Back to Journals » Journal of Inflammation Research » Volume 19
Pneumococcal Endopeptidase O Attenuates Colitis by Inhibiting the Macrophage–CCL2 Axis and Reshaping Gut Microbiota
Authors Wang H ![]() , Shao S
, Shao S ![]() , Peng Y, Xu W, Zhang J, Liu X
, Peng Y, Xu W, Zhang J, Liu X ![]() , Bian X, Huang K
, Bian X, Huang K ![]() , Zhang X
, Zhang X ![]()
Received 17 February 2026
Accepted for publication 15 May 2026
Published 27 May 2026 Volume 2026:19 601434
DOI https://doi.org/10.2147/JIR.S601434
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alberto Caminero
Hanyi Wang,1,* Shuangmei Shao,1,* Yuan Peng,1 Wenlong Xu,2 Jia Zhang,1 Xiao Liu,1 Xintong Bian,3 Kun Huang,4 Xuemei Zhang1
1Department of Laboratory Medicine, Key Laboratory of Diagnostic Medicine (Ministry of Education), Chongqing Medical University, Chongqing, People’s Republic of China; 2Department of Medical Laboratory Medicine, Chongqing University Three Gorges Hospital, Chongqing University, Chongqing, People’s Republic of China; 3Department of Medical Laboratory Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 4Department of Dermatology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xuemei Zhang, Department of Laboratory Medicine, Key Laboratory of Diagnostic Medicine (Ministry of Education), Chongqing Medical University, No. 1 Medical Hospital Road, Yuzhong District, Chongqing, 400016, People’s Republic of China, Email [email protected]
Background: Ulcerative colitis (UC) is a chronic inflammatory disease characterized by persistent immune activation and gut microbiota dysbiosis, for which current therapies remain limited by incomplete efficacy and substantial adverse effects. This study aimed to investigate the protective effects of pneumococcal endopeptidase O (PepO) on UC and its potential mechanisms of action.
Methods: The efficacy of PepO was evaluated in a dextran sulfate sodium (DSS)-induced acute colitis mouse model. Clinical phenotypes were assessed using the disease activity index (DAI), colon length, histopathological analysis, and inflammatory cytokine levels. Macrophage infiltration and key inflammatory mediators were analyzed via immunofluorescence, flow cytometry, ELISA, and qPCR. Mechanisms were further validated by macrophage depletion using clodronate liposomes and recombinant CCL2 supplementation. In addition, gut microbiota composition was analyzed via 16S rRNA sequencing, and causality was confirmed through antibiotic-induced microbiota depletion and fecal microbiota transplantation (FMT) experiments.
Results: PepO administration significantly improved disease activity, colon length, and ameliorated histopathological damage in the mice. Mechanistically, PepO directly inhibited macrophage CCL2 production, reducing macrophage infiltration and the release of pro-inflammatory cytokines in colitis. Additionally, PepO remodelled the gut microbiota and markedly increased the abundance of beneficial bacteria, such as Lactobacillus and Akkermansia. Antibiotic-induced microbiota depletion completely abolished PepO’s protective effect, while FMT successfully replicated its protective actions, including suppression of CCL2 expression and macrophage infiltration.
Conclusion: This study shows that PepO exerts potent anti-colitic effects through a dual mechanism involving direct inhibition of the macrophage-CCL2 axis and microbiota-dependent enhancement of gut immune homeostasis. The illustration compares DSS-induced colitis and PepO administration effects. On the left, DSS-induced colitis shows a decrease in MUC2, ZO-1, occludin and Ki67 in epithelial cells, with increased apoptosis. The lamina propria has elevated cytokines IL-6, IL-1 beta, TNF-alpha and increased CCL2 and F4/80 positive macrophages. On the right, PepO administration shows increased MUC2, ZO-1, occludin and Ki67, with reduced apoptosis. Akkermansia and Lactobacillus are increased in the lumen. The lamina propria shows reduced cytokines IL-6, IL-1 beta, TNF-alpha and reduced inflammation, with inhibited macrophage activation and increased CCL2 and macrophage recruitment. Arrows indicate promotion or inhibition of processes.Comparison of DSS-induced colitis and PepO administration effects on gut cells and inflammation.
Keywords: CCL2, gut microbiota, intestinal barrier, macrophages, pneumococcal endopeptidase O (PepO), ulcerative colitis
Introduction
Ulcerative colitis (UC) is an autoimmune disorder primarily marked by chronic inflammation of the bowel and is one of the most prevalent forms of inflammatory bowel disease (IBD).1 UC most commonly affects the sigmoid colon and rectum, and in severe cases, inflammation may extend throughout the entire colon, significantly impacting patients’ quality of life. While IBD was historically more prevalent in developed countries, the prevalence of UC has been steadily increasing in some Asian countries, including China, driven by shifts in lifestyle and dietary habits.2 As the precise etiology of UC remains unclear, current treatments focus on symptom management. While existing therapies can control intestinal inflammation to some extent, they fail to restore long-term immune homeostasis,3 resulting in limited clinical remission rates (30–60%)4,5 and a high risk of relapse.6 Consequently, there is an immediate need to develop alternative therapeutic strategies for UC.
The development of UC is multifactorial, involving genetic, environmental, microbial, and immune dysregulation.7 Extensive research indicates that macrophages in UC patients are activated to release multiple inflammatory mediators, which contribute significantly to disease onset and progression.8,9 In the context of chronic inflammation, their persistent and uncontrolled recruitment further amplifies the immune response, driving the persistence and progression of inflammation in UC.10,11 Therefore, modulating macrophage recruitment and activity represents a critical therapeutic target for managing UC and preventing disease progression.
As an integral element of the gastrointestinal tract, the gut microbiota is essential to host physiology, interacting with the immune system, contributing to immune homeostasis, and participating in various metabolic pathways. Dysbiosis of the gut microbiota, characterized by changes in microbial composition, reduced diversity, a loss of beneficial bacteria such as Lactobacillus, Akkermansia, Faecalibacterium, Roseburia, and Bifidobacterium, and an expansion of pathogenic species such as adherent-invasive Escherichia coli and enterotoxigenic Bacteroides fragilis, has been strongly linked to colitis.12–14 Recent studies provide strong evidence that impaired macrophage activation in response to gut microbiota may contribute to heightened susceptibility to UC.15 Increasing attention is being focused on approaches to restore gut microbiota balance, such as employing probiotics and prebiotics, to influence host health through modulation of microbial composition.16,17 Probiotics, such as Lactobacillus and Akkermansia, have been shown to have a direct link to the prevention of colitis.18,19 Therefore, addressing dysbiosis in UC patients may offer a promising therapeutic approach.
Immunomodulatory agents exert therapeutic effects in immune-mediated diseases, including UC, by either enhancing or suppressing immune responses. However, despite the widespread use of immunomodulatory-based treatment strategies, significant challenges remain, including pronounced side effects, increased risks of infections and malignancies, and the high costs associated with treatment. These agents are categorized into three types: conventional immunosuppressants, such as glucocorticoids (for acute phases) and 6-mercaptopurine (for maintaining remission);20 biological agents, chiefly anti-tumour necrosis factor (TNF) drugs, which form the core of UC therapy;21 and small-molecule drugs, such as JAK inhibitors and S1PR modulators, predominantly used as second-line treatments.22,23 However, the immunosuppressive functions of these agents pose significant risks. Anti-TNF drugs may increase the risk of tuberculosis reactivation;24 JAK inhibitors and S1PR modulators are associated with heightened risks of respiratory, urinary tract, and herpesvirus infections;25 traditional immunosuppressants (such as azathioprine) have been associated with a higher risk of certain tumors. In addition, there is a complex interaction between immunomodulators and the gut microbiota. While some drugs can alter microbiota composition, the patient’s baseline microbiota also affects drug efficacy.26 Microbiota dysbiosis, with reduced beneficial bacteria (eg, Bifidobacterium and Blautia) and increased opportunistic pathogens (eg, Escherichia and Shigella), is linked to treatment non-response.27 The persistent disruption of the “microbiota-macrophage axis” plays a crucial role in the chronicity and refractoriness of colitis, yet it is often neglected by current treatment strategies. Therefore, identifying key factors that can precisely regulate macrophages while restoring microbial homeostasis offers a novel approach to overcoming current therapeutic limitations.
In our group’s previous protein vaccine research, we identified a TLR2/4 dual-ligand protein: pneumococcal endopeptidase O (PepO). This protein is recognized by the cell surface pattern recognition receptors TLR2 and TLR4, inducing innate immune responses and trained immunity to exert broad-spectrum anti-infective effects.28,29 These findings suggest that PepO functions as an immunomodulatory enhancer targeting macrophages. Beyond its role in enhancing host defense and trained immunity, our research suggested that PepO elicits a transient inflammatory response followed by rapid resolution, indicating a potential role in immune recalibration rather than sustained immune activation.30 Given its unique capacity to directly engage macrophages while originating from a microbial context, we hypothesized that PepO may function as a molecular integrator linking immune regulation with gut microbiota remodeling. In the present investigation, we demonstrate that PepO alleviates experimental colitis by suppressing CCL2-mediated macrophage recruitment and reshaping the gut microbiota, thereby restoring intestinal immune homeostasis. The study systematically evaluates its potential as a novel protective target that can simultaneously correct immune imbalance and gut disorder. This work further enhances our understanding of the underlying mechanisms of colitis and provides a foundation for developing more targeted and lasting treatments beyond traditional immunosuppression.
Materials and Methods
Animals
All experimental mice were supplied by Tengxin Biotechnology Co., Ltd (Chongqing, China). The animals were male C57BL/6J mice under specific pathogen-free (SPF) conditions, aged 6–8 weeks, with a body weight of 18–20 g. Prior to the experiment, the animals were acclimated for one week. During the acclimation period, mice from different experimental groups were randomly co-housed to minimize cage effects and standardize environmental conditions. All animal experiments were conducted in accordance with the ethical policies and procedures approved by the Animal Care and Use Committee of Chongqing Medical University (IACUC-CQMU-2024-03125). All animal experiments conducted in this study were performed in compliance with the ARRIVE guidelines. All animals were housed at the Animal Experiment Center of Chongqing Medical University under environmental conditions of 24.0 ± 2.0 °C and 55.0 ± 5.0% humidity, with a 12–hour light/dark cycle. Animals had free access to food and water.
Murine Colitis Models
Following the acclimatization and randomization process, mice were housed in group-specific cages to ensure accurate drug administration and to prevent potential microbial cross-contamination between different treatment groups. To establish a mouse colitis model, 3% dextran sulfate sodium (DSS) was dissolved in drinking water and administered for 6 consecutive days. Based on DSS administration, the PepO administration involved an intraperitoneal injection of 200 μL (10 mg/kg) on the day of the experiment. The DSS-containing water was refreshed every other day (on days 0, 2, and 4). On day 6, the DSS water was replaced with normal drinking water, and all animals were euthanized on day 7. Euthanasia was performed as follows: deep anesthesia was first induced by an intraperitoneal injection of pentobarbital at a dose of 75 mg/kg body weight. Following confirmation of deep anesthesia by the absence of the pedal withdrawal reflex and lack of spontaneous breathing, mice were subsequently euthanized by cervical dislocation. Daily monitoring of mouse incidence and body weight recording were performed. Assessment of the clinical disease activity index (DAI) was performed according to a standard method (Supplementary Table S1), based on scores for stool consistency, weight loss, and rectal bleeding.
