Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
PVALB Was Identified as an Independent Prognostic Factor for HCC Closely Related to Immunity, and Its Absence Accelerates Tumor Progression by Regulating NK Cell Infiltration
Authors Gong Y, Zhou M, Zhu Y, Pan J, Zhou X, Jiang Y, Zeng H, Zheng H, Geng X, Huang D ![]()
Received 30 November 2023
Accepted for publication 23 April 2024
Published 8 May 2024 Volume 2024:11 Pages 813—838
DOI https://doi.org/10.2147/JHC.S450479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Yiyang Gong,1,2,* Minqin Zhou,2,* Yanting Zhu,2,* Jingying Pan,2 Xuanrui Zhou,2 Yike Jiang,2 Hong Zeng,2 Hao Zheng,2 Xitong Geng,2 Da Huang1
1Department of Thyroid Surgery; Second Affiliated Hospital of Nanchang University, Nanchang, People’s Republic of China; 2Second College of Clinical Medicine, Nanchang University, Nanchang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Da Huang, Department of Thyroid Surgery, Second Affiliated Hospital of Nanchang University, 1 Minde Road, Nanchang, Jiangxi Province, 330000, People’s Republic of China, Tel +13576113993, Email [email protected]
Purpose: Hepatocellular carcinoma is the most common primary liver cancer, with poor prognosis. Complex immune microenvironment of the liver is linked to the development of HCC. PVALB is a calcium-binding protein which has been described as a cancer suppressor gene in thyroid cancer and glioma. Nevertheless, the role of PVALB in HCC is unknown.
Materials and Methods: We obtained data from TCGA and GSE54236 datasets. MCP-counter, WGCNA and LASSO model were applied to identify PVALB. With UALCAN, MethSurv, and other websites, we probed the expression, methylation and survival of PVALB. LinkedOmics and GSEA were adopted for functional analysis, while TIMER, TISIDB, Kaplan-Meier plotter, TIDE databases were utilized to evaluate the relevance of PVALB to the tumor immune microenvironment and predict immunotherapy efficacy. TargetScan, DIANA, LncRNASNP2 databases and relevant experiments were employed to construct ceRNA network. Finally, molecular docking and drug sensitivity of PVALB were characterized by GeneMANIA, CTD, and so on.
Results: PVALB was recognized as a gene associated with HCC and NK cell. Its expression was down-regulated in HCC tissue, which lead to adverse prognosis. Besides, the hypomethylation of PVALB was related to its reduced expression. Notably, PVALB was tightly linked to immune, and its reduced expression attenuated the anticancer effect of NK cells via the Fas/FasL pathway, leading to a adverse outcome. The lnc-YY1AP1-3/hsa-miR-6735-5p/PVALB axis may regulate the PVALB expression. Finally, we found immunotherapy might be a viable treatment option.
Conclusion: In a word, PVALB is a prognostic indicator, whose low expression facilitates HCC progression by impacting NK cell infiltration.
Keywords: PVALB, hepatocellular carcinoma, immune, ceRNA, bioinformatics, prognostic factor
Graphical Abstract:

Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequent cancer worldwide, occupying approximately 90% of primary liver cancer,1 while it is also a leading cause of death related to major cancers globally.2 In contrast to the observed reduction in mortality from all other common cancers (eg, breast, lung, and prostate), the death rate from HCC continually increases by 2–3% each year.3 According to current studies, chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, and non-alcoholic fatty liver disease (NAFLD) are the most prominent risk factors for HCC.4 In recent years, tumor markers have displayed superior clinical value in assisting the diagnosis of malignant tumors, and among them, serum alpha-fetoprotein (AFP) has been the most prevalent and essential indicator for detecting HCC and monitoring the therapeutic efficacy. However, despite its overall specificity for predicting HCC as high as 80% to 94%, the sensitivity is only 25% to 65%, and there still exists a controversy about the specific threshold of AFP for survival or recurrence.5 Also, as a typical tumor related to inflammation, HCC is characterized by immune evasion during the process of development and evolution,6 which is mediated by the accumulation of immunosuppressive cells in the tumor and the activation of multiple inhibitory receptor-ligand pathways.7 Hence, it is imperative to explore alternative biomarkers for diagnosing and predicting the effectiveness of HCC treatment, while also impeding immune evasion.
The Parvalbumin (PVALB) gene is positioned on the long arm of chromosome 22 (22q13),8 which encodes a protein belongs to the EF-hand protein superfamily, and depicted as a Ca2+ buffering protein.9,10 Proteins of this family exhibit a conserved helix-loop-helix structural motif, and their combined functions encompass the modulation of numerous vital cellular processes such as gene transcription, protein phosphorylation, nucleotide metabolism, and ion transport.11–13 Under normal conditions, as a calcium-binding protein, PVALB has a high affinity for Ca2+ and functions in a variety of organs. For example, in the central nervous system, PVALB exhibits high expression level in inhibitory GABAergic interneurons, thereby managing the excitability of pyramidal cells.14 In the kidney, PVALB also modulates the expression of human sodium-chloride cotransporter and responds to extracellular ATP signaling in distal tubule cells by regulating intracellular Ca2+ in epithelial cells lining tubular subpopulations of distal nephron.15 Furthermore, as reported in previous studies, PVALB may potentially contribute to the progression of malignant tumors. In thyroid cancer, overexpression of PVALB can reduce Ca2+ influx into mitochondria, leading to modification in mitochondrial morphology, increasing the amount of mitochondria and changing subcellular localization, along with the possibility of suppressing cell proliferation and triggering cell death through the AKT / GSK-3β pathway.16 Besides, PVALB has been identified to be down-regulated in glioma and glioma patients with high expression of PVALB have a favorable progression-free survival and overall survival.17 Nevertheless, the precise function and mechanism of PVALB in HCC, as well as its connection with the prognosis of HCC patients are still unclear, which appeal to us to further study it.
In this study, we have screened PVALB by Weighted Gene Co-expression Network Analysis (WGCNA) combined with Least Absolute Shrinkage and Selection Operator (LASSO) prognostic regression and assessed the expression of PVALB in normal and HCC tissue, the impact of PVALB itself on the existence of HCC sufferers, as well as the level of PVALB promoter methylation and its correlation with the survival of HCC sufferers by several databases and samples. What’s more, we conducted functional enrichment analyses of PVALB and probed the underlying mechanism of its role in HCC from varying perspectives such as tumor immune infiltration, Competing endogenous RNA (ceRNA) regulatory network and drug sensitivity.
Materials and Methods
Data Collection and Processing
We obtained PVALB expression data from the The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov), including 374 LIHC samples and 50 normal samples. In addition, we also selected the GSE54236 dataset from the Gene Expression Omnibus database (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) which includes 81 tumor samples and 80 normal samples for our study.
Cell Culture
Human HCC cell line LM3 was transfected in 37°C DMEM (HyClone, Germany) and 10% fetal bovine serum (Gibco, USA). HCCLM3 was derived from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences and the Shanghai Institute of Cell Biology in China.
WGCNA
WGCNA performs weighted gene co-expression network analysis based on R packages, looking for interconnection among modules to facilitate the identification of candidate biomarkers or therapeutic targets.18 We have first prepared packages related to TCGA and GSE54236 cohorts. Then, β = 18 and β = 12 were selected as appropriate soft thresholds to merge similar modular genes by means of the hierarchical clustering dendrogram. Afterwards, from the result of the module-trait maps in TCGA and GSE54236 cohorts, we filtered out the purple and pink modules.
UALCAN Analysis
UALCAN (http://ualcan.path.uab.edu/) is an easy-to-use web that provides insightful analysis into the gene expression data of TCGA, which is instrumental in accelerating cancer research.19 In this work, we have explored the expression of PVALB in HCC tissue and normal tissue. Besides, UALCAN database was applied to assess the relevance of PVALB expression with clinicopathological features, as well as the association of PVALB promoter methylation level to various clinical features.
Database of Hepatocellular Carcinoma Expression Atlas (HCCDB) Analysis
The HCCDB (http://lifeome.net/database/hccdb) establishes a global differential gene expression atlas for HCC by analyzing genes that are differentially expressed in multiple datasets 20. With the help of the “Expression pattern” module in HCCDB, we performed a final validation of PVALB expression in HCC samples and adjacent samples.
MethSurv Analysis
MethSurv (https://biit.cs.ut.ee/MethSurv/) provides assistance to subscribers lacking coding ability, and dedicated to the preliminary evaluation of methylation-based cancer biomarkers.20 In our research, the methylation level of sites related to PVALB in HCC was visualized by “Gene visualization” module, and “Single CpG” module was also been adopted to examine the correlation of methylation level at the cg25625146 locus with HCC patients survival.
The Shiny Methylation Analysis Resource Tool (SMART) Analysis
SMART (http://www.bioinfo-zs.com/smartapp/) is a amiable and intuitive application according to the data from TCGA, and it could be applied to assist clients in the multidimensional exploration of DNA methylation in 33 cancer types.21 With SMART, in the “Home” module, we not only determined the distribution of methylation probes linked to PVALB on chromosomes, but also acquired more detailed information on the location of CpG genomes related to PVALB. Moreover, via the “Methylation DIY” and “Survival” modules, we evaluated the methylation level of PVALB at the cg25625146 locus in normal and HCC tissue, along with the association between the methylation level at this locus and the survivorship of HCC patients.
Kaplan Meier Plotter Analysis
Kaplan-Meier plotter database (https://kmplot.com/analysis/) introduces a web-based survival analysis tool.22 Here we initially inquired about the relation of PVALB expression with the survival of HCC sufferers through mRNA RNA-seq data, displaying survival conditions on the basis of OS (Overall Survival), PFS (Progression Free Survival), RFS (Recurrence free survival), and DSS (Disease-specific survival). More in depth, we also compared the prognostic impact of PVALB in HCC patients under varied enrichment of immune infiltrating cells.
LinkedOmics Analysis
LinkedOmics (http://www.linkedomics.org/ login.php) collects multi-omics data and clinical data from 11,158 patients with 32 cancer types, all from the TCGA project.23 With this tool, we identified the co-expressed genes of PVALB and carried out Gene Ontology (GO) pathway analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) route analysis in which pathway they are involved. Beyond that, LinkedOmics was also been utilized to create scatter plots for exploring the relationship between PVALB and CGR3A (CD16), NCAM1 (CD56) and FASLG (FasL).
TIMER Analysis
TIMER (https://cistrome.shinyapps.io/timer/) is a web server that allows a thorough study of the molecular characterization of tumor-immune interactions.24 In our study, the TIMER database was applied for investigating the relationship of PVALB expression with multiple immune cells in HCC. In addition, its upgraded TIMER2 database was further explored to learn the relevance between PVALB expression and distinct immune cell infiltration in pan-cancer under different algorithms.
TISIDB Analysis
TISIDB (http://cis.hku.hk/TISIDB/) leverages multi-omics data from TCGA to aid us in investigating the relevance of specific genes to immunity in the context of tumorigenesis.25 Building on this database, we examined the relation of PVALB expression with lymphocytes, MHCs, chemokines and chemokine receptors. In addition, the variation of PVALB expression among different subgroups was studied by “Subtype” module, as well as drugs targeting PVALB were analyzed by “Drug” module.
