Back to Journals » International Journal of Women's Health » Volume 17
Prediction of Cervical Cancer Progression Leveraging HPV16 Integration-Related Genes
Received 28 May 2025
Accepted for publication 26 September 2025
Published 22 October 2025 Volume 2025:17 Pages 3745—3760
DOI https://doi.org/10.2147/IJWH.S543345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Matteo Frigerio
Yifan Yang,1 Chaoyang Sun,1 Hui Wang2,3
1Department of Obstetrics and Gynecology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, People’s Republic of China; 2Zhejiang Provincial Key Laboratory of Precision Diagnosis and Therapy for Major Gynecological Diseases, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China; 3Department of Gynecologic Oncology, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Hui Wang, Department of Gynecologic Oncology, Women’s Hospital, Zhejiang University School of Medicine, Xueshi Road, Hangzhou, Zhejiang, 310000, People’s Republic of China, Email [email protected]
Purpose: Cervical cancer (CC) remains a significant global health burden among women, particularly in cases of advanced or recurrent disease. Current clinical parameters exhibit suboptimal accuracy in predicting disease progression. Given that HPV integration is a well-established oncogenic driver in cervical carcinogenesis, there is growing interest in leveraging HPV-related molecular signatures to improve risk stratification and guide personalized treatment strategies.
Patients and Methods: Our study design employed HPV16-postive samples from TCGA-CESC as the training set (n = 95) and a local cervical cancer cohort (n = 118) for independent validation. From differentially expressed genes (DEGs) identified in HPV16-integrated HaCaT cells, we developed a prognostic 9-gene signature through a rigorous two-stage selection process: feature reduction using LASSO regression along with 10-fold cross-validation, followed by stepwise Cox regression. The risk score’s predictive performance was systematically evaluated through Kaplan-Meier survival analysis, time-dependent ROC curves, ROC over time profiling, calibration plots, and nomogram construction. Mechanistic investigations included functional enrichment analysis, mutational profiling, and drug sensitivity prediction.
Results: The 9-gene signature (LCP1, CXCL11, NEK6, MCAM, PRRX2, NPL, PGLYRP3, SPRR3 and MMP1) demonstrated superior predictive accuracy compared to conventional clinical parameters. Mechanistic investigations revealed that the signature genes collectively influence tumor progression through two key pathways: modulation of tumor immune microenvironment and regulation of oncogenic mutation patterns. These findings were consistently supported by both functional enrichment analysis and comprehensive mutational profiling. Furthermore, pharmacological inhibition of NRF2 signaling may overcome cisplatin resistance in high-risk patients with NFE2L2-mutant tumors. While the signature shows significant clinical potential, further independent validation is required before it can be adopted into routine clinical practice.
Conclusion: We developed a robust nine-gene prognostic model for predicting Progression-Free Survival (PFS) in CC, which provides novel insights into HPV-associated oncogenesis and facilitates risk stratification and therapeutic decision-making in CC management.
Keywords: HPV16 integration, prediction model, cervical cancer, progression-free survival, risk stratification
Introduction
Cervical cancer (CC) ranks as the fourth most common malignancy in women globally, representing a major public health burden.1 While early-stage disease demonstrates favorable treatment outcomes, advanced or recurrent cases continue to exhibit poor prognosis and limited therapeutic options.2 Current clinical parameters, such as FIGO staging and lymph node metastasis status, lack sufficient discriminative power to adequately assess tumor aggressiveness or predict disease progression in CC.3,4 This critical gap in prognostic capability underscores the pressing need to develop novel biomarkers that can improve risk stratification and inform personalized treatment strategies for CC patients.
HPV integration refers to the recombination of HPV viral DNA with the host cell genome.5 After integration, the HPV genome can disrupt the genomic stability of the host cell, leading to chromosomal structural abnormalities. As a well-established driver of cervical carcinogenesis, HPV integration occurs in more than 80% of cervical cancer cases, with HPV16 being the predominant integrated subtype (58–66% of all cases).6 The clinical significance of HPV integration is twofold: First, integrated HPV forms demonstrate greater oncogenic potential than episomal HPV. Second, integration status serves as a robust prognostic biomarker, correlating with treatment response and disease outcomes.7,8 The underlying mechanisms involve both disruption of tumor suppressor loci and widespread epigenetic alterations, including chromatin remodeling and dysregulation of host gene expression networks. Notably, HPV16 integration sites show particular clinical relevance, as they frequently occur near oncogenes and tumor suppressor genes.9,10 These specific integration patterns not only influence tumor behavior but also show promise as molecular markers for patient stratification.
Currently, no consensus exists regarding a prognostic model that quantitatively assesses the contribution of HPV16 integration-related genes in predicting CC progression. To address this critical gap, we systematically investigated the expression profiles and clinical significance of HPV16 integration-associated genes in CC and developed a novel 9-gene prognostic signature derived from DEGs. The prognostic model exhibits high discriminative accuracy while maintaining straightforward clinical interpretability.
Materials and Methods
Cell Lines
Human CC cell line HeLa, SiHa and C33A were obtained from the American Type Culture Collection (ATCC). Cells were maintained at 37°C in a 5% humidified incubator using Dulbecco’s modified Eagle’s medium (DMEM, 11965092, Gibco) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin.
Data Acquisition
TGCA-CESC transcriptome profiles, genomic data and clinical information were acquired from the Cancer Genome Atlas (TCGA) database.11 We selected HPV16-positive samples (n = 95) from the TCGA-CESC dataset as the training set. Our local CC cohort comprised 118 treat-naïve tumor samples gathered at Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and Women’s Hospital, School of Medicine, Zhejiang University. Approval was granted by the Medical Ethics Committee at Tongji Hospital, Tongji Medical College at Huazhong University of Science and Technology (ID: TJ-IRB20180505), and the Medical Ethics Committee of the Women’s Hospital, School of Medicine, Zhejiang University (ID: IRB-20210085-R). Following collection, tumor tissues were subjected to bulk RNA sequencing. Our local cohort was treated as the validation set.
DEGs Identification
Transcriptome profiles of HPV16 integrated and vector HaCaT cells were obtained from a previous study.10 HPV16-integrated HaCaT cells were generated as previously described. Briefly, a CRISPR-Cas9-mediated knock-in approach was used to generate site-specific HPV16 integration at the 13q22 locus in HaCaT cells. A donor vector (pDC515-HAL-HPV16 E7 + E6 + URR-flag-Loxp + CMV-EGFP-2A-Puro-polyA + loxp-HAR) was constructed by cloning the HPV16 upstream regulatory region (URR) and E6/E7 oncogenes, flanked by homology arms (HAL/HAR), into a plasmid upstream of an EGFP-Puro reporter. HaCaT cells were co-transfected with this donor and a sgRNA/Cas9 construct targeting 13q22, followed by fluorescence-activated cell sorting (FACS) and puromycin selection. Precise integration was confirmed by whole-genome sequencing (WGS). DEGs analyzed by “limma” were identified by applying filters for an absolute fold change > 2 and adjusted P-value < 0.05.
Construction of Progression Predictive Model Based on HPV16 Integration-Related DEGs
We constructed a progression predictive model based on HPV16 integration-related DEGs as follows: first, DEGs were subjected to Least Absolute Shrinkage and Selection Operator (LASSO) regression using the “glmnet” R package. Genes remained in the model had minimum lambda values. Subsequently, these candidate genes were incorporated into stepwise Cox regression analysis. The scoring formula was as follows:
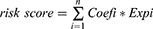
Where N denotes the number of model genes, Coefi denotes each model gene risk coefficient, and Expi denotes scaled mRNA level each model gene.
Predictive Accuracy Evaluation of the Risk Model
Time-dependent Receiver Operating Characteristic (ROC) analysis was applied to training and validation sets using the “timeROC” R package. Calibration curves were calculated using the “rms” R package. The nomogram was constructed using the “rms” R package.
Drug Sensitivity Prediction
Predicted half maximal inhibitory concentrations (IC50) of 198 common antitumor drugs in HCC were calculated using the “oncoPredict” R package.