Intestinal macrophages were depleted by intraperitoneal injection of clodronate liposomes (200 μL/animal, 5 mg/mL) at three time points: 3 days before DSS treatment (day −3), on the day of DSS initiation (day 0), and 3 days after DSS initiation (day +3). Control animals received the same volume of control liposomes (PBS).
CCL2 recombinant protein was administered via intraperitoneal injection at 200 μL/animal (10 μg/kg) on modeling day 2 and modeling day 4.
For the antibiotics (ABX) depletion assay, 6–8-week-old C57BL/6J mice received a daily oral gavage of a quadruple antibiotic cocktail (MCE) for 5 consecutive days at the following doses: ampicillin (200 mg/kg), metronidazole (200 mg/kg), neomycin sulfate (200 mg/kg), and vancomycin (100 mg/kg).
For the fecal microbiota transplantation (FMT) assay, recipient C57BL/6J mice were first rendered microbiota‑depleted using the same antibiotic pretreatment regimen as described above. Donor mice (WT and PepO-treated) were administered 3% DSS in drinking water for 6 days, followed by one day of regular water. Fecal samples were collected separately from each group. The fecal material was homogenized in sterile PBS to a final concentration of 100 mg feces/mL. The homogenized suspension was then centrifuged at 500 g for 2 minutes to obtain bacterial suspensions. The fecal suspensions were thoroughly mixed and administered at 10 μL/g body weight. Recipient WT mice, pretreated with antibiotics as described, were randomly separated into two experimental groups: the FMT-DSS group and the FMT-DSS+PepO group. Each group received a daily gavage of the corresponding fecal suspension (from DSS or DSS+PepO donors, respectively) for 6 consecutive days. Concurrent with the FMT period, all recipient mice received 3% DSS in their drinking water for 6 days to induce colitis, followed by one day of recovery with regular water.
Histopathological Analysis and Immunostaining
In the DSS-induced colitis model, 0.5 cm of distal colon tissue was harvested from each mouse for staining, with the mouse colon being transected transversely. Colon tissue was first fixed for one night using 4% paraformaldehyde. Following embedding in paraffin, tissues were sectioned at 5 μm thickness for subsequent staining with hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS). Histopathological scores were evaluated based on the extent of crypt injury, epithelial damage, and inflammatory cell infiltration. For quantitative assessment, image analysis was performed with ImageJ (National Institutes of Health, Bethesda, MD, USA).
For immunofluorescence analysis, 4-μm colonic sections were subjected to fixation in paraformaldehyde and subsequent permeabilization with Triton X-100 in phosphate-buffered saline (PBS). They were then incubated with a blocking buffer prior to antibody staining. For immunofluorescence, the sections were treated at 4°C overnight with the corresponding antibody. After PBS washing, cell nuclei were stained with DAPI. A fluorescent microscope was used to examine and take pictures. Quantitative analysis was performed using ImageJ.
Intestinal Permeability Measurement
To assess intestinal permeability, mice were fasted for 12 hours prior to the experiment, and were administered 50 mg/kg of fluorescein isothiocyanate-labeled dextran (MW 4000) via gavage. Four hours later, cardiac blood samples were collected and centrifuged to obtain serum. The supernatant was diluted and subjected to fluorescence detection (excitation at 485 nm/emission at 525 nm). The concentration of FITC-dextran was quantified against a standard curve. The curve was established using serially diluted FITC-dextran standards.
RNA Extraction and Quantitative qRT-PCR
Total RNA was isolated from both cellular and colonic tissues using TRIzol reagent, following the manufacturer’s protocol. Subsequently, the extracted RNA served as the template for complementary DNA (cDNA) synthesis, which was then performed using a PrimeScript RT reagent kit. For gene expression analysis, qRT–PCR was conducted with SYBR Premix Ex Taq in a SYBR Green-based assay, strictly adhering to the kit’s instructions. The relative mRNA expressions were determined using the ΔCt method with housekeeping gene β-actin serving as the endogenous control. Supplementary Table S5 lists the specific primer sequences used in this study.
Enzyme-Linked Immunosorbent Assay
Total protein was extracted by homogenizing frozen colon samples in RIPA lysis buffer. After homogenization, the lysates were centrifuged at 12,000 g for 15 min at 4°C, and the supernatant was collected for further analysis. Protein concentrations were determined using a bicinchoninic acid (BCA) assay kit according to the manufacturer’s instructions. Subsequently, the concentrations of key pro-inflammatory mediators (IL-6, IL-1β, TNF-α, and CCL2) in colon tissues were measured by enzyme-linked immunosorbent assay (ELISA) according to the manufacturers’ recommendations. The cytokines secreted in the supernatants from the cell cultures and mouse plasma were also measured similarly.
Isolation of Colonic Lamina Propria Cells
After excision, the mouse colon tissues were cut into 1–2 cm segments and immediately placed in ice-cold PBS. Following this, each segment was longitudinally incised to remove the luminal contents, which were then gently cleared away with several washes of PBS. To first remove colonic epithelial cells, colon fragments were incubated in Hanks’ balanced salt solution (HBSS) containing 5 mM EDTA, 1 mM DTT, and 2% FBS at 37°C for 40 min with gentle agitation. Subsequently, the remaining tissue was minced into small pieces and digested in RPMI 1640 medium supplemented with 2% FBS, 50 µg/mL DNase I, and 1 mg/mL collagenase IV at 37°C for 40 min under gentle shaking. The mixture was filtered through a 70-μm cell strainer and then centrifuged at 450 g for 10 min. The pellet was washed twice with PBS and resuspended to obtain a single-cell suspension.
Flow Cytometry
To prevent non-specific binding, cell suspensions were first incubated with an anti-mouse CD16/32 antibody. They were then stained for surface markers using a panel of specific antibodies. For cell phenotyping and sorting, we utilized BD FACSCanto Plus and FACSCanto II flow cytometers (BD Biosciences, USA). The acquired data were analyzed with the accompanying FlowJo software. Detailed information regarding the antibodies used for flow cytometry is listed in Supplementary Table S2.
Western Blotting
Colonic samples were homogenized in RIPA lysis buffer, followed by measurement of the total protein concentration. Following separation by SDS-PAGE and electrotransfer onto PVDF membranes, the membranes were blocked with 5% skim milk powder for 2 hours at room temperature. They were incubated overnight at 4 °C with primary antibodies against Cleaved caspase-3 and β‑actin, followed by incubation with HRP-conjugated secondary antibodies at room temperature for 1 h. Protein bands were detected using an ECL Western blot reagent; chemiluminescent signals were captured with a ChemiDoc XRS+ imaging system (Bio-Rad). Information on the primary and secondary antibodies used for immunoblotting is provided in Supplementary Table S3.
Bone Marrow-Derived Macrophages (BMDMs) Culture
Bone marrow cells were isolated from the tibiae and femora of C57BL/6J mice. To generate BMDMs, primary cells were cultured for 7 days in complete DMEM containing 20 ng/mL recombinant macrophage colony-stimulating factor (M-CSF). On day 7, cells were pre-treated with or without 10 μg/mL PepO for 12 h, followed by stimulation with 100 ng/mL LPS for another 12 h. The supernatants were collected and subsequently assayed by ELISA.
Transwell Assay
To evaluate the effect of PepO on BMDM migration, we performed the following assay. Conditioned medium (CM) was collected from BMDMs treated with medium only (Control), LPS (100 ng/mL), or PepO pretreatment (10 μg/mL, 12 h) followed by LPS stimulation (12 h). The CM from the PepO+LPS group was also supplemented with 50 ng/mL rCCL2 to assess the rescue effect. BMDMs were seeded into the upper chamber of Transwell inserts (8 μm pore size), and the corresponding CM was placed in the lower chamber. After overnight incubation, cells that had migrated to the lower chamber were fixed, stained with crystal violet, and counted.
Microbiota Analysis
To investigate the impact of PepO on gut microbiota, fecal samples were collected at the end of the experiment (Day 7) from mice in the Water, DSS, and DSS+PepO groups. Samples were immediately cryopreserved in liquid nitrogen and subsequently transferred to −80°C for 16S rRNA sequencing. 16S rRNA gene sequencing of the V3–V4 hypervariable region was performed on the Illumina platform by Shanghai Weihuan Biotechnology Co., Ltd. Alpha diversity indices (Chao1 and Shannon) were calculated and compared among groups using the Kruskal‑Wallis test. For beta diversity, principal coordinate analysis (PCoA) was used to visualize the microbial community structure, and PERMANOVA (Adonis) was performed to test for significant differences among groups. The Kruskal–Wallis test (for three groups) or Wilcoxon test (for two groups), along with ALDEx2 and ANCOM, was used for differential abundance analysis. For ALDEx2, significance was determined by Benjamini-Hochberg adjusted P-values (kw.eBH) < 0.05 for omnibus tests. Additionally, for pairwise comparisons, we prioritized a biological effect size > 1 to ensure robust interpretation despite the limited sample size. For ANCOM, a W-threshold > 0.6 was consistently applied as the detection criterion across all comparisons.
Statistical Analysis
Mice were enrolled in the studies in a blinded fashion and randomly allocated to their respective treatment groups. All data were analyzed with GraphPad Prism (v9.00, GraphPad Software, USA). Differences between groups were assessed for statistical significance using either one-way analysis of variance (ANOVA) or a two-tailed Student’s t-test, as appropriate. Data were shown as means ± SD. P < 0.05 was considered significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
All reagents and kits used in this study were commercially available. Detailed information including vendor names and catalog numbers is provided in Supplementary Table S4.
Results
PepO Ameliorates Symptoms of DSS-Induced Colitis
To assess the impact of PepO on colitis progression in UC mice, colitis was induced by providing 3% DSS in the drinking water for a duration of 6 days. On the first day, PepO (10 mg/kg) was administered through intraperitoneal injection (Figure 1A). Mice in the DSS-treated group displayed a range of symptoms associated with colitis, including significant diarrhea, rectal bleeding, and shortened colon length. PepO notably mitigated DSS-induced colitis, demonstrated by a significant reduction in DAI scores, which were derived from measurements of body weight reduction, stool consistency, and blood presence in the stool (Figure 1B). Additionally, colonic shortening was alleviated (Figure 1C). To investigate the impact of PepO on DSS-induced colonic damage, H&E and PAS staining were performed. In comparison to mice in the DSS group, which showed considerable loss of crypts, increased inflammatory cell infiltration, more severe damage to goblet cells, and higher histopathological scores, the DSS+PepO group showed relatively preserved colonic structure, mild mucosal damage, reduced infiltration of inflammatory cells, and lower histopathological scores (Figure 1D). PAS staining was conducted to evaluate the quantity of goblet cells. The results showed a significant decrease in the number of goblet cells in the DSS group compared to the water group. Notably, PepO mitigated the DSS-induced depletion of goblet cells (Figure 1E). Collectively, PepO significantly improves both the clinical symptoms and histopathological features of DSS-induced acute colitis.
