7
2. Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55(4):412–424. doi:10.1002/glia.20468
3. Thion MS, Ginhoux F, Garel S. Microglia and early brain development: an intimate journey. Science. 2018;362(6411):185–189. doi:10.1126/science.aat0474
4. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19(10):622–635. doi:10.1038/s41583-018-0057-5
5. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi:10.1146/annurev-immunol-051116-052358
6. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35(1):441.
7. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225–242. doi:10.1038/nri.2017.125
8. Ransohoff RM, Pan F, Ye Z, Cheng L, Liu JO. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–783. doi:10.1126/science.aag2590
9. Yu YT, Breitbart RE, Smoot LB, Lee Y, Mahdavi V, Nadal-Ginard B. Human myocyte-specific enhancer factor 2 comprises a group of tissue-restricted MADS box transcription factors. Genes Dev. 1992;6(9):1783–1798. doi:10.1101/gad.6.9.1783
10. Heidenreich KA, Linseman DA. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol. 2004;29(2):155–166. doi:10.1385/MN:29:2:155
11. Yang Q, She H, Gearing M, et al. Regulation of Neuronal Survival Factor MEF2D by Chaperone-Mediated Autophagy. Science. 2009;323(5910):124–127. doi:10.1126/science.1166088
12. Savignac M, Mellstrom B, Naranjo JR. Calcium-dependent transcription of cytokine genes in T lymphocytes. Pflugers Arch. 2007;454(4):523–533. doi:10.1007/s00424-007-0238-y
13. Pan F, Ye Z, Cheng L, Liu JO, Factor ME. 2 Mediates Calcium-dependent Transcription of the Interleukin-2 Gene in T Lymphocytes. J Biol Chem. 2004;279(15):14477–14480.
14. Gao L, She H, Li W, et al. Oxidation of Survival Factor MEF2D in Neuronal Death and Parkinson’s Disease. Antioxid Redox Signal. 2014;20(18):2936–2948. doi:10.1089/ars.2013.5399
15. Chu Y, Mickiewicz AL, Kordower JH. α-synuclein aggregation reduces nigral myocyte enhancer Factor-2D in idiopathic and experimental Parkinson’s disease. Neurobiol Dis. 2011;41(1):71–82. doi:10.1016/j.nbd.2010.08.022
16. Arosio A, Sala G, Rodriguez-Menendez V, et al. MEF2D and MEF2C pathways disruption in sporadic and familial ALS patients. Mol Cell Neurosci. 2016;74:10–17. doi:10.1016/j.mcn.2016.02.002
17. Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997;386(6622):296–299. doi:10.1038/386296a0
18. Yang S, Gao L, Lu F, et al. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J Neuroinflammation. 2015;12(1):33. doi:10.1186/s12974-015-0258-z
19. Clark Rebecca I, Tan Sharon WS, Péan Claire B, et al. MEF2 Is an In Vivo Immune-Metabolic Switch. Cell. 2013;155(2):435–447. doi:10.1016/j.cell.2013.09.007
20. Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27(2–3):229–237. doi:10.1016/0165-5728(90)90073-V
21. Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204(1):27–42. doi:10.1111/j.0105-2896.2005.00239.x
22. Chin AC. Neuroinflammation and the cGAS-STING pathway. J Neurophysiol. 2019;121(4):1087–1091. doi:10.1152/jn.00848.2018
23. Deczkowska A, Baruch K, Schwartz M. Type I/II Interferon Balance in the Regulation of Brain Physiology and Pathology. Trends Immunol. 2016;37(3):181–192. doi:10.1016/j.it.2016.01.006
24. Taniguchi T, Takaoka A. The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr Opin Immunol. 2002;14(1):111–116. doi:10.1016/S0952-7915(01)00305-3
25. Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005;15(6):407–422. doi:10.1038/sj.cr.7290309
26. Thackray LB, Duan E, Lazear HM, et al. Critical role for interferon regulatory factor 3 (IRF-3) and IRF-7 in type I interferon-mediated control of murine norovirus replication. J Virol. 2012;86(24):13515–13523. doi:10.1128/JVI.01824-12
27. Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi:10.1038/nature03464
28. Jang JS, Lee JH, Jung NC, et al. Rsad2 is necessary for mouse dendritic cell maturation via the IRF7-mediated signaling pathway. Cell Death Dis. 2018;9(8):823. doi:10.1038/s41419-018-0889-y
29. Karthikeyan A, Patnala R, Jadhav SP, Eng-Ang L, Dheen ST. MicroRNAs: key Players in Microglia and Astrocyte Mediated Inflammation in CNS Pathologies. Curr Med Chem. 2016;23(30):3528–3546. doi:10.2174/0929867323666160814001040
30. Kiernan EA, Smith SM, Mitchell GS, Watters JJ. Mechanisms of microglial activation in models of inflammation and hypoxia: implications for chronic intermittent hypoxia. J Physiol. 2016;594(6):1563–1577. doi:10.1113/JP271502
31. Ma XJ, Song TY, Wang WL, Zhang ZW. Effect of microRNA-129-5p targeting high mobility group protein box1 on regulating inflammation in lipopolysaccharide-activated primary spinal microglia cells. J Biol Regul Homeost Agents. 2020;34(4):1423–1430. doi:10.23812/20-133-L
32. Wang X, She H, Mao Z. Phosphorylation of neuronal survival factor MEF2D by glycogen synthase kinase 3beta in neuronal apoptosis. J Biol Chem. 2009;284(47):32619–32626. doi:10.1074/jbc.M109.067785
33. She H, Yang Q, Shepherd K, et al. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J Clin Invest. 2011;121(3):930–940. doi:10.1172/JCI43871
34. Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: a comprehensive review. J Autoimmun. 2017;83:1–11. doi:10.1016/j.jaut.2017.03.008
35. Nallar SC, Kalvakolanu DV. Interferons, signal transduction pathways, and the central nervous system. J Interferon Cytokine Res. 2014;34(8):559–576. doi:10.1089/jir.2014.0021
36. Reinert LS, Lopusna K, Winther H, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun. 2016;7:13348. doi:10.1038/ncomms13348
37. Baruch K, Deczkowska A, David E, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346(6205):89–93. doi:10.1126/science.1252945
38. Nazmi A, Field RH, Griffin EW, et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia. 2019;67(7):1254–1276. doi:10.1002/glia.23592
39. Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging. 2014;35(5):1012–1023. doi:10.1016/j.neurobiolaging.2013.10.089
40. Raftopoulou M. IRF-7 triggers the interferon alarm. Nat Cell Biol. 2005;7(5):459. doi:10.1038/ncb0505-459
41. Bengtsson AA, Ronnblom L. Role of interferons in SLE. Best Pract Res Clin Rheumatol. 2017;31(3):415–428. doi:10.1016/j.berh.2017.10.003
42. Farias FHG, Dahlqvist J, Kozyrev SV, et al. A rare regulatory variant in the MEF2D gene affects gene regulation and splicing and is associated with a SLE sub-phenotype in Swedish cohorts. Eur J Hum Genet. 2019;27(3):432–441. doi:10.1038/s41431-018-0297-x
43. Axtell RC, Type SL. 1 interferons cool the inflamed brain. Immunity. 2008;28(5):600–602. doi:10.1016/j.immuni.2008.04.006
44. Main BS, Zhang M, Brody KM, et al. Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson’s disease. Glia. 2016;64(9):1590–1604. doi:10.1002/glia.23028
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.