Constructing the ceRNA Network
ceRNA is a regulatory mechanism for genes. By means of the Targetscan database (http://www.targetscan.org/)26 and the DIANA (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=site/pageandview=software)27 database, we jointly predicted and filtered the target microRNA (miRNA) that was negatively correlated with the mRNA expression of PVALB, hsa-miR-6735-5p. Then, upstream of the miRNA-mRNA silencing mechanism, we further anticipated the long noncoding RNA (lncRNA) (lnc-YY1AP1-3) that might interact with hsa-miR-6735-5p using the LncRNASNP2 database (http://bioinfo.life.hust.edu.cn/LncRNASNP/),28 analyzing and modelling the HCC-associated comprehensive lncRNA-miRNA-mRNA (PVALB) ceRNA network.
GeneMANIA Analysis and Docking Analysis
Using GeneMANIA (http://www.genemania.org),29 a robust website for gene function analysis, we mapped the PPI functional network of PVALB and analyzed the proteins that physically interact with PVALB, and SREBF1 was selected through this method. The part of the “Mutations” in cBioPortal website (http://cbioportal.org)30 was used to characterize secondary structures of PVALB and SREBF1 (study ID, LIHC-TCGA-Firehouse Legacy). From the SWISS-MODEL Database (https://swissmodel.expasy.org/)31 (PDB ID: 1AM9), we retrieved SREBF1 advanced structures.31–33 Moreover, the sophisticated structure of PVALB was predicted by the AlphaFold Database (https://alphafold.com/)34 (AlphaFold ID: P20472). In the end, the HDOCK (http://hdock.phys.hust.edu.cn/) server predicted the interactive docking model of PVALB and SREBF1, and displayed them using PyMOL software.35
CTD (Comparative Toxicogenomics Database) Analysis
CTD (http://ctdbase.org/) creates an initiative digital ecosystem by linking toxicological information on chemicals, genes and diseases.36 Our study deployed cancer-related drugs interacting with PVALB from the part of the “Chemical Interactions” in CTD.
Gene Set Cancer Analysis (GSCA)
GSCA (http://bioinfo.life.hust.edu.cn/GSCA/#/) combines data from 4 public databases for 33 cancer types and carries out hypothesis checking at the genomic, pharmacogenomic and immunogenomic level.37 Employing the “Drug” module, we compared the drug sensitivity of PVALB and SREBF1 utilizing both Genomics of Drug Sensitivity in Cancer (GDSC) and The Cancer Therapeutics Response Portal (CTRP) methods.
Colony Formation Assay
After culturing the transfected cells for 48h, approximately 0.6×103 cells were carefully retrieved and seeded into 6-well plates until colonies of suitable dimensions were formed. Then, these cells were fixed with 4% paraformaldehyde for 30 minutes, followed by staining with 1.0% crystal violet for an equivalent period. Once visible colonies had formed, the number of colonies can be counted and tallied across 10 distinct fields.
Quantitative RT-PCR Analysis
Total mRNA was extracted using a standard Trizol-based protocol (Invitrogen, USA) and reverse transcription reactions were performed by the PrimeScript RT reagent kit (Invitrogen, USA). qPCR was then carried out with SYBR Premix Ex Taq (TaKaRa, China). Lastly, we conducted semi-quantitative analysis, and employed this method to detect mRNA expression of a range of cytokines.
Western Blot
Detailed instructions for the comprehensive preparation of total protein extraction for Western blot can be located in reference.38 To summarize, proteins were meticulously isolated on ice with RIPA buffer (Beyotime, Shanghai, China) from a mixture of protease and inhibitor compounds (Thermo Fisher Scientific, New York, USA). Following centrifugation, the protein concentration was closely monitored through employment of the BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA), and the proteins in the sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis were transferred to a polyvinylidene fluoride (PVDF) membrane via electroblotting. Thereafter, the primary antibody was applied and left for an overnight incubation at a temperature of 4°C, and then the closed membrane was washed three times with TBST while incubated with the secondary antibody under room temperature conditions for a duration of 1 hour. In the final step, the expression of the proteins was detected with electrochemiluminescence (ECL).
RNA Binding Protein Immunoprecipitation Assay
RIP assays were performed using the Magna RNA immune-precipitation kit (Millipore). In brief, following the use of RIP lysates for HCC cells, the lysates were incubated with anti-AGO2 (Cat#ab186733, Abcam) or IgG (Cat#ab172730, Abcam) antibody at 4°C until overnight. The above magnetic bead-antibody complexes were resuspended with 150ul Proteinase K Buffer. In the end, RNA was extracted by q-PCR.
Dual-Luciferase Reporter Gene System
The dual-luciferase reporter gene assay utilized firefly luciferase (Genepharma Company (Shanghai, China)) as the reporter gene, while the plasmid carrying the firefly luciferase gene (Genepharma Company (Shanghai, China)) was used as the control plasmid to co-transfect cells with the reporter gene. We cloned the binding site containing hsa-miR-6735-5p into pmiRGLO Vector (Genepharma Company (Shanghai, China)) (vector name). The plasmid was positively transfected into cells using Liposome 3000 and PLUS reagent (Genepharma Company (Shanghai, China)) (name of transfection reagent), and the gene expression was obtained by observing the fluorescein luminescence.
Cell Counting Kit-8(CCK-8) Assay
We determined HCC cell viability using the Cell Reagent Counting Kit-8 (manufacturer). Briefly, after incubating the cells in 96-well plates for 24 h, 10 ul of CCK8 solution was added to each well and incubated for 2 h. Subsequently, the optical density (OD) index at 450 nm was determined using an enzyme meter.
Statistical Analysis
This article utilizes R for analyzing all of the data. Initially, based on the R package “MCPcounter”, Our study computed the level of immune cell abundance and stromal cell infiltration in the TCGA-LIHC and GSE54236 cohorts. Next, the C-index was set as a filter, and we constructed the LASSO prognostic regression model by performing an analysis of the cleaned data to obtain the values of the variable lambda with the package “glmnet”. Except for the correlation chord plot, whose result was visualized with the “circlize” package, the “ggplot2” package served for all the other results. Meanwhile, the packages “stats” and “car” were designed to count the expression of PVALB in normal and HCC tissue, “pROC” was deployed for ROC analysis of the data, while the “clusterProfilerer” was employed for GO and KEGG analysis, and “msigdbr” package was additionally applied for gene set enrichment analysis (GSEA) analysis to refer the datasets. Apart from that, the correlation of PVALB with the three scores was done by the package “ESTIMATE”, while the immune infiltration of PVALB was derived from the single-sample gene set enrichment analysis (ssGSEA) algorithm provided in the R package “GSVA”. Eventually, the relationship between PVALB and microsatellite instability (MSI) was realized by the “ggstatsplot” package, and the immunotherapy efficacy was calculated by the tumor immune dysfunction and exclusion (TIDE) algorithm through the “ggpubr” package.
Results
PVALB Was Identified as a Key Gene Associated with NK Cells in HCC Patients
In an effort to screen for critical genes capable of influencing the progression of HCC, we have performed a series of studies. Primarily, on the basis of all gene expression data in the TCGA and GSE54236 datasets related to HCC, we undertook differential expression analyses, and as presented in volcano Figure 1A–B, there were 12,471 and 9395 DEGs in the two cohorts respectively. Subsequently, with the MCP-counter algorithm, we calculated the abundance of all DEGs infiltrated in immune cells from both cohorts. Interestingly, it was revealed that most immune cells specifically NK cells were infiltrating at high abundance in the tumor microenvironment (Supplementary Figure 1A and D), Which invited us to exploit the WGCNA method, incorporating adaptive immune response and NK cells into the filter conditions, so that we can further hunt for molecular targets that can potentially inhibit the progression of HCC.
 |
Figure 1 Profiling to mine potential biomarker for HCC. Volcano plots exhibited all differentially expressed genes in (A) TCGA and (B) GSE54236 cohorts. (C) Purple module in TCGA cohort and (D) pink module in GSE54236 cohort were filtered by the relationship analysis between modules and features. (E) Venn diagram displayed the 43 pivotal genes intersected by DEGs and modules. Screening of (F) LASSO regression coefficients and plotting of (G) prognostic LASSO variable trajectories based on TCGA-LIHC data. |
In the WGCNA, depending on the scale-free R2 (R2 = 0.8), we set the soft thresholds in the TCGA and GSE54236 cohorts to 18 and 12 (Supplementary Figure 1B and E), and 29 and 20 modules were acquired by average hierarchical clustering and dynamic tree clipping (Supplementary Figure 1C and F). Afterwards, by examining the correlation of each module with HCC, NK cells and adaptive immune response, we believed that the purple module in the TCGA cohort and the pink module in the GSE54236 cohort represented the most significant modules, since both modules possessed the highest negative correlation with the “HCC” column as well as the high positive correlation with the “NK” and “GOBP_ADAPTIVE_IMMUNE_RESPONSE” columns (Figures 1C and D). Moreover, at the convergence of DEGs, purple module and pink module, we have found 43 hub genes (Figure 1E). What’s more, with the importation of these 43 pivotal genes into the LASSO prognostic regression model of TCGA-LIHC, we identified three key genes affecting the prognosis of HCC patients, which were DNASE1L3, LCAT and PVALB (Figure 1F and G). Among them, the roles of DNASE1L3 and LCAT have been widely reported in HCC, so we chose PVALB as the target of this paper.
PVALB Expression Was Down-Regulated in HCC
Target at investigating the differential expression of PVALB in HCC, we have employed a variety of tools. Initially, we assessed the mRNA expression of PVALB in an entire assortment of 26 different types of tumor tissue and normal tissue, and the outcome revealed that compared to normal tissue, PVALB expression was low in 17 different types of tumor tissue, in particular it was dramatically down-regulated in HCC (Supplementary Figure 2A). Next, 374 HCC samples and 50 normal samples were retrieved via the TCGA database, and then we used R software to draw differentiated expression plot and paired differentiated expression plot. The findings indicated that the HCC tissue showed markedly lower expression of PVALB than the normal tissue (Supplementary Figure 2B–C). what’s more, as Supplementary Figure 2D–E displayed, we analyzed PVALB expression using the GSE54236 dataset downloaded from the GEO database, as well as the data from TCGA and Genotype-Tissue Expression (GTEx) databases, which both obtained the same result consistently. In addition, making use of UALCAN online website, we further determined that the mRNA and protein expression of PVALB were notably down-regulated in HCC tissue compared to normal tissue (Supplementary Figure 2F–G). Finally, we also applied the HCCDB for verification, and the result also confirm the above (Supplementary Figure 2H). In conclusion, What we found in our study was that PVALB showed lower expression level in HCC tissue compared to normal tissue.
Relationship Between the Expression of PVALB and Clinicopathologic Features in HCC Patients
Via the UALCAN website, we geared to take the association of PVALB expression with clinicopathologic features into account for further elucidation of what impact PVALB has in HCC. According to the findings, PVALB expression varied significantly depending on gender and race (Supplementary Figures 3A and B). Additionally, we observed a link between PVALB and the cancer stages of HCC patients, with stage 2 showing an increased expression level than other stages (Supplementary Figure 3C). Furthermore, there were statistically significant differences among subgroups in several histological subtypes (Supplementary Figure 3D). Overall, the findings suggest that there exists a strong association between several clinicopathologic characteristics and PVALB expression.