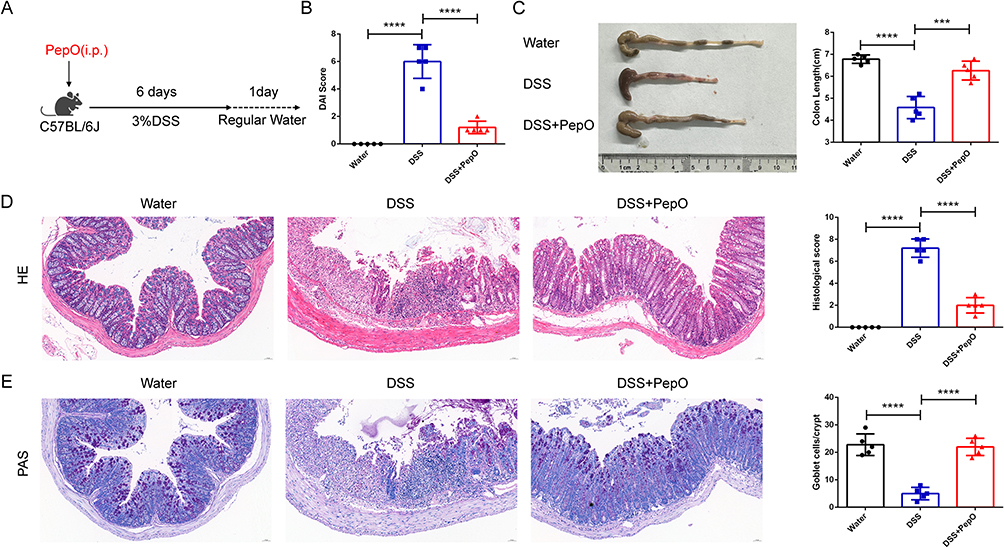 |
Figure 1 PepO effectively mitigated DSS-induced colitis. (A) Schematic of the study design, detailing the administration of PepO and induction of colitis with 3% DSS. (B) DAI on day 7 following DSS administration. (C) Representative images of colon tissues and colon length. (D) H&E staining of distal colon tissues and histopathological scores. Scale bar: 50 μm. (E) PAS staining of distal colon tissues and number of goblet cells. Scale bar: 50 μm. Individual data points for each mouse are represented by symbols: black circles for the Water group, blue squares for the DSS group, and red triangles for the DSS+PepO group. The data represent the mean ± SD, n = 5. ***P < 0.001; ****P < 0.0001. Abbreviations: i.p., intraperitoneal injection. |
PepO Protects the Intestinal Barrier by Restoring Its Structure and Epithelial Homeostasis
The integrity of the intestinal barrier is essential for maintaining intestinal homeostasis. We further investigated the impact of PepO on the intestinal mucosal barrier in colitis. On day 7, intestinal permeability was assessed in the mice through intragastric administration of FITC-dextran. Compared to the control group, mice treated with DSS exhibited significantly higher levels of FITC-dextran in the serum, whereas PepO administration notably reduced serum FITC-dextran levels (Figure 2F). These findings indicate that PepO alleviates the increased intestinal permeability caused by DSS induction.
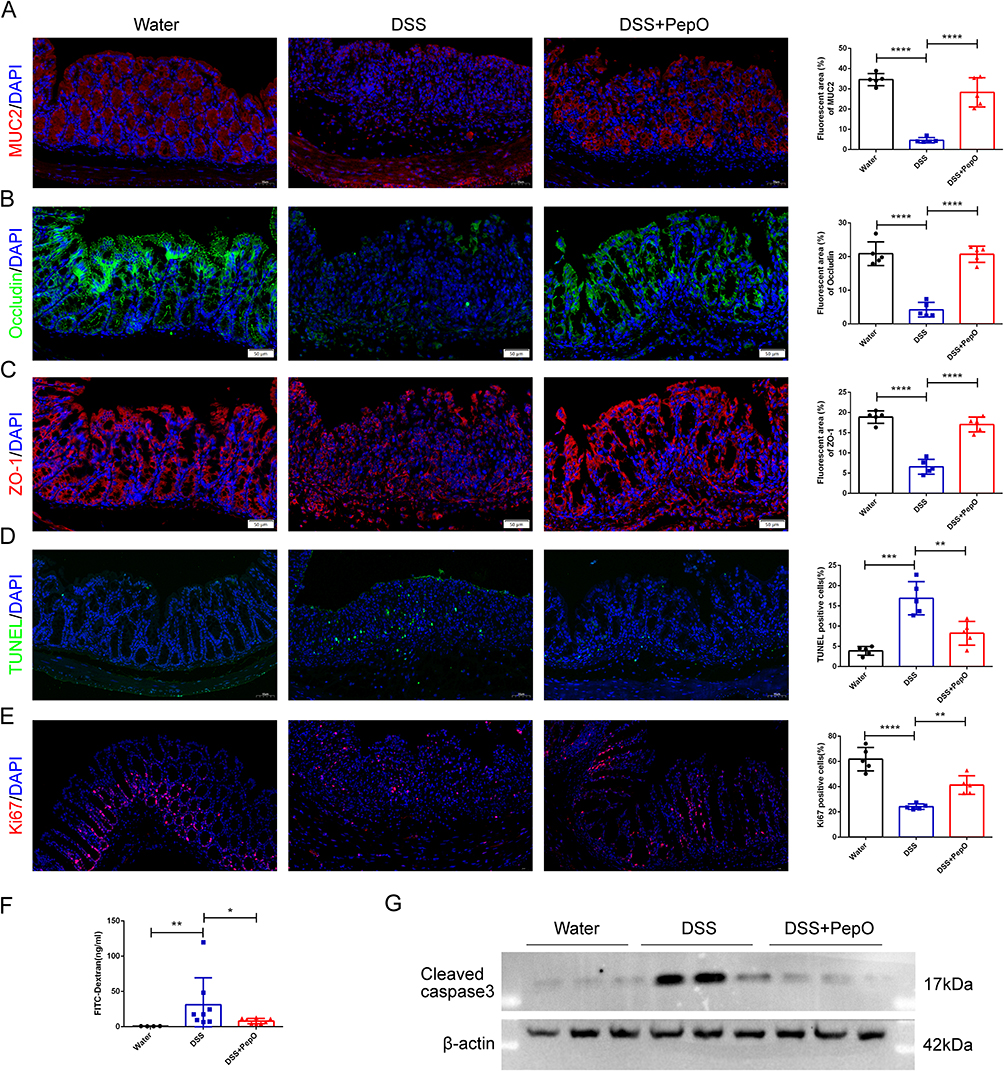 |
Figure 2 PepO maintained the homeostasis of intestinal epithelial cells in mice with DSS-induced acute colitis. (A–E) Immunofluorescent staining and quantitative analysis of MUC2, Occludin, ZO-1, TUNEL, and Ki67 in colon tissue. Scale bar: 50 μm. (F) Content of FITC-dextran in serum. (G) Western blot analysis of Cleaved caspase-3 in the colon. Individual data points for each mouse are represented by symbols: black circles for the Water group, blue squares for the DSS group, and red triangles for the DSS+PepO group. The data represent the mean ± SD, n = 3–8. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
The mucus layer that coats the epithelial cells in the colon plays a critical role in gut protection. Mucin2 (MUC2), a gel-forming mucin secreted by goblet cells, is the primary constituent of this mucus.31 Compared with the DSS group, PepO administration prevented the loss of MUC2 protein (Figure 2A).
It has been reported that decreased expression of tight junction (TJ) proteins contributes to intestinal barrier dysfunction, with ZO-1 and Occludin being the primary components of these tight junctions.32,33 Immunofluorescence staining was employed to examine the expression of TJ protein Occludin (Figure 2B) and protein ZO-1 (Figure 2C) in the colons of mice. ZO-1 expression was significantly reduced in DSS-treated mice, mirroring the decrease in occludin expression. PepO administration restored the redistribution and levels of these two proteins.
The imbalance between cell proliferation and apoptosis in the colonic crypts is a critical factor in the pathogenesis of UC in humans.34,35 To investigate this, the proliferation and apoptosis of colonic epithelial cells were evaluated using immunological techniques. A substantial number of TUNEL-positive cells were observed in the colonic tissue of DSS-treated mice, whereas the quantity of TUNEL-positive cells was markedly lower in the colon tissue of PepO-treated mice (Figure 2D). Western blot analysis was used to measure the expression of Cleaved caspase-3. The results showed a notable increase in the apoptotic protein Cleaved caspase-3 following DSS treatment (Figure 2G), whereas PepO administration markedly suppressed its expression in colonic tissue. The number of Ki67-positive cells in the colonic crypt region was significantly lower in DSS-induced mice compared to normal mice. In contrast, PepO administration led to a substantial increase in Ki67-positive cells (Figure 2E).
PepO Reduces Macrophage Infiltration and Inflammatory Cytokine Production in DSS-Induced Colitis
The excessive recruitment and subsequent activation of immune cells within intestinal tissue constitute a key characteristic in the development of ulcerative colitis, as they generate excessive pro-inflammatory cytokines and amplify inflammatory responses, ultimately leading to colonic damage.10 Immunofluorescence analysis was performed to evaluate the distribution and proportion of the macrophage surface marker F4/80 in colonic tissue. In DSS-treated mice, a significant accumulation of infiltrating macrophages was found in the lamina propria. However, PepO administration led to a significant decrease in macrophage infiltration (Figure 3A). Using the gating strategy outlined in Supplementary Figure S1A, flow cytometry analysis confirmed that PepO administration significantly decreased the infiltration of F4/80-positive macrophages within the lamina propria (Figure 3B). Furthermore, the mRNA levels of Il6, Tnf, and Il1b, as well as the protein levels of IL-6, TNF-α, and IL-1β, were markedly reduced in colonic tissue following PepO administration (Figure 3C and E). These cytokines, primarily produced by macrophages, were also significantly lower in the circulation in PepO-treated colitis mice compared to the DSS group (Figure 3D).
