Back to Journals » Drug Design, Development and Therapy » Volume 20
Phase I, Randomized, Double-Blind, Placebo-Controlled Trial Investigating the Safety, Tolerability, and Pharmacokinetics of BS1801 in Healthy Chinese Adult Subjects
Authors Chen G, Sheng G, Wu K, Jin T, Jiang Q, Guo T, Chen Y ![]() , Yin H, Zhang G, Shen Z, Zeng H
, Yin H, Zhang G, Shen Z, Zeng H
Received 19 March 2026
Accepted for publication 13 May 2026
Published 26 May 2026 Volume 2026:20 610536
DOI https://doi.org/10.2147/DDDT.S610536
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Guiling Chen,1,* Guoping Sheng,1,* Kaiqi Wu,1 Tinghan Jin,1 Qi Jiang,1 Tong Guo,1 Yifan Chen,2 Hanwei Yin,3 Guozhou Zhang,3 Zhenwei Shen,1 Huihui Zeng3
1Shulan (Hangzhou) Hospital, Shulan International Medical College, Zhejiang Shuren University, Hangzhou, 310022, People’s Republic of China; 2National Human Genetic Resources Center, National Research Institute for Family Planning, Beijing, 100081, People’s Republic of China; 3Shanghai Yuanxi Medicine Corp, Shanghai, 201203, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huihui Zeng, Email [email protected] Zhenwei Shen, Email [email protected]
Background: Fibrosis is an excessive self-repair response affecting organs like the liver and lungs. Current treatments targeting local fibroblasts have significant adverse events (AEs). BS1801 is an organoselenium compound targeting thioredoxin reductase with anti-fibrotic activities. This Phase I study evaluated its safety, tolerability, and pharmacokinetics (PK) in healthy Chinese adults.
Methods: This single-center, randomized, double-blind, placebo-controlled study enrolled 57 participants into single-dose (n=27) and multiple-dose (n=30) trials (placebo-to-drug ratios 1:3 and 1:4). Single doses of 450, 900, 1200, and 1800 mg were tested; multiple doses of 300, 450, and 600 mg twice daily (12± 0.5h apart) were investigated.
Results: All participants completed the study. Thirty-six treatment-emergent AEs were reported, including 14 in 9 participants (20%, 95% CI: 8.3– 31.7%) in single-dose, and 22 in 11 participants (24.4%, 95% CI: 11.9– 37.0%) in multiple-dose trials. All AEs were Grade I and resolved naturally; one constipation case required medication. Using BS1801-M2 as a surrogate biomarker, PK parameters increased proportionally with single doses (450– 1800 mg): Tmax 4.0– 5.0 h, Cmax 1047.3– 1730.0 ng/mL, t1/2 10.7– 18.9 h, AUC0-∞ 17.7– 42.3 μg·h/mL. In multiple-dose (steady state, 300– 600 mg twice daily): Tmax 2.2– 3.7 h, Cmax 1996.0– 2880.0 ng/mL, t1/2 9.6– 17.3 h, AUC0-∞ 46.4– 78.3 μg·h/mL, AUCss 19.2– 28.4 μg·h/mL. Serum concentration plateaued after two doses with 45%-60% peak-to-trough fluctuation. BS1801-M2 was excreted within 96 h post-last dose.
Conclusion: BS1801 is safe and well-tolerated at tested doses. Based on safety and PK profiles, the recommended dosage is 300– 450 mg twice daily. Further clinical studies are needed for evaluating the safety and efficacy of BS1801 in the target population.
Keywords: phase I trial, safety, pharmacokinetics, organoselenium, thioredoxin reductase
Introduction
Fibrosis is a common pathological outcome of dysregulated wound healing in various organs, characterized by excessive deposition of disorganized extracellular matrix (ECM), ultimately leading to organ dysfunction and failure.1 It becomes more prevalent with aging and is estimated to be responsible for up to 45% of all deaths in developed countries.1,2 The mechanisms underlying fibrosis are not yet fully understood. The pathogenesis involves complex interactions among immune cells, fibroblasts, and epithelial cells within a sustained inflammatory milieu. Due to frequently insidious onset and progression, the prevalence of fibrotic diseases is substantial but variably reported. For example, interstitial lung diseases (ILDs), often precursors to pulmonary fibrosis, have an estimated prevalence ranging from 6.3 to 76.0 per 100,000 in Europe and North America.3 Liver fibrosis prevalence varies from 0.5% to 25.7% depending on the diagnostic methodology.4 Progressive fibrotic diseases such as idiopathic pulmonary fibrosis (IPF) carry a poor prognosis, with a median survival of only 3–5 years following diagnosis.2,5–7 Current anti-fibrotic therapies, pirfenidone and nintedanib, which primarily target fibroblast proliferation and activation, can slow disease progression but are limited by frequent and sometimes significant adverse effects, including gastrointestinal disturbances, hepatotoxicity, and photosensitivity.8–11 Among IPF patients treated with pirfenidone, 70.6% required at least one dose reduction within a median of 11 months (IQR 9–12 months). Between 12 and 48 months after treatment initiation, 6.8%–12.5% of patients permanently discontinued the drug every six months, and the two-year persistence rate dropped from 67% to 47%.12–14 This highlights a significant unmet need for novel anti-fibrotic agents with improved tolerability.
BS1801 (butaselen) is a novel organoselenium compound designed as a thioredoxin reductase (TrxR) inhibitor.15–17 The TrxR/thioredoxin (Trx) system is a central regulator of cellular redox homeostasis and is implicated in key inflammatory and fibrotic signaling pathways.18,19 Preclinical studies have demonstrated that BS1801 attenuates liver and pulmonary fibrosis in animal models by modulating the NF-κB and TGF-β1/Smad pathways, underscoring its therapeutic potential. The organoselenium pharmacophore, selenazolone, as found in ebselen and its analogs including BS1801, is known to interact with the TrxR system. An intriguing paradox observed with BS1801 in preclinical studies is its sustained anti-fibrotic efficacy despite a relatively short systemic exposure. This disconnection between its pharmacokinetic profile and pharmacodynamic duration underscores the importance of first defining its fundamental human pharmacokinetic and safety characteristics. Therefore, this first-in-human phase I study was conducted to evaluate the safety, tolerability, and pharmacokinetic profile of single and multiple oral doses of BS1801 in healthy Chinese volunteers. The results will provide the essential foundation for its subsequent clinical development in fibrotic diseases.
