Back to Journals » Journal of Pain Research » Volume 15
Multiple Dose Pharmacokinetics of Tapentadol Oral Solution for the Treatment of Moderate to Severe Acute Pain in Children Aged 2 to <7 Years
Authors Jończyk R, Beuter C ![]() , Bulawa B, Buller S
, Bulawa B, Buller S ![]() , Eibl C
, Eibl C ![]() , Elling C
, Elling C ![]() , Gautrois M, Rengelshausen J
, Gautrois M, Rengelshausen J ![]() , Schmidt C
, Schmidt C ![]() , Thömmes G, Khalil F
, Thömmes G, Khalil F ![]()
Received 27 May 2022
Accepted for publication 30 August 2022
Published 30 September 2022 Volume 2022:15 Pages 3103—3114
DOI https://doi.org/10.2147/JPR.S364902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
Renata Jończyk,1 Christoph Beuter,2 Beata Bulawa,2 Stefan Buller,2 Christoph Eibl,2 Christian Elling,2 Michael Gautrois,2 Jens Rengelshausen,2 Carsten Schmidt,2 Guido Thömmes,2 Feras Khalil2
1Center for Hospital Health Care Services, Clinic of Pediatric Surgery, Subdivision of Pediatric Urology, Rzeszow, Poland; 2Grünenthal GmbH, Aachen, Germany
Correspondence: Feras Khalil, Grünenthal GmbH, Zieglerstraße 6, Aachen, 52078, Germany, Tel +49 241 569 3295, Email [email protected]
Background: This prospective, open-label trial was conducted to fulfil a post-approval commitment made to the competent authorities to extend the indication of the strong opioid analgesic tapentadol hydrochloride oral solution (OS) to the pediatric population.
Patients and Methods: The trial assessed the pharmacokinetic (PK) profile of tapentadol, tapentadol-O-glucuronide and tapentadol-O-sulfate after administration of multiple doses of tapentadol OS (1.25 mg tapentadol/kg bodyweight every 4 h for up to 72 h) in children aged 2 to < 7 years after a painful event that produces acute pain requiring treatment with a strong analgesic. The obtained PK data were integrated into a previously developed population PK (popPK) model based on single-dose data and then a model-based PK evaluation was performed. The primary trial endpoint was the area under the concentration-time curve at steady state for the dosing interval (AUCτ,ss) for tapentadol.
Results: Ten children received tapentadol OS; all completed the trial. Multiple administrations of the trial medication resulted in tapentadol serum concentrations within the concentration range predicted by the previously developed popPK model. The estimated model-based AUCτ,ss values for tapentadol ranged from 142 to 321 h•ng/mL. They were within the predicted exposure range with no higher than expected accumulation for the employed dosing regimen and also within the targeted steady state exposure range observed in adults receiving multiple doses of immediate release tapentadol 50 to 100 mg. The treatment regimen was safe and well tolerated.
Conclusion: The findings confirm the linear and predictable PK profile of tapentadol hydrochloride. The good agreement between the observed data and the model predictions shows the value of modelling and simulations in the planning and analysis of pediatric clinical trials and the ability to utilize the established PK models to predict multiple dose exposure.
Keywords: acute pain, children, multiple dosing, pharmacokinetics, popPK model, tapentadol
Introduction
The strong opioid analgesic tapentadol hydrochloride acts through a combination of μ-opioid receptor agonism and noradrenaline reuptake inhibition.1 These mechanisms of action synergistically provide analgesic effects comparable to other strong opioids but with reduced μ-opioid receptor activation (reduced µ-load).1,2 Owing to the opioid mechanism of the tapentadol molecule, opioid-related risks apply. Tapentadol provided effective pain relief in a broad range of acute and chronic pain indications in adult patients3–5 with a more favorable gastrointestinal tolerability profile compared to opioids solely acting through µ-receptor agonism. In addition, the medication has a favorable pharmacokinetic (PK) profile in adults6 with no active metabolites contributing to the analgesic effect7 and a low potential for drug–drug interaction.8,9 The main metabolic pathways are glucuronidation and sulfation.7 Up until the end of 2021, a total of 16.7 million patients had been treated globally with the medication.
The predictable PK profile and the positive benefit/risk assessment in adults made tapentadol a suitable candidate for pain treatment in children and it became the first pain medication to be comprehensively developed for the treatment of pediatric pain within a regulatory framework. The thematic pediatric tapentadol series published in this journal provides detailed information about the development program.10–18 The results of clinical trials conducted with pediatric patients suffering from acute pain led to the approval of tapentadol oral solution (OS) in the European Union. Tapentadol OS is indicated for the relief of moderate to severe acute pain in children from 2 years of age, which can be adequately managed only with opioid analgesics.19 The treatment is restricted to hospitalized children and a maximum treatment period of 3 days.
The PK profile of tapentadol in children with acute pain was determined using oral and intravenous single-dose trials with a limited number of trial participants in order to minimize medication exposure in this vulnerable population. The number of blood samples was also restricted and led to the extensive use of modelling and simulation techniques to inform the design of the conducted trials and to support the PK analysis of the collected data. Exposure data after multiple dose administrations of tapentadol OS were thus only estimated based on single-dose PK data by population PK (popPK) modelling. During the regulatory procedure to extend the indication for tapentadol OS to the pediatric population suffering from acute pain, an agreement with the competent authorities was reached to conduct a post-approval multiple dose PK clinical trial in children from 2 to <7 years of age. Results of this trial are the subject of this article.
Patients and Methods
Trial Design
This prospective, open-label, non-controlled, multicenter clinical trial was designed to assess the PK profile of tapentadol and its metabolites tapentadol-O-glucuronide and tapentadol-O-sulfate after administration of multiple doses of tapentadol OS. The trial included an enrollment period not exceeding 28 days, a treatment and evaluation period of up to 4 days, and a follow-up period of between 10 and 14 days after end of treatment (Figure 1). The painful event leading to inclusion in the trial was not considered part of the trial; patients could be enrolled before or after the event. They had to remain hospitalized during the treatment and evaluation period.
