Back to Journals » Drug Design, Development and Therapy » Volume 17
Comparative Pharmacokinetics and Bioequivalence Evaluation of Two Formulations of Pramipexole Dihydrochloride Extended-Release Tablets in Healthy Chinese Subjects Under Fasted and Fed States: A Randomized, Open-Label, Single-Dose, Two-Period Crossover Clinical Trial
Authors Yang L ![]() , Zhang L, Luo Z
, Zhang L, Luo Z
Received 8 June 2023
Accepted for publication 11 August 2023
Published 15 August 2023 Volume 2023:17 Pages 2369—2381
DOI https://doi.org/10.2147/DDDT.S421449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Frank Boeckler
Ling Yang,1,2,* Liangliang Zhang,3,* Zhu Luo2
1Department of Neurology, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 2Clinical Trial Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 3Department of Pulmonary and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhu Luo, Clinical Trial Center, West China Hospital, Sichuan University, No. 37 Guoxue Lane, Chengdu, Sichuan, 610044, People’s Republic of China, Tel +86 028 85422707, Email [email protected]
Background: Pramipexole dihydrochloride extended-release tablet is a novel long-acting form of non-ergot dopamine agonist indicated as one of main therapeutic approaches for Parkinson’s disease. However, pharmacokinetic properties of extended-release pramipexole in healthy Chinese subjects remain unclear.
Methods: A single-center, randomized, open-label, two-period crossover, single-dose study was performed to investigate comparative pharmacokinetics and evaluate bioequivalence of 0.375 mg test (Yangtze River Pharmaceutical Group Co., Ltd.) and reference (Trade name: Sifrol®, Boehringer Ingelheim Pharma GmbH & Co. KG) formulations of pramipexole dihydrochloride extended-release tablets in healthy Chinese subjects under fasted and fed states.
Results: A total of 56 subjects (28 in each dietary trial) were enrolled and randomized. After single dose of 0.375 mg test and reference formulations under fasted condition, main pharmacokinetics of pramipexole were as follows: peak concentration (Cmax) were 409.33± 95.93 and 413.77± 132.03 pg/mL; plasma area under concentration–time curve from time 0 to last measurable concentration (AUC0-t) were 8801.95± 1966.83 and 8646.37± 2600.49 h*pg/mL; AUC from time 0 to infinity (AUC0-∞) were 9469.03± 1991.61 and 9082.95± 2666.26 h*pg/mL; elimination half-life (t1/2) were 11.98± 3.91 and 9.85± 2.63 h; both time to reach Cmax (Tmax) were about 4.50 h, respectively, for test and reference formulations. The 90% confidence intervals of geometric mean ratios (test/reference) of Cmax, AUC0-t and AUC0-∞ under fasted and fed conditions were all within 80– 125%. Following administration under fed condition, Cmax and Tmax for both test and reference formulations slightly increased and prolonged to 5.0 h, respectively, but AUC approximately remained unchanged compared with dosing under fasted condition. Test and reference formulations showed similar bioequivalence and favorable safety under fasted and fed states.
Conclusion: Test and reference formulations of pramipexole dihydrochloride extended-release tablets (0.375 mg) showed similar bioequivalence and well safety and tolerability in healthy Chinese subjects under fasted and fed states, which supports further investigations of test formulation in patients with Parkinson’s disease.
Keywords: pramipexole dihydrochloride extended-release tablet, bioequivalence, pharmacokinetics, safety, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is one of the most common chronic neurodegenerative disorders characterized by progressive degeneration of dopaminergic neurons and Lewy body formation in the substantia nigra, decreased striatal dopamine transmitters, and imbalance of dopamine and acetylcholine transmitters over time, manifested as motor dysfunctions of tremor, disturbance of postural balance, bradykinesia, rigidity, as well as non-motor symptoms including sleep disorders, smell disorders, autonomic dysfunction, cognitive and psychiatric disorders.1 The major anti-Parkinsonian options available include levodopa and non-ergot dopamine agonists (NEDAs). Although levodopa is the standard therapy for PD and effectively relieves Parkinson’s symptoms, motor complications including symptom fluctuations and dyskinesias may occur along with PD progression and long-term administration of levodopa.2–4 NEDAs showed preferable efficacy and tolerability in several randomized controlled trials when applied as monotherapy for the management of early PD or as first-line adjunct treatment with levodopa for advanced PD, which showed lower incidence of dyskinesia compared with levodopa.2,3,5,6
Pramipexole is one of the extensively used NEDA, the mechanism of which as a treatment for PD is the ability to stimulate dopamine receptors in the striatum via affinity for the D2/D3 subfamily of dopamine receptors.5,7–9 Pramipexole dihydrochloride extended-release tablet is an innovative long-acting formulation of NEDA, which has been developed and prescribed in clinical practice in recent years.2,7,8,10
The majority of randomized controlled trials have assessed and compared the clinical efficacy and safety of extended-release and immediate-release formulations of pramipexole for early and advanced PD.4,11–14 When compared with three times-daily dosing of immediate-release formulation of pramipexole, extended-release tablet is administered once-daily along with more convenient dose titration regimen and reduces the fluctuation of symptoms with shortened “off” period and enhanced adherence of patients for the reason that it can reach a more stable plasma pramipexole concentration and maintain the continuous dopaminergic stimulation and efficacy over 24 h.2,7,8,15–18 However, there is no literature reporting pharmacokinetic parameters of extended-release pramipexole in healthy Chinese subjects.
In the present study, we performed a single-center, randomized, open-label, two-period crossover, single-administration trial to firstly analyze and compare the pharmacokinetics as well as bioequivalence of test (0.375 mg, Yangtze River Pharmaceutical Group Co., Ltd.) and reference (Trade name: Sifrol®, 0.375 mg, Boehringer Ingelheim Pharma GmbH & Co. KG) formulations of pramipexole dihydrochloride extended-release tablets in healthy Chinese subjects under fasted and fed states.
Subjects and Methods
Study Design
This single-center, randomized, open-label, single-dose, two-period crossover clinical study was used to explore and compare pharmacokinetic characteristics and safety and evaluate the bioequivalence of two pramipexole dihydrochloride extended-release tablet formulations in healthy Chinese subjects under fasted and fed conditions. Enrolled subjects were randomly assigned in the ratio of 1:1 to one of the two single-dose administration sequence groups: test to reference formulation (T-R) group or reference to test formulation (R-T) group under fasted and fed conditions (N=28 in each condition). The administration sequence was based on a random table generated by version 9.4 SAS software with a ratio of 1:1, and the washout time between each period was designed as 7 days, which significantly exceeded seven terminal half-lives of reference formulation according to its prescribing information and was adequate to fully eliminate the effects of drug taken in the first period. The detailed diagram of the study design is shown in Figure 1.