Materials and Methods
Study Design and Ethics
This was a single-center, randomized, double-blind, placebo-controlled, phase I dose-escalation study following a dose-escalation design. It consisted of two parts with a total of seven groups: a single-ascending-dose (SAD) part (4 cohorts) and a multiple-ascending-dose (MAD) part (3 cohorts). The primary objectives were to assess the safety, tolerability, and pharmacokinetics (PK) of BS1801 in healthy adults. The protocol was prepared and the trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines. The study was approved by the Ethics Committee of Shulan (Hangzhou) Hospital (Approval Date/Number: YW2021-004-X3). The trial is registered at https://www.chictr.org.cn (ChiCTR2100045813). All participants provided written informed consent before any study-related procedures.
Participants, Sample Size and Masking
Participants were recruited at the clinical study center. Key inclusion criteria were: 1) healthy males or females aged 18 to 55 years inclusive; 2) body mass index between 18 and 28 kg/m2 inclusive; 3) no clinically significant abnormalities in vital signs, physical examination, or laboratory tests. Major exclusion criteria included: 1) history of hypersensitivity to any drugs or food ingredients; 2) history of gastrointestinal disease that could affect drug absorption; 3) history of drug or alcohol abuse; 4) participation in another clinical trial or blood donation within a specified period prior to screening; 5) positive serology for hepatitis B, hepatitis C, syphilis, or HIV; 6) pregnant or lactating women; 7) any other condition deemed inappropriate for the study by the investigator.
A total of 57 participants were enrolled. The SAD part enrolled 24 participants into four dose cohorts, and the MAD part enrolled 30 participants into three dose cohorts. Except for the first cohort (450 mg), which was open-label to enable initial safety assessment and dose-escalation decision-making in this first-in-human study, the study employed a double-blind, randomized design. Participants were randomized to receive either BS1801 or placebo at a ratio of 3:1 or 4:1 (active:placebo) in the SAD and MAD parts, respectively. A randomization list was generated in advance by an independent statistician using SAS software (version 9.4). Treatment assignments were concealed using sequentially numbered, opaque, sealed envelopes. Investigators, study staff, participants, and personnel involved in PK sample analysis remained blinded to treatment assignment throughout the study.
Investigational Product
BS1801 is a small-molecule organoselenium compound (molecular weight: 450.26). For this study, BS1801 and matching placebo were formulated as film-coated tablets (50 mg, 200 mg, and 500 mg strengths). The placebo tablets were identical to the active tablets in appearance, weight, and taste, containing only the excipient ingredients. All investigational products were provided by Guangzhou Boji Medical Technology Co. Ltd., stored under controlled conditions (20–25°C), and administered at the clinical center.
Dosing and Dose Escalation
The starting dose for the SAD part (450 mg) was selected based on the no-observed-adverse-effect level (NOAEL) from preclinical studies, applying appropriate safety factors. Dose escalation proceeded based on the review of safety and tolerability data from the preceding cohort. A dose-limiting toxicity (DLT) was defined as any treatment-related adverse event of Grade 3 or higher (according to NCI-CTCAE version 5.0) occurring during the DLT observation period. Escalation to the next dose level was permitted if fewer than one-third of participants in a cohort experienced a DLT. The maximum tolerated dose (MTD) was defined as the highest dose at which the incidence of DLT was below this threshold. Preliminary PK data from the SAD part indicated a half-life of approximately 12–16 hours, informing the twice-daily dosing regimen (every 12 hours) in the MAD part, with total daily doses ranging from 600 mg to 1200 mg. The protocol included predefined alternative dose levels (eg, cohorts 7 and 8) for contingency planning.
Endpoints and Assessments
The primary endpoints were safety and tolerability. Safety assessments included monitoring and recording all adverse events (AEs), with severity graded by NCI-CTCAE v5.0, along with serial measurements of vital signs, physical examinations, 12-lead electrocardiograms (ECGs), and laboratory tests (hematology, clinical chemistry, coagulation, and urinalysis). All laboratory analyses were performed at certified clinical laboratories using standard reference ranges.
The secondary endpoint was the PK profile of BS1801. Blood samples for PK analysis were collected at the following time points: pre-dose (0 hour), and at 0.5,1, 2, 3, 4, 5, 6, 9, 12, 24, 48, 72, 96, and 120 hours post-dose in the single-dose part; and pre-dose on Day 1 and Day 6–7, as well as at the same post-dose intervals as above (0.5–96 hours post-dose) on Day 1 and Day 7 in the multiple-dose part. Plasma concentrations of the major metabolite M2 were quantified using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. The bioanalytical method was fully validated according to relevant guidelines (eg, FDA/EMA), demonstrating acceptable specificity, linearity, precision, accuracy, and stability. Key PK parameters included maximum plasma concentration (Cmax), time to Cmax (Tmax), terminal elimination half-life (t1/2), area under the concentration-time curve from time zero to the last quantifiable time point (AUC0-t), and AUC from time zero extrapolated to infinity (AUC0-∞). For the MAD part, steady-state parameters such as trough concentration (Cmin), AUC over a dosing interval at steady state (AUCss), accumulation index (Rac), and fluctuation index (DF) were also calculated.
Laboratory Analysis
The samples, including blood and urine, collected at designated time points in this study, were prepared according to the standard operating protocol (SOP) provided by a third-party laboratory or the clinical laboratory of the study center. This laboratory developed and validated assays to detect BS1801 and its metabolites using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). A quality assurance system was routinely employed in both the certified clinical and research laboratories. Briefly, the samples were immediately deep-frozen and stored at −80 °C until analysis.20,21
Statistical Methods
The full analysis dataset (FAS) contained information on all participants following randomization and was used to report baseline characteristics and treatment compliance (also known as the intention-to-treat dataset). The safety dataset (SS) included all participants who had received the drug, and missing safety data were not imputed. The PK dataset included all participants who had received at least one dose of the drug and had at least one concentration measurement (PKCS) or at least one eligible parameter (PKPS). Demographic and safety data were presented descriptively. SAS software (version 9.4) was used for random number generation and descriptive statistics, the Pheonix WinNonlin (version 8.10) was used to estimate non-compartmental pharmacokinetic parameters. The proportions of AEs between placebo and BS1801 were compared using the Bayesian A/B test package (bayesAB, version 1.1.3) in the R (version 4.2.2) environment. Given the phase I nature of this study, the sample size per cohort was limited (approximately 3–10 participants per active dose group). As a result, the study has limited power to detect rare or infrequent adverse events. This limitation is inherent to early-phase trials and will be addressed in subsequent larger-scale studies.
