Back to Journals » ImmunoTargets and Therapy » Volume 14
Multi-Dimensional Characterization of Programmed Cell Death Patterns for Prognostic Stratification and Therapeutic Insights in Sepsis
Received 24 July 2025
Accepted for publication 7 November 2025
Published 15 November 2025 Volume 2025:14 Pages 1313—1331
DOI https://doi.org/10.2147/ITT.S555950
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sarah Wheeler
Chen Zhou,1 Yunmeng Bai2
1Department of Laboratory Medicine, The Eighth Affiliated Hospital, Sun Yat-Sen University, Shenzhen, 518033, People’s Republic of China; 2School of Medicine, Southern University of Science and Technology, Shenzhen, 518055, People’s Republic of China
Correspondence: Yunmeng Bai, Email [email protected]
Background: Sepsis is a complex and heterogeneous syndrome characterized by dysregulated immune responses and multiple forms of programmed cell death (PCD). Comprehensive understanding of the PCD landscape may provide insights into prognosis and therapeutic targets, whereas its role in sepsis is not well-explored.
Methods: Using the microarray dataset for sepsis (GSE65682), we systematically profiled 14 PCD patterns in sepsis and stratified patients into molecular subtypes with distinct immune landscapes and clinical outcomes. PCD-related prognostic signature was developed and validated across multiple cohorts. Single-cell and multi-organ transcriptomic analyses were conducted to elucidate cellular heterogeneity and temporal dynamics. Molecular docking was used to explore interactions between active compounds of Simiao Yongan Decoction (SMYAD) and key PCD-related proteins.
Results: Two clusters with differential transcriptional programs and immune infiltration patterns were identified, in which Cluster 1 showed poorer prognosis. We then developed a seven-gene signature (ELANE, CTSG, MPO, CAMP, TFRC, IL1B, CASP5) that effectively stratified patients by survival outcomes, with robust predictive performance across independent datasets. Neutrophils, monocytes, plasma, and dendritic cells were major mediators of PCD-associated immune dysregulation, in which neutrophils showing the strongest response. Temporal transcriptomics revealed peak expression of prognostic genes in bone marrow and peripheral blood within three days post-onset, suggesting an early therapeutic window. Finally, molecular docking indicated that SMYAD compounds may target PCD proteins (MPO, ELANE, IL1B) and modulate immune responses.
Conclusion: This study delineates the multi-dimensional role of PCD in sepsis, establishes a reliable prognostic model with strong predictive value, and highlights SMYAD as a potential multi-target therapy. These findings provide new avenues for risk stratification and suggest the promise of integrating PCD biology with adjunctive immunomodulatory strategies.
Keywords: sepsis, programmed cell death, machine learning, prognostic model, Simiao Yongan decoction
Introduction
Sepsis, a life-threatening organ dysfunction caused by a dysregulated host response to infection, remains a pressing global health challenge, despite clinical advances.1 Sepsis is associated with significant morbidity and mortality, with global report in 2020 estimates indicating approximately 48.9 million cases and 11 million deaths annually, accounting for nearly 20% of the overall deaths.2 This varies considerably across regions, from 20–30% in developed countries to as high as 60% in resource-limited regions.3 From 1998 to 2023, United Kingdom sepsis-coded hospital admissions rose from 27.9 to 210.4 per 100,000 population, up to a 7.5-fold increase,4 and a total of 4,177,071 deaths were attributed to sepsis-related complications from 1999 to 2022 in the United Status,5 highlighting the growing burden of sepsis. The pathophysiology of sepsis is complex and involves excessive inflammation, immune suppression, coagulation abnormalities, and metabolic dysfunction, ultimately leading to multi-organ failure.6 This uncontrolled systemic inflammatory response, often accompanied by cytokine storms and endothelial damage, drives progression to septic shock and multiple organ dysfunction syndrome (MODS).7 Although early interventions, such as fluid resuscitation, antibiotics, and organ support have improved outcomes, mortality remains unacceptably high, especially in critically ill patients.6,8 Conventional biomarkers, such as procalcitonin, C-reactive protein, and lactate, offer limited accuracy in predicting sepsis prognosis.9 Consequently, there is an urgent need to identify reliable diagnostic and prognostic biomarkers to guide personalized treatment strategies and improve outcomes in patients with sepsis.
Programmed cell death (PCD) is a genetically regulated process essential for maintaining cellular and internal environmental homeostasis,10 and could be classified into inflammatory and non-inflammatory forms according to their immunological consequences. Inflammatory PCD, such as pyroptosis,11 necroptosis,12 netotic cell death,13 immunogenic cell death (ICD),14 alkaliptosis,15 is typically characterized by the release of damage-associated molecular patterns (DAMPs), cytokines, or extracellular traps, thereby amplifying systemic inflammation and tissue injury in sepsis. By contrast, non-inflammatory PCD generally proceeds in a more immunologically silent and context-dependent manner, including apoptosis,16 ferroptosis,17 autophagy-dependent death,18 lysosome-dependent cell death,19 parthanatos,20 mitochondrial permeability transition (MPT)-driven necrosis,21 entotic death,13 oxeiptosis,22 cuproptosis,23 etc. Increasing evidence highlights extensive crosstalk and compensatory mechanisms among PCD pathways.24 Compared with analyses focused on a single PCD type, prognostic models that integrate multiple PCD patterns provide a more comprehensive representation of disease pathophysiology and demonstrate improved performance in predicting patient outcomes and drug responsiveness.25 Previous studies mainly examined the sepsis prognostic relevance of the individual PCD type,26–28 however, a comprehensive understanding of how multiple PCD patterns collectively influence disease progression and patient outcomes remains lacking. These observations underscore the need for multi-targeted therapeutic strategies and integrated PCD-based prognostic models for effective sepsis management.
Traditional Chinese Medicine (TCM) has garnered increasing attention as a complementary approach to sepsis management because of its multitarget and multipathway therapeutic potential.29 Simiao Yongan Decoction (SMYAD), a classic TCM formula composed of Lonicerae japonicae flos, Figwort root, Angelicae sinensis radix, and Licorice, is traditionally used to treat heat-toxin syndromes and suppurative infections.30 Recent studies have shown that SMYAD possesses anti-inflammatory, immunomodulatory, antioxidative, and microcirculatory protective effects,31 which closely align with the pathophysiological mechanisms of sepsis. Experimental evidence suggests that SMYAD can attenuate systemic inflammation, inhibit excessive cytokine release, and protect against organ dysfunction,32,33 indicating its promising potential as an adjunctive therapy in sepsis treatment. However, its mechanistic relevance to PCD in sepsis remains unclear.
In this study, we aimed to elucidate how different PCD patterns collectively shape prognosis and therapeutic response in sepsis. We conducted the first multi-dimensional investigation of 14 PCD patterns in sepsis, identified molecular subtypes with distinct immune and clinical characteristics, developed and validated a robust seven-gene prognostic model, and explored the interaction between SMYAD-derived compounds and PCD-related proteins. Collectively, this framework deepens the mechanistic understanding of PCD crosstalk in sepsis, and provides a translational basis for combining biomarker-guided precision medicine with traditional Chinese medicine approaches.