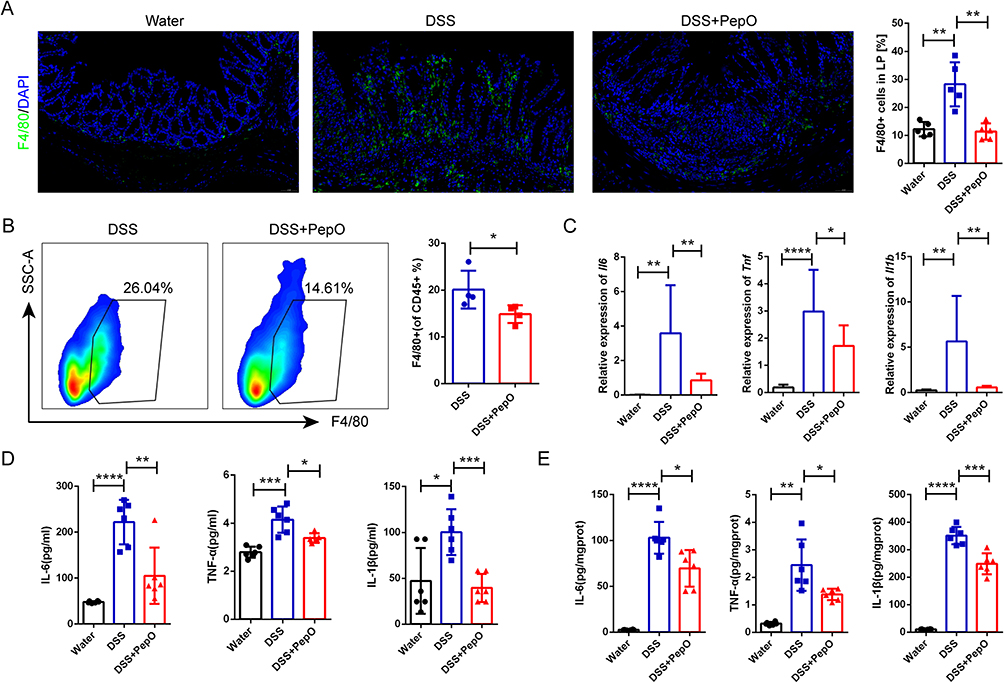 |
Figure 3 PepO reduced macrophage infiltration and inflammatory cytokine production in DSS-induced colitis. (A) Immunofluorescence staining of the macrophage marker F4/80 and quantitation in colon tissues. Scale bar: 50 μm. (B) Representative flow cytometry plots (left) and the frequency (right) of CD45⁺CD11b⁺F4/80⁺ macrophages among CD45⁺ cells in the colonic lamina propria. (C) mRNA expression of Il6, Tnf, and Il1b in colon tissues measured by qRT-PCR. (D) Plasma levels of IL-6, TNF-α, and IL-1β measured by ELISA. (E) Protein levels of IL-6, TNF-α, and IL-1β in colon tissue homogenates measured by ELISA. Individual data points for each mouse are represented by symbols: black circles for the Water group, blue squares for the DSS group, and red triangles for the DSS+PepO group. The data are presented as mean ± SD, n = 3–6. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Abbreviation: SSC-A, side scatter-area. |
Macrophage Clearance Negates the Remission-Inducing Effect of PepO on Colitis
The continuous infiltration of pro-inflammatory macrophages at the injury site disrupts immune homeostasis and sustains chronic inflammation. To further validate that macrophages are the primary target cells involved in PepO-mediated colitis remission, mice were pretreated with either control or clodronate liposomes. At the systemic level, clodronate liposomes significantly induced macrophage depletion, as confirmed by flow cytometric analysis of peritoneal macrophages (Supplementary Figure S1B). Immunofluorescence analysis revealed a significant reduction in F4/80-positive macrophages within the colonic lamina propria following clodronate liposome treatment (Figure 4C). Following macrophage clearance, PepO administration failed to reduce DAI scores (Figure 4A), colon shortening remained unchanged (Figure 4B). Histopathological examination showed that clodronate liposome treatment completely abolished the improvement conferred by PepO: colonic tissues still exhibited obvious inflammatory cell infiltration, disrupted crypt architecture, and impaired mucosal structure, similar to the DSS group (Figure 4D). Correspondingly, PAS staining demonstrated that goblet cell density was not restored by PepO after macrophage depletion (Figure 4E). The protective effect of PepO against DSS-induced colitis was abolished by clodronate liposomes, confirming that PepO exerts its anti-colitis effects through a macrophage-dependent mechanism.
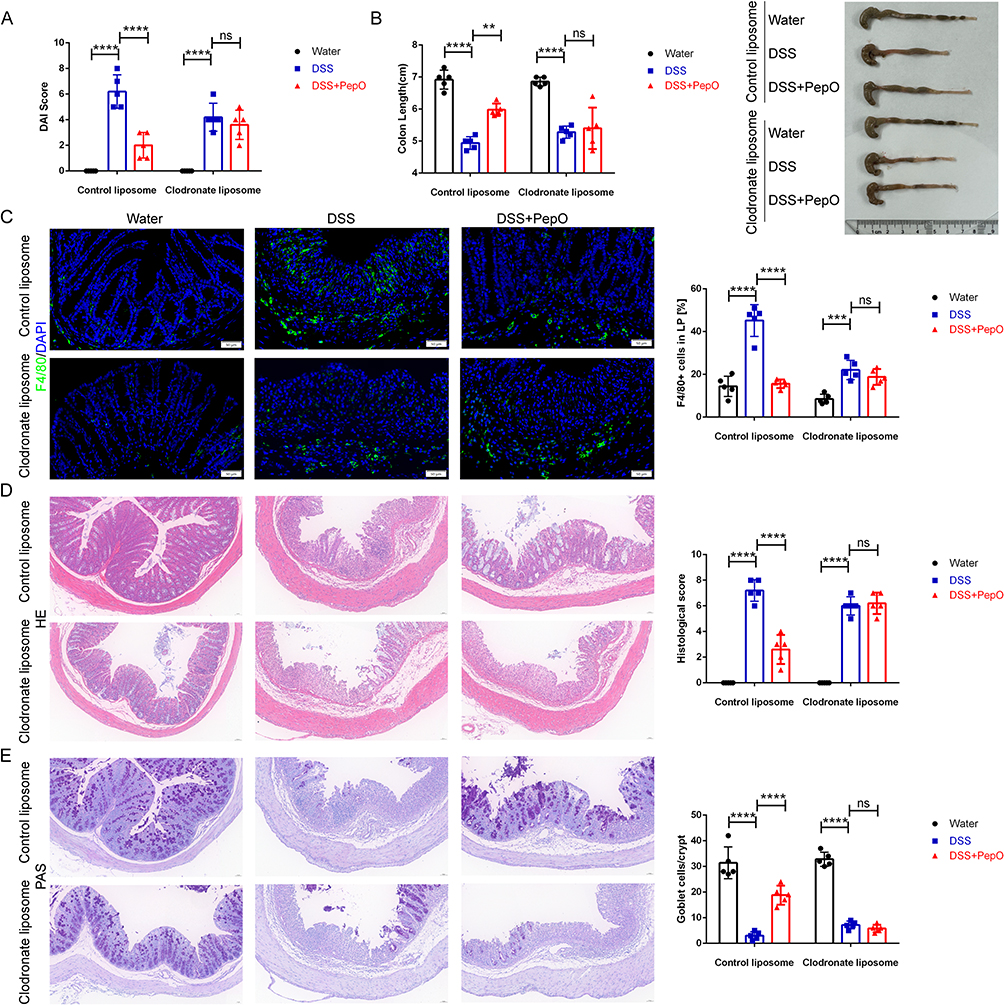 |
Figure 4 Macrophage clearance negated the remission-inducing effect of PepO on colitis. (A) DAI on day 7 post DSS drinking. (B) Representative images of colon tissues and colon length. (C) Immunofluorescence staining of the macrophage marker F4/80 and quantitation in colon tissues. Scale bar: 50 μm. (D) H&E staining of distal colon tissues and histopathological scores. Scale bar: 50 μm. (E) PAS staining of distal colon tissues and number of goblet cells. Scale bar: 50 μm. Individual data points for each mouse are represented by symbols: black circles for the Water group, blue squares for the DSS group, and red triangles for the DSS+PepO group. The data represent the mean ± SD, n = 5. **P < 0.01; ***P < 0.001; ****P < 0.0001. Abbreviation: ns, not significant. |
CCL2 Supplementation Abolishes the Inhibitory Effect of PepO on DSS-Induced Colitis
C‑C motif chemokine ligand 2 (CCL2), also referred to as monocyte chemotactic protein-1 (MCP-1), is a member of the CC chemokine family. Studies indicate that CCL2 exacerbates colitis progression by recruiting pro-inflammatory macrophages and serves as a key marker of intestinal inflammation.36,37 Therefore, we assessed its expression. PepO administration significantly reduced CCL2 levels in both colonic tissue and circulating plasma of mice with colitis (Figure 5A and B). Culture supernatants from BMDMs pretreated with PepO followed by LPS stimulation exhibited reduced CCL2 protein levels (Figure 5C). In the Transwell assay, conditioned medium from BMDMs pretreated with PepO and then stimulated with LPS recruited fewer BMDMs than conditioned medium from BMDMs treated with LPS alone. The addition of recombinant CCL2 (rCCL2) reversed this effect (Figure 5D).
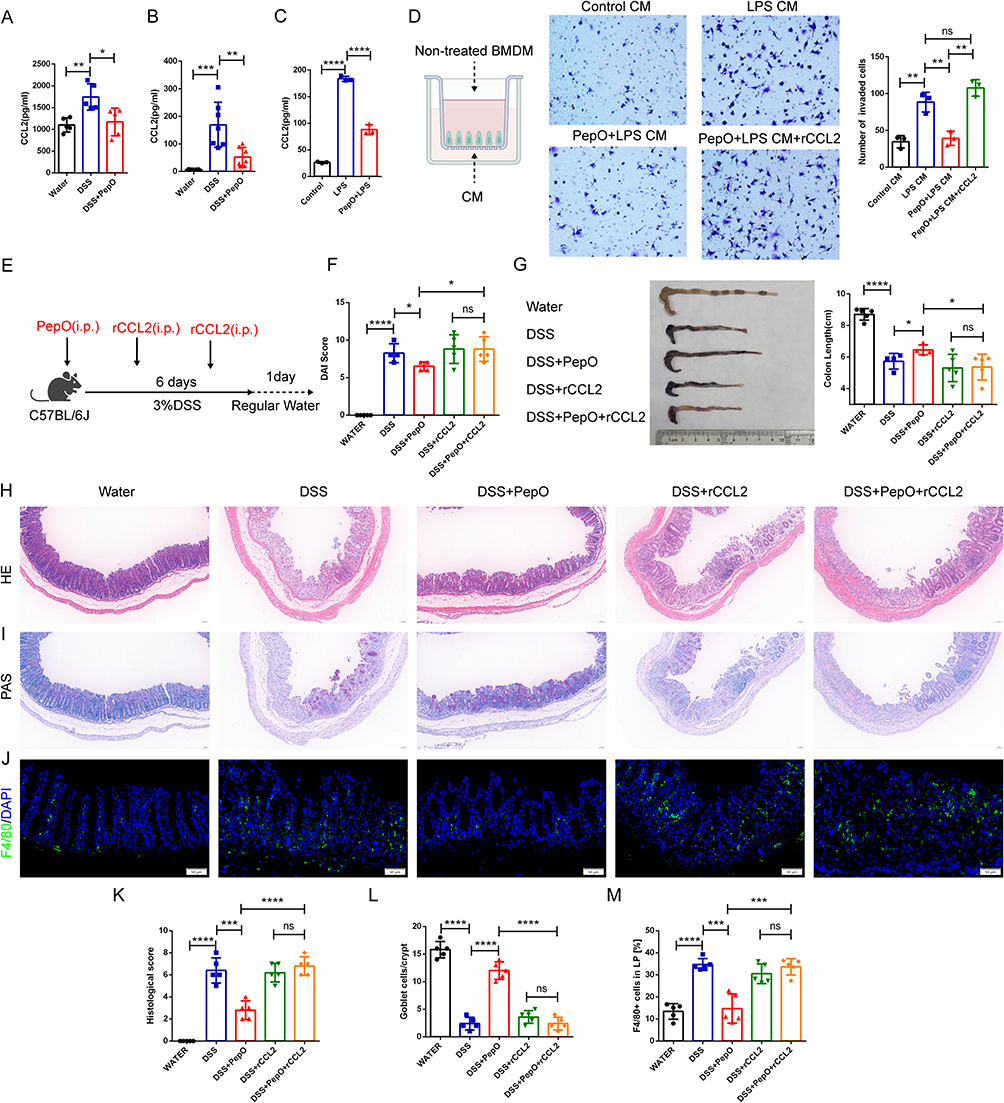 |
Figure 5 CCL2 supplementation abolished the inhibitory effect of PepO on DSS-induced colitis. (A) Protein levels of CCL2 in colon tissue measured by ELISA. (B) Plasma levels of CCL2 measured by ELISA. (C) Protein levels of CCL2 in the BMDMs supernatant measured by ELISA. (D) Representative images and counts of BMDMs in the transwell migration assay. (E) Schematic of the acute DSS-induced colitis mouse model with or without recombinant CCL2 supplementation. (F) DAI on day 7 post DSS drinking. (G) Representative images of colon tissues and colon length. (H) H&E staining of the distal colon tissues. Scale bar: 50 μm. (I) PAS staining of the distal colon tissues. Scale bar: 50 μm. (J) Immunofluorescence staining of the macrophage marker F4/80 in colon tissues. Scale bar: 50 μm. (K) Histopathological scores based on H&E staining. (L) Number of goblet cells based on PAS staining. (M) Quantitation of F4/80 in colon tissues. For animal experiments: In (A, B, F, G, K, L, M), individual mice are represented by black circles (Water), blue squares (DSS), and red triangles (DSS+PepO); additionally, in (F, G, K, L, M), green triangles (DSS+rCCL2) and yellow diamonds (DSS+PepO+rCCL2) are included. For cell-based assays: In (C), symbols represent Control (black circles), LPS (blue squares), and PepO+LPS (red triangles). In (D), symbols represent Control CM (black circles), LPS CM (blue squares), PepO+LPS CM (red triangles), and PepO+LPS CM+rCCL2 (green triangles). Data are presented as mean ± SD, n = 3–7. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Abbreviations: i.p., intraperitoneal injection; CM, conditioned medium; BMDM, bone marrow-derived macrophage; rCCL2, recombinant CCL2; ns, not significant. |
To further validate that the reduction in CCL2 played a role in PepO-mediated colitis suppression, we administered rCCL2 protein to DSS-induced colitis mice along with PepO (Figure 5E). The addition of rCCL2 negated the suppressive effect of PepO on the colitis phenotype, with no significant differences in DAI scores or colon shortening (Figure 5F and G). Histopathological analysis further confirmed this reversal effect. In H&E staining, both the DSS+rCCL2 and DSS+PepO+rCCL2 groups displayed extensive histological damage, including widespread loss of the mucosal epithelium, collapse of crypts, and dense inflammatory cell infiltration extending into the submucosal layer (Figure 5H and K). PAS staining revealed that rCCL2 supplementation completely abrogated the mucus barrier-preserving effect of PepO, with both treatment groups showing severe depletion of PAS-positive goblet cells (Figure 5I and L). Moreover, compared to mice without rCCL2 supplementation, supplementation with rCCL2 increased the number of F4/80-positive macrophages in the lamina propria of PepO-intervened colitis mice (Figure 5J and M). These findings indicate that PepO halts the progression of ulcerative colitis by reducing CCL2-mediated macrophage migration.