Determination of Drug Sensitivity
IC50 values determined by the CCK-8 assay were used to evaluate the sensitivity of cells to ML385 and cisplatin. In brief, cells were seeded in 96-well plates at a density of 4 × 10³ cells per well and cultured in medium containing various concentrations of drugs for 48 hours. Cell viability was assessed using the CCK-8 Cell Counting Kit (A311, Vazyme). For ML385 pretreatment, cells were incubated with ML385 (1 µM) for 2 hours prior to cisplatin treatment and maintained in the presence of ML385 throughout the entire 48-hour exposure period.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated using the RNA extraction kit (RC101-01, Vazyme) following the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using HiScript III RT SuperMix for qPCR (R323-01, Vazyme). Real-time PCR was performed with SYBR Green PCR Master Mix (Q711-02, Vazyme). Gene expression levels were normalized to gene β-actin and calculated using the comparative Ct method (2−ΔΔCt). All primer sequences are provided in Table S1.
Immunohistochemistry (IHC)
A section from paraffin block was deparaffinized and rehydrated, followed by boiling for 15 minutes in an antigen retrieval buffer (G1203, Servicebio). After cooling to room temperature, nonspecific binding sites were blocked with 5% bovine serum albumin (BSA) for 1 hour at room temperature. The sections were then incubated overnight at 4°C with primary antibody against NRF2 (GB113808, Servicebio). Immunodetection was carried out using an HRP-DAB polymer-based kit (G1212, Servicebio) according to the manufacturer’s instructions.
DCFH-DA Staining
DCFH-DA staining was performed according to the manufacturer’s instructions. Cells were incubated with 10 µM DCFH-DA probe (HY-D0940, MCE) for 30 minutes at 37°C, followed by detection of fluorescence intensity using flow cytometry.
Statistical Analysis
R software (v4.4.0) was employed for all statistical analyses. Continuous variables were displayed as mean ± standard error of the mean (SEM), and categorical ones as percentages. Differences between groups for continuous variables were assessed using Student’s t-test (for normally distributed data with equal variances) or the Wilcoxon test (for non-normal distributions). Mutation frequencies were compared using Fisher’s exact test. Immune cell proportions (estimated by XCELL) were analyzed using Student’s t-test or Wilcoxon test, as appropriate based on distributional assumptions. The Log rank test was used to determine survival differences using the “survival” R package. The statistical significance was established at P < 0.05.
Results
Construction of Progression Predictive Model Based on HPV16 Integration-Related DEGs
We summarized the study procedure in Figure 1A. First, HPV16-positive samples (n = 95) from the TCGA-CESC cohort were designated as the training set. Second, DEGs associated with HPV16 integration were identified using the following criteria: absolute fold change > 2 and adjusted P < 0.05. Third, LASSO regression followed by stepwise Cox regression were sequentially applied to construct a prognostic prediction model. Fourth, an independent local cohort (n = 118) was used as the validation set. Finally, to evaluate model performance, we assessed its predictive accuracy using Kaplan-Meier survival analysis, time-dependent ROC curves, ROC over time analysis, calibration curves, and a nomogram.
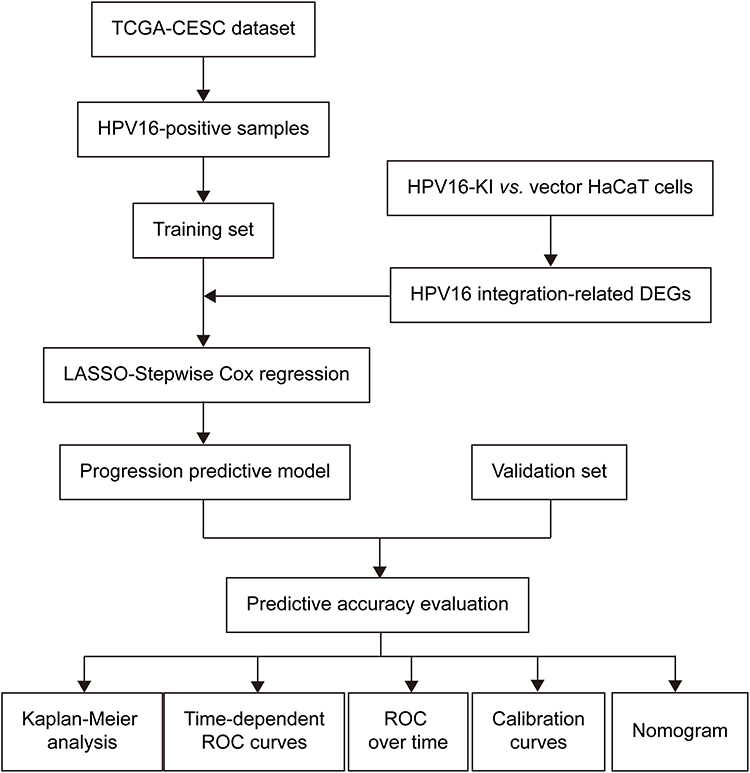 |
Figure 1 Flow chart of the study. |
To investigate the transcriptomic alterations in host cells induced by HPV16 integration at the 13q22 site, we conducted a comparative analysis between HPV16-integrated and vector control HaCaT cells using data from a previous study.10 Our analysis identified 243 DEGs, consisting of 108 up-regulated and 135 down-regulated transcripts (Figure 2A). Gene Set Variation Analysis (GSVA) of hallmark pathways revealed significant activation of two critical oncogenic programs: the unfolded protein response (UPR) (t = 2.64, P = 0.01) and epithelial-mesenchymal transition (EMT) (t = 2.28, P = 0.03) in HPV16-integrated cells (Figure 2B). These findings imply that HPV16 integration potentially contributes to enhanced malignant phenotypes. Using LASSO regression with 10-fold cross-validation, we constructed a prognostic risk model comprising 12 genes (Figure 2C, D and Table S2). Subsequent stepwise Cox regression analysis of these candidate genes refined the model to 9 robust prognostic markers, for which we calculated their weight’s coefficient (Figure 2E and Table S3). Figure 2F shows the regression analysis results for the 9 prognostic genes, including hazard ratios (HRs), 95% confident intervals (95% CIs) and statistical significance (P-values). Notably, SPRR3 emerged as a protective factor, whereas CXCL11, PRRX2, NPL, PGLYRP3, and MMP1 were identified as risk factors. The risk score was calculated using the following formula: Risk score = (0.92)*CXCL11 + (−1.45)*LCP1 + (0.71)*MCAM + (0.88)*MMP1 + (0.55)*NEK6 + (0.94)*NPL + (0.67)*PGLYRP3 + (0.87)*PRRX2 + (−2.63)*SPRR3.