Materials and Methods
Data Source and Processing
Fourteen PCD pattern genes were obtained from a previous study,34 including apoptosis, pyroptosis, necroptosis, ferroptosis, mitotic cell death, oxeiptosis, MPT-driven necrosis, parthanatos, cuproptosis, immunogenic cell death, alkaliptosis, lysosome-dependent cell death, entotic cell death, and autophagy. The microarray datasets for sepsis were downloaded from the Gene Expression Omnibus (GEO) database. GSE65682 included 802 healthy control (n = 42) and sepsis (n = 760) samples. In addition, other datasets were used to further verify the results: GSE48080 contains microarray expression patterns of peripheral blood mononuclear cells (PBMCs) from healthy volunteer (n = 3), survivor (n = 5) and non-survivor septic (n = 5) patients;35 GSE28750 is a microarray cohort of PBMCs from healthy control (n = 20), sepsis (n = 10) and post-surgical (n = 11) patients.36 GSE57065 contains patients based on the Simplified Acute Physiology Score II (SAPSII) score: SAPSII-high (n = 40) and SAPSII-low (n = 42) groups;37 GSE26440 from normal control (n = 32), survivor (n = 81), and non-survivor septic shock (n = 17) patients;38 GSE106878 comprised 84 septic shock patients treated with either Placebo or Hydrocortisone in the morning (Pre) and 24 h (Post) with different survivor status; GSE33119 of survival outcome from 20 patients before and after activated protein C (Xigis) treatment.
Correlation Analysis
Correlations among different PCD patterns and between immune cell infiltration and risk score were calculated and visualized using the corrplot package (version 0.5.9) in R. Significant correlations were defined as those with p < 0.05.
Survival Analysis
Univariate Cox proportional hazard regression analysis was performed to evaluate the prognostic significance of each PCD pattern in the sepsis cohort. Survival analysis was performed by survival package (version 3.8–3). Kaplan-Meier survival curves were generated to visualize the survival probability differences between groups, and the Log rank test was applied to assess the statistical significance of these differences. Hazard ratios (HR) and 95% confidence intervals (CI) were calculated to identify the PCD patterns associated with patient outcomes.
Consensus Clustering
Based on the activity scores of the PCD patterns with prognostic significance, we conducted consensus clustering using the ConsensusClusterPlus R package (version 1.70.0). The optimal number of clusters was determined using cumulative distribution function (CDF) curves and delta-area plots via Kvec and calcICL function. A principal component analysis (PCA) was performed by prcomp function to validate the separation of the identified clusters.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis (GSEA) was applied using the clusterProfiler package (version 4.14.6) in R. Hallmark gene sets from the Molecular Signatures Database (MSigDB) were used as the reference pathways. Statistical significance was set at a false discovery rate (FDR) < 0.05.
Differentially Expressed Genes Identification
Differentially expressed genes (DEGs) between the clusters were identified using the limma package (version 3.58.1). Gene expression values were first normalized and fitted to a linear model using lmFit function. Empirical Bayes moderation was applied via eBayes function to improve variance estimates across genes, and differential expression was assessed using moderated t-tests. P values were adjusted for multiple testing using the Benjamini-Hochberg false discovery rate (FDR < 0.05).
LASSO Regression
Prognosis-associated genes were first screened using univariate Cox regression analysis to identify genes significantly correlated with patient survival. Genes with p < 0.05 were retained for further analysis. Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression was conducted using the glmnet package (version 4.1–8) to construct a prognostic gene signature. LASSO applies an L1 penalty to the regression coefficients, shrinking some coefficients to zero, which effectively performs variable selection and reduces model overfitting. A ten-fold cross-validation was performed via cv.glmnet function to determine the optimal lambda value according to lambda.min parameter. Genes with non-zero coefficients at this lambda were retained and used to construct the final prognostic model. The coxph function from the survival package was then applied to fit a multivariate Cox proportional hazards model based on these selected genes, generating hazard ratios and coefficients for each gene. The risk score for each patient was calculated as a linear combination of the expression levels of the selected genes weighted by their Cox regression coefficients:
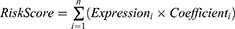
Patients were stratified into high- and low-risk groups based on the median risk score.
Receiver Operating Characteristic Curve
Time-dependent predictive performance of the prognostic model was evaluated using the timeROC R package (version 0.4). The area under the receiver operating characteristic (ROC) curve (AUC) was calculated at multiple time points (4, 7, 14, and 21 days) to assess the model’s predictive accuracy over time, providing a comprehensive assessment of its prognostic value.
Molecular Docking
The structures of prognostic target proteins were obtained from the Protein Data Bank (PDB), including MPO (PDB: 1CXP), ELANE (PDB: 5ABW), and IL1B (PDB: 1L2H). The active compounds of SMYAD were retrieved from the Traditional Chinese Medicine Systems Pharmacology (TCMSP) database, then obtained their structures from PubChem and ChemSpider database, including quercetin (PubChem CID: 5280343), oleic acid (PubChem CID: 445639), ursolic acid (PubChem CID: 64945), rutin (PubChem CID: 5280805), thymol (PubChem CID: 6989), and glycyrrhizin (ChemSpider ID: 14263). Protein structures were prepared using PyMOL (version 3.1) to remove water molecules and ligands. Molecular docking was performed using AutoDock (version 1.5.6). The docking grid was centered on the active site of each protein, with grid box dimensions set to cover the binding pocket. The Lamarckian Genetic Algorithm was applied with default parameters, and multiple docking runs were performed to ensure sampling of diverse binding poses. Docking results were ranked based on binding free energy, and the top binding conformations with the lowest energies were selected for further analysis. PyMOL was used to visualize binding poses, highlighting hydrogen bonds, hydrophobic interactions, and key residues involved in ligand recognition.
Single-Cell RNA Sequencing Analysis
Single-cell RNA sequencing (scRNA-seq) datasets (GSE16736339 and GSE17545340) were integrated via harmony method using the Seurat package (version 4.0.3) in R. Cells were remained with (1) detected genes in 200~6000; (2) read counts > 1000; (3) mitochondrial gene content < 20%. Highly variable genes were identified using SCTransform function, and PCA was performed on the scaled data, then cells were clustered using the FindClusters function. Uniform Manifold Approximation and Projection (UMAP) was used for visualization of cell clusters in two dimensions. Cells were annotated into major immune cell types based on the expression of canonical markers. PCD activity scores were estimated using AddModuleScore function for each cell type.
Multi-Organ Temporal RNA-Seq Data Analysis
Multi-organ and multi-time point RNA-seq data from a mouse sepsis model (GSE22412741) were analyzed to evaluate the spatiotemporal expression dynamics of prognostic genes. Tissues included bone marrow, peripheral blood, brain, heart, kidney, liver, skin, colon, small intestine, spleen, thymus, lymph nodes, and lungs, collected at 0, 0.25, 0.5, 1, 2, 3, and 5 d after lipopolysaccharide (LPS) injection. The mean gene expression was visualized to determine the temporal expression peaks and tissue-specific patterns. The spatial transcriptome (GSE24890441) across different organs in the control and LPS groups was subjected to the Seurat object.
Statistical Analysis
Data processing, statistical analyses and visualization were conducted using R software (version 4.1.2). Survival curve analyses (Kaplan-Meier curves) were performed via the Log rank test. Hazard ratios in the were calculated using univariate cox regression analyses. Correlations were calculated using the Pearson method. P value < 0.05 was considered statistically significant (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001).
Results
Identification of Programmed Cell Death Clusters in Sepsis
The workflow of this study was shown as Figure 1A. To systematically delineate the landscape of PCD in sepsis, we quantified the scores of 14 PCD patterns in the sepsis cohort (GSE65682), including 42 healthy controls and 760 sepsis patients. The PCD patterns were significantly different between the healthy and sepsis groups (Figure S1). Correlation analysis revealed substantial interactions among several PCD modalities. Notably, pyroptosis showed a strong positive correlation with necroptosis (r = 0.70), and necroptosis was strongly correlated with immunogenic cell death (r = 0.70) (Figure 1B), suggesting potential crosstalk among these cell death pathways within the septic microenvironment.