Results
Participant Disposition, Study Conduct, and Demographics
The first candidate was screened on April 21, 2021, and the last participant completed the study on January 24, 2022. Dose escalation in both the single-ascending-dose (SAD) and multiple-ascending-dose (MAD) parts proceeded as planned per protocol, according to the predefined algorithm illustrated in Figure 1A and B. No cohorts required the use of the predefined alternative dose levels (Cohorts 7/8). A total of 206 healthy candidates were screened, and 57 participants were enrolled and completed the study (27 in the SAD part and 30 in the MAD part) without any premature withdrawal. The participant flow is detailed in Figure 2. The placebo-to-BS1801 ratios were 1:3 and 1:4 in the SAD and MAD parts, respectively. Demographic characteristics were well-balanced across all dose cohorts within each part of the study, with no statistically significant differences (Table 1, one participant with history of appendicitis; one participant use locally applied drug to release tight bowel; none had received additional systematic medication during the trial).
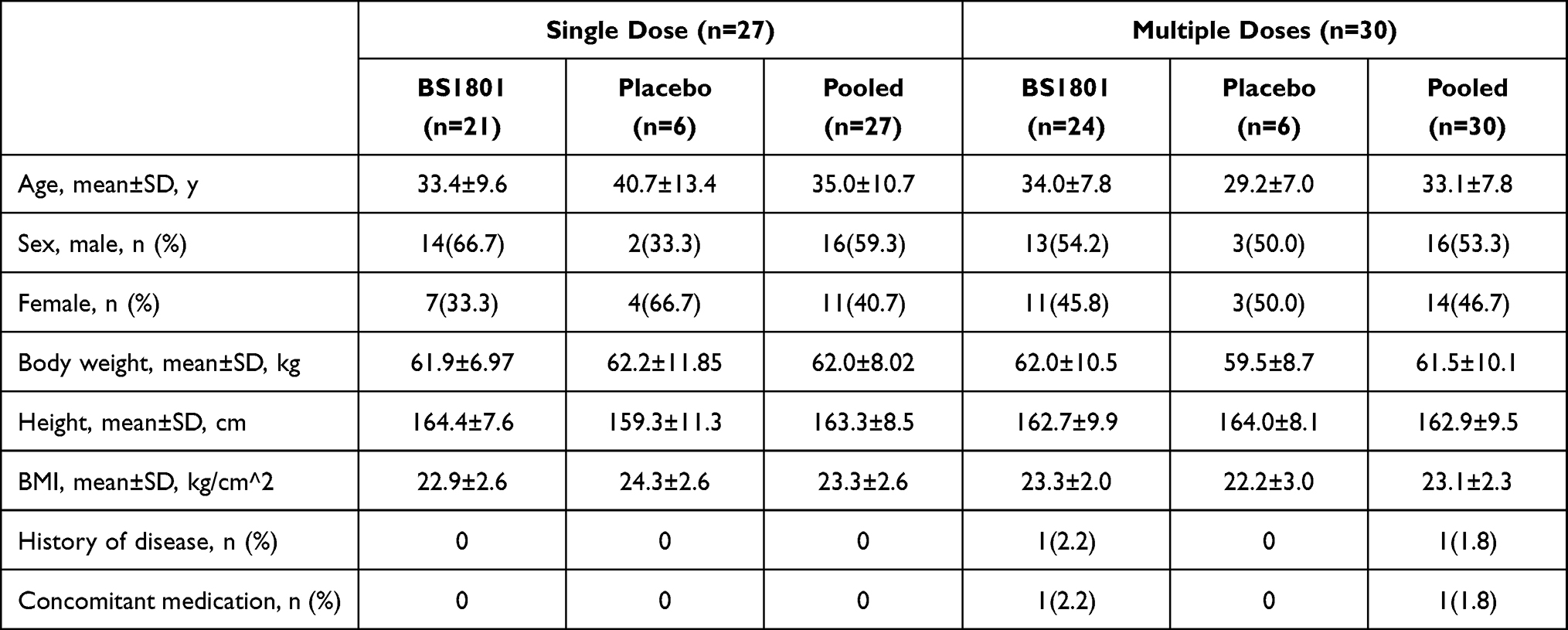 |
Table 1 The Baseline Characteristics of Trial Participants (n=57) |
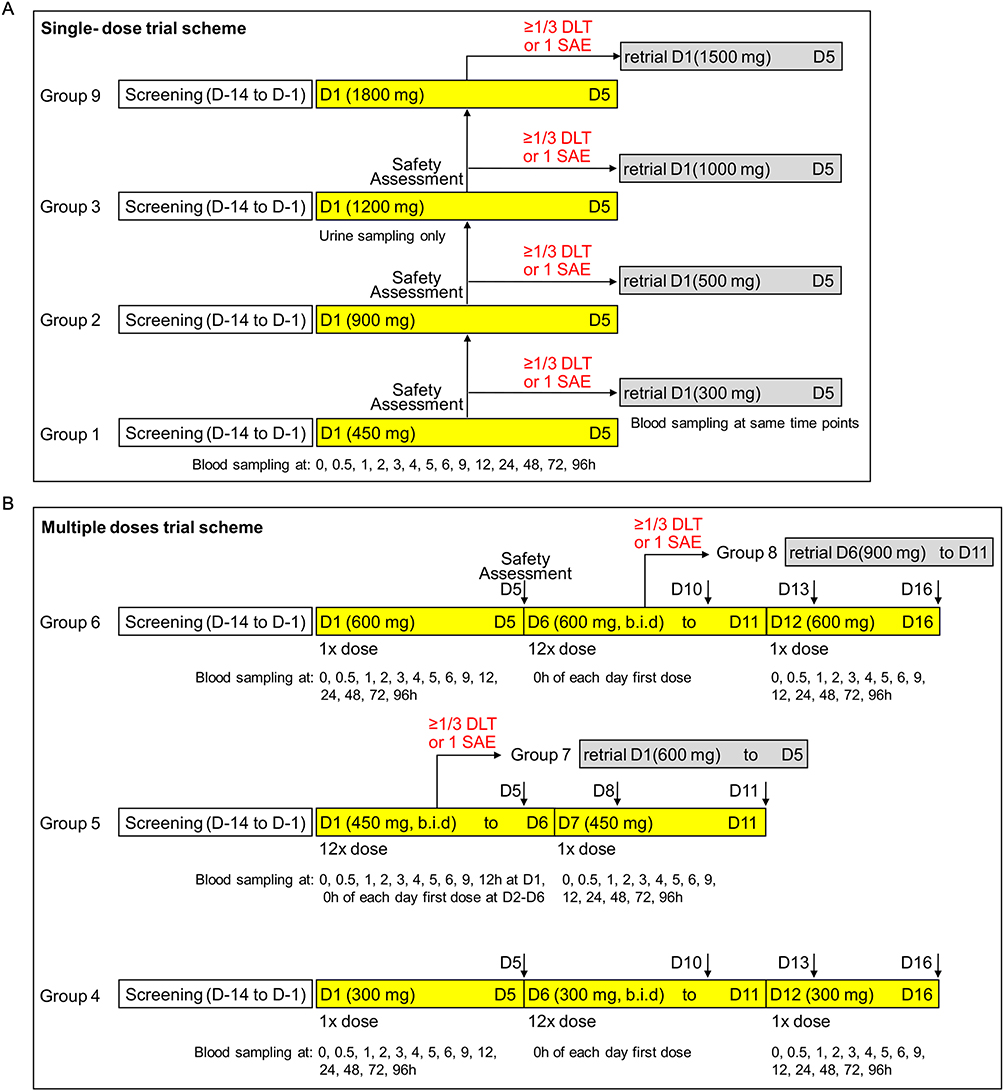 |
Figure 1 Schematic of the dose-escalation trial design. (A) The single-dose groups were tested sequentially in the order of 1, 2, and 3. Participants were closely monitored following the first dose administration in each group for safety assessment (Days 1–5). Meanwhile, blood samples were collected at designated time points. Urine samples were collected only in the 1200 mg single-dose group (Group 3) before (−2 to 0 hours) and after (0–4, 4–8, 8–24, 24–48, 48–72, 72–96, and 96–120 hours) oral administration. (B) The multiple-dose groups