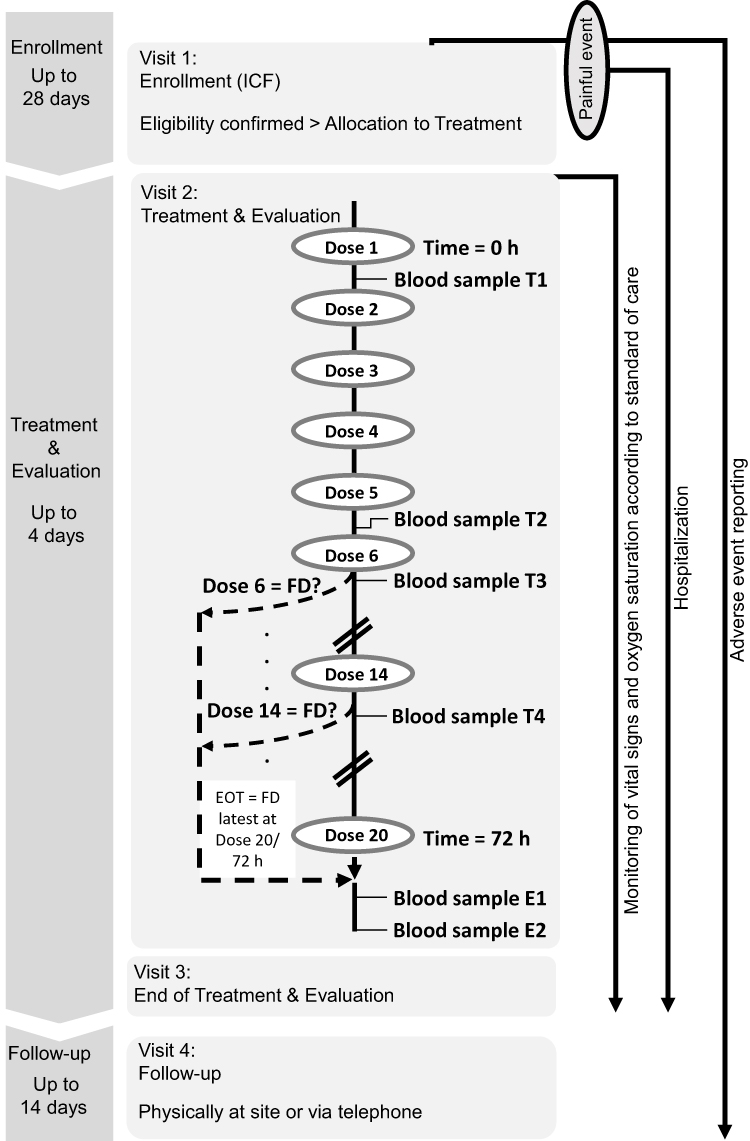 |
Figure 1 Trial design. Timepoints for blood sample collection. T1: 1.5 h (±10 min) after dose 1. T2: 3.5 h (±10 min) after dose 5. T3: 1.5 h (±10 min) after dose 6. T4: 3.5 h (±10 min) after dose 14 (if dose 14 is administered). E1: 5 h (±10 min) after final dose. E2: 10 h (±10 min) after final dose. Abbreviations: EOT, end of treatment; FD, final tapentadol dose; ICF, informed consent form. |
The trial was conducted from September 2019 to August 2020 in Poland in accordance with the Declaration of Helsinki, Good Clinical Practice, and applicable local laws and regulations. Since the trial partly overlapped with the COVID-19 pandemic, trial risk assessment was updated and additional precautionary measures regarding the trial setup were put in place. Two of the three planned trial sites enrolled patients. The protocol, amendments, and applicable informed consent/assent forms were approved by the ethics committees of these two sites (Komisja Bioetyczna przy Śląskiej Izbie Lekarskiej, Katowice, and Komisja Bioetyczna Uniwersytetu Mikołaja Kopernika w Toruniu przy Collegium Medicum im. Ludwika Rydygiera w Bydgoszczy, Bydgoszcz). Written informed consent from all parents/legal guardians as well as patient assent (where applicable) was obtained. The trial is registered as EudraCT no. 2019-000205-77.
Trial Population
The trial population included children aged 2 to <7 years who experienced a painful intervention or surgery that, in the investigator’s opinion, would reliably produce acute pain requiring treatment with a strong analgesic medication for at least 24 h following first dose administration. Further requirements were the ability to tolerate liquids, and a reliable venous vascular access or tolerance for repeated venipuncture procedures for PK blood sampling. Main exclusion criteria were a previous exposure to tapentadol, obesity or a bodyweight below 9 kg, a concomitant disease or disorder that, in the opinion of the investigator, could affect or compromise the patients’ safety during the trial, a history of seizure disorder or brain injury, renal or hepatic impairment, acute or severe bronchial asthma or hypercapnia, and a clinically relevant history of hypersensitivity, allergy, or contraindication to tapentadol, its excipients, or naloxone. Medications prohibited within 14 days prior to tapentadol OS included monoamine oxidase inhibitors, strong inducers of hepatic drug-metabolizing enzymes such as rifampicin, phenobarbital, or St John’s Wort, and strong inhibitors of uridine diphosphate transferases such as probenecid, ketoconazole, fluconazole, and meclofenamic acid.
Treatment
The OS formulation of tapentadol is available in the two concentrations 4 mg/mL and 20 mg/mL. Patients weighing ≤16 kg were administered the 4 mg/mL solution, patients weighing >16 kg received the 20 mg/mL concentration. The route of administration was oral or, if required, by orogastric, nasogastric, or gastric tube. Patients received 1.25 mg tapentadol/kg bodyweight every 4 h (±15 min) for up to 72 h (Figure 1). Treatment was discontinued either after 72 h or earlier in case treatment with a strong analgesic was no longer required.