Figure 1 Continued. Figure 1 Identification of programmed cell death clusters in sepsis. (A) The workflow of this study. (B) Correlation network of 14 programmed cell death (PCD) patterns in sepsis. (C) Prognostic analysis of each PCD pattern using Kaplan-Meier survival curves. (D) Cumulative distribution function (CDF) curve for consensus clustering. (E) The area under curve (AUC) for consensus clustering. (F) Consensus matrix heatmap at k = 2. (G) Principal component analysis (PCA) plot demonstrating transcriptional separation between the two clusters. (H) Kaplan-Meier survival of two clusters. (I) Kaplan-Meier survival of two clusters according to gender and age.
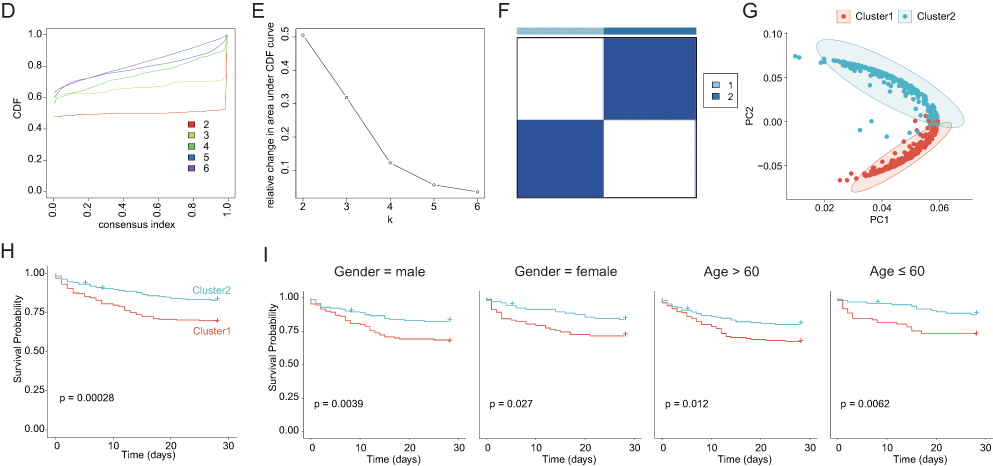
Based on the prognostic impact of each PCD pattern, we stratified the cohort into high and low groups and identified six PCD patterns with significant prognostic characteristics (p < 0.05, Figure 1C). Ferroptosis and mitotic cell death are associated with increased mortality risk, whereas lysosome-dependent cell death, MPT-driven necrosis, necroptosis, and pyroptosis are associated with improved survival outcomes. Therefore, these six PCD patterns were selected for subsequent analyses.
We next performed consensus clustering based on PCD pattern scores to identify distinct molecular subtypes of sepsis. Using the CDF and consensus matrix, we determined k = 2 was the optimal number of clusters (Figure 1D and E), resulting in the classification of sepsis patients into Cluster1 and Cluster2 with high intra-cluster average consistency (Figure 1F). PCA demonstrated a clear transcriptional separation between the two clusters (Figure 1G). Kaplan-Meier survival analysis revealed that patients in Cluster 2 had significantly better survival probabilities than those in Cluster 1 (Figure 1H), indicating that Cluster 1 was associated with a higher mortality risk. Importantly, sex and age distributions were comparable between the two clusters, thus minimizing potential demographic confounders (Figure 1I).
Characteristics of the Two Clusters in Sepsis
To elucidate the immunological characteristics of the identified clusters, we applied a single-sample gene set enrichment analysis (ssGSEA) to quantify immune cell infiltration across 28 immune-related gene sets. As shown in Figure 2A, cluster 1 exhibited markedly lower infiltration of immune cells, particularly Activated CD4 T cells, type 17 T helper cells, Activated B cells, CD56dim natural killer cells, activated/immature dendritic cells, and mast cells. In contrast, Cluster2 demonstrated significantly higher levels of immune infiltration, characterized by Activated CD8 T cells, effector memory CD8 T cells, T follicular helper cells, type 1 helper cells, Regulatory T cells, Immature B cells, myeloid-derived suppressor cells, natural killer T cells, macrophages, eosinophils, and neutrophils, suggesting a potential relationship between immune infiltration and patient outcomes. Furthermore, we observed substantial differences in the expression of immune checkpoint genes between the two clusters (Figure 2B). Cluster 2 generally exhibited higher immune checkpoint expression, indicating an active, yet regulated immune microenvironment.
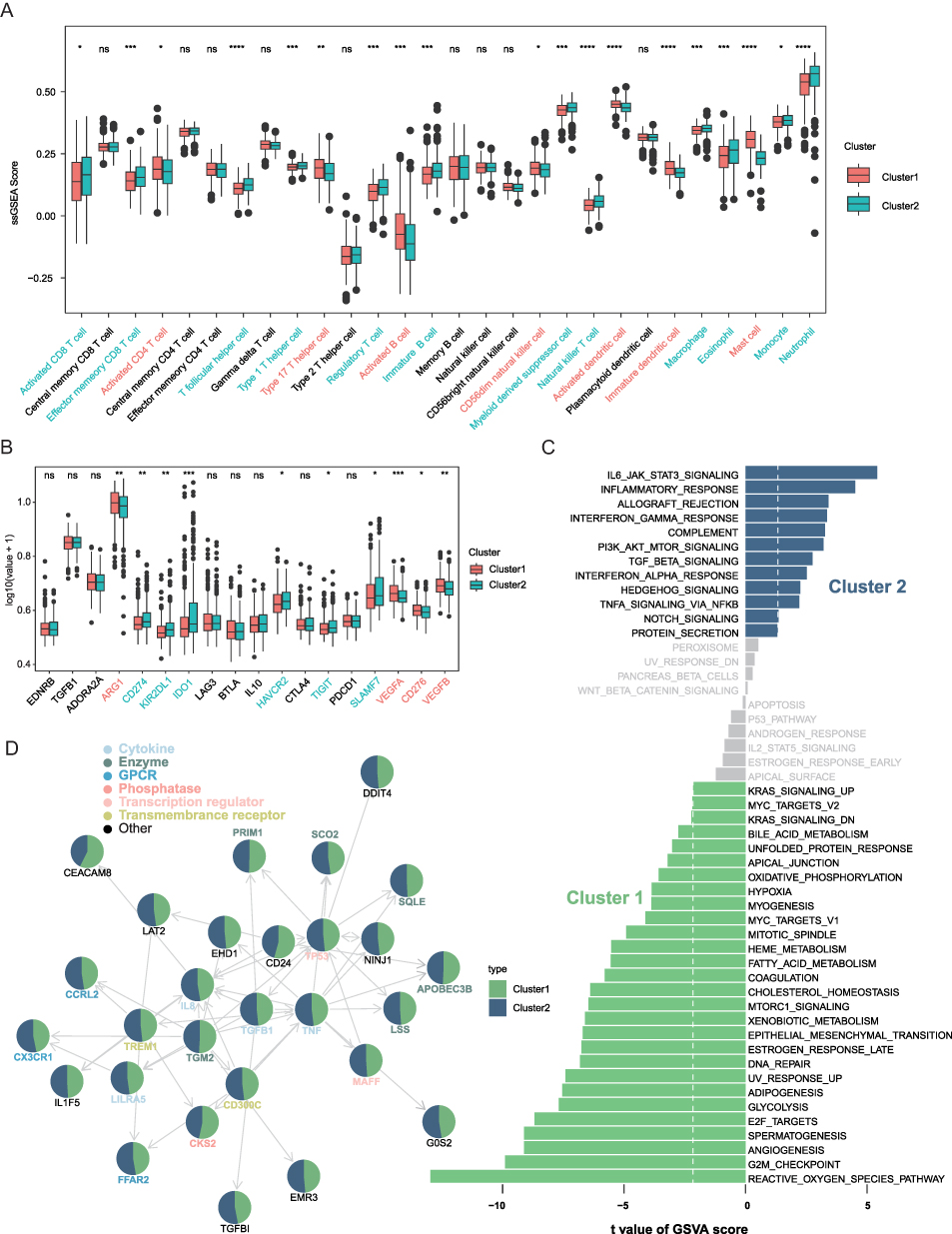 |
Figure 2 Immunological and functional characteristics of the two PCD clusters in sepsis. (A) Immune cell infiltration scores quantified by single-sample gene set enrichment analysis (ssGSEA). (B) Differential expression of immune checkpoint genes between Cluster 1 and Cluster 2. (C) Gene set variation analysis (GSVA) results of the hallmark pathways differences between clusters. (D) Distribution of candidate prognostic genes across the two clusters. ns not significant, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. |
To further investigate the biological underpinnings of the two clusters, we constructed gene set variation analysis (GSVA) to identify dysregulated hallmark pathways (Figure 2C). Cluster 1 was enriched in pathways related to hypoxia, epithelial-mesenchymal transition (EMT), glycolysis, and reactive oxygen species (ROS) signaling, suggesting a metabolic and stress-associated phenotype. In contrast, cluster 2 was predominantly enriched in immune-related pathways, including IL6-JAK-STAT3 signaling, inflammatory response, interferon-α/γ response, and complement activation, consistent with the heightened immune infiltration observed (Figure 2A). We then mapped the candidate prognostic genes42 to clusters (Figure 2D), in which cytokines, transcription regulators, and transmembrane receptors were mainly enriched in Cluster2 samples, and phosphatase CKS2, enzyme TGM2, APOBEC3B in Cluster1. These results indicate distinct molecular signatures and regulatory mechanisms between the two clusters.