were also tested in an ordinal sequence, starting with a single dose of 300 mg (group 4) or 600 mg (group 6) during the first five days (D1-D5). This was followed by six consecutive days of two doses of 300 mg or 600 mg administered 12 hours apart each day (b.i.d.) from D6 to D11, and a final single dose of 300 mg or 600 mg on the following day (D12). Participants were discharged only if no adverse events were observed or if they had recovered from mild discomfort or dysfunction. In both single-dose and multiple-dose trials, the planned alternative doses (highlighted in the light grey box, including groups 7 and 8) were not tested, as no group met the criteria. |
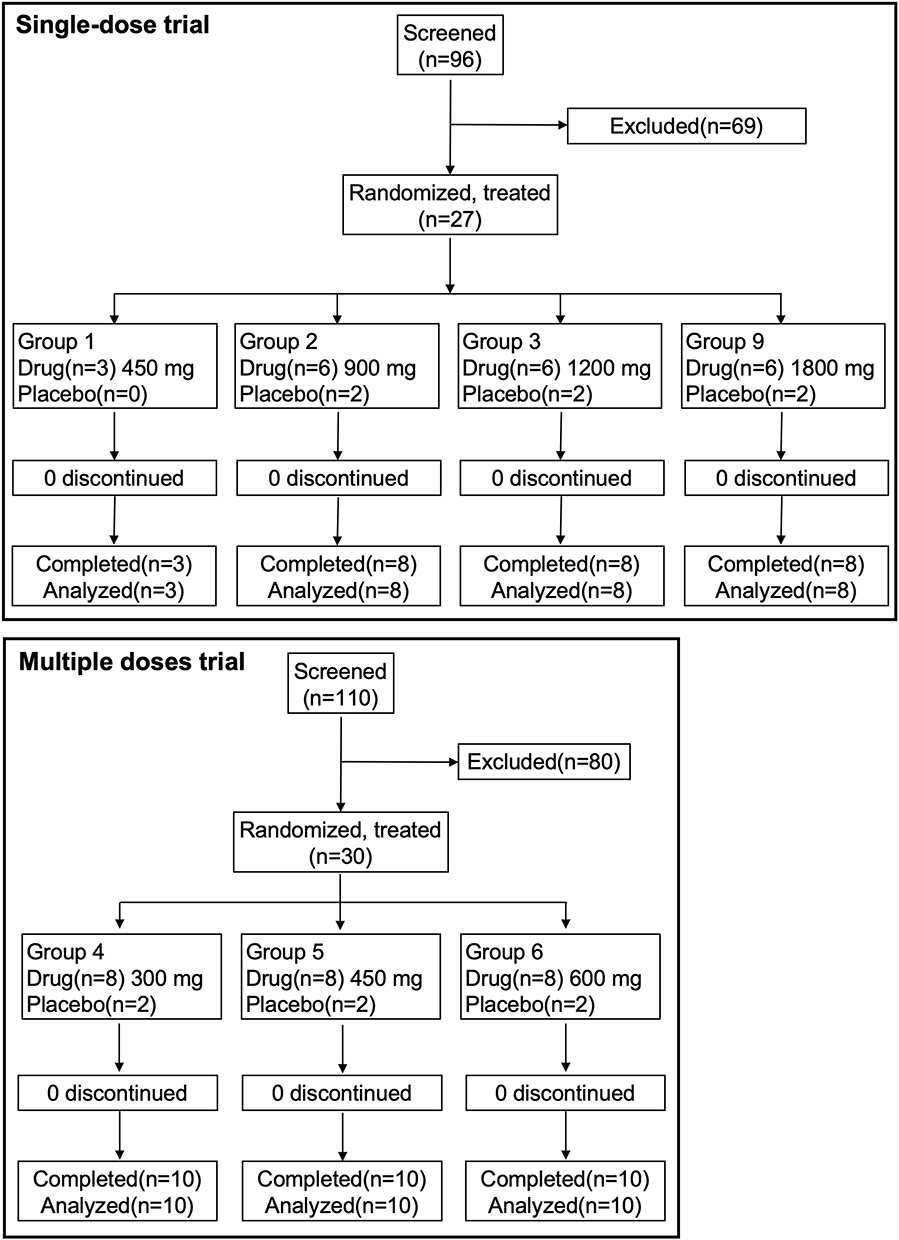 |
Figure 2 The CONSORT diagram for the single-dose and multiple-dose clinical trial. |
Safety and Tolerability
In the single-ascending-dose part, 17 AEs occurred in 10 participants, with an incidence of 22.2% (95% CI: 10.1–34.4%). In the multiple-ascending-dose part, 27 AEs occurred in 13 participants, with an incidence of 28.8% (95% CI: 15.7–42.1%). This accounted for the total of 44 AEs observed. A total of 36 treatment-emergent adverse events (TEAEs) were reported: 14 TEAEs in 9 participants in the single-ascending-dose part (incidence 20.0%, 95% CI: 8.3–31.7%) and 22 TEAEs in 11 participants in the multiple-ascending-dose part (incidence 24.4%, 95% CI: 11.9–37.0%). One AE of special interest (local use of medication to relieve constipation in the multiple-ascending-dose part) was reported in one participant. All AEs were Grade 1 in severity according to CTCAE v5.0. The proportion of participants experiencing any AE was 41.7% (5/12) in the placebo group and 51.1% (23/45) in the BS1801 groups; this difference was not statistically significant (p=0.0614). The spectrum of TEAEs is summarized