Pharmacokinetic Assessment
A maximum of 6 venous blood samples (0.5 mL/sample) for determination of serum concentrations of tapentadol, tapentadol-O-glucuronide, and tapentadol-O-sulfate were collected at the predefined times shown in Figure 1. Serum concentrations were determined using a validated liquid chromatography-tandem mass spectrometry method after protein precipitation with acetonitrile. Lower limits of quantification (LLOQ) of the bioanalytical method were 0.2 ng/mL for tapentadol, 10 ng/mL for tapentadol-O-glucuronide, and 1 ng/mL for tapentadol-O-sulfate; the corresponding correlation coefficients obtained for the three compounds during analysis of the samples were ≥0.9942, ≥0.9891 and ≥0.9929.
Pharmacometric Assessment
The concentration-time data obtained in this trial were used to validate and update an existing tapentadol popPK model, which was previously developed based on combined tapentadol OS and intravenous (IV) data from single-dose pediatric trials.15 The tapentadol model update means that the PK model structure as well as the population parameter estimates were re-visited based on an integrated analysis of all data pooled from the present trial and 4 previous pediatric trials following the administration of tapentadol IV solution or tapentadol OS (ie, n = 5 trials in total for the model update). This model was subsequently extended to a joint tapentadol parent-metabolite popPK model. The popPK models were then used to perform a model-based PK evaluation due to the sparse nature of the collected trial PK data.
However, as part of the trial data exploration and prior to any modelling update, initial simulations were performed using the previous pediatric popPK model based on single-dose tapentadol trials.15 These simulations were intended to serve as a priori confirmation for the accuracy of the prediction of tapentadol exposure and accumulation after multiple dosing from the popPK model that was previously developed based on single-dose data only. To perform the simulations, a virtual pediatric population of n = 1000 different subjects was created by randomly sampling an age (random sampling from a uniform distribution between 2 to <7 years), a gender (random sampling of gender [male/female] with equal probability), and a z-score (random sampling from a normal distribution with mean = 0 and SD = 1) for each virtual child. Subsequently, the LMS parameters reported in the Centers for Disease Control and Prevention weight-for-age growth charts20 were used to calculate the weight of each virtual child based on its age, gender, and z-score as follows:
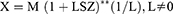
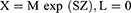
Where
- X is the weight of the subject,
- M is the median, S is the generalized coefficient of variation, and L is the power in the Box-Cox transformation values from the appropriate gender-specific weight-for-age charts table corresponding to the age in months of the child,
- Z is the simulated z-score that corresponds to the weight-for-age percentile.20
The simulation model included a maturation function on clearance (CL) that is driven by the postmenstrual age (calculated as gestational age [GA] plus postnatal age), which is important when simulating pre-term subjects. However, as all simulations were planned for children above 2 years of age, the GA was set to 40 weeks for full terms. The dosing in the simulations was set to mimic the actual dosing of the patients enrolled in the multiple dose trial to enable a direct comparison with observed data.
The popPK models were developed by means of nonlinear mixed effects modeling using the first-order method with conditional estimation and interaction; most model assumptions are detailed elsewhere.15 The final popPK model was validated by visual predictive checks and by bootstrap to ensure adequate predictive power and model stability, and to assess the uncertainty in the parameter estimates. Population and individual PK parameter estimates were assessed including model-based estimates of maximum concentration after single dose (Cmax,sd) and at steady state (Cmax,ss), minimum concentration after single dose (Cmin,sd) and at steady state (Cmin,ss), the area under the concentration-time curve for the dosing interval after single dose (AUCτ,sd) and at steady state (AUCτ,ss), and an accumulation factor (AF); parameter estimates were made in each case for tapentadol, tapentadol-O-glucuronide, and tapentadol-O-sulfate. AF was calculated as AUCτ,ss / AUCτ,sd.
Exploratory Efficacy Assessment
Efficacy was assessed in an exploratory manner using pain intensity scores determined with the age-specific Face, Legs, Activity, Cry, and Consolability (FLACC)21 scale from 0 = no pain to 10 = most severe pain. Measurements were taken within 10 min prior to first tapentadol OS administration and approx. 1.5 h after each following dose. In addition, pain intensity was assessed within 10 min prior to each PK blood sample collection. A final assessment was performed between 12 and 24 h after the final dose. Patients could be awake or asleep at the time the measurement was taken.
Safety and Tolerability Assessment
A safety laboratory assessment according to local standard of care was performed at enrollment. A physical examination was carried out at enrollment and between 12 and 24 h following the final tapentadol OS dose. Vital signs (respiratory rate, blood pressure, and heart rate) and oxygen saturation (by pulse oximetry) were assessed at enrollment and during treatment and evaluation. Adverse events were monitored throughout the trial.
Statistical Analysis
Sample size was determined based on cohort size and experience from a pediatric tapentadol IV formulation PK trial which considered eight patients per treatment group sufficient to meet a power criterion of at least 80%.17 To achieve the same power, at least 8 evaluable patients were deemed necessary for the present trial. Patients were considered evaluable if they had been treated with tapentadol OS for at least 24 h (ie, dose 6 was received before treatment was stopped) and did not vomit within 3 h after dose administration. Regulatory requirements stipulated at least 2 patients to be aged between 2 to <3 years.
The PK analysis included all patients with a quantifiable serum concentration of at least tapentadol, tapentadol-O-glucuronide, or tapentadol-O-sulfate. Concentration values below LLOQ were not considered. In case serum concentrations might have been influenced by vomiting, diarrhea, fever or other factors, the PK expert, clinical scientist, and biostatistician of the trial decided whether to exclude specific samples or subsets from the calculations. Only evaluable patients were included in the endpoint analyses. The primary trial endpoint was the AUCτ,ss for tapentadol. Secondary endpoints were the AUCτ,ss for tapentadol-O-glucuronide and tapentadol-O-sulfate, respectively.
Efficacy and safety analyses included all patients who had received at least one dose of tapentadol OS. As the trial was powered for PK measurements, efficacy assessments were exploratory.