Low Expression of PVALB Was Associated with a Worse Prognosis for HCC Patients
To discover the link between PVALB expression and prognosis in patients with HCC, we undertook a survival analysis according to the Kaplan Meier plotter database. The results indicated that OS, PFS, RFS and DSS were shorter in patients with low PVALB expression compared to those with high PVALB expression (Figure 2A–D, P<0.05). Subsequently, we ran a rate-of-occupancy curve (ROC) analysis to define the diagnostic worth of PVALB expression. It was found that the PVALB expression level could be precisely differentiated between HCC tissue and normal tissue with an AUC as high as 0.964 (Figure 2E). We examined PVALB expression in different cell lines and finally selected HCCLM3 cell line with the lowest expression for subsequent experiments (Supplementary Figure 7A). We then generated Flag-PVALB cell lines and examined them (Supplementary Figure 7B). Findings from the colony formation assay indicated that up-regulation of PVALB expression diminished the colony-forming ability of HCC cells (Figure 2F). CCK8 assay also demonstrated that overexpression of PVALB inhibited the proliferation of HCC cells (Supplementary Figure 7C). In summary, we discovered that low expression of PVALB portends a worse outcome for HCC patients and PVALB can be a valid separate predictor of survival.
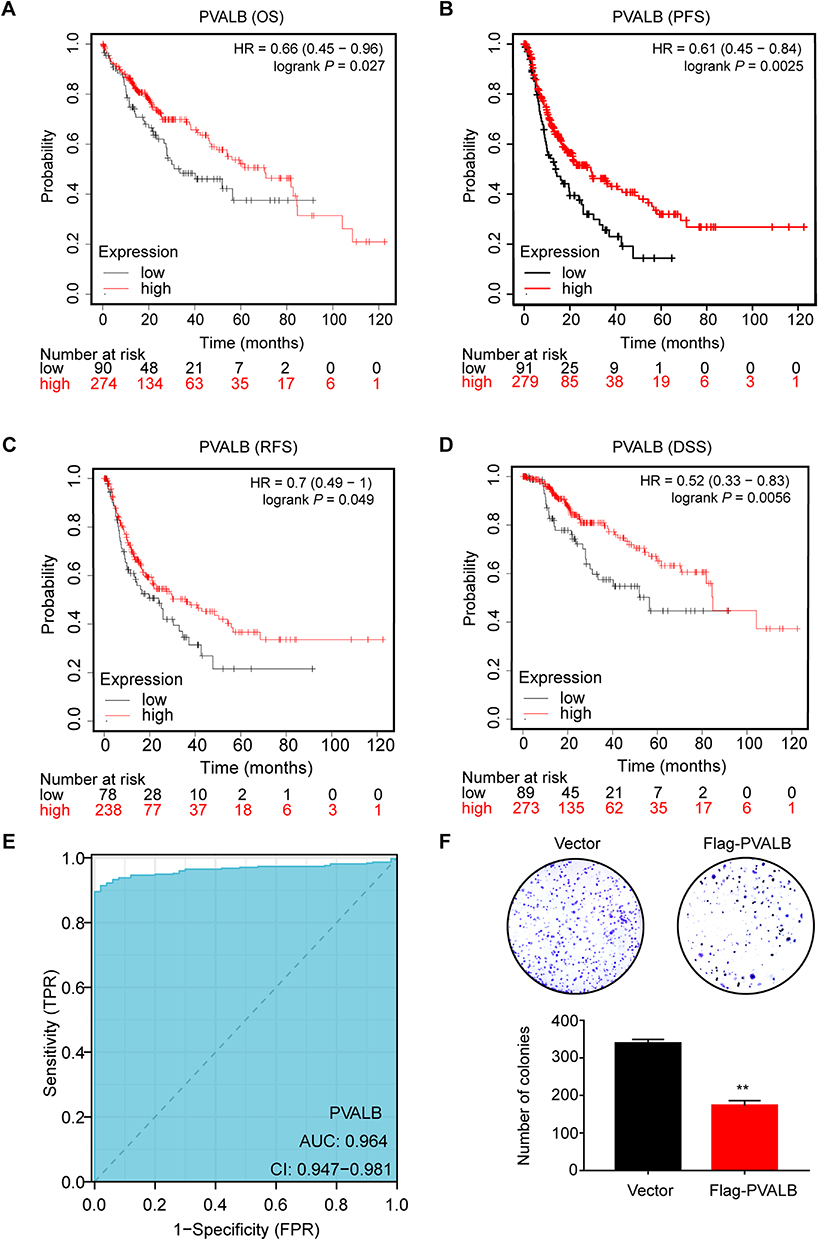 |
Figure 2 Relationship between PVALB differential expression and prognosis of HCC patients. Kaplan-Meier plotter was employed to draw (A) OS (overall survival), (B) PFS (progression-free survival), (C) RFS (relapse-free survival), and (D) DSS (disease-specific survival) survival curves. (E) Diagnostic ROC curves was used to distinguish the diagnostic value of PVALB.(F) Colonies formed by HCC cells transfected with shRNA targeting PVALB. Statistical chart shows the quantitative analysis of the result of the colony formation assay (**p < 0.01). |
DNA Methylation of PVALB in HCC
It is known that the changes in the DNA methylation level of a gene in HCC could affect its own expression level,39,40 so we started with the effect of DNA methylation on the PVALB expression, investigating the methylation level of PVALB. As a first step, we drew a heatmap using the MethSurv database and found that six sites were hypomethylated when PVALB was lowly expressed (Figure 3A). Then, through the SMART database, we explored the sites where the PVALB gene undergoes methylation and visualized the distribution of these sites on the chromosome (Figure 3B). The detailed distribution patterns of the sites were additionally displayed, with one of them on the N_shelf (cg16293983), five of them on the island (cg25625146, cg23520948, cg21789136, cg14336003, and cg03263929), six on the S_shelf (cg21385983, cg19136704, cg26244661, cg02978737, cg07856761, cg04688476) and cg06066601 was on the N_shore (Figure 3C). Also, the relationship between promoter methylation level of PVALB and various clinical features was assessed. Through the UALCAN website, we discovered that there was less PVALB promoter methylation in HCC tissue in comparison with normal tissue (Supplementary Figure 4A). Besides, the degree of PVALB promoter methylation was related to patient age, gender, presence of TP53 mutation and cancer stage according to further data. Specifically speaking, the methylation level of the PVALB promoter was strongly inversely linked with age (Supplementary Figure 4B), and it was lower in males than in females (Supplementary Figure 4C). Among them, it was captivating that TP53 mutant patients possessed a lower degree of methylation than TP53 non-mutant patients (Supplementary Figure 4D) and HCC patients at varying stages had distinct PVALB methylation (Supplementary Figure 4E). Apart from that, we once more examined the methylation of the sites and its effect on survival applying the SMART online tool, and MethSurv database. Figures 3A and E of the results indicated that cg25625146 was less methylated in HCC tissue than in normal tissue, with demethylation of this locus associated with adverse outcome in HCC patients (Figure 3D–F). All in all, hypomethylation at the cg25625146 locus might affect PVALB expression and induced poor survival in HCC patients.
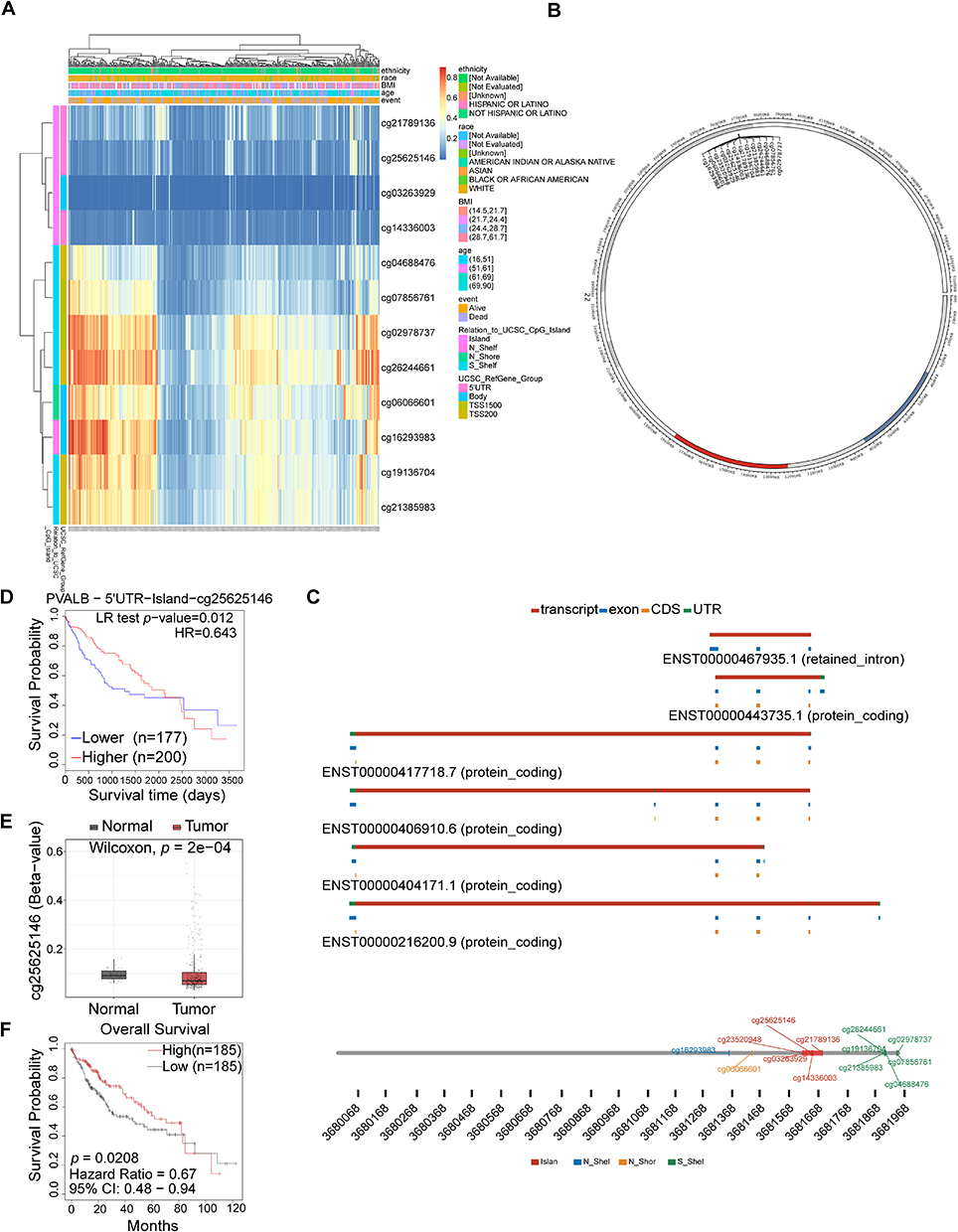 |
Figure 3 DNA methylation of PVALB in HCC. (A) Heatmap indicated the extent to which CpG sites were methylated. (B) Overall distribution for PVALB methylation sites on chromosome. (C) Localization of CpG sites linked to PVALB. (D) HCC patients had a worse prognosis when the cg25625146 locus was hypermethylated. (E) comparison of methylation level at PVALB cg25625146 site in normal and HCC tissue. (F) SMART database verified that HCC patients had a worse prognosis when hypomethylated at the cg25625146 locus. |
Enrichment Analysis of PVALB Gene Co-Expression Network in HCC
Striving to have a better understanding of the biological significance of PVALB, we attempted to pinpoint co-expressed genes with PVALB as well as analyzed the functional pathways in which they were involved. The LinkedOmics website was firstly used for the study, with volcano plot exhibiting a large number of genes significantly positively (red dots) and negatively (green dots) associated with PVALB (Figure 4A). Meanwhile two lollipop charts revealed some co-expressed genes showing the strongest plus and minus correlation with PVALB (Figure 4B and C). Following that, we performed GO (biological process) pathway profiling on these commonly expressed genes via the LinkedOmics website again, which suggested that they were majorly enriched in immune-related pathways, like type 2 immune response and adaptive immune response (Figure 4D), and the further KEGG analysis also revealed that PVALB and its related genes were engaged in immune-related pathways, such as primary immunodeficiency (Figure 4E). Within the next step, we fetched the intersection of 4519 prognostic factors from HCC with 100 genes most related to PVALB as illustrated in Figure 4B and C, and ultimately obtained 8 genes (Figure 4F). As displayed by Figure 5G, we further verified the relationship between the 8 genes at the intersection and PVALB expression, finding that when PVALB was highly expressed, the expression of genes TYROBP and CARD9 upregulated, and the remaining 6 genes were all down-regulated. Finally, we performed GO and KEGG analyses on these 8 HCC-related genes that were most correlated with PVALB as well as capable of influencing HCC patients survival, and the network diagram displayed that these PVALB co-expressed genes were functionally most relevant to the positive regulation of tumor necrosis factor superfamily cytokine production and myeloid leukocyte mediated immunity (Figure 4H). Taken together, our study revealed that genes co-expressed in PVALB were majorly enriched in pathways related to immunity.