in Table 2. The most commonly affected organ systems were the thyroid (eg, cysts/nodules, hormone level changes), cardiovascular system (eg, bradycardia, hypotension), and liver (eg, elevated bilirubin and transaminases). All AEs resolved before hospital discharge without sequelae. No dose-limiting toxicities, severe TEAEs, serious adverse events (SAEs), or deaths were observed.
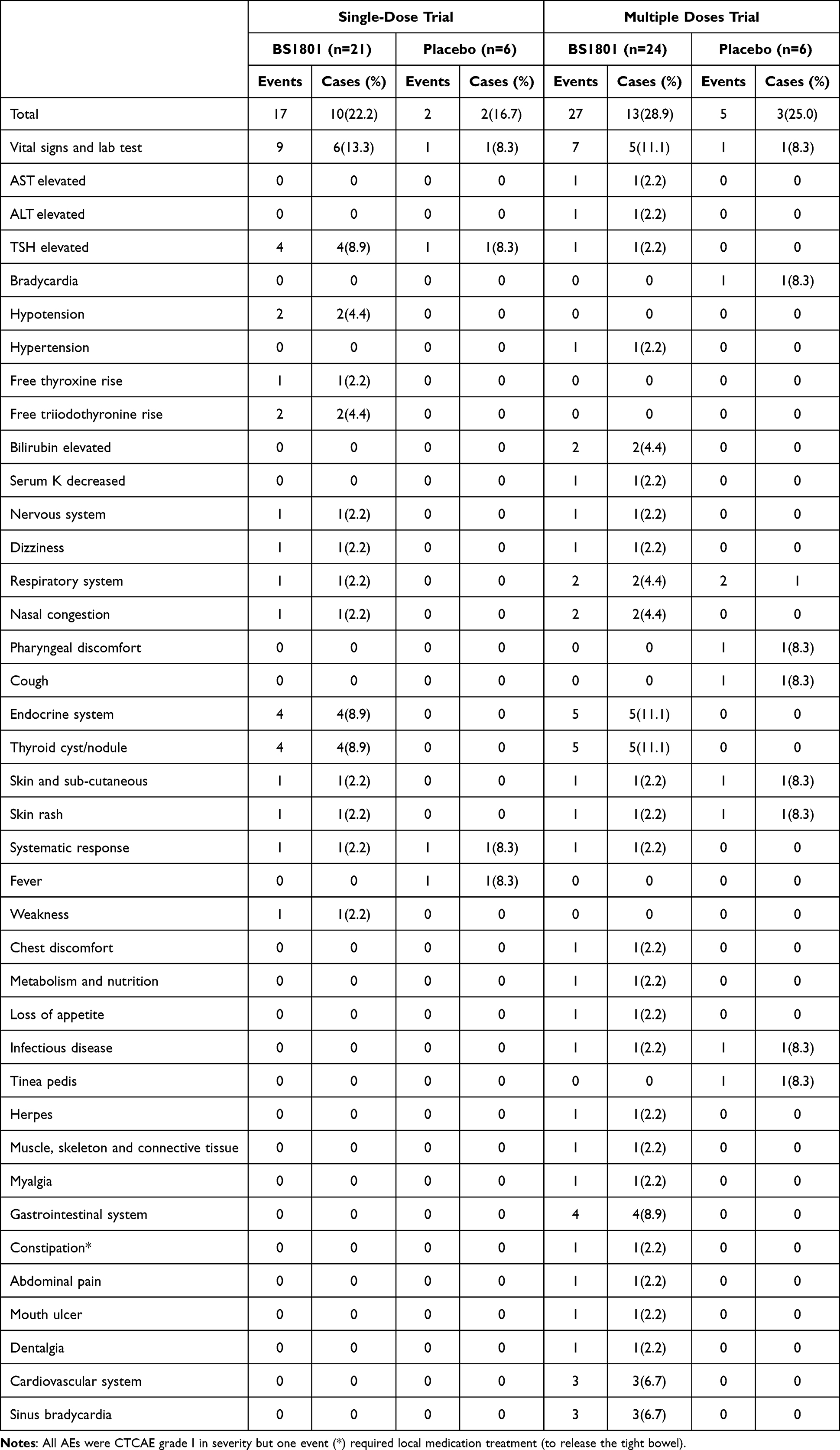 |
Table 2 The Adverse Events in the Single-Dose and Multiple-Dose BS1801 Trial |
Pharmacokinetics
The methyl conjugate of BS1801 (abbreviated as BS1801-M2) was identified as its predominant stable metabolite in humans and was used as a surrogate biomarker in this pharmacokinetic study.21 In the single-dose trial, as the dose increased, the time to peak concentration (Tmax) increased from 4.0 hours to 5.0 hours accordingly; however, the steady-state Tmax (Tmax,ss) decreased to 2.2–2.4 hours following consecutive doses. In the single-dose trial, when the dose was increased several-fold, Tmax did not significantly prolong, and the peak concentration (Cmax) showed minimal increase even when the dose was doubled. However, when the dosing frequency was changed from once to twice daily at the same total daily dose, Cmax approximately doubled, showing a similar magnitude of increase as the area under the curve (AUC), which increased 1.63- to 2.08-fold. Notably, the lower dose (300 mg vs. 450 mg twice daily) resulted in a greater increase in Cmax and AUC than the higher dose; similarly, the low dose (300 mg twice daily) showed a greater increase in Cmax and AUC than the high dose (600 mg twice daily). These data suggest that the absorption of BS1801 may occur only in a specific part of the gastrointestinal tract and that an upper limit to absorption may exist (Table 3 and Figure 3).