Statistical calculations except for the popPK analysis were performed using SAS version 9.4 (SAS Institute Inc., Cary, USA). The popPK data analysis was performed using NONMEM® (version 7.4, ICON Development Solutions, Ellicott City, MD, USA) supplemented with the PsN toolkit version 3.6.2 for model building procedure, bootstrap and visual predictive checks. The statistical software R (v3.5.1, R Foundation for Statistical Computing, Austria, 2010) was used to read and process NONMEM outputs, produce exploratory analysis and diagnostic plots, run simulations and to perform calculations. Adverse events were encoded using the Medical Dictionary for Regulatory Activities (MedDRA) version 23.0.
Results
Patients
Ten patients were enrolled; all received tapentadol OS and completed the trial. Table 1 lists their baseline characteristics. All painful events were surgical interventions (Table 1). Concomitant medications were mostly administered for painful event standard of care and included other analgesics and antipyretics for all 10 patients. Eight patients were considered evaluable as per protocol. Two patients were assessed as non-evaluable; one spat out tapentadol OS, the other regurgitated within 3 h of dose administration.
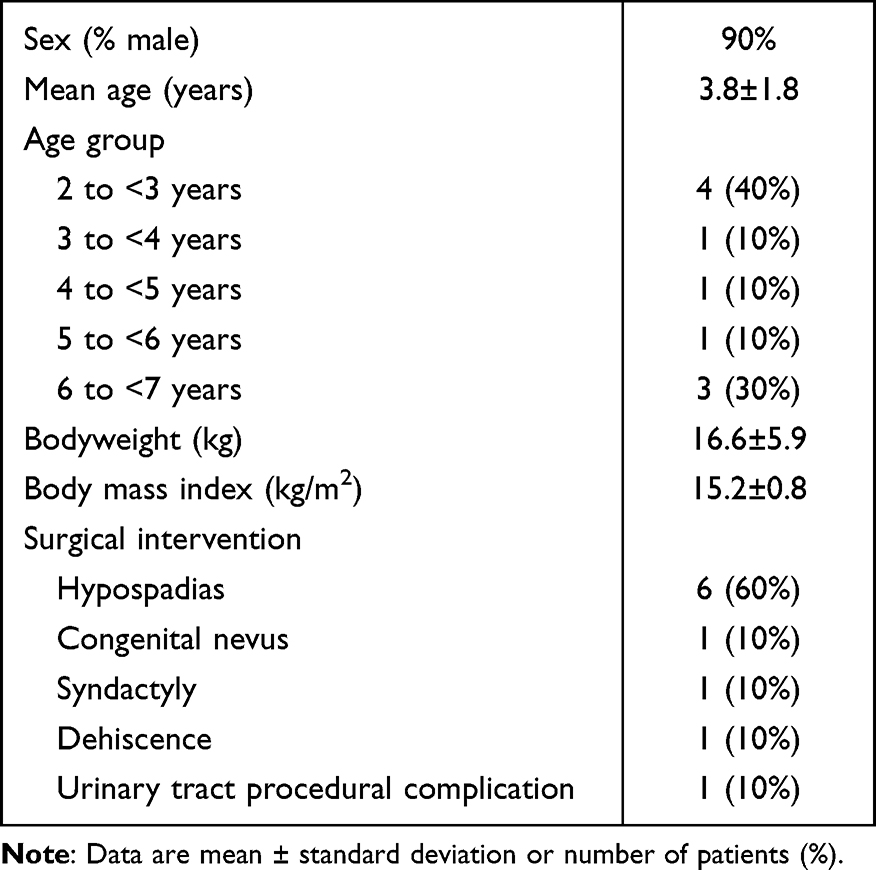 |
Table 1 Demographic Characteristics of the Trial Population (n = 10) |
Pharmacokinetics
Eight patients received six doses of tapentadol OS, one was administered 7 doses, and one patient received 14 doses. Six blood samples were excluded from analysis owing to regurgitation within 3 h of dose administration (4 samples from one patient) or spitting out part of the medication (2 samples from one patient). In addition, one sample was discarded at laboratory (1 sample), and therefore could not be used. Overall, 44 blood samples from the present trial were available for PK analysis. Table 2 shows the mean serum concentrations of tapentadol and its O-glucuronide and O-sulfate metabolites over the trial period. Mean tapentadol-O-glucuronide concentrations were substantially higher, mean tapentadol-O-sulfate concentrations were similar or slightly higher than those observed for tapentadol. Serum concentration data of all 3 compounds after the final dose were within the range of concentration data seen in earlier pediatric single-dose tapentadol trials (Figure 2).
 |
Table 2 Mean Serum Concentrations of Tapentadol, Tapentadol-O-Glucuronide, and Tapentadol-O-Sulfate (n = 10) |
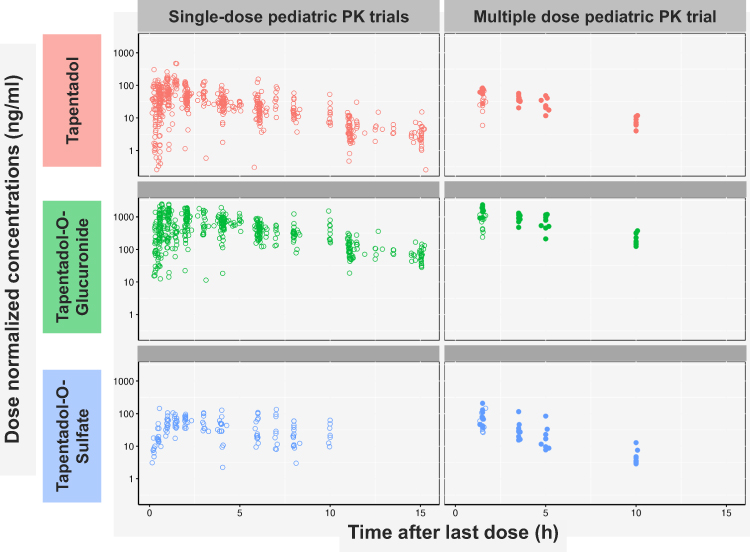 |
Figure 2 Comparison of serum concentration data of tapentadol and its two main metabolites at different time points after final tapentadol oral solution administration between this trial and earlier single-dose pediatric trials. Note: Patients in single-dose trials received either tapentadol oral solution or intravenous tapentadol.11,12,17 Serum concentrations were normalized to a dose of 1 mg/kg bodyweight. |
Pharmacometrics
Comparison of Observed Serum Concentrations with Pre-Trial Simulations
As patients received either 6, 7 or 14 doses of tapentadol OS, 3 sets of simulations were performed to mimic each of the actual dosing scenarios and thus allow a direct comparison with the individually observed concentrations. Additional graphical presentation was provided to allow a direct comparison of the mean predicted to the mean observed concentrations from all subjects taking into account that tapentadol steady state is already reached by the sixth dose and that the last two blood samples (designated E1 and E2) were collected at the same time relative to the last given dose (5 h and 10 h after the last dose). Simulations showed a very good agreement between the a priori model-based predictions and exposure data observed in this trial (Figure 3). This finding indicates that tapentadol accumulation after multiple dosing every 4 h was already well captured by the popPK model based on single-dose trials alone.