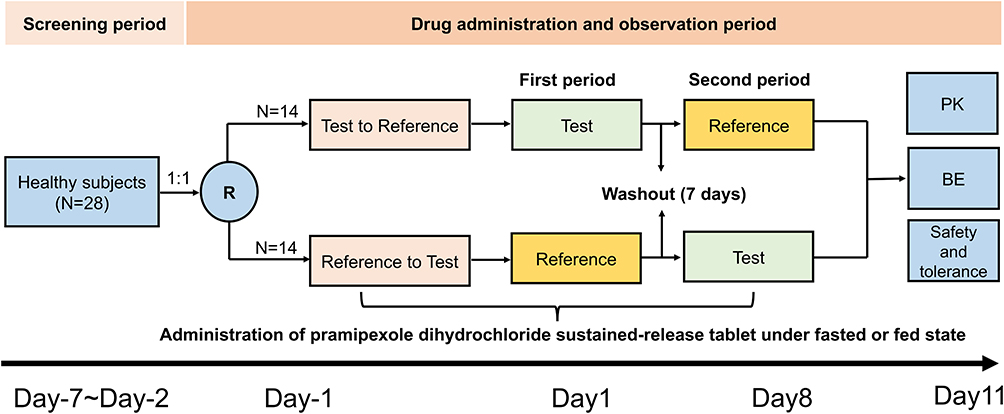 |
Figure 1 Diagram of the study design. Abbreviations: R, randomization; PK, pharmacokinetics; BE, bioequivalence analysis. Notes: Fed state: taking a high-fat meal (about 800~1000 calories and 50% from fat) 30 min before dosing. |
The study was conducted according to the Declaration of Helsinki and ICH-GCP guidelines. The study protocol and informed consent forms were reviewed and approved by the Independent Ethics Committee of West China Hospital, Sichuan University. All subjects provided written informed consents before any related procedure was conducted. In addition, the study was registered on the World Health Organization's International Clinical Trials Registry Platform (ChiCTR2000035564, date of registration: August 13, 2020). The execution dates of the trial were from August 17, 2020, to September 30, 2020.
Study Population
Healthy Chinese subjects (male and female) aged from 18 to 65 years with a body mass index (BMI) between 19 and 26 kg/m2, body weight ≥50 kg for male and ≥45 kg for female were eligible for recruitment. Subjects with any significant abnormality in medical history, laboratory tests (hematology, blood biochemistry, urinalysis, HIV antibody, hepatitis B surface antigen, hepatitis C antibody, syphilis antibody, tests for alcohol and drugs abuse, pregnancy test for female), chest X-ray, 12-lead electrocardiogram (ECG), vital signs and physical examination were excluded. Other main exclusions were as follows: allergic history of pramipexole and ingredients contained in pramipexole dihydrochloride extended-release tablet; exposure to any investigational medication including placebo within 90 days; taking any prescription or over-the-counter medication within 14 days before screening.
Test and Reference Formulation
The researched pramipexole dihydrochloride extended-release tablet (unit strength: 0.375 mg, batch number: 19081541) manufactured by Yangtze River Pharmaceutical Group Co., Ltd. (Jiangsu, China) was used as test formulation in the study. Meanwhile, the reference pramipexole dihydrochloride extended-release tablet (Trade name: Sifrol®, unit strength: 0.375 mg, batch number: 802726) was manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG (Ingelheim, Germany).
Drug Administration and Blood Sampling
All eligible subjects were admitted to the Phase I Clinical Trial Center ward of West China Hospital, Sichuan University on Day-1. The subjects under fasted status were randomized to receive a single dose of 0.375 mg pramipexole dihydrochloride extended-release tablet with 240 mL water in a sequence of T-R or R-T under fasting for at least 10 h overnight. Meanwhile, subjects under fed condition randomly received a single dose of 0.375 mg pramipexole dihydrochloride extended-release tablet with 240 mL water in T-R or R-T sequence after taking a high-fat and high-calorie meal (about 800~1000 calories and 50% from fat) 30 min before dosing. All subjects received standard meals when 4 h and 10 h after oral dosing and water intaking were not permitted 1 h before and after oral administration of drugs.
Blood samples (4mL per point) were collected for pharmacokinetic evaluation. Sampling timepoints under fasted condition were as follows: within 1 h before dosing, 1.0, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 7, 9, 12, 24, 36, 48 and 72 h after dosing, and timepoints under fed condition were as follows: within 1 h before dosing, 1, 2, 3, 4, 4.5, 5, 5.5, 6, 6.5, 7, 8, 9, 10, 12, 14, 24, 36, 48, and 72 h after dosing.
All blood samples were collected in anti-coagulation tubes containing Ethylene Diamine Tetraacetic Acid- K2 (EDTA-K2) and centrifuged (2000×g, 10 min, 4°C) within 1 h of collection. Separated plasma samples were pipetted into tubes for pharmacokinetic assessment and were stored at −80°C until assayed.
Assay of Pramipexole
Plasma concentration of pramipexole was measured using High-Performance Liquid Chromatography tandem Mass Spectrometry (HPLC-MS/MS) method developed and validated before the study. HPLC was conducted with LC-30AD (Shimadzu, Kyoto, Japan), mass spectrometric detection was carried out with Triple Quad 5500 mass spectrometer (Applied Biosystems/Sciex, MA, USA) and Welch Ultimate XB-C18 column (2.1 × 100 mm, 3 μm, Welch Materials, Maryland, USA) was applied.
For pramipexole measurements, each 200 μL plasma sample was spiked with 50 μL internal standard (IS) solution (2.00 ng/mL pramipexole-d3), and then 50.0 µL of 1 M NaOH water solution was added to all samples. After vortexed for 3 min, add 800 µL of extractant (ethyl acetate: dichloromethane, 4:1, v/v) to all samples and next vortex samples for 10 min. Samples were centrifuged at 4000 × rpm for 10 min at 8°C and 500 μL upper organic phase was moved to clean 96-well plate, which was then evaporated to dryness under a gentle stream of nitrogen at 40°C. 120 μL reconstitution solution (mobile phase A: mobile phase B, 9:1, v/v) was then added and vortexed for 10 min, which was ready for HPLC-MS/MS analysis.