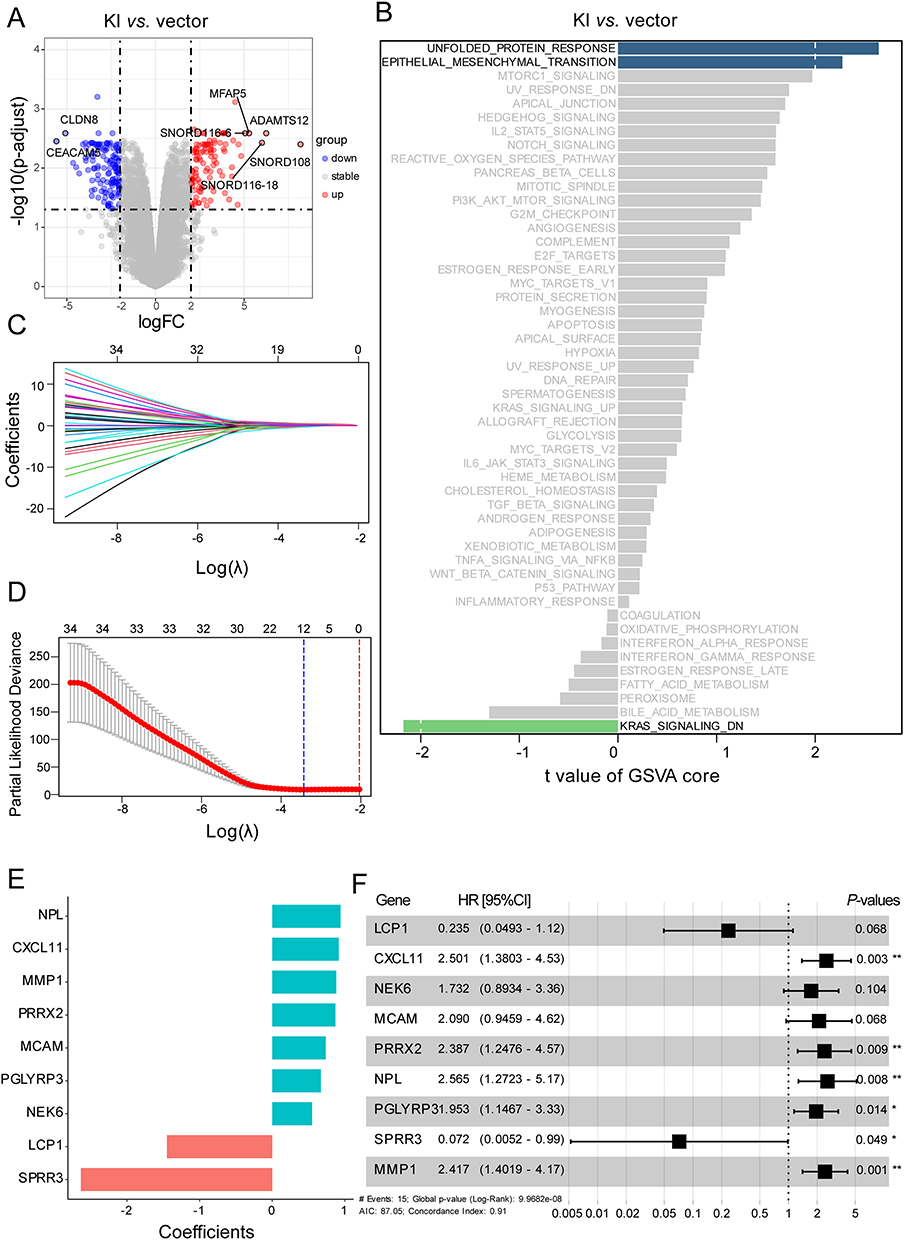 |
Figure 2 Construction of progression predictive model based on HPV16 integration-related DEGs. (A) Volcano plot showing DEGs between HPV16-KI and vector HaCaT cells. (B) Differences in Hallmark pathways between HPV16-KI and vector HaCaT cells. (C) LASSO coefficient profiles of HPV16 integration-related DEGs associated with PFS of cervical cancer. (D) LASSO regression with 10-fold cross-validation. (E) Coefficients of nine model genes identified using the StepCox model. (F) Forest plot of the StepCox model. *p< 0.05, **p< 0.01. Abbreviations: DEG, differentially expressed genes; KI, knock in; LASSO, Least Absolute Shrinkage and Selection Operator; PFS, Progression-Free Survival. |
Evaluation of Model Performance in the Training Set
Risk stratification analysis using the risk model revealed distinct clinical trajectories in the training set. Individual risk scores were calculated and systematically ranked in descending order (Figure 3A). Heatmap visualization of the 9 genes illustrated that higher risk scores implied higher expression of PRRX2, NEK6, MCAM and MMP1 (Figure S1A). The high-risk and low-risk groups were stratified according to the median risk score. The high-risk group exhibited significantly more cases with progressive status and markedly shorter PFS compared to low-risk counterparts (Figure 3B and C). The prognostic model demonstrated robust calibration accuracy, with calibration curves closely aligning with ideal predictions at 1-, 3-, and 5-year follow-ups (Figures 3D and S1B, C). To compare the predictive efficacy of the risk model with that of conventional prognostic factors such as lymph node metastasis, histologic type, FIGO stage, and age, we undertook ROC curve analyses at 1, 3, and 5 years. The results indicate that the AUC of risk scores markedly outperforms that of these individual prognostic indicators, indicating its greater accuracy in PFS prediction (Figures 3E, F and S1D). ROC over time further confirmed sustained predictive efficacy across the entire observation period (Figure 3G). To facilitate clinical implementation, we developed a multivariate nomogram incorporating the risk score with conventional clinical parameters such as histologic type, lymph node metastasis and age (Figure 3H). Collectively, these findings demonstrate that the 9-gene risk model exhibits robust discrimination performance in the training set, effectively stratifying patients into distinct risk groups with significant differences in clinical outcomes. The model’s strong predictive accuracy, as evidenced by its high sensitivity and specificity, suggests its potential utility as a prognostic tool in clinical settings.
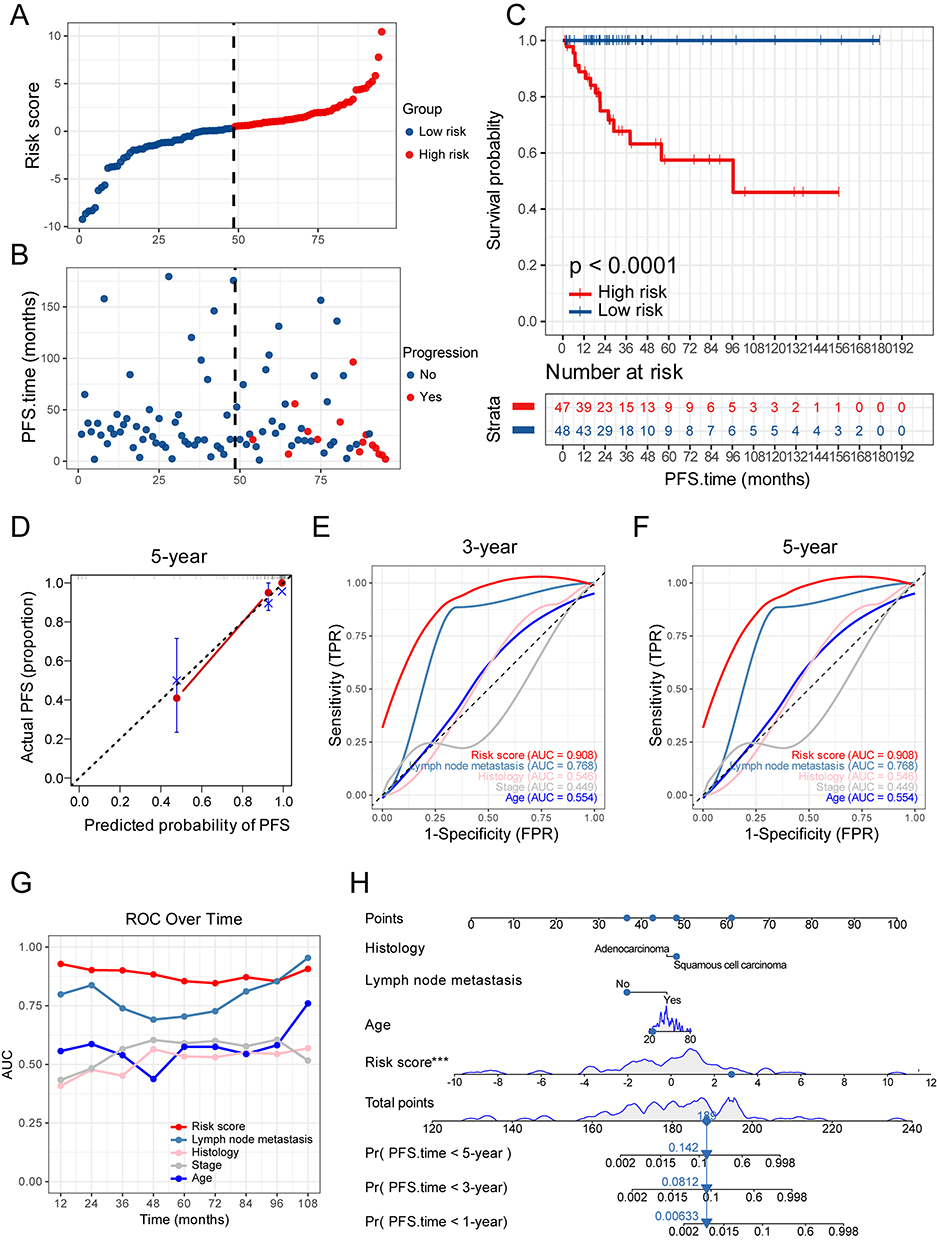 |
Figure 3 Evaluation in the training set. (A) Distribution of risk score in the training set. Red dots indicate high risk samples and blue dots indicate low risk samples. (B) The outcome of progression status and time of patients in the training set. The ordinate represents survival time. Red dots indicate patients with progression and blue dots indicate progression-free cases. (C) Survival curves of high- and low-risk groups in the training set. (D) Calibration plots of the model for 5-year PFS. (E) ROC curves of risk scores and conventional clinical factors for predicting 3-year PFS in the training set. (F) ROC curves of risk scores and conventional clinical factors for predicting 5-year PFS in the training set. (G) AUC values for different time periods. (H) Nomogram integrating conventional clinical factors and risk scores to forecast 1-, 3-, and 5-year PFS. ***p< 0.001. Abbreviations: ROC, Receiver Operating Characteristic; AUC, Area Under the Curve. |
Validation of the Prognostic Risk Model
To rigorously confirm the clinical generalizability and translational applicability of the prognostic risk model, we performed independent validation using a local cohort comprising 118 treatment-naïve CC patients. The validation process assessed the model’s performance in risk stratification, prognostic accuracy, and clinical utility, ensuring its robustness beyond the initial training dataset. Patients were first stratified into high- and low-risk groups based on tertiles of the risk score, with the upper third designated as the high-risk group and the remaining two-thirds as the low-risk group. Consistent with results in the training set, the gene expression heatmap of the validation set illustrated that higher risk scores implied upregulated expression levels of MMP1, PRRX2, NEK6 and MCAM (Figure 4A). The correlations of the variables included in the risk model are presented in Figure 4B. Aligning with the results in the training set, CXCL11, MCAM, MMP1, NEK6 and PRRX2 maintained their status as risk factors in the validation set, while SPRR3 emerged as a robust protective factor (Figure 4C). Survival analysis demonstrated significant clinical utility of the risk model, with high-risk patients exhibiting shorter PFS compared to their low-risk counterparts, consistent with the results observed in the training set (Figure 4D). The predictive accuracy was assessed through time-dependent ROC curve analysis, revealing AUC value of 0.708 for predicting 3-year survival rates. The performance of risk scores was markedly better than that of conventional prognostic factors such as lymph node metastasis (AUC = 0.451), histologic type (AUC = 0.463) and FIGO stage (AUC = 0.535) (Figure 4E). ROC over time further confirmed the high predictive performance of the risk model throughout the whole observation period (Figure 4F). The consistent discriminative capacity across both training and validation cohorts underscores the robustness of this molecular signature. These findings collectively establish the risk model as a superior prognostic tool that transcends traditional clinicopathological parameters, with potential clinical utility for personalized surveillance strategies.