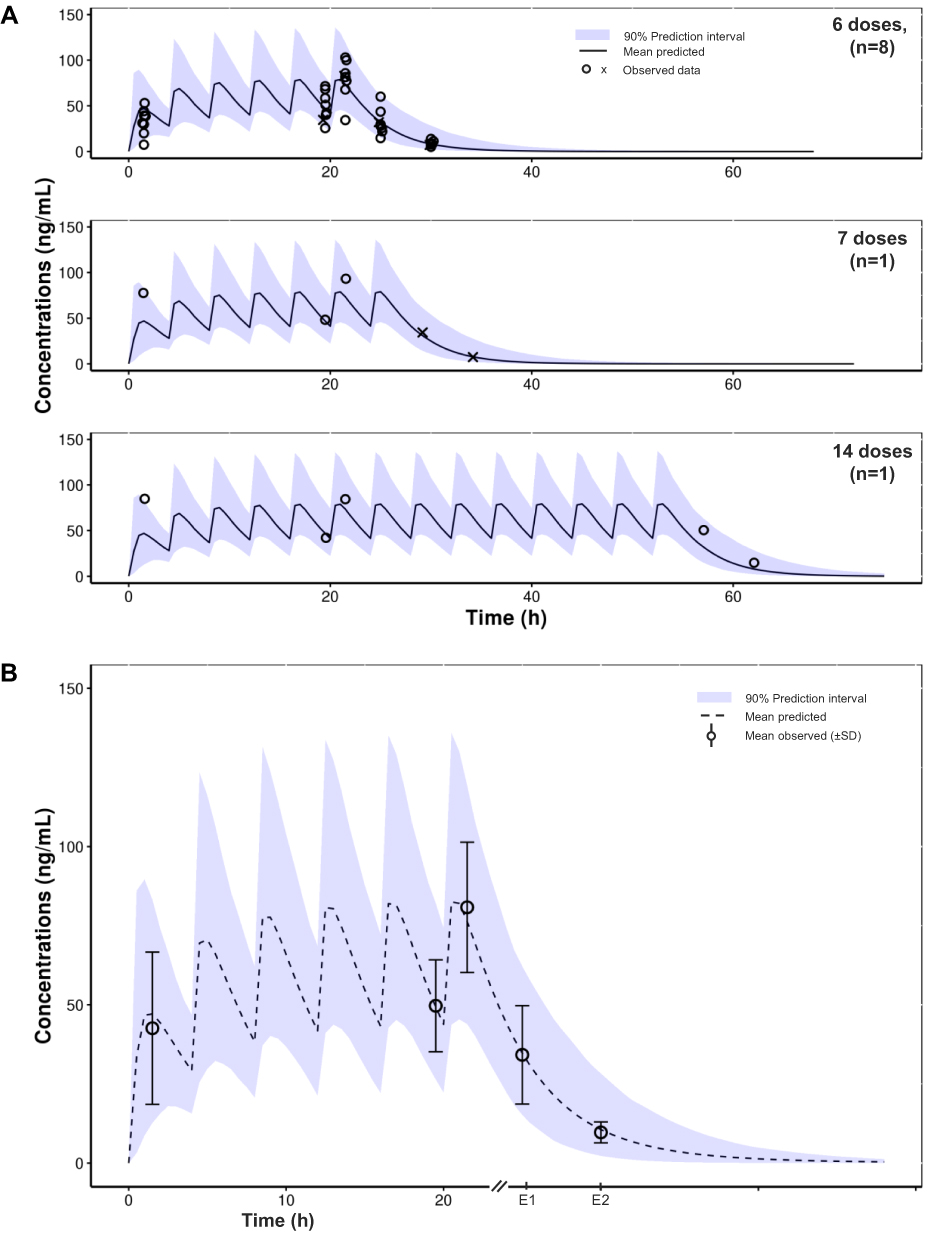 |
Figure 3 Tapentadol concentrations observed in the present trial after multiple dosing compared with simulations using the previously developed pediatric population pharmacokinetic model based on single-dose pediatric trials. (A) Concentration-time data for individual patients, stratified by number of administered doses. (B) Mean observed data (± standard deviation); data from evaluable patients only. Abbreviation: SD, standard deviation. Notes: E1 and E2 refer to sample collection timepoints 5 and 10 h after final dose. X refers to data following an incidence of regurgitation or spitting out of tapentadol oral solution dose, which were excluded from the calculated descriptive statistics presented in (B) and from the population pharmacokinetic analysis. |
Updated Tapentadol and Metabolite popPK Models
Tapentadol PK was best described by a 1-compartment model with linear absorption including absorption lag time, linear elimination, interindividual variability on CL, volume (V), and oral absorption rate constant (Ka), and a combined error model. The model also included allometric scaling on the disposition parameters CL and V, as well as maturation functions on oral bioavailability (F) and CL. Tapentadol mature CL and V at a reference weight of 70 kg were estimated to be 93.7 L/h (= CL/F of 272 L/h) and 414 L (= V/F of 1203 L), respectively. This model was subsequently extended to a joint tapentadol parent-metabolite model in order to allow the characterization of tapentadol-O-glucuronide and tapentadol-O-sulfate PK profiles. The population estimates of the systemic metabolite CL and V at a reference weight of 70 kg were 10.5 L/h and 28.1 L for tapentadol-O-glucuronide, and 30.9 L/h and 16.8 L for tapentadol-O-sulfate, respectively. The fraction of tapentadol that is metabolized to glucuronide and to sulfate was assumed to be 0.55 and 0.15, respectively, based on information obtained in adults.7
Further details about estimated model parameters and visual predictive checks for the developed popPK models are provided as supplementary material (Tables S1 and S2, Figures S1 and S2).