The Ameliorative Effect of PepO on Colitis is Dependent on the Gut Microbiota
Gut microbiota is essential for digestion, inflammation, immune regulation, and therapeutic responses. To explore whether PepO influences the gut microbiota, 16S rRNA sequencing was conducted on fecal samples from each group of mice. Alpha diversity analysis showed no significant differences in Chao1 index across groups. However, the Shannon index revealed a significant difference between the DSS and DSS+PepO groups, with the DSS+PepO group showing lower diversity (Figure 6A). Regarding beta diversity, PERMANOVA (Adonis) confirmed significant differences in microbial community structure among the three groups (all adjusted P < 0.01; Supplementary Table S6). Consistent with this, principal coordinate analysis (PCoA) plots indicated that PepO administration attenuated disease-induced gut microbiota dysbiosis. The DSS group was distinctly separated from the control group, while PepO administration re-established a healthier microbial composition (Figure 6B).
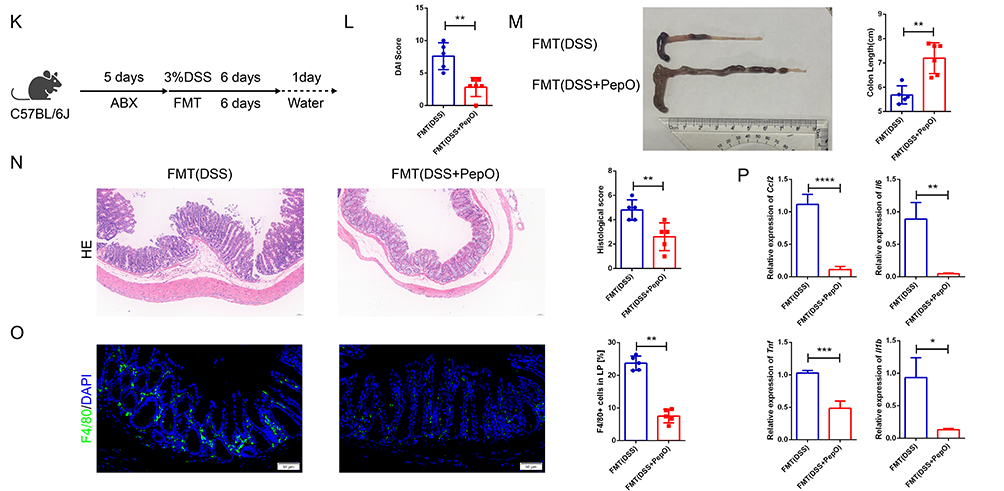
Figure 6 The ameliorative effect of PepO on colitis is dependent on the gut microbiota. (A) Community alpha diversity was evaluated using the richness (Chao) and diversity (Shannon) indices. (B) Principal coordinate analysis plot based on Bray-Curtis distance for beta diversity. (C) Relative abundance bar chart of the top 20 genera. (D) Key genera with significant intergroup differences in relative abundance. (E) Schematic of the antibiotic treatment experiment. (F) DAI scores. (G) Representative images of colon tissues and colon length. (H) H&E staining of the distal colon tissues and histopathological scores. Scale bar: 50 μm. (I) Immunofluorescence staining of the macrophage marker F4/80 and quantitation in colon tissues. Scale bar: 50 μm. (J) mRNA expression of Ccl2, Il6, Tnf, and Il1b in colon tissues measured by qRT-PCR. (K) Schematic diagram of FMT experimental treatments. (L) DAI scores. (M) Representative images of colon tissues and colon length. (N) H&E staining of the distal colon tissues and histopathological scores. Scale bar: 50 μm. (O) Immunofluorescence staining of the macrophage marker F4/80 and quantitation in colon tissues. Scale bar: 50 μm. (P) mRNA expression of Ccl2, Il6, Tnf, and Il1b in colon tissues measured by qRT-PCR. In (E–J), individual data points for each mouse are represented by symbols: black circles for the ABX+Water group, blue squares for the ABX+DSS group, and red triangles for the ABX+DSS+PepO group. In (K–P), individual data points for each mouse are represented by symbols: blue squares for the FMT(DSS) group and red triangles for the FMT (DSS+PepO) group. Data are presented as mean ± SD, n = 3–6. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Abbreviations: i.p., intraperitoneal injection; ABX, antibiotics; FMT, fecal microbiota transplantation; ns, not significant. Figure 6 continued.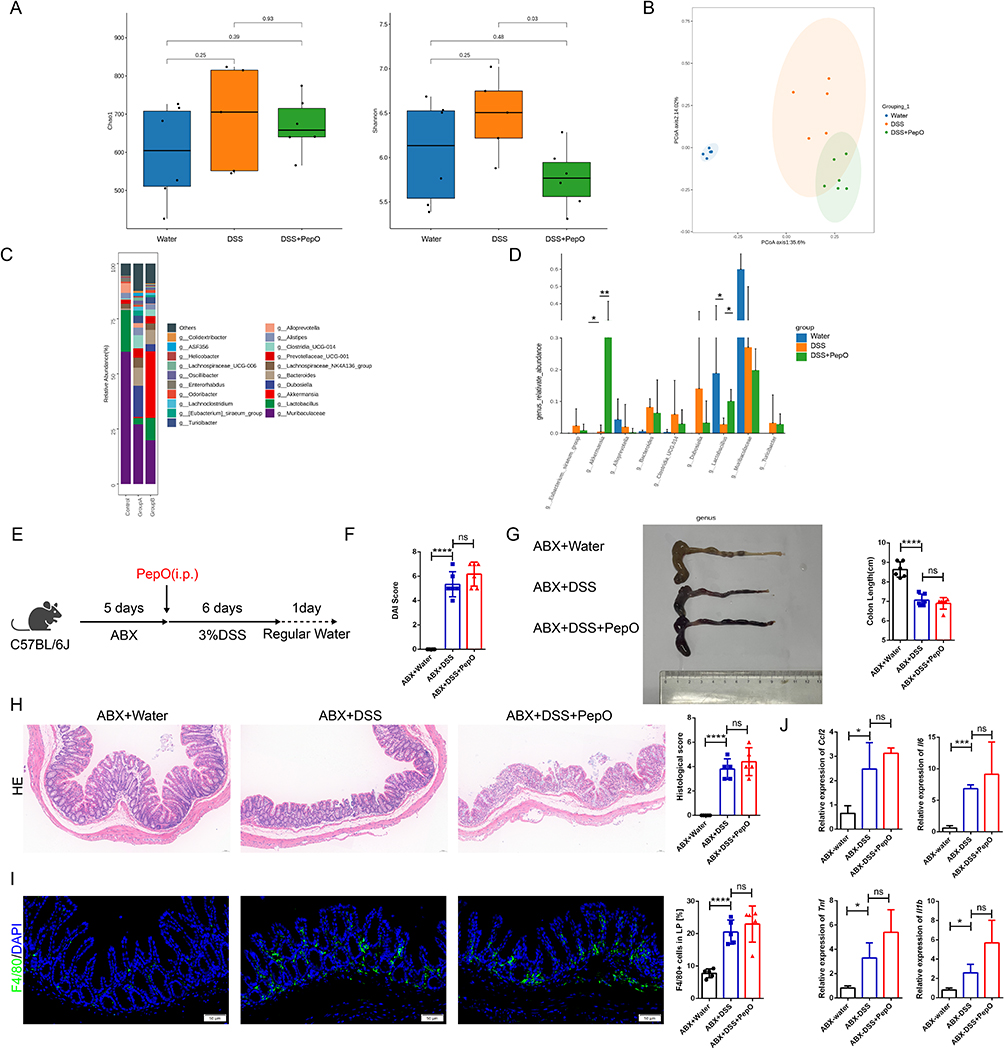
Differential abundance analysis using ALDEx2 and ANCOM revealed that PepO treatment significantly enriched a cluster of beneficial microbes. Notably, Akkermansia emerged as a key biomarker highly responsive to PepO administration, demonstrating a markedly higher biological effect compared to the DSS group (ALDEx2 effect size = 5.72; ANCOM W > 0.6; Supplementary Tables S7 and S8). Regarding Lactobacillus, PepO administration exerted a significant biological effect compared to the DSS group, with an effect size greater than 1 in the ALDEx2 analysis (Supplementary Table S7). Moreover, Lactobacillus was identified as a significantly altered feature in the omnibus test (three-group comparison) by both ALDEx2 (kw.eBH = 0.0397) and ANCOM (W > 0.6) (Supplementary Tables S9 and S10). Consistently, the relative abundance barplots confirmed that PepO administration markedly altered the gut microbiota of DSS-treated mice, and significantly increased the relative abundance of probiotics including Lactobacillus and Akkermansia (Figure 6C and D).