The mobile phases, water solution containing 0.1% Formic Acid and 20 mM NH4Ac (Phase A) and 100% Methanol (Phase B), were used in gradient program. The flow rate and column temperature were at 0.300 mL/min and 40°C, respectively. The transitions for Multiple Reaction Monitor (MRM) mode were at m/z 212.1/153.2 (pramipexole) and 215.2/153.2 (IS, pramipexole-d3). The parameters of MS detection were optimized: ion spray voltage, 1500 V; curtain gas, 25 psi; gas 1, 60 psi; gas 2, 60 psi; temperature, 600°C; pause between mass: 20.00 ms; collision-activated dissociation, 7 units; dwell time, 150 ms; declustering potential, 60 V; entrance potential, 6 V. The collision energy and collision cell exit potential were 20 V for pramipexole and 23 V and 9 V for IS, respectively. Data were analyzed through AB Sciex Analyst 1.6.3 quantitative software (Applied Biosystems Inc., Foster City, CA, USA).
The linear calibration curve of plasma pramipexole was over the range of 15.0–800 pg/mL. Typical chromatograms of pramipexole and IS are presented in Supplementary Figure 1. The intra- and inter-run relative standard deviation (RSD) values of quality control (QC) samples at different concentrations (low, medium and high levels) and lower limit of quantification (LLOQ) were <5.9%, <7.9%, <5.3% and <9.9%, respectively. The intra- and inter-run accuracy relative error (RE) values of QC samples at different concentrations (low, medium and high levels) and LLOQ were −9.5%~-0.4%, −13.3%~3.3%, −5.8%~-1.7% and −3.3%, respectively.
Safety and Tolerability
All subjects were under medical monitoring in the Phase I clinical trial center of West China Hospital of Sichuan University, during the study. Safety and tolerability were evaluated by monitoring Adverse Events (AEs), and main indicators included laboratory tests (hematology, blood biochemistry and urinalysis), vital signs, physical examination and 12-lead ECG. AEs were recorded after eligible healthy Chinese subjects were randomized and the analysis of AEs focused on treatment-emergent adverse events (TEAEs), which were AEs that occurred after the start of drug administration. The severity degrees of AEs were judged according to Common Terminology Criteria for Adverse Events (CTCAE) v5.0, and all laboratory examinations were performed in the clinical laboratory of West China Hospital, Sichuan University, authenticated by College of American Pathologists (CAP).
Pharmacokinetic and Statistical Analysis
Pharmacokinetic parameters were calculated using Phoenix WinNonlin Version 8.2 (Pharsight, Mountain View, CA, USA) using non-compartmental method, and other statistical analyses were conducted through SAS software Version 9.4 (SAS Institute Inc., Cary, NC, USA). Pharmacokinetic characteristics in the study included peak concentration (Cmax), plasma area under concentration–time curve from time 0 to last measurable concentration (AUC0-t), plasma area under concentration–time curve from time 0 to infinity (AUC0-∞), time to reach Cmax (Tmax), elimination half-life (t1/2) and terminal elimination rate constant (λz).
Pharmacokinetic concentration and parameter set (PKCS and PKPS) were analyzed in randomized subjects with at least one plasma concentration and pharmacokinetic parameter, respectively, and the safety analysis set (SS) referred to randomized subjects who had recorded safety data after receiving the study drugs. Subjects who completed at least one period of dosing with at least one pharmacokinetic parameter were included in the bioequivalence analysis set (BES).
For bioequivalence analysis set, the geometric mean ratios (test/reference) and their 90% CIs (confidence intervals) of Cmax, AUC0-t and AUC0-∞ were determined through an analysis of variance (ANOVA) mixed effect model with dosing period, dosing sequence and preparation factors as fixed effects, but subjects as the random effect after Cmax, AUC0-t and AUC0-∞ were natural logarithm transformed. The test and reference formulations of pramipexole dihydrochloride extended-release tablets were considered to be bioequivalent, when the 90% CIs of the geometric mean ratios (test/reference) of Cmax, AUC0-t and AUC0-∞ between the test and reference formulation were within 80%~125%. Moreover, signed rank test was used to compare the difference of Tmax between the two formulations. p< 0.05 was statistically significant for tests in the study.
Results
Study Population
A total of 44 subjects and 37 subjects were screened in the fasted trial and in the fed trial, respectively, and ultimately, 28 eligible subjects in each dietary trial were enrolled and randomized. The detailed demographic characteristics of enrolled subjects are demonstrated in Table 1, which were not significantly different between the fasted and fed trials (all p > 0.05).
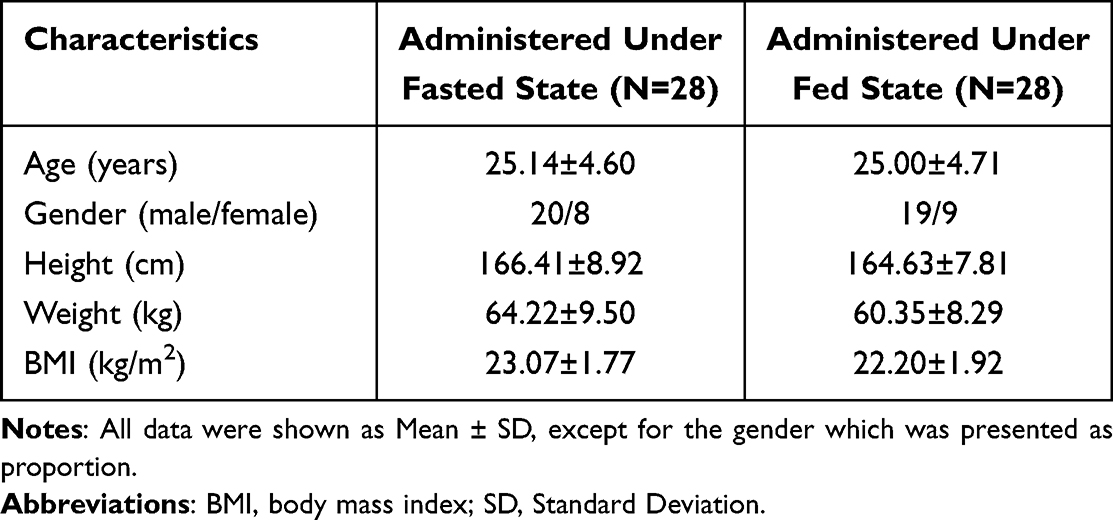 |
Table 1 Demographic Characteristics of Enrolled Subjects |
A total of 56 eligible subjects in the fasted and fed trial were enrolled and randomized. In the fasted trial, one subject in the T-R sequence withdrew after completing the first period of administration and blood collection, and one subject in the T-R sequence vomited within 24 h after dosing in the second period, thus only their data in first period were included for analysis. Besides, the plasma concentration at the first point reached Cmax after the first period of dosing for one subject in the T-R sequence, so merely the results in second period were analyzed. Other subjects in the fasted trial and all subjects in the fed trial completed the study in line with protocol and were included in the analysis sets of pharmacokinetics, bioequivalence and safety. The distribution of the study subjects under fasted and fed states are shown in Figure 2.