Model-Based Pharmacokinetic Evaluation
Table 3 shows the model-based estimated PK parameters of tapentadol and its two metabolites. The estimated model-based primary endpoint (AUCτ,ss) values calculated for tapentadol ranged from 142 h•ng/mL to 321 h•ng/mL with an estimated median AF of 1.73. These estimates were within the exposure range predicted for children aged 2 to <7 years using the popPK model based on single-dose data (138 h•ng/mL to 457 h•ng/mL; 95% prediction interval), without any indication of a higher accumulation than expected for the dosing regimen. The AUCτ,ss values were also within the targeted steady state exposure range established from the adult therapeutic doses of 50–100 mg tapentadol immediate release (IR) tablets given every 4 h (130.7–706 h•ng/mL).13
 |
Table 3 Model-Based Estimated Pharmacokinetic Parameters of Tapentadol, Tapentadol-O-Glucuronide, and Tapentadol-O-Sulfate |
AUCτ,ss values of the two secondary trial endpoints ranged from 4501 h•ng/mL to 7617 h•ng/mL for tapentadol-O-glucuronide and from 180 h•ng/mL to 590 h•ng/mL for tapentadol-O-sulfate. The estimated median AF for tapentadol-O-glucuronide was 1.84 and 1.28 for tapentadol-O-sulfate.
Efficacy (exploratory)
The median pain intensity levels assessed by the FLACC decreased from 5.0 at baseline to 0.5 1.5 h after administration of first dose and remained at 0.0 at later assessments (Table 4). Timepoints after dose 7 are not shown, as the number of patients with FLACC assessments had declined.
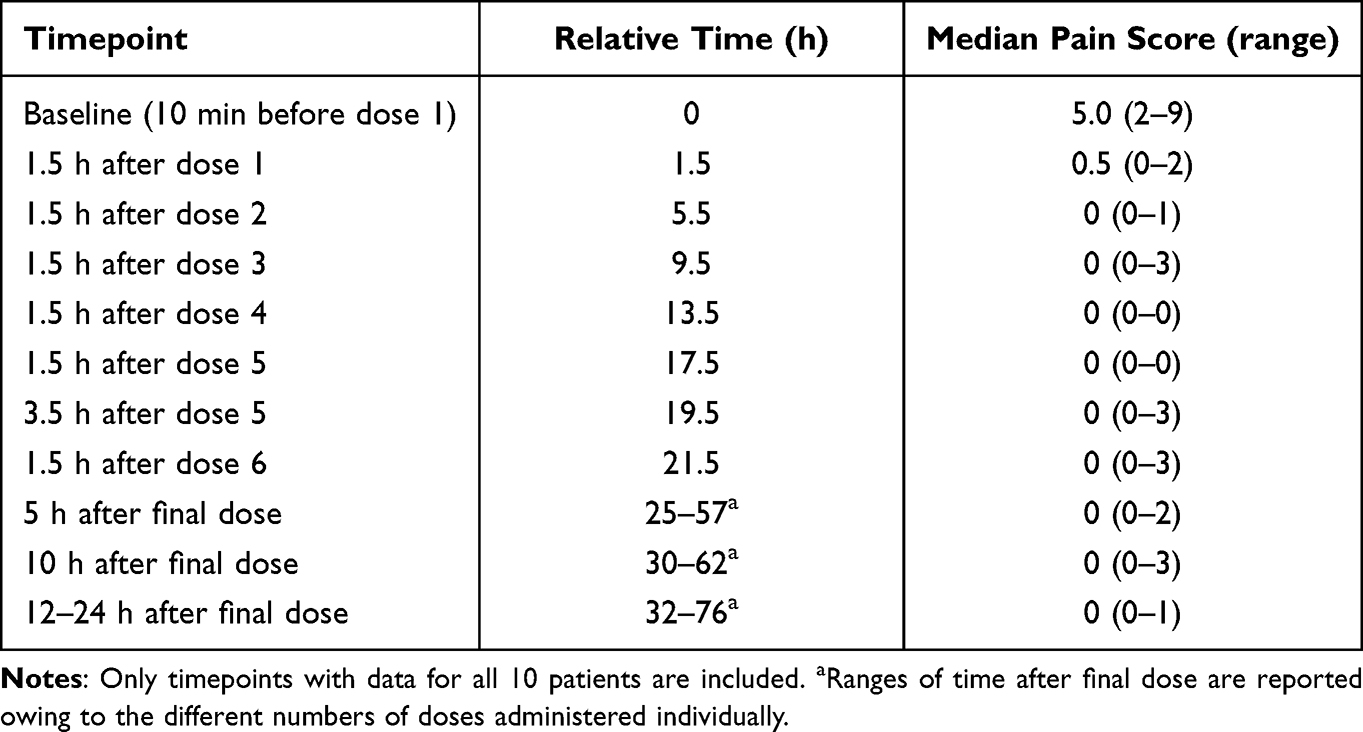 |
Table 4 Median Pain Intensity Over Time Rated with the Observational Face, Legs, Activity, Cry, and Consolability Scale |
Safety and Tolerability
Five patients (50%) experienced 7 treatment-emergent AEs (TEAEs) during the trial. Nausea occurred in 2 patients, constipation, vomiting, regurgitation, and hyperhidrosis in one patient each. All TEAEs except for regurgitation were considered as being probably/likely related to tapentadol OS treatment by the investigators. All TEAEs were mild in intensity with the exception of a moderate event of vomiting; all TEAEs resolved. Death, serious TEAEs, or treatment discontinuation due to TEAEs did not occur. There were no clinically meaningful changes in physical examination findings nor clinically meaningful abnormal vital signs or oxygen saturation values at any assessment. No new safety concerns were identified.