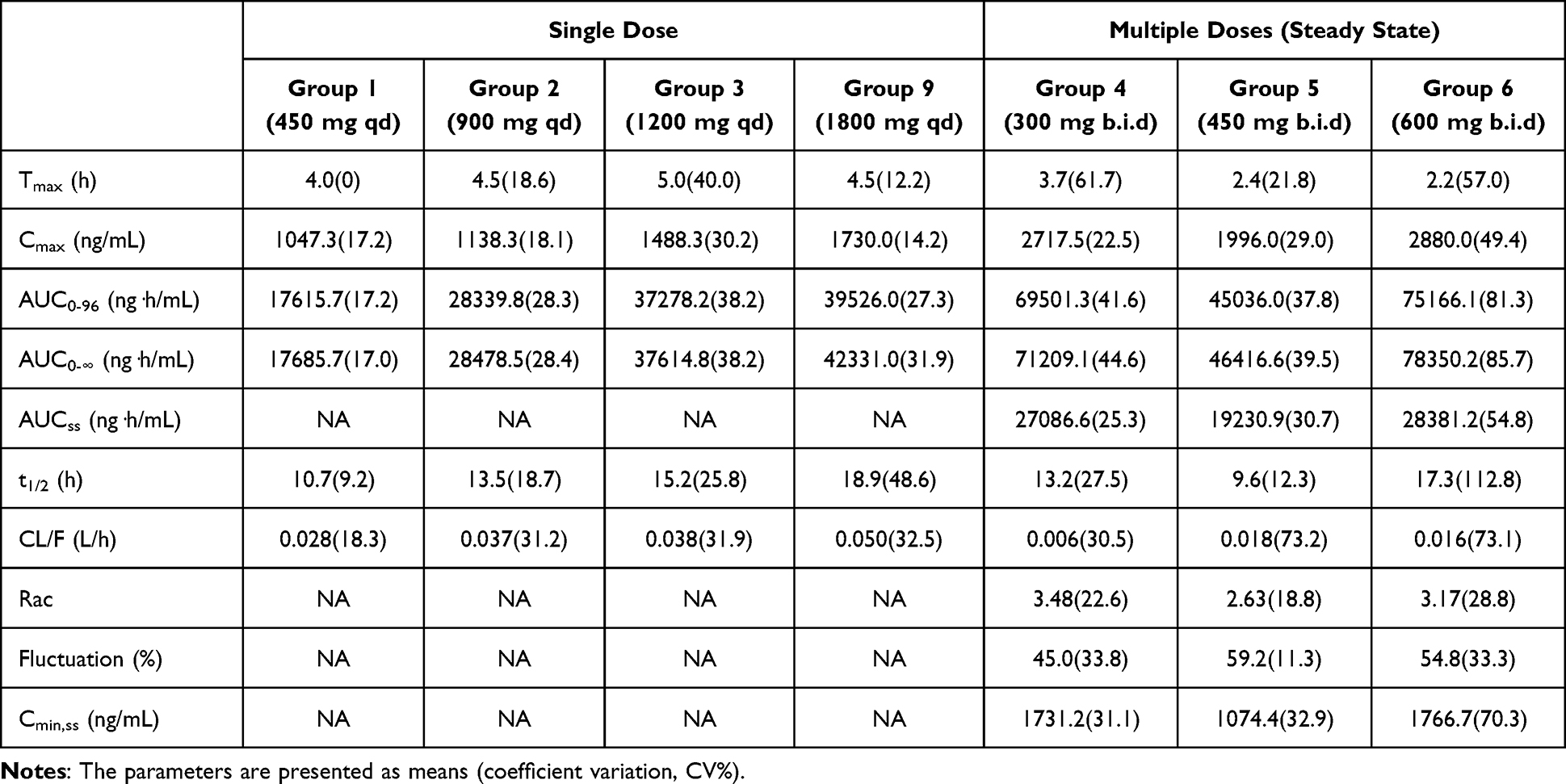 |
Table 3 Pharmacokinetic Parameters Estimated from Single-Dose and Multiple-Dose Trials |
 |
Figure 3 The pharmacokinetic profile of BS1801-M2 following single and multiple dose administrations. (A) A single dose of BS1801 was administered to six groups, namely Groups 1, 2, 3, 4, 6, and 9. As the dose increased, the Cmax also increased, although not proportionally to the dose increments. The concentration of BS1801-M2 in the low-dose groups (300 mg in Group 4; 450 mg in Groups 1 and 5) declined rapidly, nearly reaching baseline within the first two days. In contrast, the concentration of BS1801-M2 in the high-dose groups (900 mg in Group 2; 1200 mg in Group 3; 1800 mg in Group 9) decreased at an approximately constant rate over the first four days. The inset displays the concentration profile of the low-dose groups on a logarithmic time scale. (B) Following two consecutive doses, BS1801-M2 reached its plateau and remained nearly constant over the subsequent six days. The low-dose groups (300 mg or 450 mg b.i.d.) exhibited nearly identical pharmacokinetic profiles. After a brief extension, withholding BS1801 resulted in a nearly identical rate of decline. (C) The logarithmic time scale of the last dose in the multiple-dose trial. |
As the dose increased, the half-life (t1/2) was prolonged from approximately 10 hours to 18 hours, paralleling the increase in Cmax following single-dose administration. This suggests that the metabolism of BS1801 may have reached its maximal capacity in this trial and indicates the presence of rate-limiting step(s) in BS1801 methylation and/or demethylation. After consecutive doses, withholding drug administration led to rapid clearance of BS1801 within 96 hours (Figure 3 and Table 3).