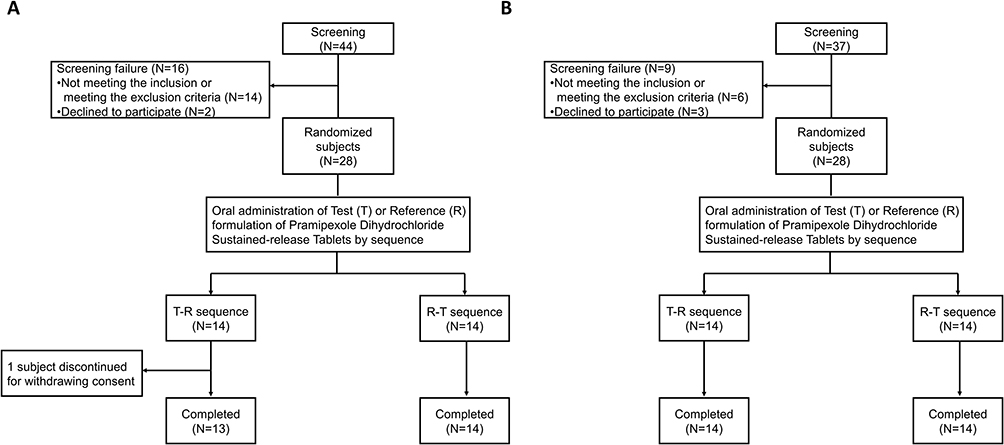 |
Figure 2 Distribution of the study subjects under (A) fasted and (B) fed states. |
Pharmacokinetic Properties
Main pharmacokinetic profiles of test and reference formulations of 0.375 mg pramipexole dihydrochloride extended-release tablets in healthy Chinese subjects under two dietary conditions are displayed in Table 2. In addition, the mean plasma concentration–time curves of pramipexole after a single oral administration of 0.375 mg test and reference formulations under fasted and fed conditions are shown in Figures 3 and 4 (A, linear scale; B, semi-logarithmic scale), respectively.
 |
Table 2 Pharmacokinetic Parameters of Pramipexole After Administration of 0.375 Mg Test and Reference Drugs in Healthy Chinese Subjects Under Fasted and Fed States |
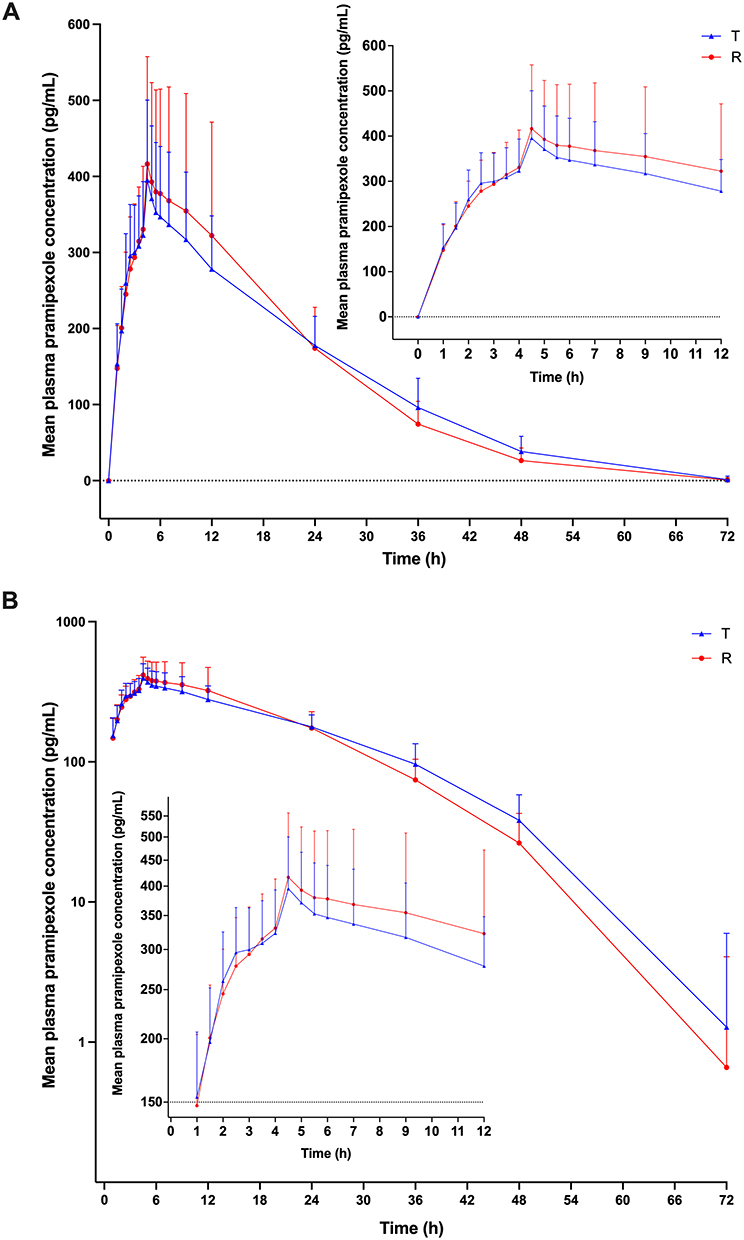 |
Figure 3 Mean plasma concentration–time curves of pramipexole after oral administration of 0.375 mg test (T) and reference (R) formulations of pramipexole dihydrochloride sustained-release tablet under fasted condition (A, linear scale; B, semi-logarithmic scale). Abbreviation: SD, standard deviation. Notes: Data were presented as Mean±SD. |
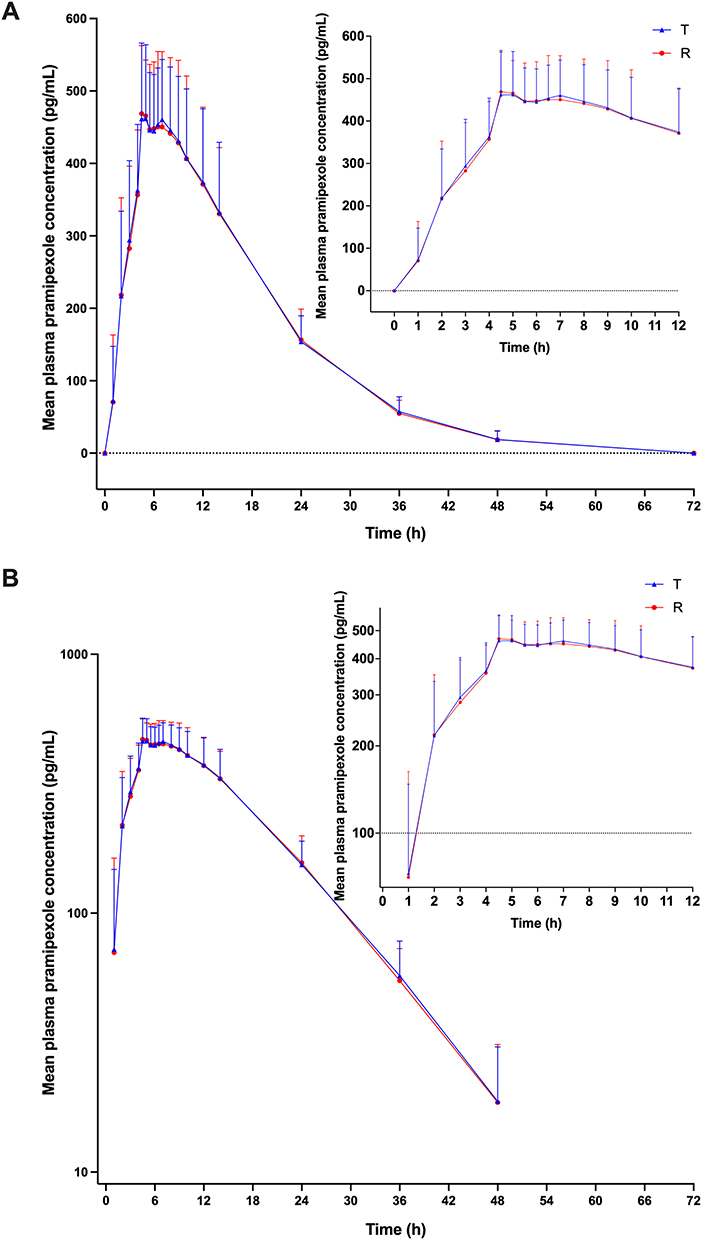 |
Figure 4 Mean plasma concentration–time curves of pramipexole after oral administration of 0.375 mg test (T) and reference (R) formulations of pramipexole dihydrochloride sustained-release tablet under fed condition (A), linear scale; (B), semi-logarithmic scale). Notes: Data were presented as Mean±SD. |
After single oral dosing of 0.375 mg test and reference formulations of pramipexole dihydrochloride extended-release tablets under fasted state, the mean ± standard deviation (SD) of Cmax of plasma pramipexole were 409.33 ± 95.93 pg/mL and 413.77 ±132.03 pg/mL; AUC0-t were 8801.95 ± 1966.83 h*pg/mL and 8646.37 ± 2600.49 h*pg/mL; AUC0-∞ were 9469.03 ± 1991.61 h*pg/mL and 9082.95 ± 2666.26 h*pg/mL; t1/2 were 11.98 ± 3.91 h and 9.85 ± 2.63 h; median (range) values of Tmax were 4.50 (2.00, 9.00) h and 4.50 (3.00, 12.02) h, respectively, for