Establishment and Validation of a PCD-Related Prognostic Model
To further refine the transcriptional differences between clusters, we identified DEGs (Figure 3A) and intersected them with prognosis-associated genes derived from univariate Cox regression analysis. Using LASSO Cox regression, we constructed a robust seven-gene prognostic signature comprising ELANE, CTSG, MPO, CAMP, TFRC, IL1B and CASP5 (Figure 3B and C). The risk score was calculated using the following formula: risk score = ELANE*0.36917+CTSG*0.22875+MPO*(−0.46844)+CAMP*(−0.31068)+TFRC*(0.28079)+IL1B*(0.10971)+CASP5*(−0.14144). These seven genes showed higher expression levels in the sepsis than healthy samples in GSE65682 cohort (Figure 3D). Stratification based on the median risk score revealed that patients in the high-risk group exhibited significantly worse survival outcomes than those in the low-risk group (Figure 3E), and the 6 PCD patterns were differentially expressed between the two groups (Figure S2). Moreover, the risk scores were significantly elevated in Cluster 1, which was also associated with poor prognosis (Figure 3F). Sex and age were evenly distributed between the high- and low-risk groups, further supporting the prognostic independence of this model (Figure 3G). Subgroup analysis confirmed that the survival probability differences remained consistent across the sex and age groups (Figure 3H).
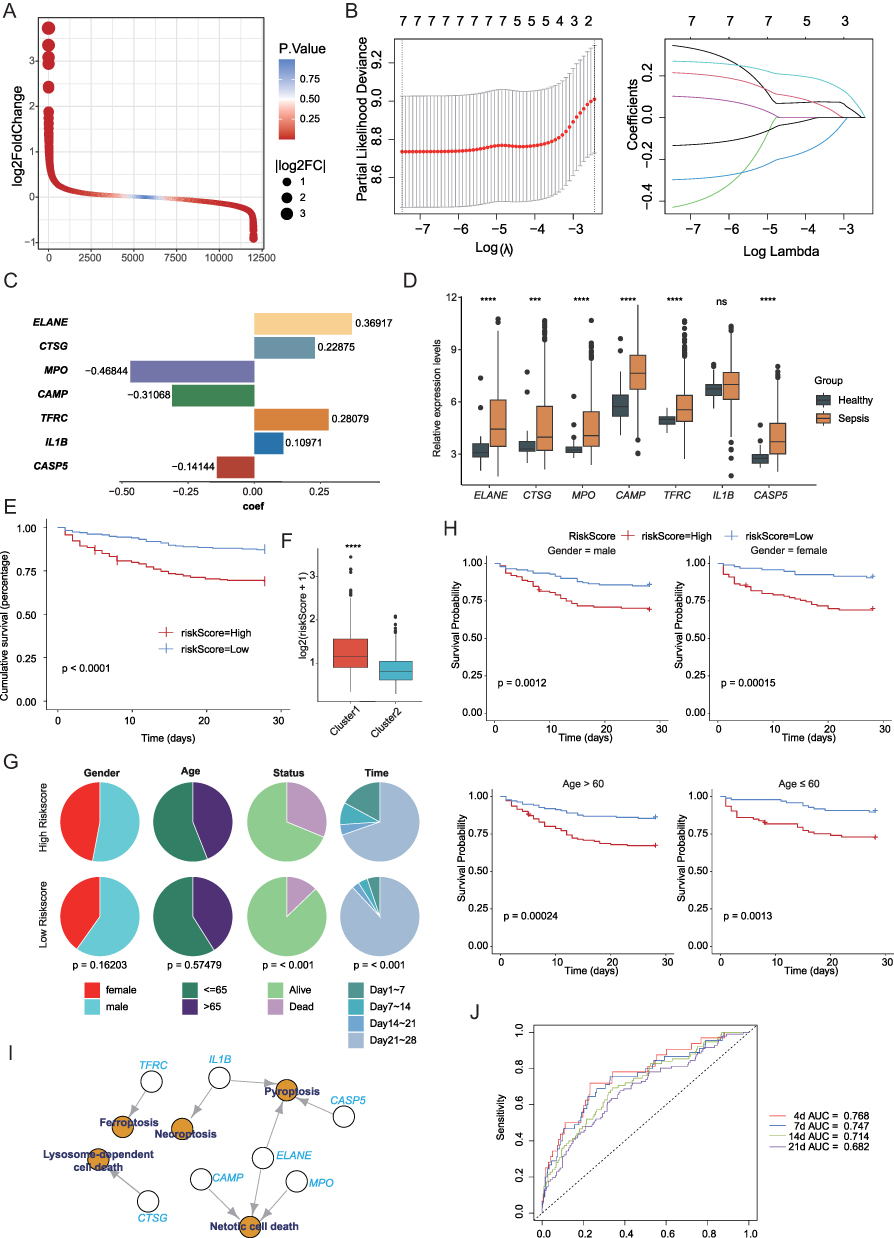 |
Figure 3 Construction of a PCD-related prognostic model. (A) Volcano plot of differentially expressed genes (DEGs) between clusters. (B) Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression to identify key prognostic genes. (C) Risk score formula based on seven PCD-related genes. (D) Expression profiles of the seven prognostic genes in healthy and sepsis samples. (E) Kaplan-Meier survival curves showing worse survival in the high-risk group. (F) Distribution of risk scores in Cluster 1 and Cluster 2. (G) Gender and age distribution between high- and low-risk groups. (H) Subgroup survival analysis by gender and age. (I) The correlation of the seven genes with PCD patterns. (J) Time-dependent receiver operating characteristic (ROC) curves demonstrating the prognostic performance of the risk model at 4, 7, 14, and 21 days. ns not significant, *** p < 0.001, **** p < 0.0001. |
Functionally, these seven prognostic genes were closely associated with ferroptosis, necroptosis, pyroptosis, mitotic cell death, and lysosome-dependent cell death (Figure 3I), underscoring their relevance in the cell death regulatory network of sepsis. Time-dependent ROC curve analysis demonstrated good predictive performance of the model, with AUC values of 0.768, 0.747, 0.714, and 0.682 at 4, 7, 14, and 21 days, respectively (Figure 3J).