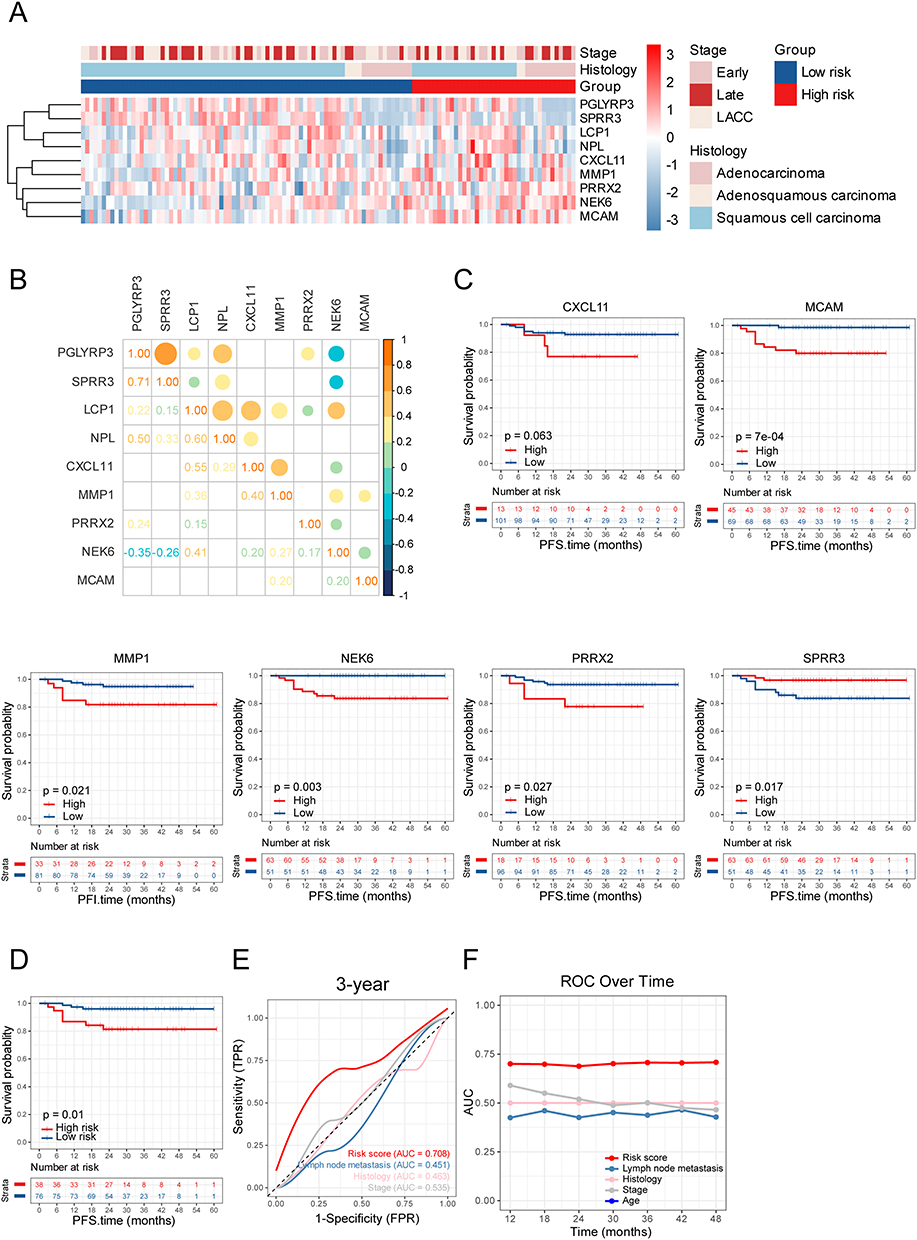 |
Figure 4 Validation in the validation set. (A) Expression levels of the 9 genes of model in the validation set. (B) Heatmap represents the correlations within genes of the model in the validation set, with rho values rounded to two decimals. (C) Survival curves of high and low groups of indicated genes (optimal cut-off) in the validation set. (D) Survival curves of high- and low-risk groups in the validation set. (E) ROC curves of risk scores and conventional clinical factors for predicting 3-year PFS in the validation set. (F) AUC values for different time periods. |
Analysis of Immune-Cell Infiltration Between Two Risk Groups
To explore the different molecular characteristics between two risk groups, we performed GSVA to assess enrichment patterns of hallmark pathways (Figure 5A). The low-risk group demonstrated significant activation of immune-related pathways, including “Allograft rejection”, “Inflammatory response”, and “Interferon gamma response”, suggesting enhanced immunogenic potential. This immunologically active phenotype was corroborated by XCELL-based deconvolution of tumor microenvironment composition (Figure 5B).12 Specifically, the low-risk group showed elevated infiltration of immune cells with antitumor effects, such as CD8+ T cells (CD8+ T cells, CD8+ central memory T cells and CD8+ effector memory T cells), CD4+ T cells (CD4+ memory T cells, CD4+ non-regulatory T cells and CD4+ effector memory T cells), NK cells, professional antigen-presenting cells (myeloid dendritic cells, activated dendritic cells, M1-like macrophages and plasmacytoid dendritic cells. To mechanistically decipher the immunological difference between the two groups, we further systematically quantified the association between risk scores and tumor microenvironment composition through Spearman correlation analysis. Notably, most adaptive immune cells, such as CD8+ T cells, CD4+ T and B cells showed a striking negative correlation with the risk scores (Figure 5C). The observed anti-tumor immune bias in low-risk patients provides a mechanistic explanation for their superior clinical outcomes.