test and reference formulations. For the fed trial, the mean ± SD of Cmax of plasma pramipexole were 514.79 ± 77.42 pg/mL and 526.07 ± 92.92 pg/mL; AUC0-t were 9025.35 ± 1476.70 h*pg/mL and 8963.48 ± 1580.62 h*pg/mL; AUC0-∞ were 9347.24 ± 1469.11 h*pg/mL and 9289.73 ± 1569.19 h*pg/mL; t1/2 were 8.52 ± 1.55 h and 8.32 ± 1.45 h; median (range) values of Tmax were 5.00 (4.00, 12.00) h and 5.00 (2.00, 10.00) h, respectively, for test and reference formulations. Based on the above results, the pharmacokinetic features of the test and reference formulations were similar under both fasted and fed conditions (all p > 0.05).
Bioequivalence Evaluation
Furthermore, bioequivalence analysis of pharmacokinetic parameters of test and reference drugs after administration is revealed in Table 3. The geometric mean ratios (test/reference) and 90% CIs of Cmax, AUC0-t and AUC0-∞ of pramipexole after administration of 0.375 mg test and reference drugs under fasted state were 98.27% (90.47–106.74%), 101.92% (95.74–108.51%) and 104.91% (98.24–112.04%), respectively, and the geometric mean ratios and 90% CIs of Cmax, AUC0-t and AUC0-∞ under fed state were 98.31% (92.62–104.36%), 100.95% (97.23–104.81%) and 100.86% (97.19–104.67%), respectively. As indicated in Table 3, 90% CIs of the geometric mean ratios of Cmax, AUC0-t and AUC0-∞ under both fasted and fed conditions were all within 80%–125%, suggesting test and reference formulations of pramipexole dihydrochloride extended-release tablets were bioequivalent. Meanwhile, the Tmax between test and reference drugs under both fasted and fed conditions showed no significant difference (p = 0.14 and 0.44, respectively).
 |
Table 3 Bioequivalence Analysis of Main Pharmacokinetic Parameters of Pramipexole After Administration of 0.375 Mg Test and Reference Drugs in Healthy Chinese Subjects Under Fasted and Fed States |
Safety and Tolerability
All TEAEs of test and reference formulations of 0.375 mg pramipexole dihydrochloride extended-release tablets under fasted and fed states are summarized in Table 4.