We validated the prognostic robustness of the model across independent external genes (Figure 4A–G). The risk scores were higher in patients with sepsis than in healthy volunteers (Figure 4A), and patients with higher-grade sepsis exhibited higher risk scores than those with lower-grade sepsis (Figure 4C). Survivor patients consistently displayed lower risk scores than non-survivors (Figure 4A, D, and G). Additionally, surgical interventions were associated with reduced risk scores (Figure 4B, E, F), whereas xigris treatment (activated protein C) did not significantly alter the risk scores.
 |
Figure 4 External validation of the PCD-based prognostic model. (A–G) Validation of the risk model across multiple independent GEO datasets. (H) Correlation between the prognostic risk score and immune cell infiltration. (I) Correlation heatmap of individual prognostic genes with various immune cell types. (J) Comparison of immune cell abundance between high- and low-risk groups. ns not significant, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. |
Further correlation analysis revealed a significant positive association between the risk score and infiltration of activated CD4+ T cells, whereas neutrophils exhibited a strong negative correlation (Figure 4H). At the individual gene level, strong correlations were observed with natural killer T cells, myeloid-derived suppressor cells, monocytes, and neutrophils (Figure 4I). Immune cell abundance across the risk groups further highlighted the immunological divergence associated with the prognostic model (Figure 4J). In summary, the established seven-gene prognostic model provides a reliable survival prediction and offers insights into the immune landscape and mechanisms of cell death in sepsis.
scRNA-Seq and Multi-Organ Temporal Transcriptomics Reveal Cellular Heterogeneity and Dynamic Changes in Sepsis
To further investigate the cellular heterogeneity of sepsis, we integrated scRNA-seq data from human PBMCs in datasets GSE167363 and GSE175453, and obtained 102,689 cells in total, which derived from 7 healthy control (HC, n = 39,204) and 14 sepsis (n = 63,485) samples. Based on the expression of canonical markers, the cells were divided into eight cell types (Figure 5A and B): B cells (CD79A+), dendritic cells (DC, CD1D+CD1C+), monocytes (CD14+VCAN+), neutrophils (CSF3R+MMP9+), plasma (JCHAIN+), platelets (PPBP+), red blood cells (RBC, HBA1+), and T/NK cells (IL7R+). Cellular proportions varied across different samples in the two datasets (Figure S3), in which monocytes and neutrophils mostly increased in abundance, whereas DC were reduced in sepsis samples, suggesting a shift in the immune cell composition during disease progression.
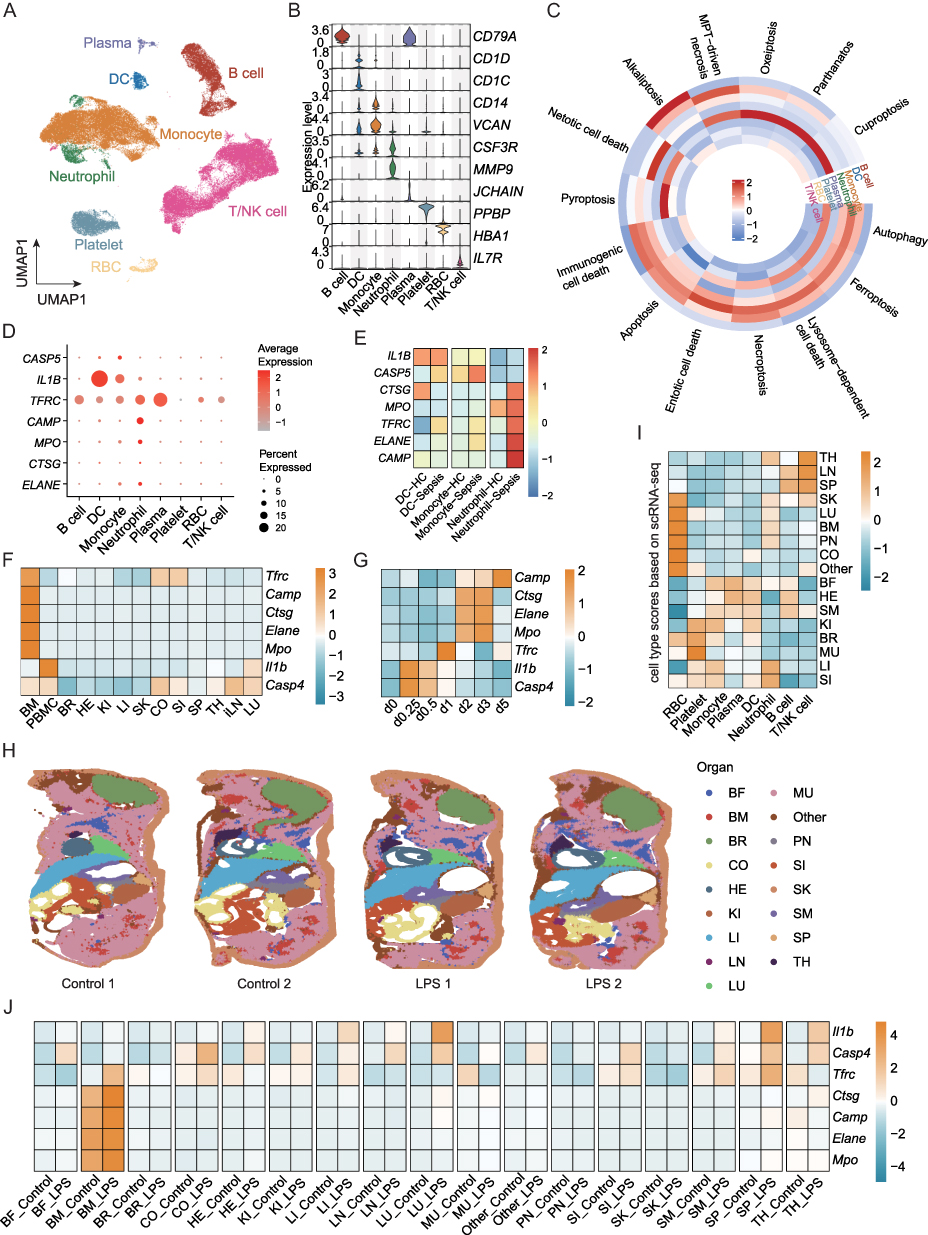 |
Figure 5 scRNA-seq and multi-organ temporal transcriptomics data analysis. (A) UMAP visualization of 8 cell types. (B) Violin plots show the relative expression levels of canonical markers. (C) Heatmap shows the activities of PCD patterns in each cell type. (D) The expression of seven prognosis genes in the main cell types. (E) The expression of seven prognosis genes in different groups. (F and G) The expression of seven prognosis genes in different tissues (F) and time points (G) after lipopolysaccharide (LPS) injection. (H) The spatial distribution of different organs of ST-seq dataset. (I) The cell type module score from scRNA-seq dataset in different organs of ST-seq dataset. (J) The expression of seven prognosis genes in different organs between control and LPS groups. Abbreviations: PMBC, peripheral blood mononuclear cell; MU, muscle; LU, lung; BF, brown fat; BM, bone marrow; LI, liver; SK, skin; SI, small intestine; BR, brain; CO, colon; KI, kidney; SM, stomach; HE, heart; SP, spleen; PN, pancreas; LN, lymph node; TH, thymus; DC, dendritic cell; RBC, red blood cell. |
We then quantified the activity scores of the 14 PCD patterns in the major cell types and found that PCD was predominantly active in plasma, DC, monocytes, and neutrophils, suggesting their central role in driving immune dysfunction and organ damage in sepsis (Figure 5C). Seven prognostic genes showed elevated expression in sepsis samples, particularly in DC, monocytes, and neutrophils (Figure 5D). Notably, these genes exhibited the highest expression levels in neutrophils of sepsis samples (Figure 5E), emphasizing the pivotal role of neutrophil-mediated immune dysregulation and cell death in sepsis.