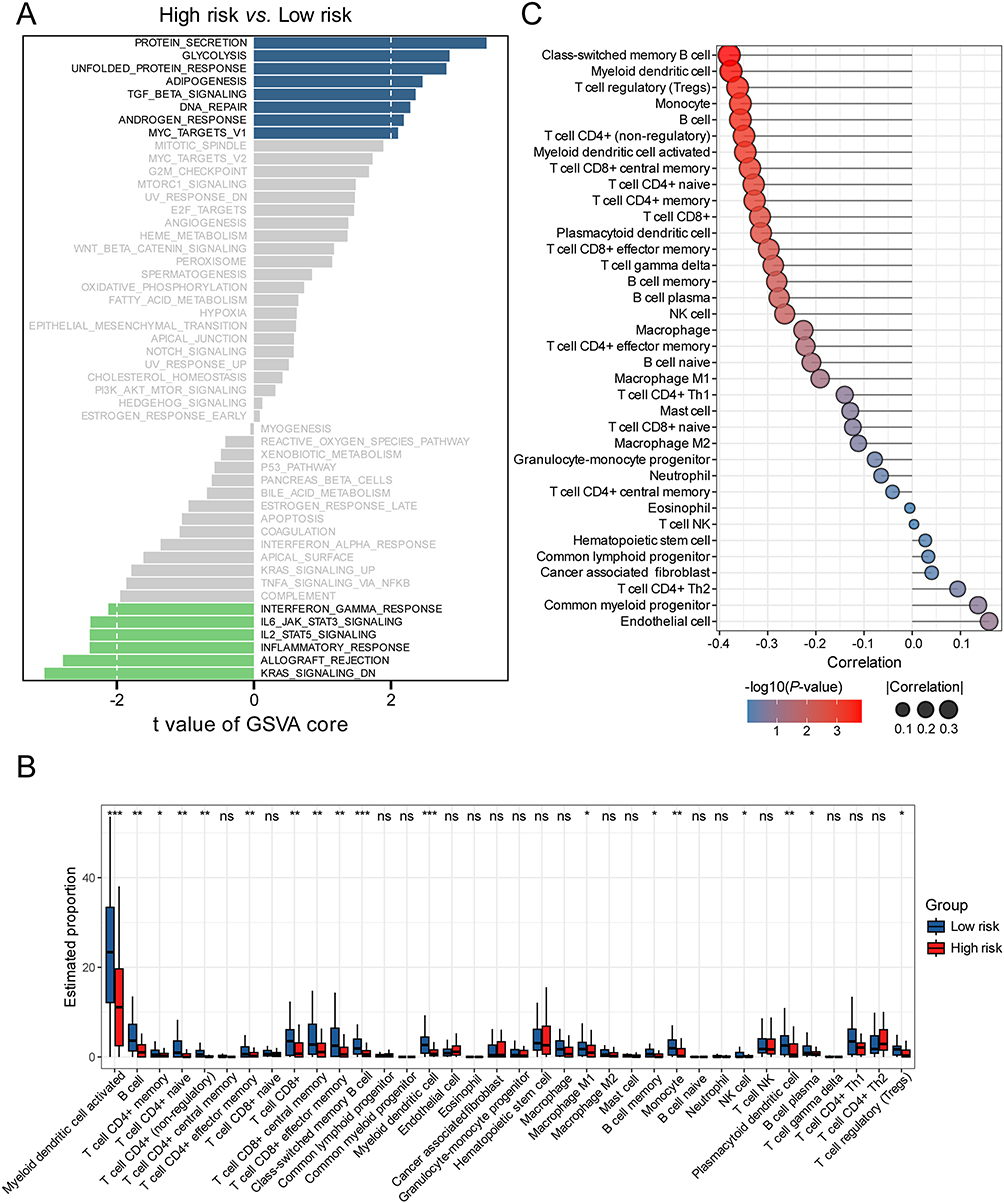 |
Figure 5 Analysis of immune-cell infiltration between two risk groups in the training set. (A) Differences in Hallmark pathways between high- and low-risk groups. (B) Comparison of immune-cell infiltration scores between two risk groups analyzed by the XCELL algorithm. (C) Correlation of risk scores wit immune cell types. Correlation, Spearman correlation coefficient; p-value, P-values for Spearman correlation. *p< 0.05, **p< 0.01, ***p< 0.001. |
The High-Risk Group Exhibits Reduced Sensitivity to Cisplatin, Likely Due to Activation of the NRF2 Pathway
To explore the therapeutic implications of our risk stratification, we employed the OncoPredict algorithm to estimate chemosensitivity. High-risk patients exhibited significantly higher predicted cisplatin IC50 values compared to low-risk patients (Figure 6A), with a positive correlation between risk scores and predictive drug sensitivity of cisplatin (Figure S2A). This suggests that platinum-based regimens may have limited efficacy in the high-risk group, highlighting the need for alternative treatment strategies. Computational drug repositioning also identified the Polo-like kinase 1 (Plk1) inhibitor BI-2536 as a promising candidate, showing greater sensitivity in high-risk tumors (Figure S2B). Given the established role of HPV integration in promoting host genome destabilization,5 we performed comprehensive genomic profiling to characterize intergroup instability patterns (Figure 6B). Although the total tumor mutation burden did not differ significantly (Figure S2C), we observed distinct patterns in oncogenic driver mutations. Notably, PIK3CA (35% vs 24%), KMT2C (20% vs 14%), and NFE2L2 (15% vs 7%) mutations were enriched in high-risk tumors.13–15 We compared the mutation frequencies of frequently altered genes between the low- and high-risk groups. The Fisher’s exact test revealed no statistically significant differences in individual gene mutation rates after multiple testing correction. While the difference in NFE2L2 mutation frequency did not reach statistical significance (15% vs 7%; P = 0.246)—likely due to moderate sample size—NFE2L2 is recurrently mutated in cervical cancer and has been consistently associated with platinum resistance and poor prognosis in multiple cancer types, including head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC).16,17 NFE2L2, which encodes the NRF2 transcription factor, is a master regulator of the cellular antioxidant response. Gain-of-function mutations, particularly within the KEAP1-binding Neh2 domain, impair NRF2 ubiquitination and proteasomal degradation, resulting in its nuclear accumulation and constitutive activation of downstream genes such as NQO1 and HMOX1.18 This persistent antioxidant activity can mitigate chemotherapy-induced oxidative stress, thereby promoting drug resistance. Given this well-established role in therapy resistance, we prioritized NFE2L2 for further investigation despite the marginal statistical difference in mutation frequency. Survival analysis revealed that patients harboring NFE2L2 mutations had significantly shorter PFS (Figure 6C), underscoring its clinical significance. Furthermore, we detected a significant co-occurrence between FLG and NFE2L2 mutations (Figure S2D), suggesting potential synergistic oncogenic effects. Indeed, FLG-mutant patients also exhibited reduced PFS (Figure S2E), reinforcing the clinical relevance of this mutational pattern. To validate NRF2 pathway activation in high-risk tumors, we assessed its expression at both protein and transcript levels. IHC showed increased NRF2 protein expression in high-risk tumor tissues (Figure 6D), accompanied by elevated mRNA levels of downstream targets NQO1 and HMOX1 as measured by RT-qPCR (Figure 6E and S2F). Next, we employed the selective NRF2 inhibitor ML385 to investigate whether pharmacological inhibition of the NRF2 signaling pathway could sensitize CC cells to cisplatin treatment. Among the CC cell lines used, SiHa is HPV16-positive, HeLa is HPV18-positive, and C33A is HPV-negative (Figure S3A). Cell viability assays demonstrated that 1 μM ML385 exhibited minimal toxicity, with over 90% cell viability maintained after 48 hours of exposure (Figure S3B), indicating its suitability for functional studies. Notably, a 2-hour pretreatment with ML385 significantly enhanced the cytotoxicity of cisplatin in all tested CC cell lines (Figure 7A and B), suggesting that NRF2 inhibition potentiates chemosensitivity of cisplatin. To evaluate the functional efficacy of ML385, we examined the expression of canonical NRF2 target genes NQO1 and HMOX1 by RT-qPCR. As expected, cisplatin treatment alone induced upregulation of these antioxidant genes, consistent with NRF2 activation in response to drug-induced stress. However, this induction was markedly suppressed upon co-treatment with ML385 (Figure 7C and S3C), confirming effective inhibition of NRF2 transcriptional activity. Given the well-established role of NRF2 in maintaining redox homeostasis through the regulation of antioxidant genes and the fact that elevated intracellular reactive oxygen species (ROS) are closely linked to increased cisplatin sensitivity, we next investigated whether NRF2 inhibition modulates oxidative stress in CC cells. Using the DCFH-DA fluorescent probe, we detected a significant increase in intracellular ROS levels following ML385 treatment (Figure 7D). This finding indicates that blocking NRF2 signaling impairs the cellular antioxidant defense system, leading to ROS accumulation. Collectively, these results demonstrate that pharmacological inhibition of NRF2 not only disrupts redox balance but also enhances the efficacy of cisplatin in CC cells. This synergistic effect likely arises from the inability of cancer cells to counteract cisplatin-induced oxidative damage when NRF2 is inhibited. These findings highlight the NRF2-ROS axis as a promising therapeutic target and suggest that combining cisplatin with NRF2 inhibitors such as ML385 may represent a viable strategy to overcome chemoresistance, particularly in high-risk patient populations exhibiting hyperactive NRF2 signaling.