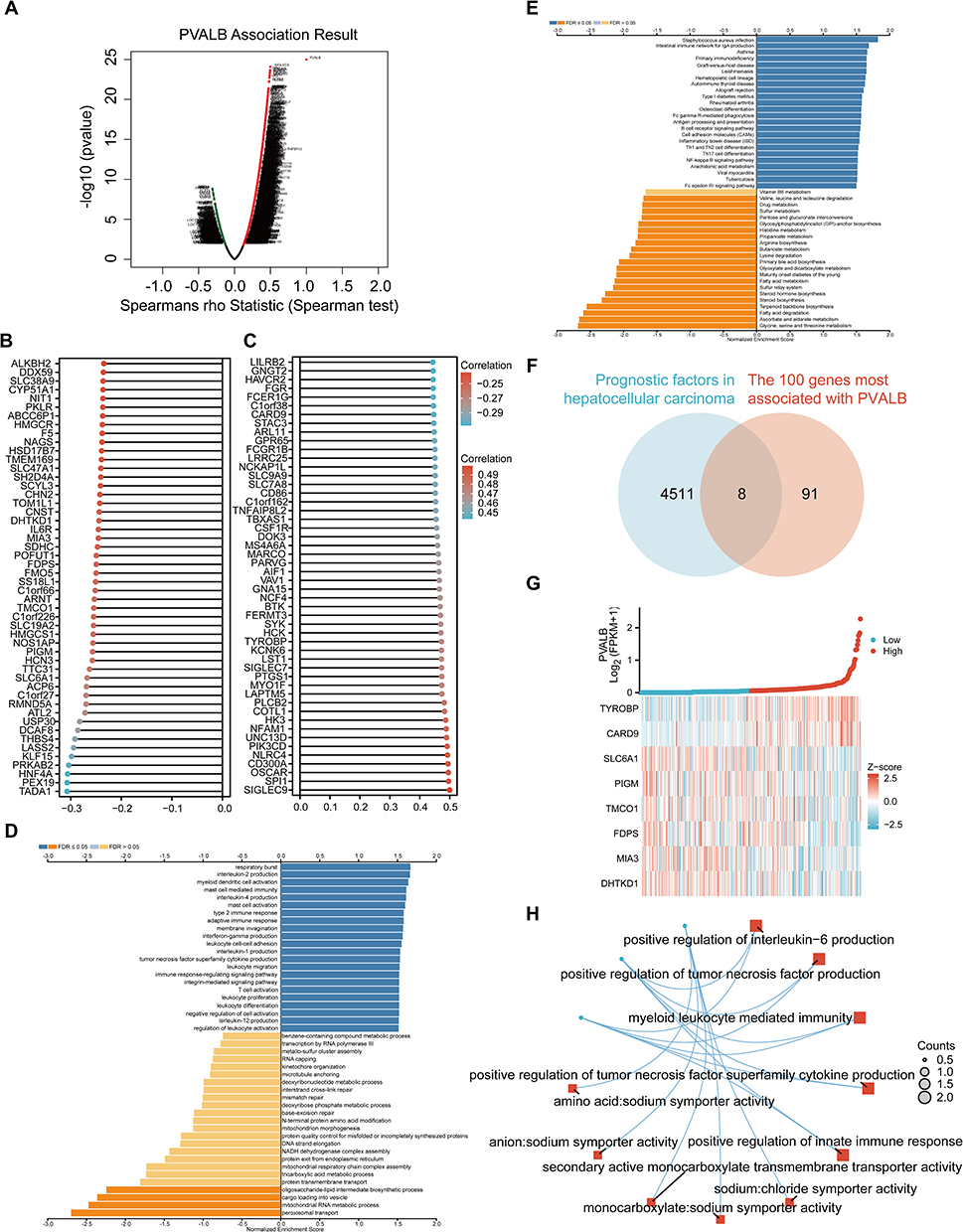 |
Figure 4 Enrichment analysis of PVALB gene co-expression network in HCC. (A) Volcano plot showed co-expressed genes associated with PVALB expression. (B–C) Lollipop charts showed the top 50 co-expressed genes each with the strongest positive and strongest negative correlation with PVALB expression. (D) Enrichment analysis of Gene Ontology (GO) terms for PVALB co-expressed genes (GO-BP). (E) Enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) terms for PVALB co-expressed genes. (F) Eight genes were identified at the intersection of 4519 HCC prognosis factors and 100 genes with the strongest positive and negative correlations with PVALB. (G) Co-expression heatmap displayed the relationship between PVALB and 8 genes. (H) Significant enrichment of the GO and KEGG pathways of the 8 genes. |
 |
Figure 5 Relationship between PVALB and immune infiltration in HCC. (A–C) Correlation of PVALB expression level with StromalScore, ImmuneScore and ESTIMATEScore was demonstrated by scatter plots based on TCGA-LIHC data. Correlation of PVALB expression with multiple immune cell infiltration based on (D) TCGA-LIHC data and (E) TIMER database. (F) Grouped comparison plot demonstrated the enrichment scores of different immune cells in high and low PVALB expression groups. (***p < 0.001, **p < 0.01, *p < 0.05, ns: non-significant). |
The Link Between PVALB and the Immune Microenvironment of HCC
According to previous findings, the tumor-infiltrating lymphocytes and the cytokines it generates have been shown to remodel the tumor immune microenvironment, thus exerting an effect on the existence of tumor sufferers.41 Besides, based on the results of enrichment analysis, it was observed that PVALB and its co-expressed genes were mostly engaged in pathways related to immune (Figure 4), and this encouraged us to further probe the connection between PVALB and the immune microenvironment in HCC. First, on the basis of TCGA-LIHC data, we computed the association of PVALB expression with StromalScore, ImmuneScore and ESTIMATEScore in HCC by applying the ESTIMATE algorithm, and it pointed a strong positive relationship of PVALB expression with three types of scores, which were 0.35, 0.40 and 0.41, respectively (Figures 5A–C). Then, we independently examined the correlation of PVALB expression with the different immune cells infiltration using the data from TCGA and TIMER databases. In HCC, both findings indicated that PVALB was most highly correlated with macrophages infiltration, while PVALB expression also exhibited a significantly positive correlation with the infiltration of various immune cells such as NK cells, B cells, CD8+ T cells, neutrophils, dendritic cells, TFH etc. (Figure 5D and E). Additionally, the infiltration of specific immune cell in each tumor on the basis of various algorithms was demonstrated in Supplementary Figure 5. At the same time, classifying PVALB into high and low expression groups, an R-based ssGSEA analysis on HCC revealed significantly higher immune cell infiltration in the PVALB high expression group across a wide range of immune cells (Figure 5F).
Next, to validate the above outcomes, we assessed the relationship between tumor-infiltrating lymphocytes (TILs) and the expression of PVALB by utilizing the TISIDB database, and the finding displayed that almost all lymphocytes were positively correlated with PVALB expression in HCC, of which macrophages and MDSC cells had the highest relevance at 0.432 and 0.441, respectively (Figure 6A). Alternatively, after further characterizing the association of PVALB expression with major histocompatibility complex (MHC), we similarly discovered that PVALB maintained a positive association with most of the MHCs, especially HLA-DPB1 (rho=0.399, p=1.68e-16) and HLA-DQB1 (rho=0.396, p=1.02e-15) (Figure 6B). Lastly, we comprehensively explored the association of PVALB expression with all cytokines, particularly chemokines and chemokine receptors capable of recruiting circulating NK cells to the site of tumorigenesis in HCC, and the outcomes showed a considerable correlation between PVALB expression and the chemokines CXCL12 and CX3CL1, as well as the chemokine receptors CXCR3 (rho=0.252, p=8.72e-07), CXCR4 (rho=0.26, p=3.86e-07), CCR7 (rho=0.198, p=0.000126), CX3CR1 (rho=0.136, p=0.00858) (Figure 6C and D). Apart from that, it was noted that PVALB expression was highest in iCluster1 and C2 (IFN-gamma dominant) subtypes when resolving the expression of PVALB in diverse molecular and immune subtypes (Figures 6E and F). These findings suggested that PVALB was closely associated with the immune microenvironment in HCC and may play a role in regulating tumor immunity.