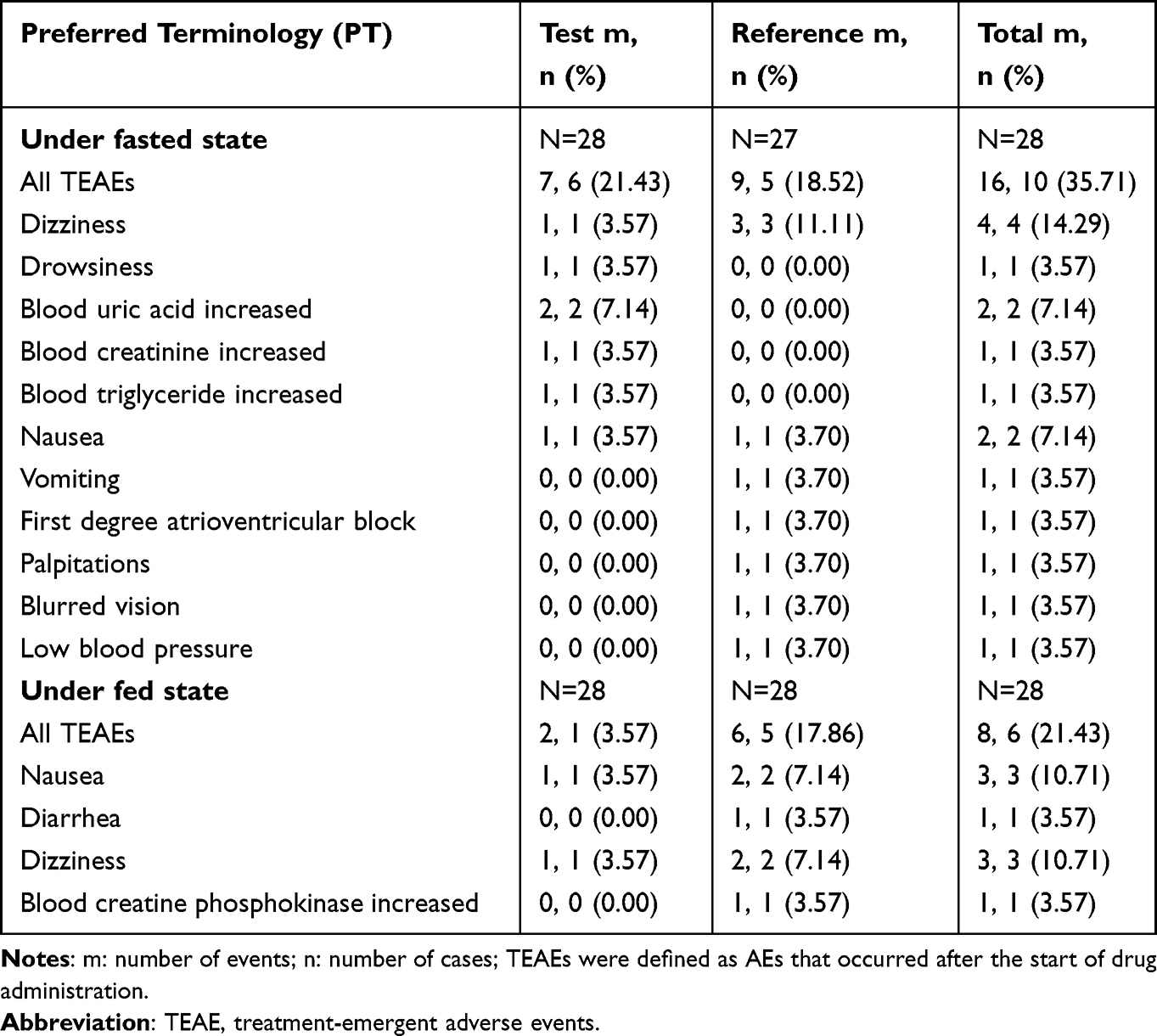 |
Table 4 Summary of All the Treatment-Emergent Adverse Events (TEAEs) After Administration of 0.375 Mg Test and Reference Drugs in Healthy Chinese Subjects Under Fasted and Fed States |
Among the 28 subjects in the fasted trial, 10 subjects experienced 16 TEAEs after administration, among which, 7 adverse events occurred in 6 subjects (21.43%) after receiving test formulation and merely 3 TEAEs (including dizziness, drowsiness and nausea) in 2 subjects (7.14%) were judged to be drug-related, while 9 adverse events happened in 5 subjects (18.52%) after taking reference drugs, all of which were considered to be drug-related including dizziness, nausea, vomiting, first degree atrioventricular block, palpitations, blurred vision and low blood pressure. For the fed trial, six subjects had eight TEAEs after dosing. A total of two adverse events happened in one subject (3.57%) for test formulation, all of which were drug-related such as nausea and dizziness, and 6 adverse events occurred in 5 subjects (17.86%) for reference formulation and 5 TEAEs (including nausea, diarrhea and dizziness) in 4 subjects (14.29%) were drug-related.
Grades of all TEAEs were ≤2 in severity and recovered without any intervention. No Grade ≥3 AE or AE resulting in discontinuation, serious AE and deaths occurred under both fasted and fed conditions during the study.
Discussion
Pramipexole is an approved and effective drug known as monotherapy for early PD or as an adjunctive approach to levodopa for advanced PD, showing less development of dyskinesia compared with levodopa treatment.2,11,19 Furthermore, novel long-acting formulation of pramipexole is designed and applied as a continuous delivery system, which not only exhibits similar efficacy and safety properties as immediate-release form,5,7,17 but also has advantages of less frequent administration, more stable plasma pramipexole concentration, convenient up-titration phase initiated at the lowest dosage strength 0.375 mg given once daily, and improved medication compliance over immediate-release pramipexole.2,15–17,20
However, pharmacokinetic properties of extended-release pramipexole in healthy Chinese subjects remained unclear. Therefore, a single-center, randomized, open-label, two-period crossover, single-administration study was to first investigate the pharmacokinetics of extended-release pramipexole and evaluate bioequivalence of 0.375 mg test and reference formulations of pramipexole dihydrochloride extended-release tablets in healthy Chinese subjects under fasted and fed states. For all eligible subjects in the study, plasma concentration of pramipexole before dosing in the second period was lower than the lower limit of quantification (15.0 pg/mL), furtherly verifying that the 7-day washout time was adequate to fully eliminate the effects of drug taken in the first period.