Discussion
Multiple administrations of tapentadol OS using the approved and recommended dose of 1.25 mg/kg bodyweight every 4 h in children aged 2 to <7 years resulted in tapentadol serum concentrations within the concentration range predicted by the popPK model based on single-dose PK data.15 The observed model-based exposure estimates for AUCτ,ss of tapentadol were within the predicted exposure range with no higher than expected accumulation for this dosing regimen. The estimated exposure values were also within the targeted steady state exposure range established from the adult therapeutic doses of 50 mg to 100 mg tapentadol IR tablets every 4 h.13 Furthermore, the estimated mean accumulation ratio of 1.84 (median 1.73) was also close to the range of mean values observed for adults (1.4–1.7).19 These results confirm that a dosing regimen of 1.25 mg/kg every 4 h in children aged 2 to <7 years results in exposure distributions that stay within the targeted efficacious exposure range. This also confirms the linear and predictable PK profile of tapentadol and shows the value of modelling and simulations in the planning and analysis of pediatric clinical trials and the ability to utilize established PK models to predict multiple dose exposure based on single-dose data. The very good agreement between the observed data in this trial and the model predictions illustrate the potential of modelling and simulation for reducing the number of clinical trials or subjects required to be enrolled, particularly in vulnerable populations such as children.
The integration of the serum concentration-time tapentadol data obtained in the present trial into the previous popPK model resulted in almost identical structure and parameter estimates of the updated tapentadol popPK model to the previous one based on single-dose data.15 The PK parameter estimates for tapentadol at full maturation were also in good agreement with those previously reported in the adult popPK model for tapentadol immediate release.22 The estimated values for tapentadol CL/F and V/F from the pediatric tapentadol popPK model for a 70 kg reference weight (272 L/h and 1203 L, respectively) compare very well to the reported CL/F of 214 L/h and V/F of 1170 L in the published adult model.22 Furthermore, the estimated value for tapentadol Ka of 2.2 h−1 in the pediatric model agrees well with the value reported in the adult model (Ka = 2.06 h−1).
Although the two tapentadol metabolites tapentadol-O-glucuronide and tapentadol-O-sulfate do not contribute to the analgesic activity of tapentadol, both were included in the evaluation for comparison with data obtained in single-dose pediatric trials. Mean tapentadol-O-glucuronide concentrations were substantially higher than those observed for tapentadol which is consistent with observations in adult healthy volunteers23 and in previous single-dose pediatric trials.11,12 For tapentadol-O-sulfate, mean serum concentrations were similar or slightly higher than those observed for tapentadol, again in line with earlier data after single dose oral administration in younger children.17 The subsequent extension of the updated model to a joint tapentadol parent-metabolite model allowed for the first time to simultaneously describe the PK of tapentadol and its O-glucuronide and O-sulfate metabolites.
The trial was primarily designed to assess the PK profile of tapentadol and two of its metabolites after administration of multiple doses of tapentadol OS. Small patient numbers and the lack of a placebo control only permitted an exploratory efficacy analysis which showed a reduction in pain intensity following tapentadol OS administration. The efficacy of multiple doses of tapentadol OS in the treatment of acute pain was demonstrated in a randomized placebo-controlled trial with more than one hundred children aged 2 to <18 years.14 In that trial, tapentadol OS was significantly more efficacious than placebo; the result was confirmed by sensitivity analyses and supported by secondary efficacy analyses. The trial also assessed palatability and acceptability of the medication which were considered adequate to ensure intake compliance in this pediatric population.
The administered tapentadol dose of 1.25 mg/kg bodyweight every 4 h was well tolerated. The observed TEAEs were mostly expected and reflected the known safety profile of tapentadol. This is in agreement with data from pediatric patients in a randomized placebo-controlled trial14 where multiple doses of tapentadol OS were generally well tolerated. As is the case for every treatment, warnings and contraindications as described in the SmPC19 need to be considered by the treating physician.
Limitations
Compared to dense blood sampling in adults, only limited blood samples could be drawn in this vulnerable pediatric population and a popPK modelling approach was required to support the PK evaluation. The exposure assessments might thus not have been as precise as with a dense sampling approach. In addition, the design of the trial limits the possibility to draw conclusions about the efficacy of tapentadol OS.
Conclusions
The findings of this trial confirm the linear and predictable PK profile of tapentadol. The good agreement between the observed data and the model predictions shows the value of modelling and simulations in the planning and analysis of pediatric clinical trials and the ability to utilize the established PK models to predict multiple dose exposure based on single-dose data.
Abbreviations
AF, accumulation factor; AUC, area under the curve; CL, clearance; F, oral bioavailability; FLACC, Face, Legs, Activity, Cry, and Consolability; GA, gestational age; IR, immediate release; IV, intravenous; Ka, oral absorption rate constant; LLOQ, lower limit of quantification; OS, oral solution; PK, pharmacokinetic; pop, population; SD, standard deviation; TEAE, treatment-emergent adverse event; V, volume.
Data Sharing Statement
Any request for data should be submitted via the following website: https://www.grunenthal.com/en/research-and-development/clinical-trials/data-sharing-clinical-trials
Acknowledgments
The authors thank all patients, parents/legal guardians, investigators, and site teams involved in this investigation. The trial was funded by Grünenthal GmbH. Writing and editorial assistance was provided by Elke Grosselindemann and Martin Brett and was paid for by Grünenthal GmbH.