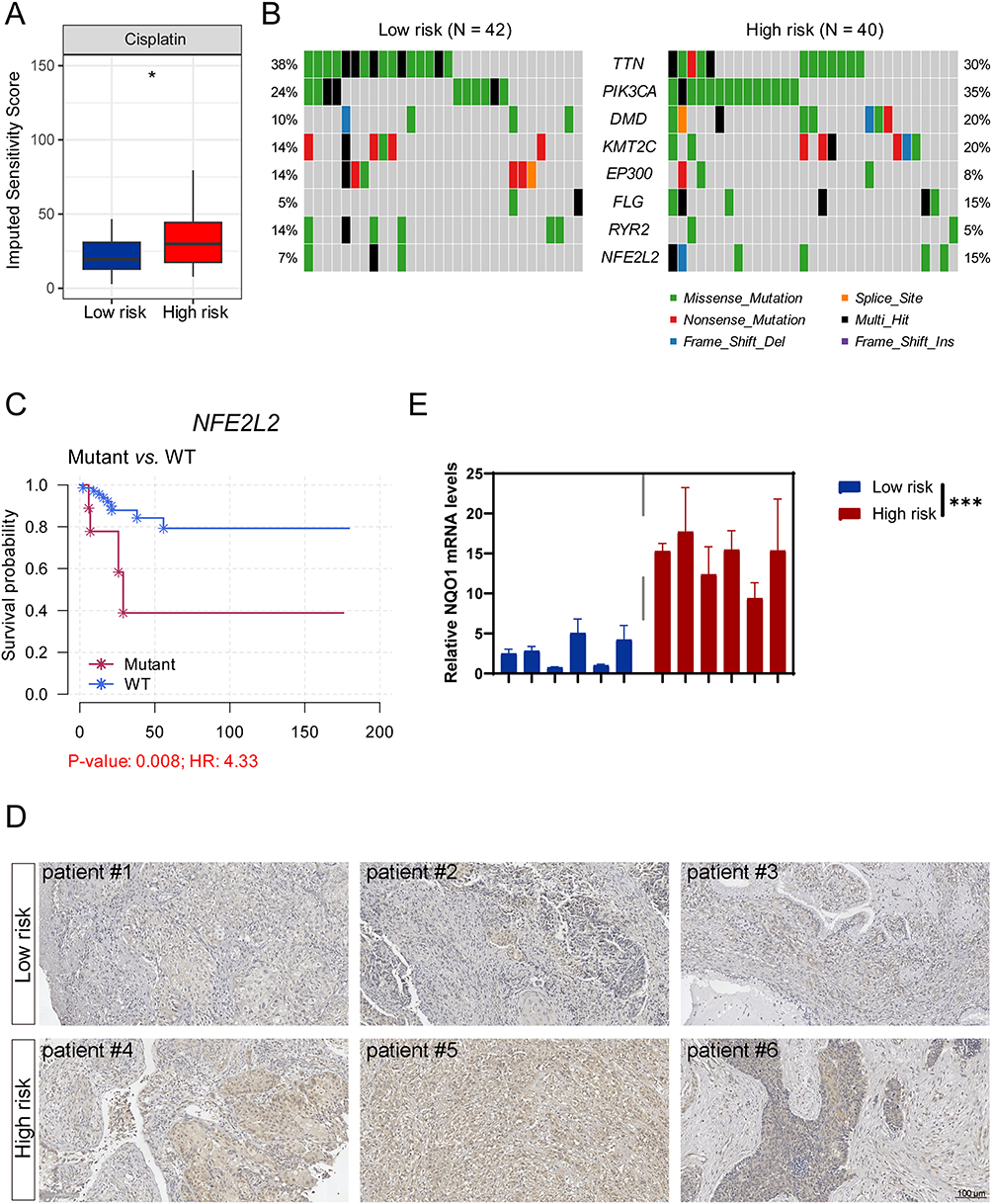 |
Figure 6 The high-risk group is less sensitive to cisplatin, with concomitant activation of the NRF2 pathway. (A) Predicted drug sensitivity of cisplatin between two risk groups analyzed by the OncoPredict algorithm. (B) Genomic alteration landscape of the most frequently mutated genes across the high- and low-risk groups. (C) Survival curves according to NFE2L2 mutation status. (D) Representative immunohistochemical images showing NRF2 expression in CC tumor tissues from the high- and low-risk groups. (E) RT-qPCR showing the mRNA expression of NQO1 in CC tumor tissues from the high- and low-risk groups. The data are representative of 3 biological replicates. *p< 0.05, ***p< 0.001. |
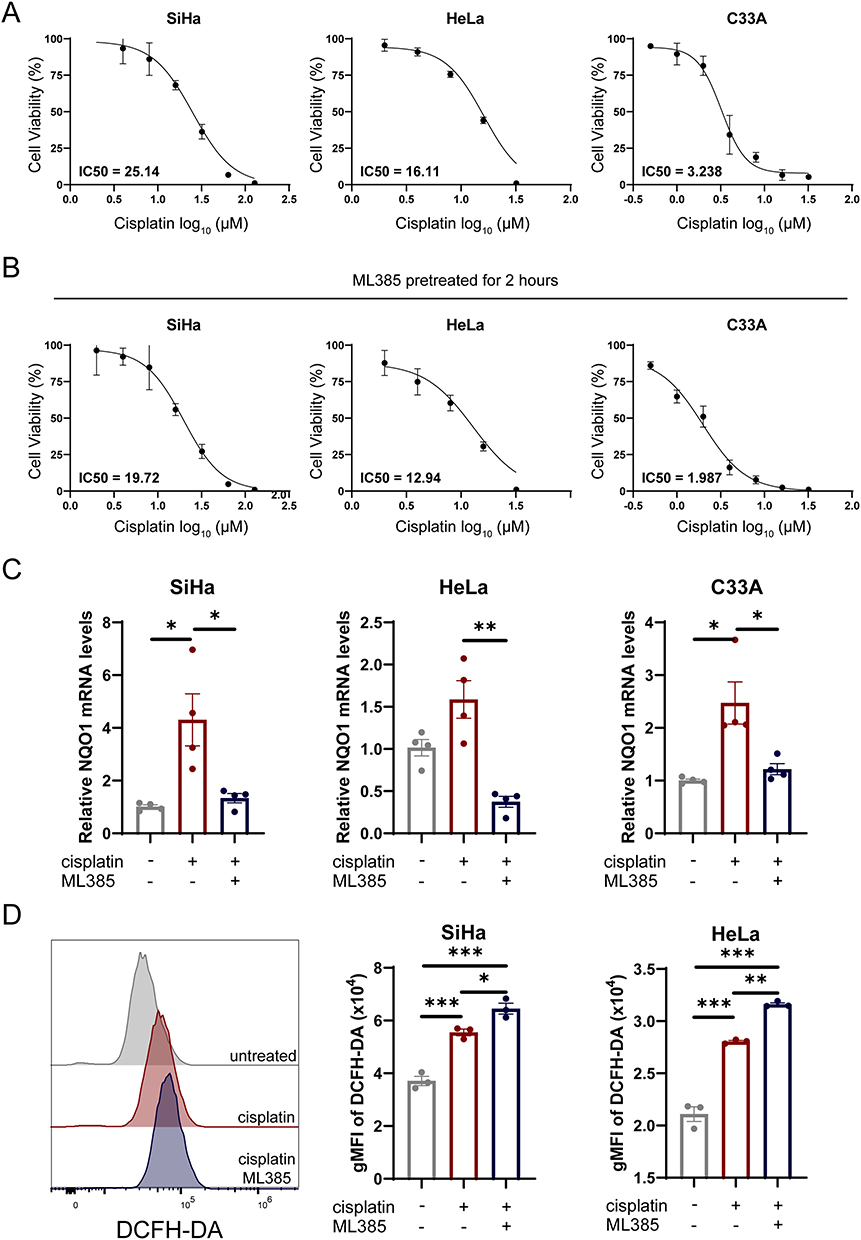 |
Figure 7 NRF2 signaling inhibition sensitizes CC cells to cisplatin treatment. (A) IC50 values of HeLa, SiHa and C33A cells treated with cisplatin for 48 h. The data are representative of 3 biological replicates. (B) IC50 values of HeLa, SiHa and C33A cells treated with cisplatin for 48 h. Cells were pretreated with ML385 (1 µM) for 2 hours prior to cisplatin exposure, with ML385 maintained throughout the entire treatment period. The data are representative of 3 biological replicates. (C) RT-qPCR showing the mRNA expression of NQO1 in CC cell lines. Cells were pretreated with ML385 (1 µM) for 2 hours prior to cisplatin exposure. The data are representative of 3 biological replicates. (D) Representative histograms and gMFI of DCFH-DA in SiHa and HeLa cells. The data are representative of 3 biological replicates. *p< 0.05, **p< 0.01, ***p< 0.001. Abbreviations: CC, cervical cancer; IC50, half maximal inhibitory concentrations. |
Discussion
Advances in systemic chemotherapy and radiotherapy have significantly reduced the incidence and mortality of CC.19 Nevertheless, in developing countries, recurrence rates and disease-related deaths remain alarmingly high.20 Given the limited predictive accuracy of conventional clinical parameters such as FIGO staging and lymph node metastasis status in forecasting tumor progression and treatment response,3,4 we sought to develop a robust prognostic signature. The advent of high-throughput technologies has revolutionized gene expression profiling, offering new opportunities to identify clinically relevant molecular biomarkers for CC progression and prognosis.21–23 In this study, we conducted comprehensive transcriptomic analyses using data from both the TCGA-CESC cohort and a local cohort. Building upon transcriptomic data, we developed and validated a novel transcriptome-based risk stratification model with superior predictive performance for CC progression.