In our study, we showed that, after a single dose of 0.375 mg test and reference formulations under fasted condition, mean Cmax of plasma pramipexole were 409.33 and 413.77 pg/mL; AUC0-t were 8801.95 and 8646.37 h*pg/mL; AUC0-∞were 9469.03 and 9082.95 h*pg/mL; t1/2 were 11.98 and 9.85 h; both median Tmax were 4.50 h, respectively, for test and reference formulations. According to the data of Study 248.560 (NCT02260024) provided by US Food and Drug Administration (FDA) with regard to the reference formulation, the mean Cmax is 268 pg/mL; AUC0-∞ is 6610 h*pg/mL; t1/2 is approximate 9.38 h and the median Tmax is about 9.98 h in healthy male Caucasian subjects following a single dose of 0.375 mg extended-release pramipexole tablet under fasted condition.21,22 As we can see, the main pharmacokinetic characteristics of extended-release pramipexole in healthy Chinese subjects were different from results in healthy male Caucasian subjects. When compared to healthy male Caucasians, the Cmax and AUC0-∞ of pramipexole for both reference and test formulations were approximately increased by 50% and 40%, respectively, and average Tmax was reduced to 4.5 h for healthy Chinese subjects after administration at same dose under fasted state. Above tendencies resemble the pharmacokinetic differences between Study 248.607 (NCT02264132) and Study 248.530 (NCT02261103), in which, the Cmax and AUC of pramipexole are approximately 40–50% and 12–22% higher in healthy male Japanese subjects, respectively, when compared with data in healthy male Caucasians after oral administration of multiple ascending doses (0.375 mg to 1.5 mg, once-daily) of extended-release pramipexole tablets.22 The differences are most likely caused by the difference in body weight for the two studies, with the mean body weight of 60.2 kg in Japanese, while 79.3 kg in Caucasians.22 Because detailed demographic characteristics of Caucasian subjects receiving a single dose of 0.375 mg extended-release pramipexole tablet under fasted condition in Study 248.560 (NCT02260024) are not revealed, thus we cannot directly further analyze the reasons for the pharmacokinetic difference between healthy Chinese and Caucasians. However, we find that the main pharmacokinetic parameters of 0.375 mg extended-release pramipexole in healthy Chinese subjects of the study are similar to results in healthy male Japanese subjects, and the mean body weight of enrolled subjects in the study is also about 60 kg. In addition, although Cmax and AUC0-∞ of pramipexole are increased in healthy Chinese after single dose of 0.375 mg extended-release pramipexole in comparison with results in Caucasians, several randomized controlled trials have verified well safety and clinical efficacy of extended-release pramipexole in Chinese PD patients,5,23 so no special dose adjustment is required for Chinese.
The pharmacokinetic properties of the test and reference formulations were similar under both fasted and fed conditions. In fasted and fed states, the intra-individual variation rates of Cmax, AUC0-t and AUC0-∞ were all ≤30%, and power degrees of Cmax, AUC0-t and AUC0-∞ were all >99%, in addition, the geometric mean ratios (test/reference) and 90% CIs of Cmax, AUC0-t and AUC0-∞ were all within 80%–125% and Tmax between two formulations was not significantly different, furtherly verifying the bioequivalence of test and the reference formulations under fasted and fed conditions.
Besides, the food effects of reference formulation in healthy male Caucasians demonstrate that, for the highest strength 4.5 mg and lowest strength 0.375 mg, no food effects are observed for AUC of pramipexole after high-fat meal, although Cmax is increased by about 20% and 24% for 4.5 mg and 0.375 mg strengths, respectively, and steady-state Tmax is slightly prolonged by approximate 2 h for 4.5 mg strength under steady-state conditions, while the Tmax for 0.375 mg single-dose is shortened by about 4 h, compared with parameters under fasted state. The above differences are considered to be not clinically relevant for similar exposure under fasted and fed states.21,22 Therefore, a reference formulation of pramipexole dihydrochloride extended-release tablets is suggested to be taken once daily with or without food.22,24 In our study, the Cmax of test and reference formulations increased by 25% and 27.4%, respectively, and Tmax delayed by about 0.5 h, but the AUC of two formulations approximately remained unchanged after high-fat meal when compared with the data under fasted state in healthy Chinese subjects, which was consistent with the tendency of reference formulation in healthy male Caucasians22,24 and the increased Cmax was also deemed as no clinical significance for the preferable safety profiles and similar exposures of two formulations under fed and fasted conditions, thus the test formulation of pramipexole dihydrochloride extended-release tablet could also be administered under either fasted or fed state.
For safety assessments throughout the study, a single dose of 0.375 mg test and reference formations of pramipexole dihydrochloride extended-release tablets were both well tolerated and exhibited preferable safety profiles in healthy Chinese subjects. All TEAEs recovered without any treatment and the drug-related TEAEs with higher incidence were as follows: dizziness (T drug 3.57%, R drug 11.11% under fasted state and T drug 3.57%, R drug 7.14% under fed state) and nausea (T drug 3.57%, R drug 3.70% under fasted condition and T drug 3.57%, R drug 7.14% under fed state), which was in agreement with the commonly reported drug-regarding TEAEs of prescribing information of reference formulation.22,24
Some limitations of our study should be pointed out. The pharmacokinetics, efficacy and safety of test formulation in patients were not explored and remained unknown in the study, which might be different from healthy subjects. Therefore, further clinical studies regarding test formulation of pramipexole dihydrochloride extended-release tablets in patients with PD are needed, especially the incidences of impulse control disorder should be emphasized and noticed in PD patients while taking extended-release pramipexole according to the prescribing information of reference formulation.24
Conclusion
The test formulation of 0.375 mg pramipexole dihydrochloride extended-release tablet manufactured by Yangtze River Pharmaceutical Group Co., Ltd. (Jiangsu, China) was bioequivalent to 0.375 mg reference formulation (Trade name: Sifrol®) under both fasted and fed conditions, and two formulations could be administered under either fasted or fed state. In addition, a single oral administration of 0.375 mg of the two formulations was well tolerated with favorable safety profiles in healthy subjects. These results in the study support further subsequent clinical studies of test formulation of pramipexole dihydrochloride extended-release tablets in patients with Parkinson’s disease.
Data Sharing Statement
The data in the study are available by contacting the corresponding author upon reasonable request for research purpose.
Acknowledgments
We would like to thank the support from Yangtze River Pharmaceutical Group Co., Ltd. (Jiangsu, China) and express our appreciation to all the study nurses in Phase I clinical trial center ward for sampling. We would like to acknowledge all the human subjects for participation in the study.
Funding
The study was funded by Yangtze River Pharmaceutical Group Co., Ltd. (Jiangsu, China).