Disclosure
RJ was the international coordinating investigator of the trial. BB was an employee of Grünenthal GmbH at the time the trial was conducted. All other authors are employees of Grünenthal GmbH. The authors report no other conflicts of interest in this work.
References
1. Tzschentke TM, Christoph T, Kögel BY. The mu-opioid receptor agonist/noradrenaline reuptake inhibition (MOR-NRI) concept in analgesia: the case of tapentadol. CNS Drugs. 2014;28(4):319–329. doi:10.1007/s40263-014-0151-9
2. Raffa RB, Elling C, Tzschentke TM. Does ‘strong analgesic’ equal ‘strong opioid’? Tapentadol and the concept of ‘μ-load’. Adv Ther. 2018;35(10):1471–1484. doi:10.1007/s12325-018-0778-x
3. Xiao JP, Li AL, Feng BM, Ye Y, Wang GJ. Efficacy and safety of tapentadol immediate release assessment in treatment of moderate to severe pain: a systematic review and meta-analysis. Pain Med. 2017;18(1):14–24. doi:10.1093/pm/pnw154
4. Viscusi E, Allard R, Sohns M, Eerdekens M. Tapentadol immediate release for moderate to severe acute post-surgery pain. J Opioid Manag. 2019;15(1):51–67. doi:10.5055/jom.2019.0486
5. Baron R, Eberhart L, Kern KU, et al. Tapentadol prolonged release for chronic pain: a review of clinical trials and 5 years of routine clinical practice data. Pain Pract. 2017;17(5):678–700. doi:10.1111/papr.12515
6. Göhler K, Brett M, Smit JW, Rengelshausen J, Terlinden R. Comparative pharmacokinetics and bioavailability of tapentadol following oral administration of immediate- and prolonged-release formulations. Int J Clin Pharmacol Ther. 2013;51(4):338–348. doi:10.5414/CP201722
7. Terlinden R, Kogel BY, Englberger W, Tzschentke TM. In vitro and in vivo characterization of tapentadol metabolites. Methods Find Exp Clin Pharmacol. 2010;32(1):31–38. doi:10.1358/mf.2010.32.1.1434165
8. Kneip C, Terlinden R, Beier H, Chen G. Investigations into the drug-drug interaction potential of tapentadol in human liver microsomes and fresh human hepatocytes. Drug Metab Lett. 2008;2(1):67–75. doi:10.2174/187231208783478434
9. Smit JW, Oh C, Rengelshausen J, et al. Effects of acetaminophen, naproxen, and acetylsalicylic acid on tapentadol pharmacokinetics: results of two randomized, open-label, crossover, drug-drug interaction studies. Pharmacotherapy. 2010;30(1):25–34. doi:10.1592/phco.30.1.25
10. Eerdekens M, Beuter C, Lefeber C, van den Anker J. The challenge of developing pain medications for children: therapeutic needs and future perspectives. J Pain Res. 2019;12:1649–1664. doi:10.2147/JPR.S195788
11. Finkel J, Goldberg J, Rosenburg R, et al. First evaluation of tapentadol oral solution for the treatment of moderate to severe acute pain in children aged 6 to <18. J Pain Res. 2019;12:1925–1936. doi:10.2147/JPR.S197348
12. Muse D, Tarau E, Lefeber C, et al. Pharmacokinetics, safety, and efficacy of tapentadol oral solution for treating moderate to severe pain in pediatric patients. J Pain Res. 2019;12:1777–1790. doi:10.2147/JPR.S197039
13. Watson E, Khandelwal A, Freijer J, van den Anker J, Lefeber C, Eerdekens M. Population pharmacokinetic modelling to facilitate dose selection of tapentadol in the pediatric population. J Pain Res. 2019;12:2835–2850. doi:10.2147/JPR.S208454
14. Beuter C, Volkers G, Radic T, Goldberg J, van den Anker J. Efficacy and safety of multiple doses of tapentadol oral solution in the treatment of moderate to severe acute pain in children aged 2 to <18 years – a randomized, double-blind, placebo-controlled trial. J Pain Res. 2019;12:3099–3112. doi:10.2147/JPR.S207010
15. Khalil F, Choi SL, Watson E, et al. Population pharmacokinetics of tapentadol in children from birth to <18 years. J Pain Res. 2020;13:3107–3123. doi:10.2147/JPR.S269549
16. Howard R, Radic T, Sohns M, Eerdekens M, Waßmuth A. Tapentadol prolonged release for long-term treatment of pain in children. J Pain Res. 2020;13:3157–3170. doi:10.2147/JPR.S272751
17. Eissa A, Tarau E, Beuter C, et al. Tapentadol for the treatment of moderate-to-severe acute pain in children under the age of two years. J Pain Res. 2021;14:229–248. doi:10.2147/JPR.S269530
18. Eerdekens M, Radic T, Sohns M, Khalil F, Bulawa B, Elling C. Outcomes of the pediatric development plan of tapentadol. J Pain Res. 2021;14:249–261. doi:10.2147/JPR.S290487
19. Electronic Medicines Compendium. Palexia oral solution 20 mg/mL. Available from: https://www.medicines.org.uk/emc/product/5346/smpc.
20. National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion. Available from: http://www.cdc.gov/growthcharts/who_charts.htm.
21. Merkel SI, Voepel-Lewis T, Shayevitz JR, Malviya S. The FLACC: a behavioral scale for scoring postoperative pain in young children. Pediatr Nurs. 1997;23(3):293–297.
22. Xu XS, Smit JW, Lin R, Stuyckens K, Terlinden R, Nandy P. Population pharmacokinetics of tapentadol immediate release (IR) in healthy subjects and patients with moderate or severe pain. Clin Pharmacokinet. 2010;49(10):671–682. doi:10.2165/11535390-000000000-00000
23. Terlinden R, Ossig J, Fliegert F, Lange C, Göhler K. Absorption, metabolism, and excretion of 14C-labeled Tapentadol HCL in healthy male subjects. Eur J Drug Metab Pharmacokinet. 2007;32(3):163–169. doi:10.1007/BF03190478
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.