To assess whether the gut microbiota contributes to the protective effect of PepO against colon inflammation, mice were pretreated with broad-spectrum antibiotics (ABX) to deplete the microbiota prior to DSS administration (Figure 6E). This ABX regimen abolished the protective effect of PepO in DSS-induced ulcerative colitis. PepO administration failed to reduce DAI scores (Figure 6F), and colon shortening remained unchanged (Figure 6G). Histopathological observation showed no obvious differences in mucosal structure or inflammatory cell infiltration between the ABX+DSS and ABX+DSS+PepO groups, with no noticeable improvement conferred by PepO treatment (Figure 6H). Furthermore, no difference in infiltrating macrophages was observed between the ABX+DSS+PepO and ABX+DSS groups (Figure 6I), nor were there differences in the transcriptional levels of Ccl2, Il1b, Tnf, and Il6 in colonic tissue (Figure 6J).
To further examine whether the gut microbiota remodeled by PepO provides protective effects against colitis, mice were pretreated with ABX, followed by oral gavage of intestinal microbiota collected from DSS-induced colitis or PepO-treated mice with DSS-induced colitis. At the same time, acute colitis was induced in the recipient mice through DSS treatment (Figure 6K). The FMT (DSS+PepO) group, which received microbiota from the PepO-treated DSS group, showed significant improvements in colitis, as indicated by longer colon lengths and lower DAI scores (Figure 6L and M), compared to the FMT (DSS) group, which received microbiota from the DSS group. Histopathological analysis showed that the FMT (DSS+PepO) group exhibited a more organized colonic architecture compared to the FMT (DSS) group, with better-preserved crypt structures, a more continuous epithelial lining, and reduced inflammatory cell density in the colonic mucosa (Figure 6N). The FMT (DSS+PepO) group reduced Ccl2 expression, decreased F4/80-positive macrophage infiltration in the lamina propria, and significantly alleviated tissue inflammation (Figure 6O and P). These findings indicate that PepO confers protective effects against DSS-induced colitis by remodeling the composition of the gut microbiota.
Discussion
Ulcerative colitis is a multifactorial disease influenced by host, microbial, and environmental factors. We found that PepO directly inhibited macrophage production of the chemokine CCL2, thereby reducing their pathological infiltration into the colon. Crucially, we demonstrated that this protective effect was dependent on the ability of PepO to remodel the gut microbiota. Collectively, this study demonstrates that PepO mediates its protective effects in colitis through dual mechanisms: direct immune regulation and microbiota remodeling. These findings offer new insights for developing therapies targeting the microbiota‑immune axis.
The DSS-induced ulcerative colitis model is widely used for studying UC.38 In this model, PepO administration significantly alleviated clinical manifestations in colitis-affected mice. This protective effect was evidenced by attenuated colon shortening, reduced disease activity scores, diminished inflammatory infiltration, and restored goblet cell numbers.
The intestinal barrier is crucial for preventing the invasion of bacterial pathogens into intestinal tissues.39 Research has confirmed that enhanced intestinal barrier permeability is a key pathogenic mechanism in colitis, closely linked to the presence of mucins and the integrity of tight junction proteins.40 MUC2 is a critical protein that constitutes and protects the intestinal mucus layer, playing a crucial role in preserving the integrity of the intestinal barrier.41 ZO-1 and occludin are key barrier proteins, and alterations in their expression and distribution directly compromise the mechanical barrier function of the intestinal epithelium.42 Following PepO administration, the expression of MUC2 and TJ proteins ZO-1 and occludin was significantly upregulated. These findings suggest that PepO protects intestinal barrier integrity by increasing mucin expression and enhancing tight junction protein levels, thereby mitigating pathological damage to colonic tissue. Previous studies have shown markedly elevated intestinal epithelial cell apoptosis in patients with ulcerative colitis.43 Inhibiting intestinal epithelial cell apoptosis is essential for enhancing compromised intestinal barrier function in patients with ulcerative colitis.44 Following DSS treatment, we observed increased expression of cleaved caspase-3, an apoptotic effector enzyme, along with multiple clinical and histological characteristics resembling human UC. Moreover, Ki67 expression was significantly lower in the colonic crypts of the DSS-treated group. However, PepO effectively alleviated these pathological changes, suggesting its potential as a protective agent for ulcerative colitis.
The gut is one of the body’s largest immune organs, with macrophages representing the predominant population of immune cells.45 Macrophage infiltration has been demonstrated to play a vital role in the progression of ulcerative colitis.46 In comparison to the DSS model group, PepO administration notably decreased the infiltration of F4/80+ macrophages in the colonic lamina propria, concurrently downregulating levels of key pro-inflammatory factors—TNF-α, IL-6, and IL-1β—primarily produced by activated macrophages. These findings unequivocally indicate that PepO effectively suppresses macrophage-driven inflammation during colitis. Building upon this observation and our group’s prior discovery of PepO’s significant regulatory effects on macrophages,28,29,47 we assessed the necessity of macrophages in PepO-mediated colitis remission via macrophage depletion experiments. Results demonstrated that following macrophage depletion using clodronate liposomes, the protective effect of PepO against DSS-induced colitis was completely abolished, indicating that macrophages are an indispensable cellular target for PepO’s protective efficacy.
To further investigate the molecular mechanism by which PepO regulates macrophages, we focused on the key chemokine CCL2. CCL2 plays a central role in recruiting monocytes/macrophages to the inflamed intestine, thereby driving and amplifying the inflammatory cascade.36,37 Consistent with our hypothesis, we found that PepO administration significantly reduced CCL2 levels in both colonic tissue and the circulation of DSS-induced colitis mice in vivo. In vitro studies revealed that PepO directly inhibits CCL2 production in BMDMs and attenuates its chemotactic effect on macrophages, an effect reversible by exogenous CCL2. Crucial in vivo experiments confirmed that CCL2 supplementation completely abolished PepO’s protective effect against colitis. Collectively, these results demonstrate that PepO attenuates colitis by inhibiting CCL2-mediated macrophage recruitment, in line with previous studies showing that targeting CCL2 reduces pro-inflammatory macrophage infiltration and alleviates colitis.48–50 This conclusion appears to contradict a report by Maharshak et al, which suggested that exogenous CCL2 at extremely low concentrations (pM levels) can improve colitis by inhibiting T-cell migration.51 However, the two studies are not completely contradictory, as the function of CCL2 is highly dependent on the environment, cell type, and concentration, with effects that may be opposite in direction.52 Therefore, reducing excessively elevated endogenous CCL2 in pathological conditions and supplementing with extremely low doses of exogenous CCL2 represent two entirely different intervention strategies, together confirming the dual role of CCL2 in intestinal inflammation.
Increasing evidence suggests that gut microbiota dysbiosis plays a key role in the pathogenesis of ulcerative colitis. DSS-treated mice also display dysbiosis, characterized by reduced microbial diversity, a decline in probiotic species, and increased levels of pathogenic bacteria.53,54 This study further investigates the essential role of the gut microbiota in mediating the protective effects of PepO. 16S rRNA sequencing analysis revealed that PepO intervention significantly induced structural remodeling of the gut microbiota. The analysis of gut microbiota diversity revealed that the Shannon index was significantly lower in the DSS+PepO group compared to the DSS group. Although it is generally accepted that the resolution of colitis is associated with an increase in microbiota diversity, the Shannon index reflects both species richness and evenness. In this study, the decrease in the Shannon index may not indicate a worsening of dysbiosis but rather reflect the targeted expansion of specific beneficial microbial groups in the gut following PepO administration, resulting in a change in community evenness. At the genus level, the PepO-treated group showed significant enrichment of beneficial bacteria, particularly with substantial increases in the relative abundance of Lactobacillus and Akkermansia. Akkermansia has been extensively recognized as an essential commensal bacterium that enhances intestinal mucosal barrier integrity and promotes mucus secretion,55 while also being involved in immune regulation.56,57 Meanwhile, Lactobacillus produces metabolites such as short-chain fatty acids58 and antimicrobial peptides,59 regulates immune cell populations,60 modulates macrophage phenotypes,61 and is vital for maintaining epithelial homeostasis.62
To validate the causal relationship between microbiota alterations and protective effects, we conducted gut microbiota depletion experiments. Results showed that following broad-spectrum antibiotic-induced gut microbiota depletion, PepO’s protective effect against colitis was completely abolished, and its benefits in ameliorating clinical symptoms and histopathological damage were lost. This crucial evidence suggests that an intact gut microbial ecosystem is a prerequisite for PepO to exert its biological effects in vivo. Further fecal transplantation experiments confirmed that transferring microbiota from PepO-treated donor mice to DSS-induced colitis recipient mice successfully reproduced a phenotype similar to that observed with direct PepO administration. This included significant improvement in clinical symptoms and histopathology. In summary, these findings imply that the protective effect of PepO on experimental colitis is closely associated with its remodeling of the gut microbiota.
Notably, a direct link between gut microbiota remodeling and the downregulation of the key chemokine CCL2 was identified: reduced Ccl2 expression was observed in recipient mice transplanted with microbiota from the PepO-treated DSS group. These beneficial bacteria may exert their effects through two main pathways: first, by producing immunomodulatory metabolites such as short-chain fatty acids (eg, butyrate);63 second, by fortifying the intestinal barrier function to reduce the translocation of pathogen-associated molecular patterns (eg, LPS)64 After improving the microbiota, PepO creates an anti-inflammatory environment that further suppresses CCL2 expression, thereby reducing macrophage recruitment This process may create a positive feedback loop with changes in the microbiota, enhancing protective efficacy. This positive feedback loop amplifies PepO’s protective effect, leading to more sustained remission. Further investigation is needed to determine whether PepO regulates the microbiota directly through its enzymatic or antimicrobial activity, or indirectly via immune modulation (eg, macrophage function), or a combination of both Although research suggests that macrophages influence the microbiota by secreting cytokines and antimicrobial peptides,65–68 further studies are needed to clarify how PepO reduces macrophage recruitment and alters their phenotype, subsequently affecting the microbiota and promoting a beneficial microbial composition. Understanding this bidirectional mechanism will improve our knowledge of PepO’s synergistic effects and open new directions for future research.
Of note, helminths and their derivatives also exert protective effects against colitis. Helminths and their derivatives have been shown to induce a Th2-type immune response, promote M2 macrophage polarization, restore gut microbiota diversity, and reduce colonic pro-inflammatory chemokines, thereby alleviating colitis.69–71 These effects are similar to those of PepO, which also involves immune regulation and gut microbiota modulation. However, helminths and their derivatives pose risks such as pathogenicity, ethical concerns, treatment dependency, and nonspecific immunosuppression.72–74 In contrast, PepO targets CCL2 and macrophages, offering protective efficacy with a reduced risk of immunosuppression. Additionally, PepO’s ability to reshape the gut microbiota provides broad potential for supporting gut health, with better safety and clinical controllability since it does not rely on live microorganisms.