The integration of HPV DNA into the host genome is a key oncogenic event that compromises genomic stability. This genomic instability subsequently induces extensive transcriptomic dysregulation, resulting in a substantial number of differentially expressed genes DEGs.24–26 These DEGs are enriched in critical oncogenic pathways, including cell cycle progression, DNA damage repair, and immune-regulated mechanisms, which collectively contribute to cervical carcinogenesis.27–29 The resulting gene expression signature not only reflects HPV-driven malignant transformation but also serves as a potential biomarker for disease progression and therapeutic response.30,31 To identify robust prognostic biomarkers derived from HPV16 integration, we employed LASSO Cox regression—a method particularly suited for feature selection in high-dimensional data as it shrinks regression coefficients toward zero, effectively eliminating non-predictive variables. Using 10-fold cross-validation, we narrowed down the candidate genes and identified 12 with the strongest prognostic significance. These were further refined through stepwise Cox regression analysis, ultimately yielding a 9-gene risk model for predicting PFS in CC.
To assess the predictive accuracy of our risk model, we initially performed Kaplan-Meier survival analysis, which revealed statistically significant differences in survival outcomes between the two risk groups. To further evaluate the model’s discriminative ability, we conducted time-dependent ROC analysis and calculated the AUC at sequential time points. Time-dependent ROC curve analysis demonstrated consistent predictive accuracy across various clinical timepoints. Furthermore, ROC over time analysis revealed that our 9-gene risk model maintained robust prognostic value throughout the clinical follow-up period. Notably, comparative analyses showed that our transcriptome-based model significantly outperformed conventional clinical parameters in predicting disease progression, as evidenced by higher sensitivity and specificity metrics. Additionally, calibration analysis showed excellent agreement between predicted and observed survival probabilities. The superior performance of our model suggests that transcriptome-based approaches may offer valuable complementary information to traditional clinicopathological factors in prognostic evaluation.
The biological roles of the nine genes, while diverse, align with several established pathways in cervical carcinogenesis. For instance, the interplay between immune modulation (eg, CXCL11, NPL, PGLYRP3), epithelial-mesenchymal plasticity (SPRR3, MMP1, PRRX2, MCAM), and cell cycle progression (NEK6) provides a compelling multi-mechanistic rationale for the aggressive phenotype observed in the high-risk group. This reinforces that our signature captures a cohesive biological program beyond a mere statistical association. To elucidate the biological functions of the nine-gene signature, we conducted comprehensive functional enrichment analysis. The results demonstrated significant enrichment in immune-related pathways, particularly those governing immune cell infiltration and activation. Subsequent correlation analysis showed a strong inverse relationship between risk scores and adaptive immune cell populations, which may explain the poorer survival outcomes observed in high-risk group.
Consistent with the disruptive role of HPV integration in host cell genome stability, we observed significantly elevated mutation frequencies in key proto-oncogenes such as PIK3CA, KMT2C, and NFE2L2 in the high-risk group. Notably, patients harboring NFE2L2 mutations demonstrated markedly poorer PFS compared to wild-type cases. Drug sensitivity analysis further predicted that high-risk patients showed reduced responsiveness to cisplatin, corroborating established associations between NFE2L2 mutations and platinum-based chemotherapy resistance. Mechanistically, NFE2L2 encodes the NRF2 transcription factor, and gain-of-function mutations lead to constitutive activation of the NRF2-mediated antioxidant pathway, which can attenuate cisplatin-induced oxidative stress and limit therapeutic efficacy. Importantly, we found that pharmacological inhibition of NRF2 with ML385 further increased intracellular ROS levels in CC cell lines, thereby enhancing their sensitivity to cisplatin. These findings suggest that targeting the NRF2 pathway may overcome cisplatin resistance in high-risk patients. Thus, combining cisplatin with NRF2 inhibitors such as ML385 represents a promising therapeutic strategy, particularly for tumors exhibiting NFE2L2 mutations or hyperactivation of the NRF2 signaling axis. Collectively, our risk stratification system shows potential clinical utility in guiding therapeutic decisions, as evidenced by its ability to predict response to platinum-based chemotherapy. These findings position our model as a promising tool for personalized patient management in CC care.
This study has several limitations. First, HPV18—the second most common high-risk HPV type—was not experimentally included due to the lack of appropriate integrated cell line models, which may affect the generalizability of our findings to all HPV-related cervical cancers. Second, direct comparison with established prognostic models (eg, the EMC2 clinicopathological model or HPV methylation-based signatures) was not feasible due to incomplete clinical annotations and lack of methylation data in our cohort. Third, although the sample size of our local cohort (n=118) is comparable to many prior biomarker studies, it remains relatively limited for robust subgroup analyses and may constrain the statistical power of multivariable models. Finally, the absence of in vivo validation warrants caution in interpreting the therapeutic implications of targeting the proposed pathway. Future studies involving larger, multi-center cohorts, prospective validation, and suitable in vivo models are warranted to confirm the generalizability and clinical utility of the signature across diverse HPV subtypes.
Conclusion
We established a robust 9-gene prognostic signature that effectively predicts PFS in CC. This model offers novel molecular insights into HPV-associated oncogenic mechanisms and enables improved risk stratification to guide clinical decision-making in CC management.