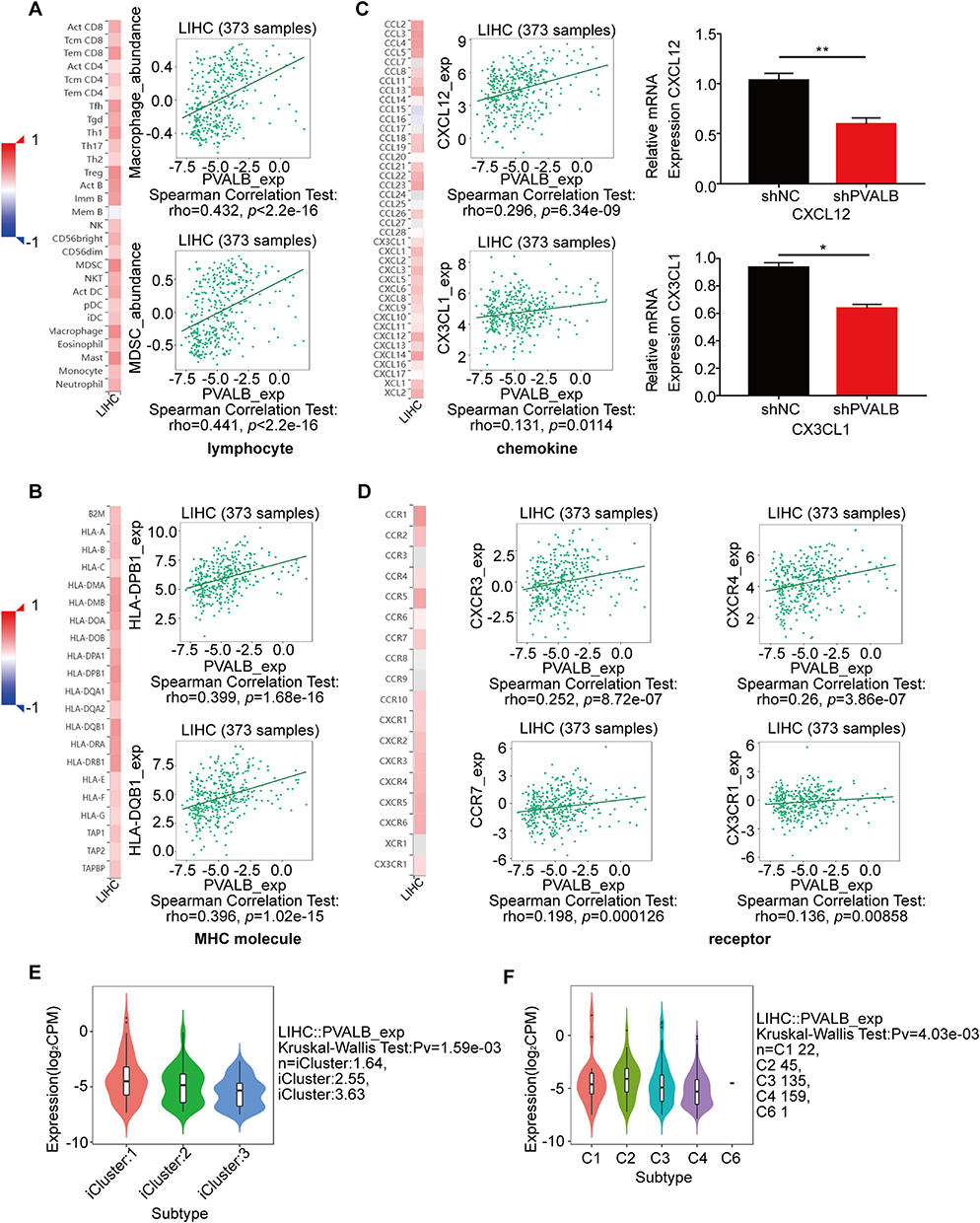 |
Figure 6 Relationship of PVALB with lymphocytes, MHCs and cytokines as well as their approximate expression level. Correlation of PVALB expression level with (A) lymphocytes (B) MHCs (C) chemokines (D) chemokine receptors in HCC. Expression of PVALB in different (E) molecular subtypes and (F) immunosubtypes in HCC.(**p < 0.01, *p < 0.05). |
PVALB Expression May Impact the Survival of HCC Sufferers Through Immune Cell Infiltration
Given the confirmed association between PVALB expression and the prognosis of HCC patients, as well as immune cell infiltration, it is plausible to suggest that PVALB may potentially impact HCC prognosis through specific immune cells. To test this conjecture, we examined the impact of PVALB on HCC patients survival utilizing the Kaplan-Meier plotter database, comparing outcomes for patients with enriched or decreased immune-infiltrating lymphocytes in the tumor immune microenvironment. Strikingly, when Natural killer T-cells and Type 1 T-helper cells were decreased, the prognosis of HCC patients with low PVALB expression was worse, whereas when Natural killer T-cells and Type 1 T-helper cells were enriched, the prognosis of HCC patients were not affected, regardless of the high or low expression of PVALB (Figure 7D and E). Meanwhile, the survival situation of the HCC patients would not be influenced when the infiltration of other immune cell change, no matter how PVALB expressed (Figure 7A–7C, 7F–7I). Therefore, this suggests us that PVALB may influence the survival of HCC sufferers by Natural killer T-cells and Type 1 T-helper cells.
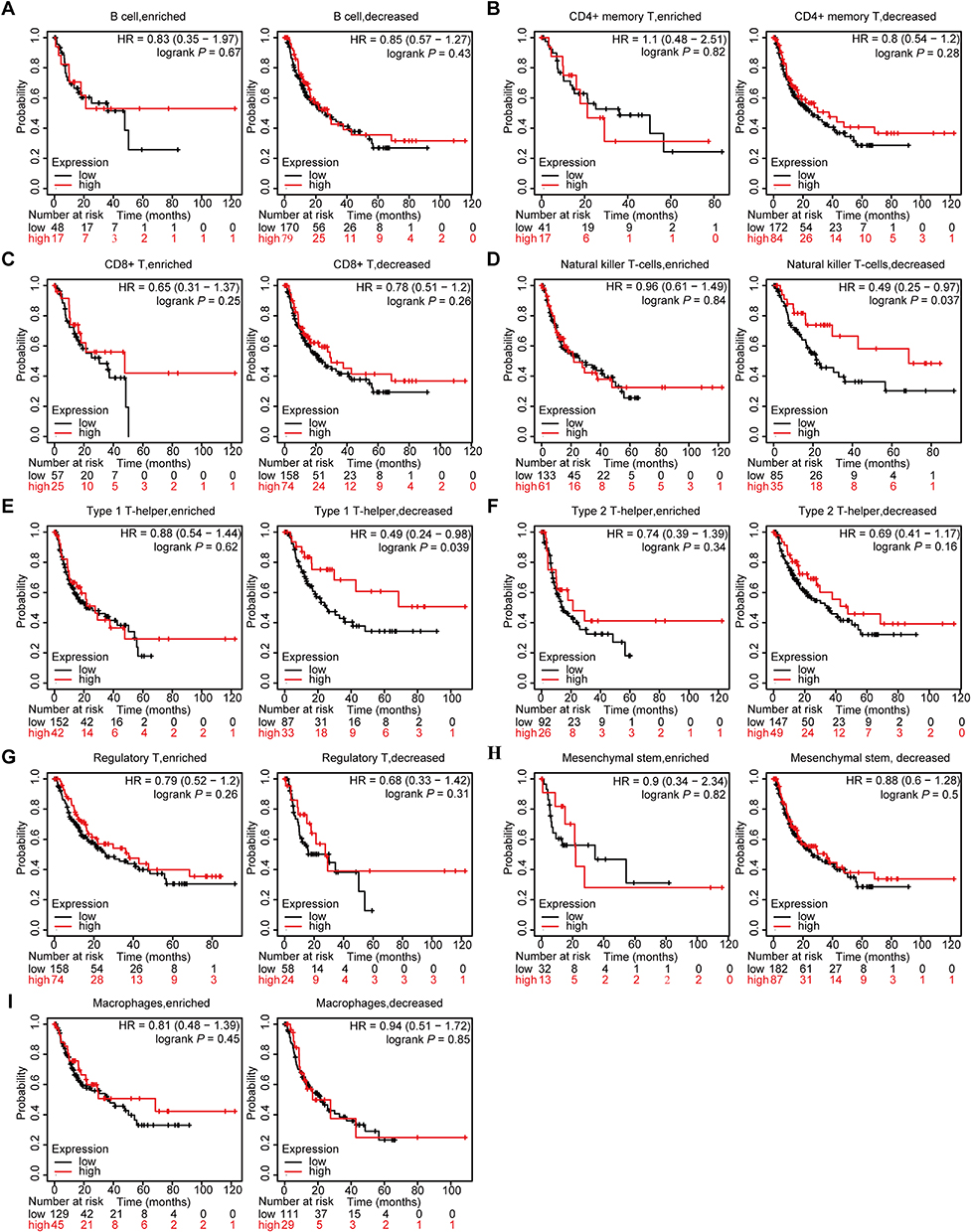 |
Figure 7 Relationship between PVALB expression and prognosis of HCC patients based on different immune cell subgroups. (A) B cell; (B) CD4+ memory; (C) CD8+ T-cell; (D) Natural killer T-cell; (E) Type 1 T-helper cell; (F) Type 2 T-helper cell; (G) Regulatory T cell; (H) Mesenchymal stem cell; (I) Macrophage. |
PVALB May Affect NK Cell Infiltration Through Specific Pathways in HCC
Considering the effect of NK cell enrichment on the survival of HCC patients, we would like to investigate the relation between PVALB and the Fas pathway from a bioinformatic perspective in depth. We initially ran a GSEA study, and the mountain range plot displayed that PVALB was enriched in five related pathways including the Fas pathway (Supplementary Figure 6A), with more detailed enrichment analyses as shown in Supplementary Figure 6B–F. These plots suggested PVALB was enrichment in the Tnfrelated Weak Inducer of Apoptosis Tweak Signaling Pathway, Pi3k akt Signaling Pathway, Fas Pathway, WNT Pathway and Adaptive Immune System, which may exert vital biological functions. Following this, we created scatter plots to investigate the correlation between PVALB and NK cell surface markers as well as ligands for Fas, discovering that when the expression of the PVALB was down-regulated, FCGR3A (CD16), NCAM1 (CD56) and FASLG (FasL) were all decreased (Supplementary Figure 6G–6I). Combined with the analysis of our previous results, we hypothesized that low expression of PVALB would affect the expansion content of some key proteins in the Fas pathway by influencing NK cell infiltration, and then activate or inhibit this pathway in order to reduce apoptosis, which ultimately promotes the immune escape of HCC cells to boost their proliferation.
The Lnc-YY1AP1-3/Hsa-miR-6735-5p/PVALB Axis May Modulate the Progression of HCC
Recently, there has been a growing amount of researches demonstrating that the lncRNA-miRNA-mRNA regulatory network had a pivotal role in driving the progression and pathogenesis of HCC.42,43 Consequently, we constructed a ceRNA regulatory network focused on PVALB in HCC and attempted to elucidate the underlying mechanism of PVALB from another perspective. Via TargetScan and DIANA databases, we jointly predicted 16 miRNAs potentially binding to PVALB (Figure 8A), while only one of them (hsa-miR-6735-5p) was identified to satisfy a negative correlation with PVALB (Figure 8B). Utilizing the TargetScan database once again, we also pinpointed promising sites on PVALB mRNA that bind to hsa-miR-6735-5p and subsequently introduced mutation at these sites (Figure 8C). We performed RIP-qPCR and demonstrated that PVALB could bind to hsa-miR-6735-5p (Figure 8D). Based on the discovery of a dual-luciferase reporter gene system, we ascertained that the hsa-miR-6735-5p mimic could reduce the luciferase activity of cells that transfected with wild-type PVALB (Figure 8E). Additionally, as seen in the comparison with the mock group, the hsa-miR-6735-5p mimic was capable of inhibiting the expression of PVALB in HCCLM3 cells, whereas the hsa-miR-6735-5p inhibitor has conversely increased the expression level of PVALB (Figures 8F–G). Afterwards, exploiting the LncRNASNP2 database, we delved deeper into the potential lncRNAs that might bind to hsa-miR-6735-5p, and identified the only one of these lncRNAs (lnc-YY1AP1-3) that conformed to negative correlation with hsa-miR-6735-5p and positive correlation with PVALB according to the experimental results. Similarly, the prospective binding sites of hsa-miR-6735-5p to lnc-YY1AP1-3 were displayed in Figure 8H, and the binding sites of hsa-miR-6735-5p were further mutated. Alternatively, the result of the dual-luciferase reporter gene system revealed that the lnc-YY1AP1-3 mimic reduced luciferase activity of cells that transfected with the hsa-miR-6735-5p wild type, whereas this did not occur of cells that transfected with the mutant-type hsa-miR-6735-5p (Figure 8I). Besides, it was not difficult to notice from Figure 8J that lnc-YY1AP1-3 mimic can suppress the expression of hsa-miR-6735-5p, and the overexpression of lnc-YY1AP1-3 can also elevate the expression level of PVALB in HCCLM3 cells (Figure 8K). Thereafter, relying on a rescue experiment, we also observed that lnc-YY1AP1-3 overexpression was able to rescue the reduction in PVALB expression level caused by hsa-miR-6735-5p overexpression (Figure 8L). Ultimately, as indicated in the colony formation assay, hsa-miR-6735-5p could facilitate the proliferation of HCCLM3 cells, in direct contrast to the effect of lnc-YY1AP1-3. Notably, hsa-miR-6735-5p overexpression had the ability to rescue the attenuation of the cellular proliferative capacity caused by lnc-YY1AP1-3 overexpression (Figure 8M). Altogether, our findings suggest that the lnc-YY1AP1-3/hsa-miR-6735-5p/PVALB axis may be involvement in modulating PVALB expression, with potential implication for influencing the proliferation of HCC cells.