A sensitivity analysis was conducted by excluding data from five participants in the multiple-dose trial with outlying concentrations. The changes in all estimated pharmacokinetic parameters remained within ‑20.4% to +19.1% of the original values, which was considered acceptable relative to the coefficients of variation of the primary analysis (Table 3).
Discussion
This first-in-human study demonstrated that BS1801 was safe and well-tolerated in healthy adults at single doses up to 1200 mg and multiple doses up to 600 mg administered twice daily. The pharmacokinetic profile revealed complex, non-linear characteristics suggestive of saturable processes. The integrated safety and pharmacokinetic data support advancing BS1801 into Phase II clinical trials.
The most salient pharmacokinetic finding of this study is the evidence suggestive of saturable absorption. The less-than-dose-proportional increase in systemic exposure (Cmax and AUC), the disproportionate increase in exposure when the same total daily dose is administered in divided portions, and the observation that a lower dose (300 mg, twice daily) yielded a greater relative increase than higher doses are collectively indicative of a capacity-limited absorption process. This phenomenon may be explained by the physicochemical and metabolic properties of BS1801. Consistent with preclinical absorption, distribution, metabolism, and excretion studies, after oral administration, BS1801 is extensively metabolized during first-pass. The parent compound is detected only in feces, while the methylated metabolite M2 (BS1801-M484, generated via N-Se bond reduction and Se-methylation) is reported as the predominant drug-related component in plasma both in rats and in humans.21 The saturation observed in humans may therefore occur at the intestinal absorption site and/or during this extensive, potentially rate-limited first-pass metabolism. Furthermore, the shortening of Tmax at steady-state (Tmax,ss) compared to single-dose administration may reflect adaptive changes in gastrointestinal transit or pre-systemic metabolism with repeated dosing under such saturable conditions.
The safety profile of BS1801 in this healthy volunteer population is favorable and warrants its further development for fibrotic diseases. All reported adverse events were mild (Grade 1), transient, and resolved without sequelae, with no dose-limiting toxicities or serious adverse events observed. The most common treatment-emergent adverse events involved the thyroid, cardiovascular, and hepatic systems. The rapid onset of thyroid parameter changes and sonographic findings is noteworthy. While the formation of structural thyroid nodules is typically chronic, rapid biochemical and functional alterations can occur and may represent a direct pharmacodynamic effect related to BS1801’s mechanism of action as a thioredoxin reductase inhibitor, given the role of redox systems in thyroid hormone metabolism. However, the precise mechanisms underlying the observed thyroid and cardiovascular events remain unclear, as no specific preclinical or clinical evidence is currently available to elucidate these findings. Future pharmacodynamic and translational studies are warranted to investigate the potential causal pathways. These findings, while not dose-limiting in this study, define important safety parameters for monitoring in future patient trials. In this study, the majority of treatment-emergent adverse events occurred within 12 hours to 14 days following drug administration. Thyroid parameter changes were observed as early as 4 days post-dose, while gastrointestinal and cardiovascular events typically manifested within 24 hours of dosing. All adverse events resolved spontaneously before hospital discharge without intervention, except for one case of constipation requiring topical medication.
Integrating the pharmacokinetic and safety data allows for a scientifically grounded recommendation for phase II studies. The observed saturable absorption and a half-life (~12 hours) supporting twice-daily administration directly inform the dosing regimen. A twice-daily schedule mitigates high peak-to-trough fluctuations and optimizes systemic exposure within the apparent saturable range. Based on the safety margins observed (no dose-limiting toxicities up to 600 mg twice daily) and the pharmacokinetic principle of maintaining exposure within the linear or predictable range, a starting dose range of 300 mg to 450 mg administered twice daily is recommended for phase II efficacy trials in patient populations. This range is expected to provide clinically relevant exposure while prioritizing safety.
This study has limitations inherent to early-phase trials. The sample size per cohort was small, the study was conducted in healthy volunteers, and the treatment duration was short. Consequently, the pharmacokinetic variability observed and the long-term safety profile, particularly regarding the thyroid effects, require further evaluation in larger, longer-duration patient studies. In addition, the small cohort sizes limit the ability to detect rare adverse events that may occur at frequencies below approximately 5–10%, which should be a focus of post-marketing surveillance.
In conclusion, BS1801 exhibited a favorable safety profile and a pharmacokinetic pattern consistent with saturable absorption in healthy subjects. The data robustly support the progression of BS1801 into phase II trials. Future studies should investigate the efficacy of the proposed 300–450 mg twice daily regimen in patients with fibrotic diseases, include focused monitoring of thyroid and hepatic function, and incorporate translational research to elucidate the relationship between exposure to the active metabolite M2, target engagement at thioredoxin reductase, and clinical outcomes.
Conclusion
In summary, in this first-in-human clinical trial in healthy Chinese adults, BS1801 was safe and well tolerated within the tested dose range. All observed adverse events were mild (Grade 1) and transient, with no dose-limiting toxicities or serious adverse events reported. Based on the safety profile and PK parameters, the recommended dosage may range from 300 mg to 450 mg twice daily. Further clinical studies will be conducted to evaluate the safety and efficacy of BS1801 in the target population.
Data Sharing Statement
The datasets used and/or analyzed during the current study can be obtained from the corresponding author on reasonable request.
Funding
This work was sponsored by Yuanxi Med, Shanghai; Chinese Clinical Trial Registry number: ChiCTR2100045813.