Nevertheless, several limitations of the present study should be acknowledged. First, as PepO was administered on the same day as colitis induction, our data primarily support a protective role rather than a therapeutic effect for established disease. Future studies involving delayed PepO administration are needed to further explore its potential in managing established colitis. In addition, although the fecal inocula were standardized by weight and volume to ensure consistency, the lack of specific CFU quantification remains a technical limitation of this assay. Addressing these limitations in future research will provide a more comprehensive understanding of the clinical potential of PepO.
In summary, this study demonstrates that PepO offers protective effects in colitis. We reveal a dual protective pathway: first, it directly acts on macrophages to inhibit CCL2 production, thereby blocking macrophage recruitment to the gut and the inflammatory cascade; second, its overall efficacy depends on reshaping and regulating the gut microbiota. Notably, PepO offers potential benefits in alleviating ulcerative colitis. Although current therapies control inflammation, they increase the risk of infection and colitis-associated cancer.75,76 In contrast, its antimicrobial and anti-tumor properties may reduce inflammation and mitigate these risks.28–30,47 The dual effects of PepO open new possibilities for immunomodulatory treatments and provide fresh targets for using microbial proteins in inflammatory bowel disease.
Conclusion
This study demonstrates that PepO alleviates DSS-induced colitis by inhibiting CCL2 production and macrophage infiltration. We have established that this protective effect is dependent on the remodeling of the gut microbiota. Our findings reveal a dual-pathway mechanism: PepO directly suppresses macrophage CCL2 expression while simultaneously modulating the gut microbiota to foster a favorable immune environment. These results highlight PepO as a promising protective agent and support its dual-pathway mechanism for the alleviation of IBD.
Data Sharing Statement
The 16S rRNA gene sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1405891. The authors declare that any other supporting data or material associated with this original research is available from the corresponding author, Xuemei Zhang, upon reasonable request.
Author Contributions
Hanyi Wang.: Investigation, Methodology, Data curation, Visualization, Writing-original draft, review and editing. Shuangmei Shao.: Investigation, Methodology, Validation, Formal analysis, Writing-review and editing. Yuan Peng.: Investigation, Methodology, Validation, Writing-review and editing. Wenlong Xu.: Investigation, Methodology, Validation, Funding acquisition, Writing—review and editing. Jia Zhang.: Investigation, Methodology, Validation, Writing-review and editing. Xiao Liu.: Investigation, Methodology, Validation, Writing-review and editing. Xintong Bian.: Supervision, Data curation, Formal analysis, Writing-review and editing. Kun Huang.: Investigation, Methodology, Data curation, Validation, Writing-review and editing. Xuemei Zhang.: Supervision, Investigation, Formal analysis, Validation, Funding acquisition, Writing-review and editing.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Youth Innovation in Future Medicine [grant numbers W0148, Xuemei Zhang], the National Natural Science Foundation of China [grant numbers 81571622, Xuemei Zhang] and the Scientific Research Project of the Joint Medical Department of Kewei [grant numbers wzwjw-kw2024014, Wenlong Xu].
Disclosure
Professor Xuemei Zhang, Miss Shuangmei Shao, and Dr. Hanyi Wang report a patent for Streptococcus pneumoniae endopeptidase O (PepO) for the treatment of ulcerative colitis (Pub. No. CN120695168A) pending to the China National Intellectual Property Administration.
References
1. Gilliland A, Chan JJ, De Wolfe TJ, Yang H, Vallance BA. Pathobionts in inflammatory bowel disease: origins, underlying mechanisms, and implications for clinical care. Gastroenterology. 2024;166(1):44–20. doi:10.1053/j.gastro.2023.09.019
2. Wan J, Zhou J, Wang Z, et al. Epidemiology, pathogenesis, diagnosis, and treatment of inflammatory bowel disease: insights from the past two years. Chinese Med J. 2025;138(7):763–776. doi:10.1097/CM9.0000000000003542
3. Yue N, Hu P, Tian C, et al. Dissecting innate and adaptive immunity in inflammatory bowel disease: immune compartmentalization, microbiota crosstalk, and emerging therapies. J Inflamm Res. 2024;17:9987–10014. doi:10.2147/JIR.S492079
4. Mak WY, Zhao M, Ng SC, Burisch J. The epidemiology of inflammatory bowel disease: east meets west. J Gastroenterol Hepatol. 2020;35(3):380–389. doi:10.1111/jgh.14872
5. Targownik L, Afif W, Singh S, et al. Treatment patterns for advanced therapies in Canadians with moderate-to-severe inflammatory bowel disease: a retrospective cohort analysis. J. Can. Assoc. Gastroentero. 2025;8(1):21–30. doi:10.1093/jcag/gwae040
6. Zhang B, Gulati A, Alipour O, Shao L. Relapse from deep remission after therapeutic de-escalation in inflammatory bowel disease: a systematic review and meta-analysis. J Crohn’s Colitis. 2020;14(10):1413–1423. doi:10.1093/ecco-jcc/jjaa087
7. de Souza HS, Fiocchi C, de Souza HSP. Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016;13(1):13–27. doi:10.1038/nrgastro.2015.186
8. Hegarty LM, Jones GR, Bain CC. Macrophages in intestinal homeostasis and inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2023;20(8):538–553. doi:10.1038/s41575-023-00769-0
9. Luo M, Zhao F, Cheng H, Su M, Wang Y. Macrophage polarization: an important role in inflammatory diseases. Front Immunol. 2024;15:1352946. doi:10.3389/fimmu.2024.1352946
10. Pan X, Zhu Q, Pan -L-L, Sun J. Macrophage immunometabolism in inflammatory bowel diseases: from pathogenesis to therapy. Pharmacol Ther. 2022;238:108176. doi:10.1016/j.pharmthera.2022.108176
11. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017;79:593–617. doi:10.1146/annurev-physiol-022516-034356
12. Nishino K, Nishida A, Inoue R, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol. 2018;53(1):95–106. doi:10.1007/s00535-017-1384-4
13. Schaubeck M, Clavel T, Calasan J, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65(2):225–237. doi:10.1136/gutjnl-2015-309333
14. Qian G, Chen X, Liu G, et al. Exploring the etiology of colitis: insights from gut microbiota research. Gut Microbes. 2025;17(1):2512010. doi:10.1080/19490976.2025.2512010
15. Ai L, Ren Y, Zhu M, et al. Synbindin restrains proinflammatory macrophage activation against microbiota and mucosal inflammation during colitis. Gut. 2021;70(12):2261–2272. doi:10.1136/gutjnl-2020-321094
16. Martyniak A, Medyńska-Przęczek A, Wędrychowicz A, Skoczeń S, Prebiotics TPJ, Probiotics S. Paraprobiotics and Postbiotic Compounds in IBD. Biomolecules. 2021;11(12):1903. doi:10.3390/biom11121903
17. Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. 2020;145(1):16–27. doi:10.1016/j.jaci.2019.11.003
18. Li C, Peng K, Xiao S, Long Y, Yu Q. The role of Lactobacillus in inflammatory bowel disease: from actualities to prospects. Cell Death Discovery. 2023;9(1):361. doi:10.1038/s41420-023-01666-w
19. Rodrigues VF, Elias-Oliveira J, Í S P, et al. Akkermansia muciniphila and gut immune system: a good friendship that attenuates inflammatory bowel disease, obesity, and diabetes. Front Immunol. 2022;13:934695. doi:10.3389/fimmu.2022.934695
20. Chande N, Townsend CM, Parker CE, MacDonald JK. Azathioprine or 6-mercaptopurine for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2016;10(10):Cd000545. doi:10.1002/14651858.CD000545.pub5
21. Neurath MF. Strategies for targeting cytokines in inflammatory bowel disease. Nat Rev Immunol. 2024;24(8):559–576. doi:10.1038/s41577-024-01008-6
22. Sandborn WJ, Su C, Panes J. Tofacitinib as induction and maintenance therapy for ulcerative colitis. New Engl J Med. 2017;377(5):496–497.
23. Sandborn WJ, Feagan BG, D’Haens G, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. New Engl J Med. 2021;385(14):1280–1291. doi:10.1056/NEJMoa2033617
24. Zhang Z, Fan W, Yang G, et al. Risk of tuberculosis in patients treated with TNF-α antagonists: a systematic review and meta-analysis of randomised controlled trials. BMJ open. 2017;7(3):e012567. doi:10.1136/bmjopen-2016-012567
25. Szekanecz Z, Buch MH, Charles-Schoeman C, et al. Efficacy and safety of JAK inhibitors in rheumatoid arthritis: update for the practising clinician. Nat Rev Rheumatol. 2024;20(2):101–115. doi:10.1038/s41584-023-01062-9
26. Becker HEF, Demers K, Derijks LJJ, Jonkers D, Penders J. Current evidence and clinical relevance of drug-microbiota interactions in inflammatory bowel disease. Front Microbiol. 2023;14:1107976. doi:10.3389/fmicb.2023.1107976
27. Liu L, Liang L, Liang H, et al. Microbiome-metabolome generated bile acids gatekeep infliximab efficacy in Crohn’s disease by licensing M1 suppression and Treg dominance. J Adv Res. 2025.
28. Yao H, Zhang H, Lan K, et al. Purified Streptococcus pneumoniae Endopeptidase O (PepO) Enhances Particle Uptake by Macrophages in a Toll-Like Receptor 2- and miR-155-Dependent Manner. Infect Immun. 2017;85(4). doi:10.1128/IAI.01012-16.
29. Shu Z, Yuan J, Wang H, et al. Streptococcus pneumoniae PepO promotes host anti-infection defense via autophagy in a Toll-like receptor 2/4 dependent manner. Virulence. 2020;11(1):270–282. doi:10.1080/21505594.2020.1739411
30. Xu W, Yuan Y, Shu Z, et al. Streptococcus pneumoniae endopeptidase O induces trained immunity and confers protection against various pathogenic infections. Clin Immunol. 2024;263:110226. doi:10.1016/j.clim.2024.110226
31. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105(39):15064–15069. doi:10.1073/pnas.0803124105
32. Yu M, Wang Q, Ma Y, et al. Aryl hydrocarbon receptor activation modulates intestinal epithelial barrier function by maintaining tight junction integrity. Int J Bio Sci. 2018;14(1):69–77. doi:10.7150/ijbs.22259
33. Shil A, Olusanya O, Ghufoor Z, Forson B, Marks J, Chichger H. Artificial sweeteners disrupt tight junctions and barrier function in the intestinal epithelium through activation of the sweet taste receptor, T1R3. Nutrients. 2020;12(6):1862. doi:10.3390/nu12061862
34. Jones BA, Gores GJ. Physiology and pathophysiology of apoptosis in epithelial cells of the liver, pancreas, and intestine. A J Physiol. 1997;273(6):G1174–1188.