 |
Figure 8 Construction of the ceRNA regulatory network of PVALB in HCC. (A) Venn diagram showed miRNAs jointly predicted by TargetScan and DIANA databases. (B) Correlation of PVALB with hsa-miR-6735-5p. (C) Potential binding sites of PVALB to hsa-miR-6735-5p predicted by TargetScan database. (D) RIP-qPCR showed the enrichment of hsa-miR-6735-5p detected by the PVALB. (E) Potential binding sites of PVALB to hsa-miR-6735-5p validated by the dual-luciferase reporter gene system. (F) mRNA expression level of PVALB under the influence of hsa-miR-6735-5p mimic and (G) hsa-miR-6735-5p inhibitor. (H) Potential binding sites of lnc-YY1AP1-3 to hsa-miR-6735-5p predicted by the LncRNASNP2 database and (I) validated by the dual-luciferase reporter gene system. (J) Expression level of hsa-miR-6735-5p under the influence of lnc-YY1AP1-3 mimic. (K) Protein expression level of PVALB in mock and lnc-YY1AP1-3 groups were explored by Western blot. (L) Changes in protein expression level of PVALB under four different conditions. (M) Changes in the number of colonies in four different groups.(***p < 0.001, **p < 0.01, *p < 0.05, ns: non-significant). |
PVALB Expression Could Serve as a Prognostic Marker for Immunotherapy Response in HCC Patients
Over the past few years, immunotherapy has gradually come into the limelight and is rapidly growing as a robust clinical strategy for the healing of cancer.44 In this article, we carried out a lot of works with the aim of further elucidating the role of PVALB in immunotherapy (mainly ICB). Initially, employing TCGA-LIHC data, an association between PVALB expression and immune checkpoint genes was investigated, and we observed significant correlation between PVALB expression and a majority of these factors (Figure 9A). Specifically, it was meaningfully and positively linked to 9 immune checkpoint genes except TGFBR1 (Figure 9B). In addition, the 3 most correlated genes were presented in scatter plots, which were CD244 (R=0.403, p<0.001), TIGIT (R=0.400, p<0.001), CD96 (R=0.375, p<0.001) (Figure 9C–E). As a critical tumor feature, MSI may be connected with the reaction to immunotherapy,45 therefore, we probed the link between PVALB expression and MSI score in HCC. It was revealed that the two maintained a negative correlation, suggesting that the probability of mutation was higher when PVALB was lowly expressed in HCC, at which time a better result was achieved with immunotherapy (Figure 9F). Simultaneously, the TIDE score also implied that lower score may be received when PVALB was lowly expressed, which hinted us that immune checkpoint blockade therapy was more therapeutic when the expression of PVALB was low in HCC (Figure 9G). All of these findings indicated that PVALB may be instrumental in forecasting the response to immunotherapy for HCC patients, and better efficacy may be harvested with immunotherapy in the presence of low PVALB expression.
 |
Figure 9 Role of PVALB in immunotherapeutic response. (A) Chordal plot demonstrated the overall connection between PVALB and immune checkpoint genes (B) Heatmap displayed the specific relationship between PVALB expression and 10 immune checkpoint genes (C–E) Scatter plots showed the correlation between PVALB expression and CD244, TIGIT, CD96 (F) Association between PVALB expression and MSI score (G) Box plot demonstrates the difference in TIDE score between high and low PVALB expression groups.(****p < 0.0001, *p < 0.05). |
Network and Drug Sensitivity Analysis Across PVALB and Its Interacting Genes
In search of other related proteins that possibly interact with PVALB, we constructed the interaction network with GeneMANIA online tool for PVALB, in which PVALB had an explicit physical interaction with SREBF1 (Figure 10A). Then, we have further downloaded the secondary structure of some proteins, which were encoded by these two genes, and analyzed their structural domains. The analysis gave the results that PVALB had an EF-hand_7 structural domain and SREBF1 possessed an HLH structural domain (Figure 10B). Subsequently, we obtained the spatial structure of SREBF1 from the SWISS-MODEL database and made a prediction of the spatial structure of PVALB with the AlphaFold database, then utilizing HDOCK Server to predict the docking model of its protein-protein interaction (Figure 10C). Besides, by modeling the PVALB-SREBF1 molecular docking, we also observed that it contains 11 hydrogen bonds (Figure 10D). Moreover, in order to better probe the clinical value of PVALB for HCC patients, we thus examined the relevance of PVALB expression to multiple drugs related to cancer via the CTD, finding that 8 drugs could affect PVALB expression, of which copper could boost its expression level (Figure 10E). Afterwards, we mapped using the TISIDB and recognized PVALB as one of the targets of the drug DB01942, also known as formic acid (Figure 10F). On a final note, the GSCA database was employed to perform drug sensitivity analysis, as shown in Figure 10G. To our surprise, with low PVALB expression, cells were only resistant to Navitoclax, while sensitive to the other 16 drugs. Whereas, in the CTRP drug sensitivity analysis, cells with low PVALB expression were sensitive to CAL-101, TGX-221, dasatinib and lovastatin (Figure 10H). In conclusion, our findings provide new and alternative treatment options for HCC patients accompanied by low PVALB expression.
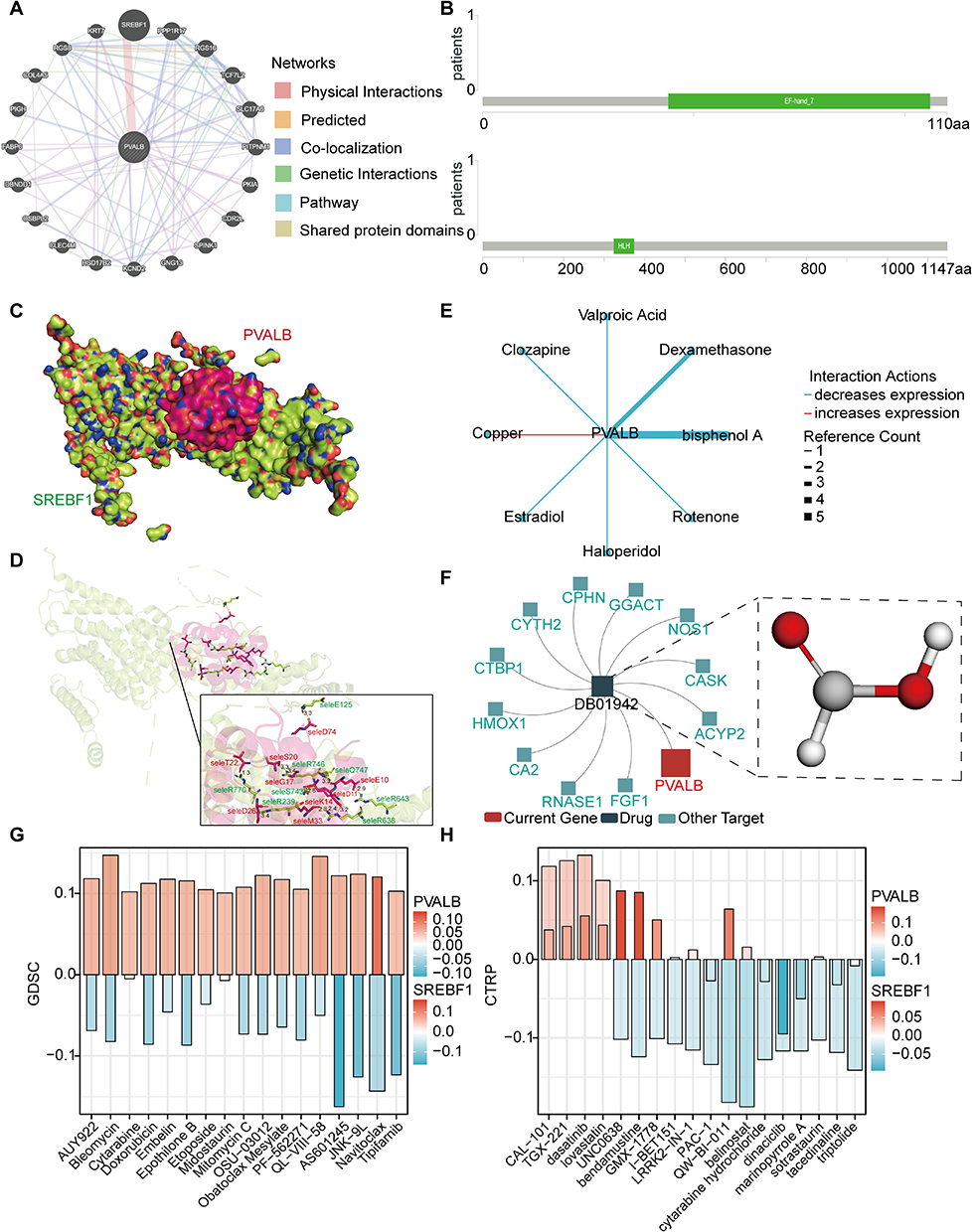 |
Figure 10 Molecular docking analysis of PVALB and its drug sensitivity analysis. (A) Construction of the interaction network of PVALB with other genes by GeneMANIA database. (B) Protein secondary structures underlying PVALB and SREBF1. (C–D) Molecular docking model of PVALB-SREBF1 (E) Cancer-related drugs from CTD that interact with PVALB. (F) Drugs targeting PVALB studied by TISIDB. Pharmacosensitivity analysis of PVALB and SREBF1 based on the (G) GDSC and (H) CTRP databases (The large column represent PVALB and the small columns represent SREBF1). |
Discussion
HCC is the fifth largest cancer in the world,46 with high malignancy, poor long-term prognosis and easy recurrence. The five-year survival rate of HCC is only 18%. At present, the treatment of HCC is still mainly based on surgical treatment, supplemented by radiotherapy, chemotherapy and ablation.47–49 In addition, there are other novel approaches, such as TACE, minimally invasive interventional therapy, molecular targeted therapy, immune checkpoint inhibition, etc,50–53 which are also the research hotspots of HCC treatment. Some existing molecular biomarkers, such as AFP, are used for the auxiliary diagnosis of HCC in clinical practice, but their sensitivity and accuracy are still insufficient. Finding a more reliable biomarker is therefore essential for the diagnosis, therapy and prognostic assessment of HCC patients. In this research, an array of rigorous bioinformatic analyses and experimental validation identified PVALB as an independent prognostic factor and a prospective therapeutic candidate that can regulate the HCC immune microenvironment and affect HCC progression.
Immunological escape is a key factor in the refractoriness of HCC, which possesses a complicated immunological microenvironment.54,55 The liver has long been recognized as the site of immune tolerance. This property results from the interaction between para-hepatic cells and peripheral leukocytes, involving high expression of immune checkpoints, IL-10 and TGFβ mediated immune suppression microenvironment, etc. NK cells in the liver can directly kill stress cells and tumor cells, and mediate antibody-dependent cytotoxicity (ADCC) with the participation of CD16.56 Once carcinogenesis occurs, these regulatory mechanisms are destroyed, the immune microenvironment of the liver will also be disordered, the infiltration of immune cells will be changed, the infiltration of dendritic cells, tumor-associated macrophages, regulatory CD4+T cells and other cells will be increased,57–59 CD8+T cells will be functionally depleted.60,61 NK cells will also show a depleted phenotype, and express high level of programmed death ligand 1 (PD-L1).62,63 The secretion of immunosuppressive cytokines such as IL-10 causes immune escape of tumor cells, and HCC develops in the direction of malignancy. Therefore, it is important to explore the immune microenvironment of HCC and the mechanism of immunosuppression. In our study, MCP-counter findings also suggested the importance of exploring the immune microenvironment in HCC.