To extend our findings to tissue-level and temporal dynamics, we analyzed the multi-organ and multi-timepoint transcriptomic profiles of LPS-induced sepsis mice (GSE224127), covering bone marrow (BM), PBMC, brain (BR), heart (HE), kidney (KI), liver(LI), skin (SK), colon (CO), small intestine (SI), spleen (SP), thymus (TH), inguinal lymph node (iLN), lung (LU) at 0, 0.25, 0.5, 1, 2, 3, and 5 days after sublethal LPS injection. The expression of the prognostic genes was predominantly enriched in the BM and PBMC samples (Figure 5F), and their levels gradually increased over time (Figure 5G). Importantly, the transcriptional activity of these genes plateaued around day 5, consistent with previous studies reporting transcriptional stabilization in non-lymphoid tissues after sepsis onset,41 suggesting that the most active inflammatory and cell death responses occur within the initial 3–5 days.
In addition, we utilized the spatial transcriptome across different organs in control and LPS-treated mice (Figure 5H). The cell type abundance in each organ was estimated using scRNA-seq-derived markers (Figure 5I). For example, TH, LN, and SP showed more T/NK and B cells and SK, LU, BM, Pancreas (PN)-enriched RBC, followed by neutrophils. Similarly, prognostic genes were mainly expressed in the BM and were elevated in the LPS group (Figure 5J). Taken together, single-cell and spatial transcriptomic analyses highlighted the central role of bone marrow and neutrophil-mediated cell death in sepsis progression, providing insights into the spatiotemporal dynamics of immune dysregulation.
Exploration of Potential Therapeutic Targets
To investigate potential therapeutic avenues, we integrated prognostic genes with active components from the SMYAD, a traditional Chinese medicine widely used for its anti-inflammatory and detoxifying properties. Based on the Traditional Chinese Medicine Systems Pharmacology Database, six active compounds with high bioavailability (quercetin, oleic acid, glycyrrhizin, ursolic acid, rutin, and thymol) were identified as candidate molecules that target prognostic genes (Figure 6A and B). Specifically, MPO and IL1B shared targets across multiple herbal components of Lonicerae japonicae flos, Figwort root, and Licorice, while ELANE was targeted by thymol from Lonicerae japonicae flos.
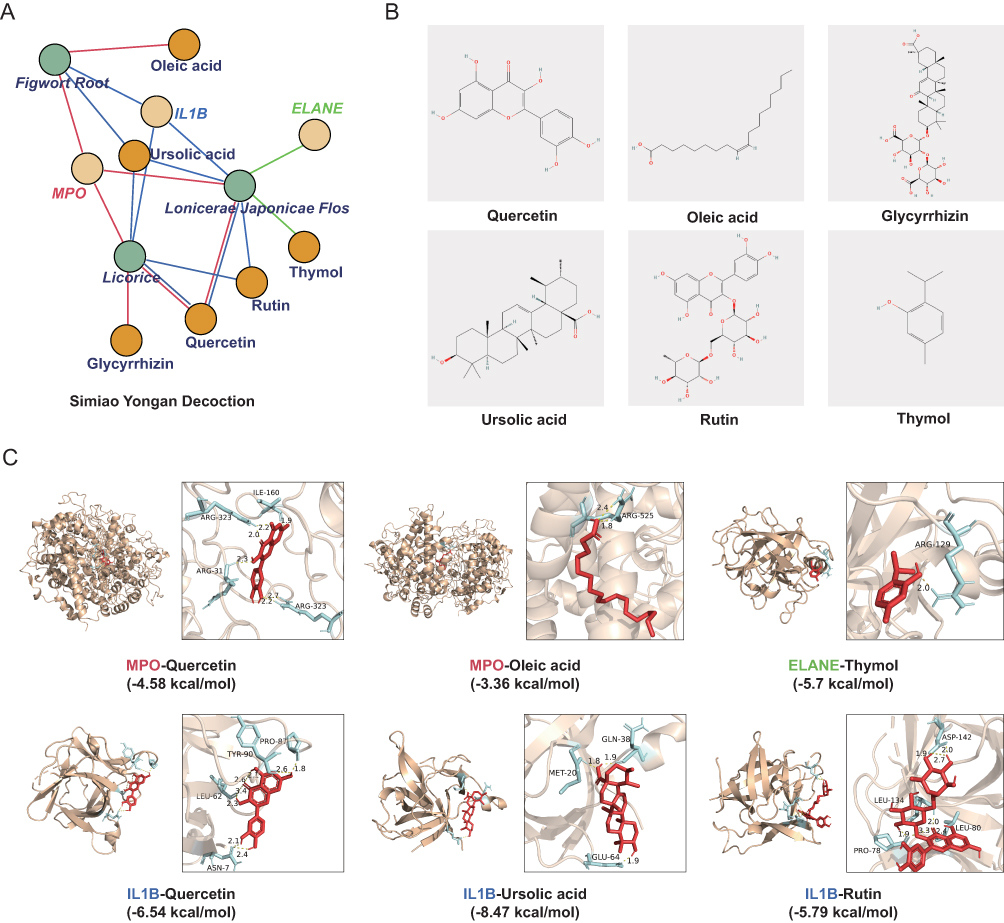 |
Figure 6 Molecular docking of active compounds from Simiao Yongan Decoction with prognostic target proteins. (A) The network of predicted interactions between Simiao Yongan Decoction (SMYAD) active components and prognostic genes. (B) The structures of active components. (C) Docking results showing the binding energies of each compound-protein complex. |
To further explore the binding affinities and potential mechanisms of SMYAD, we performed a molecular docking analysis of the target proteins (MPO, IL1B, ELANE) with the six hub compounds in Figure 6B. We prioritized the docked conformations with favorable hydrogen-bonding interactions, selecting those with the lowest binding energies, ranging from −8.47 kcal/mol (IL1B-Ursolic acid) to −3.36 kcal/mol (MPO-Oleic acid) (Figure 6C). Key amino acid residues were identified as potential binding sites for the small molecules. For example, ARG323 of MPO interacts with quercetin, ARG525 of MPO interacts with oleic acid, and ARG129 of ELANE interacts with thymol. In IL1B, MET20 binds to ursolic acid, and LEU134 interacts with rutin. More importantly, the docking energies of the latter four (Figure 6C) were less than −5 kcal/mol. These residues are likely to contribute to ligand-binding specificity and complex stability.
Discussions
Sepsis is a life-threatening disease that is characterized by a dysregulated host response to infection.43 Currently, clinical assessment of sepsis prognosis mainly relies on scoring systems such as the Acute Physiology and Chronic Health Evaluation (APACHE) II44 and Sequential Organ Failure Assessment (SOFA)45 but fails to fully reflect clinical complexity or accurately predict disease severity. Previous studies have demonstrated that multiple forms of PCD, including ferroptosis,46 cuproptosis,23 and pyroptosis,28 are involved in sepsis, exhibiting extensive interactions and crosstalk with one another. However, comprehensive investigation of the overall PCD landscape in sepsis is lacking.