Disclosure
All authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
1. Obeso JA, Stamelou M, Goetz CG, et al. Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the Shaking Palsy. Mov Disord. 2017;32(9):1264–1310. doi:10.1002/mds.27115
2. Fox SH, Katzenschlager R, Lim SY, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018;33(8):1248–1266. doi:10.1002/mds.27372
3. Study Group P. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA. 2000;284(15):1931–1938. doi:10.1001/jama.284.15.1931
4. Stocchi F, Torti M, Fossati C. Advances in dopamine receptor agonists for the treatment of Parkinson’s disease. Expert Opin Pharmacother. 2016;17(14):1889–1902. doi:10.1080/14656566.2016.1219337
5. Wang Y, Sun S, Zhu S, et al. The efficacy and safety of pramipexole ER versus IR in Chinese patients with Parkinson’s disease: a randomized, double-blind, double-dummy, parallel-group study. Transl Neurodegener. 2014;3:11. doi:10.1186/2047-9158-3-11
6. Chen XT, Zhang Q, Wen SY, Chen FF, Zhou CQ. Efficacy and safety of non-ergot dopamine-receptor agonists as an adjunct to levodopa in advanced Parkinson’s disease: a network meta-analysis. Eur J Neurol. 2023;30(3):762–773. doi:10.1111/ene.15635
7. Frampton JE. Pramipexole extended-release: a review of its use in patients with Parkinson’s disease. Drugs. 2014;74(18):2175–2190. doi:10.1007/s40265-014-0322-5
8. Salawu FK. Patient considerations in early management of Parkinson’s disease: focus on extended-release pramipexole. Patient Prefer Adherence. 2012;6:49–54. doi:10.2147/ppa.S11841
9. Hametner EM, Seppi K, Poewe W. Role and clinical utility of pramipexole extended release in the treatment of early Parkinson’s disease. Clin Interv Aging. 2012;7:83–88. doi:10.2147/cia.S11829
10. Hauser RA, Schapira AH, Barone P, et al. Long-term safety and sustained efficacy of extended-release pramipexole in early and advanced Parkinson’s disease. Eur J Neurol. 2014;21(5):736–743. doi:10.1111/ene.12375
11. Schapira AH, Barone P, Hauser RA, et al. Extended-release pramipexole in advanced Parkinson disease: a randomized controlled trial. Neurology. 2011;77(8):767–774. doi:10.1212/WNL.0b013e31822affdb
12. Poewe W, Rascol O, Barone P, et al. Extended-release pramipexole in early Parkinson disease: a 33-week randomized controlled trial. Neurology. 2011;77(8):759–766. doi:10.1212/WNL.0b013e31822affb0
13. Hauser RA, Schapira AH, Rascol O, et al. Randomized, double-blind, multicenter evaluation of pramipexole extended release once daily in early Parkinson’s disease. Mov Disord. 2010;25(15):2542–2549. doi:10.1002/mds.23317
14. Antonini A, Calandrella D. Once-daily pramipexole for the treatment of early and advanced idiopathic Parkinson’s disease: implications for patients. Neuropsychiatr Dis Treat. 2011;7:297–302. doi:10.2147/ndt.S10097
15. Shen T, Ye R, Zhang B. Efficacy and safety of pramipexole extended-release in Parkinson’s disease: a review based on meta-analysis of randomized controlled trials. Eur J Neurol. 2017;24(6):835–843. doi:10.1111/ene.13303
16. Schapira AH, Barone P, Hauser RA, et al. Patient-reported convenience of once-daily versus three-times-daily dosing during long-term studies of pramipexole in early and advanced Parkinson’s disease. Eur J Neurol. 2013;20(1):50–56. doi:10.1111/j.1468-1331.2012.03712.x
17. Perez Lloret S, Rascol O. Pramipexole extended-release (once-daily formulation) for the treatment of Parkinson’s disease. Expert Opin Pharmacother. 2010;11(13):2221–2230. doi:10.1517/14656566.2010.510515
18. Antonini A, Calandrella D. Pharmacokinetic evaluation of pramipexole. Expert Opin Drug Metab Toxicol. 2011;7(10):1307–1314. doi:10.1517/17425255.2011.614232
19. Tayarani-Binazir KA, Jackson MJ, Rose S, Olanow CW, Jenner P. Pramipexole combined with levodopa improves motor function but reduces dyskinesia in MPTP-treated common marmosets. Mov Disord. 2010;25(3):377–384. doi:10.1002/mds.22960
20. Shen Z, Kong D. Meta-analysis of the adverse events associated with extended-release versus standard immediate-release pramipexole in Parkinson disease. Medicine. 2018;97(34):e11316. doi:10.1097/md.0000000000011316
21. Jenner P, Könen-Bergmann M, Schepers C, Haertter S. Pharmacokinetics of a once-daily extended-release formulation of pramipexole in healthy male volunteers: three studies. Clin Ther. 2009;31(11):2698–2711. doi:10.1016/j.clinthera.2009.10.018
22. Co.KG BIPG. Sifrol® (pramipexole dihydrochloride extended-release tablets) clinical pharmacology and biopharmaceutics review(s); 2008. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022421s000ClinPharmR.pdf.
23. Ma JF, Wan Q, Hu XY, et al. Efficacy and safety of pramipexole in Chinese patients with restless legs syndrome: results from a multi-center, randomized, double-blind, placebo-controlled trial. Sleep Med. 2012;13(1):58–63. doi:10.1016/j.sleep.2011.03.021
24. Co.KG BIPG. Sifrol® (pramipexole dihydrochloride extended-release tablets) prescribing information; 2021. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/022421s022lbl.pdf.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of Pharmacokinetics and Safety with Bioequivalence of Ibuprofen Sustained-Release Capsules of Two Formulations, in Chinese Healthy Volunteers: Bioequivalence Study
Huang C, Yin Z, Yang Y, Mo N, Yang H, Wang Y
Drug Design, Development and Therapy 2023, 17:1881-1888
Published Date: 23 June 2023
Effect of Food on the Pharmacokinetics of Tenofovir Amibufenamide: A Phase I, Randomized, Open-Label, Two-Period Crossover Trial in Healthy Adult Subjects

Liu J, Wu M, Kai J, Lin M, Zheng Y, Jiang Y, Huang Q, Zhai Y, Qiu Y
Drug Design, Development and Therapy 2023, 17:3061-3072
Published Date: 9 October 2023
Evaluation of Olaparib Tablet Safety and Pharmacokinetics in Healthy Chinese Male Subjects
Dong R, Chen J, Guo N, Yang Y, Wu J, Wang X, Song Y, Zhang X
Drug Design, Development and Therapy 2024, 18:5529-5539
Published Date: 3 December 2024
Pharmacokinetics and Bioequivalence of Two Fixed-Dose Combination Tablets of Valsartan/Amlodipine (80/5 Mg) in Healthy Chinese Subjects
Tian M, Huang J, Chen Y, Jin Q, Jiang H, Shi C, Mei J, Xu M, Yu X, Yang S
Drug Design, Development and Therapy 2025, 19:11-22
Published Date: 3 January 2025
Pharmacokinetics and Safety with Bioequivalence of Isosorbide Mononitrate Sustained-Release Tablets in Chinese Healthy Volunteers: Bioequivalence Study
Wu B, Wang W, Zhang Q, Yu G, Lin J, Zhang T, Ye L, Wang K, Zhang W, Wu X
Drug Design, Development and Therapy 2025, 19:6025-6035
Published Date: 14 July 2025