In order to find the key factors that can prevent the development of immunosuppressive microenvironment and immune escape of HCC, we used WGCNA, based on TCGA and GEO databases, to screen out the tumor suppressor modules closely related to NK cells and adaptive immunity. Through LASSO regression analysis, combined with the background report of the gene, we finally selected PVALB. Our results firstly indicated that HCC patients with elevated PVALB expression were inclined to exhibit better prognostic outcomes, and low expression of PVALB was a biomarker indicating a unfavorable outcomes in HCC patients. We also found that PVALB was hypomethylated, and cg25625146 was hypomethylated in multiple databases, which predicted poor prognosis of HCC. Thus, we identified PVALB as a reliable prognostic biomarker in HCC.
After exploring the reasons why PVALB expression is low in HCC, we turned to explore the biological functions involved in PVALB. Pathway enrichment analysis showed that PVALB was strongly linked to immunity. We sought to analyze how PVALB affects the immune microenvironment of HCC. Numerous researches have shown that change of immune cells infiltration is an important part of the changes in the immune microenvironment, and it is also a major contributor to the adverse prognosis of HCC. It has been reported that BRCA1-PALB2 interaction can induce immunosuppression in HCC, improve T cell infiltration and lead to poor prognosis.64 Exosomal UHRF1 secreted by cancer cells induces NK cell dysfunction, forms immunosuppression, and then leads to anti-PD1 immunotherapy resistance in HCC patients.65 We addressed the relevance of PVALB expression to immune cells in HCC, and detected the infiltration of various immune cells under the condition of low expression of PVALB. It could be seen that the infiltration level of macrophages, iDC, T cells, NK cells and other cells was positively correlated with PVALB. In other words, when the expression of tumor suppressor gene PVALB is low, the infiltration of immune cells is generally reduced and the ability to resist tumor is decreased. Therefore, it is reasonable to speculate that the low expression of PVALB will cause immunosuppression. Besides, we also detected the expression of MHCs, chemokines and chemokine receptors, finding that most of them were also positively correlated with PVALB expression, and their expression was inhibited when PVALB expression was low. Maybe there are some difference between our results and other literature, this is because the infiltration of immune cells in HCC can be affected by many other pathways, and the immune microenvironment contains not only immune cells, but also stromal cells and tumor cells, which will also affect the formation of tumor microenvironment in HCC. We cannot deny that the infiltration of immune cells is influenced by factors other than PVALB.
To further clarify which immune cell infiltration changes affect the survival of HCC patients, we resolved the survival of HCC patients with or without enrichment of various immune cells when the expression of PVALB was low. Ultimately we found that patients with low PVALB expression had poor survival when NK cells were low enriched, whereas the survival of patients with low PVALB expression was not different from that of the high expression group when NK cells were enriched. This suggests that low infiltration of NK cells is a crucial link in the poor prognosis of HCC induced by low expression of PVALB. Similar results were found in Type-1 T-helper cells. However, the enrichment of other immune cells does not affect the survival of HCC patients. Therefore, we hypothesized that the low expression of PVALB contributes to the poor prognosis of HCC by affecting the low infiltration of NK cells and Type-1 T-helper cells. However, combined with our previous analysis of the association between PVALB expression and immune cell infiltration, the positive correlation between PVALB and NK cells is more significant, while the correlation with Type-1 T-helper cells is positive but weak. Therefore, we further identified NK cells as the most critical part for PVALB-mediated poor prognosis in HCC.
There are numerous reports which confirmed that the failure and loss of NK cells in HCC is an important causes of the incidence and progression of HCC. Decreased cytotoxicity of NK cells in HCC patients and weakened killing effect on HCC cells, leading to the progression of HCC.66 By restoring NK cell activity, it can inhibit the progression of HCC and even treat HCC. It has been reported that reversal of NK cell depletion by blocking CD96-CD155 interaction restores immunity to HCC.62 Serum cholesterol buildup in NK cells triggers their activity on HCC cells, augmenting the anti-tumor function of NK cells.67
It is known that HCC cells can avoid the attack of immune cells through a series of pathways,68 invading and destroying normal cells or tissues of the human body. NK cells are important anti-tumor immune cells and can also perform immune surveillance on malignant tumors.69–71 Its function is mainly to actuate apoptosis in tumor cells by releasing cytotoxic particles or cell surface synthesized proteins, and also to secrete a series of pro-inflammatory factors and chemokines to enhance the anti-tumor effect.72–74 The main pathway engaged in NK cell-mediated killing of tumor cells is the Fas/FasL pathway. Activated NK cells express FasL and bind to Fas on the surface of tumor cells to activate apoptosis of target cells.72,75 In the HCC environment, the low infiltration of NK cells will prevent the FasL on the surface of NK cells from combining with the Fas on the surface of tumor cells to induce the apoptosis of HCC. In addition, some studies have confirmed that while the infiltration of NK cells is low, HCC cells also highly express FasL, which binds to Fas on the surface of lymphocytes to induce apoptosis of lymphocytes and induce immune escape of HCC cells.76 Our results found that PVALB was positively related to NK cell infiltration and FasL expression. And when the expression of PVALB is low, the NK cell infiltration and FasL expression are also decreased. We guessed that it attenuates the Fas/FasL pathway mediated apoptosis, which leads to the progression and poor prognosis of HCC. This suggests that restoring the high expression of PVALB to restore the high infiltration of NK cells to enhance Fas/FasL pathway mediated apoptosis, and improving the tumor-killing ability of NK cells to kill HCC cells may be a feasible idea for treating HCC. Recently, CAR-NK methods have been developed to enhance the apoptosis of tumor cells induced by NK cells through the Fas/FasL pathway, and improve the selectivity and effectiveness of killing cancer cells.77 This shows that our thinking is scientific, meaningful and correct.
ceRNA refers to a subset of non-coding RNAs that aggressively bind to and cross-regulate each other through shared microRNAs at the post-transcriptional level.78 ceRNA hypothesis has been used to explain the altered expression of certain genes in various cancers like gastric carcinoma, gallbladder carcinoma. Therefore, we investigated the ceRNA regulation mechanism of PVALB.79,80 Based on the results, lnc-YY1AP1-3/hsa-miR-6735-5p/PVALB axis was excavated, which perfectly explains why PVALB is down-regulated in HCC.
In addition, we further explored the impact of PVALB knockdown on immunotherapy in HCC. MSI results suggest that when PVALB expression is low, MSI scores are high, indicating that errors are more likely to occur in the process of DNA replication and the mutation rate is high. Clinical studies have shown that MSI-H tumors are related to immune checkpoint inhibitor sensitivity, and patients will benefit from immunotherapy. TIDE score directly predicted better immunotherapy efficacy in patients with low PVALB expression. The findings indicated that copper could increase PVALB expression, suggesting that we can find suitable copper formulations to restore the expression of PVALB and its tumor suppressive effect in the future.
Conclusions
In conclusion, this research firstly demonstrates the functional role of PVALB in HCC and the underlying mechanism, which might offer novel insights into the therapy of HCC.
Abbreviations
HCC, Hepatocellular carcinoma; PVALB, Parvalbumin; OS, Overall Survival; PFS, Progression Free Survival; RFS, Recurrence free survival; DSS, Disease-specific survival; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; GSEA, gene set enrichment analysis; ssGSEA, single-sample gene set enrichment analysis; MSI, microsatellite instability.
Data Availability
Publicly available datasets were analyzed in this study. The data are accessible in TCGA and GEO databases. Further inquiries can be directed to the corresponding author.
The datasets analysed during the current study are available in the TCGA (https://portal.gdc.cancer.gov) and GEO databases, (https://www.ncbi.nlm.nih.gov/geo/).
Ethics Approval and Informed Consent
According to item 1 and 2 of Article 32 of “the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects”, which was reviewed by the National Science and Technology Ethics Committee, approved by the State Council of China, and jointly promulgated by the National Health Commission, the Ministry of Education, the Ministry of Science and Technology and the State Administration of Traditional Chinese Medicine on Feb. 18, 2023. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by grants from Science and Technology Project of Jiangxi Provincial Health Commission(202210611) and Science and Technology project of traditional Chinese medicine in Jiangxi province(2023Z030) and Jiangxi Provincial Administration of Traditional Chinese Medicine Science and Technology Plan Project (2023B1181) and Science and Natural Science Foundation of Jiang xi Province Youth Science Foundation (No:20224BAB216055) and National Natural Science Foundation of China(82260472).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Asafo-Agyei KO, Samant H. Hepatocellular carcinoma. In: StatPearls. StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC.; 2023.
2. Piñero F, Dirchwolf M, Pessôa MG. Biomarkers in hepatocellular carcinoma: diagnosis, prognosis and treatment response assessment. Cells. 2020;9(6). doi:10.3390/cells9061370
3. Wang W, Wei C. Advances in the early diagnosis of hepatocellular carcinoma. Genes Dis. 2020;7(3):308–319. doi:10.1016/j.gendis.2020.01.014
4. Mak LY, Cruz-Ramón V, Chinchilla-López P, et al. Global epidemiology, prevention, and management of hepatocellular carcinoma. Am Soc Clin Oncol Educ Book. 2018;38:262–279. doi:10.1200/edbk_200939
5. Bruix J, Castells A, Bosch J, et al. Surgical resection of hepatocellular carcinoma in cirrhotic patients: prognostic value of preoperative portal pressure. Gastroenterology. 1996;111(4):1018–1022. doi:10.1016/s0016-5085(96)70070-7
6. Shlomai A, de Jong YP, Rice CM. Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin Cancer Biol. 2014;26:78–88. doi:10.1016/j.semcancer.2014.01.004
7. Villalba M, Rathore MG, Lopez-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol. 2013;45(1):106–113. doi:10.1016/j.biocel.2012.04.024
8. Ritzler JM, Berchtold MW. An STS in the human parvalbumin gene (PVALB). Nucleic Acids Res. 1992;20(6):1428. doi:10.1093/nar/20.6.1428-A
9. Wang W, Martindale J, Metzger JM. Parvalbumin: targeting calcium handling in cardiac diastolic dysfunction. Gen Physiol Biophys. 2009;28:F3–6.