In this study, we systematically characterized 14 distinct PCD patterns in sepsis, identified two distinct molecular subtypes, and developed a robust seven-gene prognostic model that was validated using external datasets. Using single-cell and multi-organ transcriptomic data, we emphasized the cellular heterogeneity and dynamic molecular changes associated with sepsis progression. Finally, we explored the potential therapeutic value of SMYAD through a molecular docking analysis.
The seven-gene signatures ELANE, CTSG, MPO, CAMP, TFRC, IL1B, and CASP5 were significantly associated with poor survival outcomes in patients with sepsis. These genes are not only prognostically relevant but also functionally involved in various PCD pathways that contribute to the pathophysiology of sepsis. ELANE, which encodes neutrophil elastase, is a serine protease released by activated neutrophils that plays a critical role in pyroptosis47 and netotic cell death.48 Its overexpression can lead to excessive neutrophil extracellular trap (NET) formation, resulting in uncontrolled inflammation, tissue damage, and microvascular thrombosis, which are hallmarks of sepsis-induced organ dysfunction.49 CTSG is another neutrophil-derived protease implicated in lysosome-dependent cell death, and has been reported to exacerbate inflammation by promoting proteolytic tissue injury and cytokine release in sepsis.50 MPO, encoding myeloperoxidase, is a key enzyme in neutrophils responsible for ROS production. MPO is a central regulator of mitotic cell death,51 and elevated MPO activity has been linked to endothelial damage and increased disease severity in septic patients.52 CAMP, which encodes the cathelicidin antimicrobial peptide, not only exhibits potent antimicrobial activity, but also contributes to netotic cell death by modulating neutrophil activation and degranulation,53 further amplifying inflammatory responses during sepsis. TFRC is an essential regulator of cellular iron uptake, and its dysregulation contributes to ferroptosis,54 an iron-dependent form of PCD driven by lipid peroxidation. Increased TFRC expression during sepsis may exacerbate oxidative stress and ferroptosis in parenchymal and immune cells, thereby promoting organ injury.55 IL1B, a classical pro-inflammatory cytokine and canonical marker of pyroptosis and necroptosis,56 is activated and released in response to inflammasome signaling. It directly drives systemic inflammation and vascular leakage in septic shock.57 CASP5, a member of the inflammatory caspase family, functions as a critical effector of noncanonical pyroptosis. Upon sensing intracellular LPS, CASP5 activates gasdermin D and initiates pyroptotic membrane rupture, promoting inflammatory cell death and immune dysregulation.58 Together, these gene sets embody the convergence of inflammatory amplification and programmed cell death, leading to poor sepsis outcomes.
scRNA-seq data revealed that DCs, monocytes, and neutrophils exhibited high susceptibility to the prognostic gene signature, with neutrophils showing the strongest response to sepsis. In patients with sepsis, the abundance of DCs is reduced via apoptosis and pyroptosis,59,60 accompanied by impaired maturation and diminished antigen-presenting capacity, ultimately leading to suppression of innate immune responses. Monocyte counts and percentages are significantly decreased in the early stage of sepsis, accompanied by the upregulation of apoptosis- and pyroptosis-related genes.61,62 Continuous stimulation impairs the ability of monocytes to produce proinflammatory cytokines while promoting anti-inflammatory cytokine expression,63 leading to sepsis-associated immunosuppression. Neutrophil dysfunction during sepsis is particularly notable These cells exhibit impaired chemotaxis,64 delayed apoptosis,59 and excessive formation of neutrophil extracellular traps (NETs),65 resulting in a hyperinflammatory and cytotoxic phenotype. This persistent activation and dysregulated cell death contribute to immunopathology, endothelial dysfunction, and ultimately multi-organ failure.66 NETs are web-like extracellular structures decorated with antimicrobial components such as neutrophil elastase (NE, encoded by ELANE), myeloperoxidase (MPO) and histones.67 Notably, both ELANE and MPO were present in our prognostic gene signature, underscoring their potential involvement in sepsis-associated immune dysregulation.
Furthermore, the multi-organ, multi-time point transcriptomic dataset revealed that the expression of prognostic genes was predominantly elevated in bone marrow and PBMCs. As a dynamic immune organ, the bone marrow undergoes profound dysregulation during sepsis, leading to significant alterations in hematopoiesis and immune cell function.68 Notably, sepsis shifts myeloid progenitors toward emergency myelopoiesis,69 resulting in aberrant production of neutrophils and monocytes. PBMCs, as key mediators of the systemic inflammatory response,70 show high expression of these prognostic genes, suggesting their critical involvement in peripheral immune activation and immune imbalance during sepsis. Importantly, the gene expression patterns exhibited a progressive increase, peaking around day 3 following LPS-induced sepsis, aligning with transcriptional homeostasis in non-lymphoid tissues after sepsis onset,41 suggesting that the most active inflammatory and cell death responses occur within the initial days, highlighting a critical therapeutic window during the early stages of sepsis.71 Morever, this temporal expression pattern was consistent with the kinetics of neutrophil activation and NETs formation,72 further supporting the contribution of neutrophil-driven pathology in early sepsis.
In parallel, we explored the pharmacological relevance of SMYAD, a classical TCM formula used for conditions resembling heat, toxin, and blood stasis syndromes in TCM theory.73,74 Linear-trap-LC/MSN identified 113 chemical compounds in the extract and medicated serum of SMYAD,30 in which vitexin could inhibit the release of pro-inflammatory cytokines (TNF-α, IL-1β) from macrophages to effectively reduce leukocyte migration in septic mice;75 Ferulic acid modulates inflammatory responses, autophagy and apoptosis during sepsis via NF-κB and Nrf2 pathways, thereby alleviating sepsis-related organ injury.76,77 Furthermore, compounds such as Lonicerae japonicae flos and Figwort root are rich in flavonoids and organic acids, which can markedly decrease TNF-α and IL-6 levels, to reduce cytokine storm and disease exacerbation.78,79 Clinically, SMYAD combined with antibiotics has been reported to further reduce TNF-α and IL-8 levels compared with antibiotics alone;80 and combination with Imipenem/Cilastatin restore cytokine balance (eg, IL-2, TNF-α, IL-4) and attenuate systemic inflammation in septic rats.30 In our study, molecular docking revealed that key SMYAD-derived compounds exhibited strong binding affinities to MPO, IL1B, and ELANE, which are central regulators of PCD pathways in sepsis. These interactions suggest that SMYAD may exert anti-inflammatory and immunomodulatory effects by directly targeting PCD-related proteins,81 supporting its potential as an adjunctive therapy for sepsis.