Data Sharing Statement
Raw sequencing data generated in this study are deposited in the National Genomics Data Center (https://ngdc.cncb.ac.cn/) with accession number HRA005334. The conclusions in the paper can be evaluated using data available in the paper and the Supplementary Materials.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from all participants prior to their enrollment in the study. The study protocol was reviewed and approved by the Medical Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Approval No. TJ-IRB20180505) and by the Medical Ethics Committee of the Women’s Hospital, School of Medicine, Zhejiang University (Approval No. IRB-20210085-R).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Key Research and Development Program of China (No.2021YFC2701204 to H.W.), the National Natural Science Foundation of China (No.82373260 to H.W.), the “Jianbing” and “Lingyan” R&D programs of Zhejiang province (No.2022C03013 to H.W.).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Gennigens C, Jerusalem G, Lapaille L, et al. Recurrent or primary metastatic cervical cancer: current and future treatments. ESMO Open. 2022;7(5):100579. doi:10.1016/j.esmoop.2022.100579
3. Wright JD, Matsuo K, Huang Y, et al. Prognostic performance of the 2018 international federation of gynecology and obstetrics cervical cancer staging guidelines. Obstet Gynecol. 2019;134(1):49–57. doi:10.1097/AOG.0000000000003311
4. Fleming ND, Frumovitz M, Schmeler KM, et al. Significance of lymph node ratio in defining risk category in node-positive early stage cervical cancer. Gynecol Oncol. 2015;136(1):48–53. doi:10.1016/j.ygyno.2014.11.010
5. Hu Z, Zhu D, Wang W, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015;47(2):158–163. doi:10.1038/ng.3178
6. Matovina M, Sabol I, Grubisic G, Gasperov NM, Grce M. Identification of human papillomavirus type 16 integration sites in high-grade precancerous cervical lesions. Gynecol Oncol. 2009;113(1):120–127. doi:10.1016/j.ygyno.2008.12.004
7. Molina MA, Steenbergen RDM, Pumpe A, Kenyon AN, Melchers WJG. HPV integration and cervical cancer: a failed evolutionary viral trait. Trends Mol Med. 2024;30(9):890–902. doi:10.1016/j.molmed.2024.05.009
8. Karimzadeh M, Arlidge C, Rostami A, Lupien M, Bratman SV, Hoffman MM. Human papillomavirus integration transforms chromatin to drive oncogenesis. Genome Biol. 2023;24(1):142. doi:10.1186/s13059-023-02926-9
9. Zhao JW, Fang F, Guo Y, et al. HPV16 integration probably contributes to cervical oncogenesis through interrupting tumor suppressor genes and inducing chromosome instability. J Exp Clin Cancer Res. 2016;35(1):180. doi:10.1186/s13046-016-0454-4
10. Li X, Ren C, Huang A, et al. PIBF1 regulates multiple gene expression via impeding long-range chromatin interaction to drive the malignant transformation of HPV16 integration epithelial cells. J Adv Res. 2024;57:163–180. doi:10.1016/j.jare.2023.04.015
11. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi:10.1038/nature07385
12. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi:10.1186/s13059-017-1349-1
13. Arafeh R, Samuels Y. PIK3CA in cancer: the past 30 years. Semin Cancer Biol. 2019;59:36–49. doi:10.1016/j.semcancer.2019.02.002
14. Jiao Y, Lv Y, Liu M, et al. The modification role and tumor association with a methyltransferase: KMT2C. Front Immunol. 2024;15:1444923. doi:10.3389/fimmu.2024.1444923
15. Cui Y, Chen H, Xi R, et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res. 2020;30(10):902–913. doi:10.1038/s41422-020-0333-6
16. Frank R, Scheffler M, Merkelbach-Bruse S, et al. Clinical and pathological characteristics of KEAP1- and NFE2L2-mutated non-small cell lung carcinoma (NSCLC). Clin Cancer Res. 2018;24(13):3087–3096. doi:10.1158/1078-0432.CCR-17-3416
17. Islam SS, Qassem K, Islam S, et al. Genetic alterations of Keap1 confers chemotherapeutic resistance through functional activation of Nrf2 and Notch pathway in head and neck squamous cell carcinoma. Cell Death Dis. 2022;13(8):696. doi:10.1038/s41419-022-05126-8
18. Zhang DD. Thirty years of NRF2: advances and therapeutic challenges. Nat Rev Drug Discov. 2025;24(6):421–444. doi:10.1038/s41573-025-01145-0
19. Burmeister CA, Khan SF, Schafer G, et al. Cervical cancer therapies: current challenges and future perspectives. Tumour Virus Res. 2022;13:200238. doi:10.1016/j.tvr.2022.200238
20. Lemp JM, De Neve JW, Bussmann H, et al. Lifetime prevalence of cervical cancer screening in 55 low- and middle-income countries. JAMA. 2020;324(15):1532–1542. doi:10.1001/jama.2020.16244
21. Garg P, Krishna M, Subbalakshmi AR, et al. Emerging biomarkers and molecular targets for precision medicine in cervical cancer. Biochim Biophys Acta Rev Cancer. 2024;1879(3):189106. doi:10.1016/j.bbcan.2024.189106
22. Reza MS, Harun-Or-Roshid M, Islam MA, et al. Bioinformatics screening of potential biomarkers from mRNA expression profiles to discover drug targets and agents for cervical cancer. Int J Mol Sci. 2022;23(7):3968. doi:10.3390/ijms23073968
23. Lizano M, Carrillo-Garcia A, De La Cruz-Hernandez E, Castro-Munoz LJ, Contreras-Paredes A. Promising predictive molecular biomarkers for cervical cancer (Review). Int J Mol Med. 2024;53(6):50. doi:10.3892/ijmm.2024.5374
24. Tian R, Huang Z, Li L, et al. HPV integration generates a cellular super-enhancer which functions as ecDNA to regulate genome-wide transcription. Nucleic Acids Res. 2023;51(9):4237–4251. doi:10.1093/nar/gkad105
25. Zhou L, Qiu Q, Zhou Q, et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat Commun. 2022;13(1):2563. doi:10.1038/s41467-022-30190-1
26. Liu M, Han Z, Zhi Y, et al. Long-read sequencing reveals oncogenic mechanism of HPV-human fusion transcripts in cervical cancer. Transl Res. 2023;253:80–94. doi:10.1016/j.trsl.2022.09.004
27. Fan J, Fu Y, Peng W, et al. Multi-omics characterization of silent and productive HPV integration in cervical cancer. Cell Genom. 2023;3(1):100211. doi:10.1016/j.xgen.2022.100211
28. Bruyere D, Roncarati P, Lebeau A, et al. Human papillomavirus E6/E7 oncoproteins promote radiotherapy-mediated tumor suppression by globally hijacking host DNA damage repair. Theranostics. 2023;13(3):1130–1149. doi:10.7150/thno.78091
29. Guo C, Qu X, Tang X, et al. Spatiotemporally deciphering the mysterious mechanism of persistent HPV-induced malignant transition and immune remodelling from HPV-infected normal cervix, precancer to cervical cancer: integrating single-cell RNA-sequencing and spatial transcriptome. Clin Transl Med. 2023;13(3):e1219. doi:10.1002/ctm2.1219
30. Huang F, He L, Li W, et al. HPV integration: a precise biomarker for detection of residual/recurrent disease after treatment of CIN2-3. Infect Agent Cancer. 2024;19(1):36. doi:10.1186/s13027-024-00600-8
31. Kamal M, Lameiras S, Deloger M, et al. Human papilloma virus (HPV) integration signature in Cervical Cancer: identification of MACROD2 gene as HPV hot spot integration site. Br J Cancer. 2021;124(4):777–785. doi:10.1038/s41416-020-01153-4
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Construction of a Nomogram Prediction Model for Individualized Prediction of Cervical Cancer Risk: A Retrospective Study
Zhou W, Wang Y, Long H, Wang J
International Journal of Women's Health 2025, 17:4435-4446
Published Date: 13 November 2025
Long-Term Survival Outcomes of HPV DNA–Negative Cervical Cancer Compared with HPV DNA–Positive Disease in a Thai Tertiary Cohort
Phisalmonhkhon T, Kantathavorn N, Wetcho T, Samrarn J, Kittikhun R, Sukjariangporn H, Thongaram M
International Journal of Women's Health 2026, 18:601452
Published Date: 24 April 2026
Construction of a Risk Identification Model Based on Electrolytes and Related Factors for Nausea and Vomiting in Pregnancy: A Secondary Analysis of Clinical Trial Data
Gao Z, Feng J, Liu Y, Gao Q, Yu J, Liu Y, Li Z, Duan Y, Jiang S, Wu X, Gao J
International Journal of Women's Health 2026, 18:618603
Published Date: 15 July 2026
A Machine Learning-Based Model for Cirrhosis Risk Stratification Incorporating Noninvasive Markers and Clinical Variables
Wang Y, Liang H, Si C, Li A, Wu H, Chen B
International Journal of General Medicine 2026, 19:602749
Published Date: 23 July 2026