10. Schwaller B. Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol. 2010;2(11):a004051. doi:10.1101/cshperspect.a004051
11. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–529. doi:10.1038/nrm1155
12. Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405(2):199–221. doi:10.1042/bj20070255
13. Grabarek Z. Structural basis for diversity of the EF-hand calcium-binding proteins. J Mol Biol. 2006;359(3):509–525. doi:10.1016/j.jmb.2006.03.066
14. Schwaller B, Tetko IV, Tandon P, et al. Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures. Mol Cell Neurosci. 2004;25(4):650–663. doi:10.1016/j.mcn.2003.12.006
15. Olinger E, Schwaller B, Loffing J, Gailly P, Devuyst O. Parvalbumin: calcium and magnesium buffering in the distal nephron. Nephrol Dial Transplant. 2012;27(11):3988–3994. doi:10.1093/ndt/gfs457
16. Mendes TB, Nozima BH, Budu A, et al. PVALB diminishes [Ca2+] and alters mitochondrial features in follicular thyroid carcinoma cells through AKT/GSK3β pathway. Endocr Relat Cancer. 2016;23(9):769–782. doi:10.1530/erc-16-0181
17. Chen YT, Tu JJ, Kao J, Zhou XK, Mazumdar M. Messenger RNA expression ratios among four genes predict subtypes of renal cell carcinoma and distinguish oncocytoma from carcinoma. Clin Cancer Res. 2005;11(18):6558–6566. doi:10.1158/1078-0432.Ccr-05-0647
18. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi:10.1186/1471-2105-9-559
19. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
20. Modhukur V, Iljasenko T, Metsalu T, Lokk K, Laisk-Podar T, Vilo J. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 2018;10(3):277–288. doi:10.2217/epi-2017-0118
21. Li Y, Ge D, Lu C. The SMART App: an interactive web application for comprehensive DNA methylation analysis and visualization. Epigenet Chromatin. 2019;12(1):71. doi:10.1186/s13072-019-0316-3
22. Lánczky A, Győrffy B. Web-based survival analysis tool tailored for medical research (KMplot): development and implementation. J Med Internet Res. 2021;23(7):e27633. doi:10.2196/27633
23. Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956–d963. doi:10.1093/nar/gkx1090
24. Li T, Fan J, Wang B, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e110. doi:10.1158/0008-5472.Can-17-0307
25. Ru B, Wong CN, Tong Y, et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019;35(20):4200–4202. doi:10.1093/bioinformatics/btz210
26. Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18(10):1139–1146. doi:10.1038/nsmb.2115
27. Alexiou P, Maragkakis M, Papadopoulos GL, Simmosis VA, Zhang L, Hatzigeorgiou AG. The DIANA-mirExTra web server: from gene expression data to microRNA function. PLoS One. 2010;5(2):e9171. doi:10.1371/journal.pone.0009171
28. Miao YR, Liu W, Zhang Q, Guo AY. lncRNASNP2: an updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018;46(D1):D276–d280. doi:10.1093/nar/gkx1004
29. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–20. doi:10.1093/nar/gkq537
30. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
31. Waterhouse A, Bertoni M, Bienert S, et al. Swiss-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–w303. doi:10.1093/nar/gky427
32. Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with Swiss-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(Suppl 1):S162–73. doi:10.1002/elps.200900140
33. Bienert S, Waterhouse A, de Beer TA, et al. The Swiss-MODEL repository-new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–d319. doi:10.1093/nar/gkw1132
34. David A, Islam S, Tankhilevich E, Sternberg MJE. The alphafold database of protein structures: a biologist’s guide. J Mol Biol. 2022;434(2):167336. doi:10.1016/j.jmb.2021.167336
35. Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9(2):173–175. doi:10.1038/nmeth.1818
36. Davis AP, Grondin CJ, Johnson RJ, et al. Comparative Toxicogenomics Database (CTD): update 2021. Nucleic Acids Res. 2021;49(D1):D1138–d1143. doi:10.1093/nar/gkaa891
37. Liu CJ, Hu FF, Xie GY, et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 2023;24(1). doi:10.1093/bib/bbac558
38. Kirby ED, Kuwahara AA, Messer RL, Wyss-Coray T. Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc Natl Acad Sci U S A. 2015;112(13):4128–4133. doi:10.1073/pnas.1422448112
39. Ahn HR, Baek GO, Yoon MG, et al. Hypomethylation-mediated upregulation of the WASF2 promoter region correlates with poor clinical outcomes in hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):158. doi:10.1186/s13046-022-02365-7
40. Hernandez-Meza G, von Felden J, Gonzalez-Kozlova EE, et al. DNA methylation profiling of human hepatocarcinogenesis. Hepatology. 2021;74(1):183–199. doi:10.1002/hep.31659
41. Gao F, Xie K, Xiang Q, et al. The density of tumor-infiltrating lymphocytes and prognosis in resectable hepatocellular carcinoma: a two-phase study. Aging. 2021;13(7):9665–9678. doi:10.18632/aging.202710
42. Wang J, Liu X, Wu H, et al. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38(16):5366–5383. doi:10.1093/nar/gkq285
43. Huang G, Liang M, Liu H, et al. CircRNA hsa_circRNA_104348 promotes hepatocellular carcinoma progression through modulating miR-187-3p/RTKN2 axis and activating Wnt/β-catenin pathway. Cell Death Dis. 2020;11(12):1065. doi:10.1038/s41419-020-03276-1
44. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–821. doi:10.1038/s41423-020-0488-6
45. Bai Y, Cao K, Zhang P, Ma J, Zhu J. Prognostic and Immunological Implications of FAM72A in pan-cancer and functional validations. Int J Mol Sci. 2022;24(1). doi:10.3390/ijms24010375
46. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi:10.1002/ijc.29210
47. Roayaie S, Jibara G, Tabrizian P, et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology. 2015;62(2):440–451. doi:10.1002/hep.27745
48. Bruix J, Takayama T, Mazzaferro V, et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a Phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015;16(13):1344–1354. doi:10.1016/s1470-2045(15)00198-9
49. Au KP, Chok KSH. Multidisciplinary approach for post-liver transplant recurrence of hepatocellular carcinoma: a proposed management algorithm. World J Gastroenterol. 2018;24(45):5081–5094. doi:10.3748/wjg.v24.i45.5081
50. Xu Z, Xie H, Zhou L, Chen X, Zheng S. The combination strategy of transarterial chemoembolization and radiofrequency ablation or microwave ablation against hepatocellular carcinoma. Anal Cell Pathol. 2019;2019:8619096. doi:10.1155/2019/8619096
51. Nordness MF, Hamel S, Godfrey CM, et al. Fatal hepatic necrosis after nivolumab as a bridge to liver transplant for HCC: are checkpoint inhibitors safe for the pretransplant patient? Am J Transplant. 2020;20(3):879–883. doi:10.1111/ajt.15617
52. Liang W, Wang D, Ling X, et al. Sirolimus-based immunosuppression in liver transplantation for hepatocellular carcinoma: a meta-analysis. Liver Transpl. 2012;18(1):62–69. doi:10.1002/lt.22441
53. Lencioni R, de Baere T, Soulen MC, Rilling WS, Geschwind JF. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: a systematic review of efficacy and safety data. Hepatology. 2016;64(1):106–116. doi:10.1002/hep.28453
54. Das S, Camphausen K, Shankavaram U. Cancer-specific immune prognostic signature in solid tumors and its relation to immune checkpoint therapies. Cancers. 2020;12(9). doi:10.3390/cancers12092476
55. Li T, Yan Z, Wang W, et al. SEMA6B overexpression predicts poor prognosis and correlates with the tumor immunosuppressive microenvironment in colorectal cancer. Front Mol Biosci. 2021;8:687319. doi:10.3389/fmolb.2021.687319
56. Mossanen JC, Kohlhepp M, Wehr A, et al. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4(+) T-cell-dependent control of senescence. Gastroenterology. 2019;156(6):1877–1889.e4. doi:10.1053/j.gastro.2019.01.247
57. Zhang Q, Lou Y, Yang J, et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut. 2019;68(11):2019–2031. doi:10.1136/gutjnl-2019-318912
58. Ally A, Balasundaram M, Carlsen R, et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–1341.e23. doi:10.1016/j.cell.2017.05.046
59. Chew V, Lai L, Pan L, et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A. 2017;114(29):E5900–e5909. doi:10.1073/pnas.1706559114
60. Itoh S, Yoshizumi T, Yugawa K, et al. Impact of immune response on outcomes in hepatocellular carcinoma: association with vascular formation. Hepatology. 2020;72(6):1987–1999. doi:10.1002/hep.31206
61. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829–845.e20. doi:10.1016/j.cell.2019.10.003
62. Sun H, Huang Q, Huang M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. 2019;70(1):168–183. doi:10.1002/hep.30347
63. Zhou J, Peng H, Li K, et al. Liver-resident NK cells control antiviral activity of hepatic T Cells via the PD-1-PD-L1 Axis. Immunity. 2019;50(2):403–417.e4. doi:10.1016/j.immuni.2018.12.024
64. Ma H, Kang Z, Foo TK, Shen Z, Xia B. Disrupted BRCA1-PALB2 interaction induces tumor immunosuppression and T-lymphocyte infiltration in HCC through cGAS-STING pathway. Hepatology. 2023;77(1):33–47. doi:10.1002/hep.32335
65. Zhang PF, Gao C, Huang XY, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19(1):110. doi:10.1186/s12943-020-01222-5
66. Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807. doi:10.1002/hep.23054
67. Qin WH, Yang ZS, Li M, et al. High serum levels of cholesterol increase antitumor functions of nature killer cells and reduce growth of liver tumors in mice. Gastroenterology. 2020;158(6):1713–1727. doi:10.1053/j.gastro.2020.01.028
68. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
69. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6(12):940–952. doi:10.1038/nri1983
70. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–1036. doi:10.1038/ni.3518
71. Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. 2019;16(5):415–422. doi:10.1038/s41423-019-0224-2
72. Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol. 2019;105(6):1319–1329. doi:10.1002/jlb.Mr0718-269r
73. Prager I, Liesche C, van Ooijen H, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. 2019;216(9):2113–2127. doi:10.1084/jem.20181454
74. Sordo-Bahamonde C, Lorenzo-Herrero S, González S, López-Soto A. A cytofluorimetric assay to evaluate intracellular cytokine production by NK cells. Methods Enzymol. 2020;631:343–355. doi:10.1016/bs.mie.2019.05.049
75. Que ZJ, Yao JL, Zhou ZY, et al. Jinfukang inhibits lung cancer metastasis by upregulating CX3CL1 to recruit NK cells to kill CTCs. J Ethnopharmacol. 2021;275:114175. doi:10.1016/j.jep.2021.114175
76. Strand S, Hofmann WJ, Hug H, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells--A mechanism of immune evasion? Nat Med. 1996;2(12):1361–1366. doi:10.1038/nm1296-1361
77. Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–553. doi:10.1056/NEJMoa1910607
78. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi:10.1186/1471-2105-12-77
79. Liu XH, Sun M, Nie FQ, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92. doi:10.1186/1476-4598-13-92
80. Wang SH, Ma F, Tang ZH, et al. Long non-coding RNA H19 regulates FOXM1 expression by competitively binding endogenous miR-342-3p in gallbladder cancer. J Exp Clin Cancer Res. 2016;35(1):160. doi:10.1186/s13046-016-0436-6
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
An Analysis Regarding the Association Between the Nuclear Pore Complex (NPC) and Hepatocellular Carcinoma (HCC)
Huang P, Hu YD, Liu YJ, Li JP, Zhang YH
Journal of Hepatocellular Carcinoma 2023, 10:959-978
Published Date: 22 June 2023
Integrated Analysis of Immune Infiltration and Hub Pyroptosis-Related Genes for Multiple Sclerosis
Zhang S, Ma Y, Luo X, Xiao H, Cheng R, Jiang A, Qin X
Journal of Inflammation Research 2023, 16:4043-4059
Published Date: 13 September 2023
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024
Comprehensive Analysis of the Role of Heat Shock Proteins in the Immune Microenvironment and Clinical Significance of Hepatocellular Carcinoma
Xiao H, Wang B, Xiong S, Li C, Ding Y, Chao D, Mei B, Shen N, Luo G
Journal of Hepatocellular Carcinoma 2025, 12:325-342
Published Date: 17 February 2025
Integrated Multi-Omics Profiling Identifies PDZ-Binding Kinase (PBK) as a Novel Prognostic Biomarker in Hepatocellular Carcinoma
Zhang J, Xu Y, Ni X, Mao Z, Xiao H, Chen M, Lin Y, Pan J, Zhang B, Zhang L, Zheng X, Song G, Ge N
Journal of Hepatocellular Carcinoma 2025, 12:1453-1469
Published Date: 17 July 2025