Disclosure
Hanwei Yin, Guozhou Zhang, and Huihui Zeng are employees of Shanghai Yuanxi Medicine Corp, which developed BS1801. This relationship has the potential to introduce bias in the reporting of results and conclusions. The study design, data collection, analysis, and interpretation were conducted in accordance with rigorous scientific standards to minimize potential bias. In addition, Huihui Zeng has a patent for butaselen (US 11,952,356 B2) licensed. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587(7835):555–12. doi:10.1038/s41586-020-2938-9
2. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
3. Olson A, Hartmann N, Patnaik P, et al. Estimation of the prevalence of progressive fibrosing interstitial lung diseases: systematic literature review and data from a physician survey. Adv Ther. 2021;38(2):854–867. doi:10.1007/s12325-020-01578-6
4. Harris R, Harman DJ, Card TR, Aithal GP, Guha IN. Prevalence of clinically significant liver disease within the general population, as defined by non-invasive markers of liver fibrosis: a systematic review. Lancet Gastroenterol Hepatol. 2017;2(4):288–297. doi:10.1016/S2468-1253(16)30205-9
5. Fernandez Perez ER, Daniels CE, Schroeder DR, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137(1):129–137. doi:10.1378/chest.09-1002
6. Nunes H, Schubel K, Piver D, et al. Nonspecific interstitial pneumonia: survival is influenced by the underlying cause. Eur Respir J. 2015;45(3):746–755. doi:10.1183/09031936.00148613
7. Samarelli AV, Tonelli R, Heijink I, et al. Dissecting the role of mesenchymal stem cells in idiopathic pulmonary fibrosis: cause or solution. Front Pharmacol. 2021;12:692551. doi:10.3389/fphar.2021.692551
8. Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-beta-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. doi:10.1016/j.ejps.2014.02.014
9. King TE, Bradford WZ, Castro-Bernardini S, et al. A Phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. doi:10.1056/NEJMoa1402582
10. Richeldi LCU, Selman M, Kim DS, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. doi:10.1056/NEJMoa1103690
11. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349(2):209–220. doi:10.1124/jpet.113.208223
12. Kang J, Chung MP, Park MS, et al. Clinical outcomes of dose modification during pirfenidone treatment for IPF: a nationwide post-marketing surveillance study. Front Pharmacol. 2022;13:1025947. doi:10.3389/fphar.2022.1025947
13. Levra S, Guida G, Sprio AE, et al. Long-term safety of antifibrotic drugs in IPF: a real-world experience. Biomedicines. 2022;10(12):3229. doi:10.3390/biomedicines10123229
14. Santoleri F, Auriemma L, Spacone A, et al. Adherence, persistence, and effectiveness in real life. Multicenter long-term study on the use of pirfenidone and nintedanib in the treatment of idiopathic pulmonary fibrosis. J Pharm Pract. 2022;35(6):853–858. doi:10.1177/08971900211008625
15. He J, Li D, Xiong K, et al. Inhibition of thioredoxin reductase by a novel series of bis-1,2-benzisoselenazol-3(2H)-ones: organoselenium compounds for cancer therapy. Bioorg Med Chem. 2012;20(12):3816–3827. doi:10.1016/j.bmc.2012.04.033
16. Amporndanai K, Meng X, Shang W, et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat Commun. 2021;12(1):3061. doi:10.1038/s41467-021-23313-7
17. Zheng X, Chen Y, Bai M, et al. The antimetastatic effect and underlying mechanisms of thioredoxin reductase inhibitor ethaselen. Free Radic Biol Med. 2019;131:7–17. doi:10.1016/j.freeradbiomed.2018.11.030
18. Jiao W, Bai M, Yin H, et al. Therapeutic effects of an inhibitor of thioredoxin reductase on liver fibrosis by inhibiting the transforming growth factor-beta1/Smads pathway. Front Mol Biosci. 2021;8:690170. doi:10.3389/fmolb.2021.690170
19. Chen Y, Yin H, Sun J, Zhang G, Zhang Y, Zeng H. TrxR/Trx inhibitor butaselen ameliorates pulmonary fibrosis by suppressing NF-κB/TGF-β1/Smads signaling. Biomed Pharmacother. 2023;169:115822. doi:10.1016/j.biopha.2023.115822
20. Tian Q, Jiang J, Yin H, et al. Investigating the metabolic mechanisms of butaselen, An Ebselen analog. Curr Drug Metab. 2022;23(11):928–939. doi:10.2174/1389200223666220520115014
21. Tian Q, Jiang J, Yin H, et al. Quantification of the major circulating metabolite of BS1801, an ebselen analog, in human plasma. J Pharm Biomed Anal. 2022;212:114638. doi:10.1016/j.jpba.2022.114638
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of Pharmacokinetics and Safety with Bioequivalence of Ibuprofen Sustained-Release Capsules of Two Formulations, in Chinese Healthy Volunteers: Bioequivalence Study
Huang C, Yin Z, Yang Y, Mo N, Yang H, Wang Y
Drug Design, Development and Therapy 2023, 17:1881-1888
Published Date: 23 June 2023
Comparative Pharmacokinetics and Bioequivalence Evaluation of Two Formulations of Pramipexole Dihydrochloride Extended-Release Tablets in Healthy Chinese Subjects Under Fasted and Fed States: A Randomized, Open-Label, Single-Dose, Two-Period Crossover Clinical Trial
Yang L, Zhang L, Luo Z
Drug Design, Development and Therapy 2023, 17:2369-2381
Published Date: 15 August 2023
Effect of Food on the Pharmacokinetics of Tenofovir Amibufenamide: A Phase I, Randomized, Open-Label, Two-Period Crossover Trial in Healthy Adult Subjects

Liu J, Wu M, Kai J, Lin M, Zheng Y, Jiang Y, Huang Q, Zhai Y, Qiu Y
Drug Design, Development and Therapy 2023, 17:3061-3072
Published Date: 9 October 2023
The Pharmacokinetics, Safety and Tolerability of Aclidinium Bromide 400 μg Administered by Inhalation as Single and Multiple (Twice Daily) Doses in Healthy Chinese Participants
Li W, Daoud SZ, Trivedi R, Lukka PB, Jimenez E, Molins E, Stewart C, Bharali P, Garcia-Gil E
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:2725-2735
Published Date: 27 November 2023
Pharmacokinetics and Bioequivalence of Two Fixed-Dose Combination Tablets of Valsartan/Amlodipine (80/5 Mg) in Healthy Chinese Subjects
Tian M, Huang J, Chen Y, Jin Q, Jiang H, Shi C, Mei J, Xu M, Yu X, Yang S
Drug Design, Development and Therapy 2025, 19:11-22
Published Date: 3 January 2025