35. Watson AJ, Pritchard DM. Lessons from genetically engineered animal models. VII. Apoptosis in intestinal epithelium: lessons from transgenic and knockout mice. Am J Physiol Gastrointest Liver Physiol. 2000;278(1):G1–5. doi:10.1152/ajpgi.2000.278.1.G1
36. Yang Y, Li L, Xu C, et al. Cross-talk between the gut microbiota and monocyte-like macrophages mediates an inflammatory response to promote colitis-associated tumourigenesis. Gut. 2020;70(8):1495–1506. doi:10.1136/gutjnl-2020-320777
37. Begum J, Fatima A, Rimmer P, et al. P0130 Assessment of macrophage types in the intestinal tissue of treatment naïve Inflammatory Bowel Disease patients in relation to their disease and activation of the CCL2/CCR2 pathway. J Crohn’s Colitis. 2025;19(Supplement_1):i517–i517. doi:10.1093/ecco-jcc/jjae190.0304
38. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014;104:15.25.11–15.25.14. doi:10.1002/0471142735.im1525s104
39. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target?. Nat. Rev. Gastroenterol. Hepatol. 2017;14(1):9–21. doi:10.1038/nrgastro.2016.169
40. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124(1):3–20. doi:10.1016/j.jaci.2009.05.038
41. Gustafsson JK, Johansson MEV. The role of goblet cells and mucus in intestinal homeostasis. Nat Rev Gastroenterol Hepatol. 2022;19(12):785–803. doi:10.1038/s41575-022-00675-x
42. Kuo WT, Odenwald MA, Turner JR, Zuo L. Tight junction proteins occludin and ZO-1 as regulators of epithelial proliferation and survival. Ann. N.Y. Acad. Sci. 2022;1514(1):21–33. doi:10.1111/nyas.14798
43. Garcia-Carbonell R, Yao SJ, Das S, Guma M. Dysregulation of intestinal epithelial cell RIPK pathways promotes chronic inflammation in the IBD gut. Front Immunol. 2019;10:1094. doi:10.3389/fimmu.2019.01094
44. Subramanian S, Geng H, Tan XD. Cell death of intestinal epithelial cells in intestinal diseases. Sheng li xue bao. Acta physiologica Sinica. 2020;72(3):308–324.
45. Delfini M, Stakenborg N, Viola MF, Boeckxstaens G. Macrophages in the gut: masters in multitasking. Immunity. 2022;55(9):1530–1548. doi:10.1016/j.immuni.2022.08.005
46. Zhang M, Li X, Zhang Q, Yang J, Liu G. Roles of macrophages on ulcerative colitis and colitis-associated colorectal cancer. Front Immunol. 2023;14:1103617. doi:10.3389/fimmu.2023.1103617
47. Liu B, Huang J, Xiao J, et al. The Streptococcus virulence protein PepO triggers anti-tumor immune responses by reprograming tumor-associated macrophages in a mouse triple negative breast cancer model. Cell Biosci. 2023;13(1):198. doi:10.1186/s13578-023-01153-w
48. Mackos AR, Galley JD, Eubank TD, et al. Social stress-enhanced severity of Citrobacter rodentium-induced colitis is CCL2-dependent and attenuated by probiotic Lactobacillus reuteri. Mucosal Immunology. 2016;9(2):515–526. doi:10.1038/mi.2015.81
49. Yang X, Yabe-Wada T, Han J, et al. PCBP1 acts as a regulator of CCL2 expression in macrophages to induce recruitment of monocyte-derived macrophages into the inflamed colon. Int. Immunol. 2023;35(6):287–299. doi:10.1093/intimm/dxad003
50. Mei C, Meng F, Wang X, et al. CD30L is involved in the regulation of the inflammatory response through inducing homing and differentiation of monocytes via CCL2/CCR2 axis and NF-κB pathway in mice with colitis. Int Immunopharmacol. 2022;110:108934. doi:10.1016/j.intimp.2022.108934
51. Maharshak N, Hart G, Ron E, et al. CCL2 (pM levels) as a therapeutic agent in inflammatory bowel disease models in mice. Inflamm. Bowel Dis. 2010;16(9):1496–1504. doi:10.1002/ibd.21254
52. Gschwandtner M, Derler R, Midwood KS. More Than Just Attractive: how CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol. 2019;10:2759. doi:10.3389/fimmu.2019.02759
53. Caruso R, Lo BC, Núñez G. Host-microbiota interactions in inflammatory bowel disease. Nat Rev Immunol. 2020;20(7):411–426. doi:10.1038/s41577-019-0268-7
54. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation?. Nat. Rev. Gastroenterol. Hepatol. 2017;14(10):573–584. doi:10.1038/nrgastro.2017.88
55. Wade H, Pan K, Duan Q, et al. Akkermansia muciniphila and its membrane protein ameliorates intestinal inflammatory stress and promotes epithelial wound healing via CREBH and miR-143/145. J Biomed Sci. 2023;30(1):38. doi:10.1186/s12929-023-00935-1
56. Chen T, Wang R, Duan Z, et al. Akkermansia muciniphila protects against psychological disorder-induced gut microbiota-mediated colonic mucosal barrier damage and aggravation of colitis. Front Cell Infect Microbiol. 2021;11:723856. doi:10.3389/fcimb.2021.723856
57. Zhao Q, Yu J, Hao Y, et al. Akkermansia muciniphila plays critical roles in host health. Crit. Rev. Microbiol. 2023;49(1):82–100. doi:10.1080/1040841X.2022.2037506
58. Gu Q, Xia C, Liu N, Chen Z, Zhou Q, Li P. Lactobacillus plantarum ZJ316 alleviates ulcerative colitis by inhibiting inflammation and regulating short-chain fatty acid levels and the gut microbiota in a mouse model. Food Funct. 2023;14(9):3982–3993. doi:10.1039/D2FO02567A
59. Mokoena MP. Lactic acid bacteria and their bacteriocins: classification, biosynthesis and applications against uropathogens: a mini-review. Molecules. 2017;22(8):1255. doi:10.3390/molecules22081255
60. Liu HY, Gu F, Zhu C, et al. Epithelial heat shock proteins mediate the protective effects of limosilactobacillus reuteri in dextran sulfate sodium-induced colitis. Front Immunol. 2022;13:865982. doi:10.3389/fimmu.2022.865982
61. Dias AMM, Douhard R, Hermetet F, et al. Lactobacillus stress protein GroEL prevents colonic inflammation. J Gastroenterol. 2021;56(5):442–455. doi:10.1007/s00535-021-01774-3
62. Shi J, Xie Q, Yue Y, et al. Gut microbiota modulation and anti-inflammatory properties of mixed lactobacilli in dextran sodium sulfate-induced colitis in mice. Food Funct. 2021;12(11):5130–5143. doi:10.1039/D1FO00317H
63. Serrano-Fernández V, Carmona-Torres JM, López-Fernández-Roldán Á, et al. Short-chain fatty acids in the treatment of ulcerative colitis. Systematic review and meta-analysis. Inflamm. Res. 2025;74(1):142. doi:10.1007/s00011-025-02112-6
64. Jiang Y, Jiang M, Zhu J, et al. Christensenella tenuis alleviates endotoxemia and metabolic disorders via inhibition of intestinal lipopolysaccharide translocation. Sci China Life Sci. 2025;68(12):3711–3727. doi:10.1007/s11427-025-3014-6
65. Li CX, Wang YM, Zhang WJ, et al. IL-10-dependent effect of chinese medicine abelmoschus manihot on alleviating intestinal inflammation and modulating gut microbiota. Am J Chin Med. 2023;51(6):1527–1546. doi:10.1142/S0192415X23500696
66. Yang Y, Gharaibeh RZ, Newsome RC, Jobin C. Amending microbiota by targeting intestinal inflammation with TNF blockade attenuates development of colorectal cancer. Nat Cancer. 2020;1(7):723–734. doi:10.1038/s43018-020-0078-7
67. Zhou X, Li W, Wang S, et al. YAP aggravates inflammatory bowel disease by regulating m1/m2 macrophage polarization and gut microbial homeostasis. Cell Rep. 2019;27(4):1176–1189.e1175. doi:10.1016/j.celrep.2019.03.028
68. Zou Y, Pu L, Guo A, et al. Helminth reshapes host gut microbiota and immunoregulation by deploying an antimicrobial program of innate immunity. Gut Microbes. 2025;17(1):2496447. doi:10.1080/19490976.2025.2496447
69. Jamtsho T, Loukas A, Wangchuk P. Pharmaceutical potential of remedial plants and helminths for treating inflammatory bowel disease. Pharmaceuticals. 2024;17(7):819. doi:10.3390/ph17070819
70. Prosberg MV, Halkjær SI, Lo B, et al. Probiotic treatment of ulcerative colitis with Trichuris suis ova: a randomised, double-blinded, placebo-controlled clinical trial [the PROCTO trial]. J Crohn’s Colitis. 2024;18(11):1879–1893. doi:10.1093/ecco-jcc/jjae095
71. Long SR, Shang WX, Zhang HR, et al. Trichinella-derived protein ameliorates colitis by altering the gut microbiome and improving intestinal barrier function. Int Immunopharmacol. 2024;127:111320. doi:10.1016/j.intimp.2023.111320
72. Helmby H. Human helminth therapy to treat inflammatory disorders - where do we stand? BMC immunol. 2015;16:12. doi:10.1186/s12865-015-0074-3
73. Pastille E, Frede A, McSorley HJ, et al. Intestinal helminth infection drives carcinogenesis in colitis-associated colon cancer. PLoS Pathogens. 2017;13(9):e1006649. doi:10.1371/journal.ppat.1006649
74. Nembot Fogang BA, Debrah LB, Owusu M, et al. Helminth coinfections modulate disease dynamics and vaccination success in the era of emerging infectious diseases. Vaccines. 2025;13(5):436. doi:10.3390/vaccines13050436
75. Piovani D, Danese S, Peyrin-Biroulet L, Nikolopoulos GK, Bonovas S. Systematic review with meta-analysis: biologics and risk of infection or cancer in elderly patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2020;51(9):820–830. doi:10.1111/apt.15692
76. Zhang L, Zhang X, Su T, Xiao T, Xu H, Zhao S. Colorectal cancer risk in ulcerative colitis: an updated population-based systematic review and meta-analysis. EClinicalMedicine. 2025;84:103269. doi:10.1016/j.eclinm.2025.103269
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Single-Cell RNA-Sequencing Analysis of Colonic Lamina Propria Immune Cells Reveals the Key Immune Cell-Related Genes of Ulcerative Colitis
Hua R, Qiao G, Chen G, Sun Z, Jia H, Li P, Zhang B, Qi F
Journal of Inflammation Research 2023, 16:5171-5188
Published Date: 11 November 2023
Portulaca Oleracea L. as a Potential Therapeutic Drug Intervention in Ulcerative Colitis: Mechanisms of Action and Clinical Studies
Liu XY, Liu ZX, Tan WW, Zhang WB, Zhang YL, Zheng L, Que RY, Wen HZ, Dai YC
Drug Design, Development and Therapy 2024, 18:5931-5946
Published Date: 11 December 2024
Coix Seed Oil Alleviates DSS-Induced Ulcerative Colitis via Intestinal Barrier Repair and Ferroptosis Regulation
Zeng YX, Li NR, Deng BY, Gu YF, Lu SF, Liu Y
Journal of Inflammation Research 2025, 18:2557-2581
Published Date: 20 February 2025
Si-Ni Decoction as a Potential Treatment for Ulcerative Colitis: Modulation of Gut Microbiota and AKT1 Inhibition Through Network Pharmacology and in vivo Validation
Shi L, Chen L, Jin G, Yang Y, Zhu F, Zhou G
Journal of Inflammation Research 2025, 18:6263-6280
Published Date: 14 May 2025
Targeting the Gut Microbiota with Herbal Compounds from Traditional Chinese Medicine: A Mechanistic Synthesis of a Novel Therapeutic Approach for Ulcerative Colitis
Chen J, Chen J, Wang Z
Journal of Inflammation Research 2026, 19:590456
Published Date: 29 June 2026