Previous studies typically focused on individual PCD pathways in sepsis. For example, Liang et al developed a six-gene (GZMB, CHMP7, NLRP1, MYD88, ELANE, and AIM2) prognostic model related with pyroptosis;28 ferroptosis-related siguratures included TFRC, DPP4 and PRKCA;46 hub genes in apoptosis contained BCL2, FASLG, IRF9 and JAK3;82 cuproptosis identified a five-gene signature (PTPRJ, FCN1, SVIP, LILRB1 and LST1);83 necroptosis highlighted BACH2, GATA3, LEF1, and BCL2;84 and PANoptosis, referring to the interplay of pyroptosis (P), apoptosis (A), and necroptosis (N), involved CD14, GSDMD, IL1B, and FAS.85 These studies provided valuable insights, whereas leaving the broader landscape of PCD crosstalk in sepsis unexplored. Notably, accumulating evidence suggests that distinct cell death programs are not isolated but can interact and influence one another,86 and the comprehensive analysis promote to uncover the potential therapeutic targets.87 Our work systematically integrates 14 distinct PCD patterns into a unified analytical framework, emphasizing the pivotal roles of pyroptosis, ferroptosis, necroptosis, lysosome-dependent cell death, and netotic cell death. Combining previous findings with the relationship between prognostic genes and PCD patterns, we propose that in sepsis, distinct PCD programs form an interconnected network. Activated CASP5 cleaves gasdermin D (GSDMD) to generate a functional N-terminal fragment capable of pore formation on the plasma membrane,88 enabling IL1B release into the extracellular space. This process triggers pyroptosis,11 as well as necroptotsis,89 thereby amplifying inflammatory responses. The cleavage of GSDMD can also be mediated by ELANE.90 During sepsis, excessive inflammatory cytokines upregulate transferrin receptor (TFRC) expression on the cell surface,91 promoting Fe uptake and triggering ferroptosis.54 Loss of lysosomal membrane integrity leads to the release of enzymes such as CTSG, causing lysosome-dependent cell death, meanwhile lysosomal dysfunction further disrupts Fe homeostasis and modulates ferroptosis.92 Moreover, pro-inflammatory cytokines activate neutrophils,93 inducing nuclear translocation of MPO and ELANE, which compromise the nuclear envelope and facilitate the formation of NETs (composed of MPO, ELANE, CAMP, etc.), resulting in netotic cell death.94 Collectively, these interconnected programmed cell death pathways exacerbate systemic inflammation and tissue damage, driving the progression of sepsis. Although these mechanisms are not directly tested in our study, they provide a plausible biological framework linking PCD heterogeneity with sepsis outcomes, suggesting that coordinated regulation of multiple PCD programs may be critical for therapeutic intervention.
However, there are several limitations should be acknowledged. First, our model was developed using retrospective datasets, potential confounding factors, including patient age, comorbidities, and sample heterogeneity, could influence the observed associations between PCD patterns and clinical outcomes. Second, while our molecular docking results offer promising targets, in vitro and in vivo validation of these compound-protein interactions is necessary. Finally, as our analyses are primarily observational and computational, causality cannot be directly inferred, and model-based predictions may be affected by inherent limitations of the datasets.
Conclusion
This study comprehensively delineated the landscape of 14 PCD patterns in sepsis. We identified two distinct subtypes, and established a robust seven-gene prognostic signature (including ELANE, CTSG, MPO, CAMP, TFRC, IL1B, and CASP5) with poor outcome, which was significantly elevated in sepsis and decreased after treatment in the validation cohorts. Moreover, SMYAD might interact with key PCD proteins according to the molecular docking analysis, supporting its potential as a multi-target adjunctive therapy. Collectively, these findings provide a framework for early risk stratification and biomarker-guided interventions in sepsis. Future research should validate these results in larger and prospective cohorts, and assess the therapeutic efficacy of SMYAD through preclinical and clinical trials.
Abbreviations
PCD, programmed cell death; ICD, immunogenic cell death; DAMPs, damage-associated molecular patterns; SMYAD, Simiao Yongan Decoction; MODS, multiple organ dysfunction syndrome; MPT, mitochondrial permeability transition; TCM, Traditional Chinese Medicine; GEO, Gene Expression Omnibus; PBMCs, peripheral blood mononuclear cells; SAPSII, Simplified Acute Physiology Score II; CDF, cumulative distribution function; PCA, principal component analysis; GSEA, Gene Set Enrichment Analysis; MSigDB, Molecular Signatures Database; FDR, false discovery rate; DEGs, differentially expressed genes; LASSO, Least Absolute Shrinkage and Selection Operator; ROC, receiver operating characteristic; AUC, area under curve; HR, hazard ratios; CI, confidence intervals; PDB, Protein Data Bank; TCMSP, Traditional Chinese Medicine Systems Pharmacology; scRNA-seq, single-cell RNA sequencing; UMAP, Uniform Manifold Approximation and Projection; LPS, lipopolysaccharide; neutrophil extracellular traps, NETs; ssGSEA, single-sample gene set enrichment analysis; GSVA, gene set variation analysis; EMT, epithelial-mesenchymal transition; ROS, reactive oxygen species; DC, dendritic cells; RBC, red blood cells; BM, bone marrow; BR, brain; HE, heart; KI, kidney; LI, liver; SK, skin; CO, colon; SI, small intestine; SP, spleen; TH, thymus; iLN, inguinal lymph node; LU, lung; PN, pancreas; APACHE, Acute Physiology and Chronic Health Evaluation; SOFA, Sequential Organ Failure Assessment; NET, neutrophil extracellular trap; NE, neutrophil elastase; MPO, myeloperoxidase.
Data Sharing Statement
Publicly available datasets were analyzed in this study. These data are available from the GEO database (https://www.ncbi.nlm.nih.gov/).
Ethics Approval and Consent to Participate
The public data used in this study were obtained from previously published research, and all datasets had already received approval from the respective institutional review committees. According to Article 32 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects dated February 18, 2023, China, certain studies involving human subjects might be exempt from ethical review if they pose no harm to human beings, no involvement of sensitive personal information, and no involvement of commercial interests. Specifically, items 1 and 2 point that researches (1) using legally obtained public data or data generated through observation that does not interfere with public behaviors; (2) using anonymized information and data, might qualify for exemption. As our study fully meets these criteria, it falls within the exemption scope stipulated by the Measures.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet. 2020;395(10219):200–211. doi:10.1016/s0140-6736(19)32989-7
3. Shankar-Hari M, Harrison DA, Rowan KM, Rubenfeld GD. Estimating attributable fraction of mortality from sepsis to inform clinical trials. J Crit Care. 2018;45:33–39. doi:10.1016/j.jcrc.2018.01.018
4. Allen VB, Bechman K, Russell MD, et al. Rising rates of sepsis in England: an ecological study. Infection. 2025. doi:10.1007/s15010-025-02601-0
5. Morrissey R, Lee J, Baral N, et al. Demographic and regional trends of sepsis mortality in the United States, 1999-2022. BMC Infect Dis. 2025;25(1):504. doi:10.1186/s12879-025-10921-7
6. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. doi:10.1136/bmj.i1585
7. Zhang H, Chen S, Wang Y, et al. Neutrophil-based single-cell sequencing combined with transcriptome sequencing to explore a prognostic model of sepsis. Sci Rep. 2024;14(1):29856. doi:10.1038/s41598-024-80791-7
8. Jiang S, Zhang W, Lu Y. Development and validation of novel inflammatory response-related gene signature for sepsis prognosis. J Zhejiang Univ Sci B. 2022;23(12):1028–1041. doi:10.1631/jzus.B2200285
9. Pierrakos C, Velissaris D, Bisdorff M, et al. Biomarkers of sepsis: time for a reappraisal. Crit Care. 2020;24(1):287. doi:10.1186/s13054-020-02993-5
10. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21(11):678–695. doi:10.1038/s41580-020-0270-8
11. Zheng X, Chen W, Gong F, et al. The role and mechanism of pyroptosis and potential therapeutic targets in sepsis: a review. Front Immunol. 2021;12:711939. doi:10.3389/fimmu.2021.711939
12. Hu S, Huang M, Mao S, et al. Serinc2 deficiency exacerbates sepsis-induced cardiomyopathy by enhancing necroptosis and apoptosis. Biochem Pharmacol. 2023;218:115903. doi:10.1016/j.bcp.2023.115903
13. Florey O, Kim SE, Overholtzer M. Entosis: cell-in-cell formation that kills through entotic cell death. Curr Mol Med. 2015;15(9):861–866. doi:10.2174/1566524015666151026100042
14. Li G, Tian X, Wei E, et al. Immunogenic cell death biomarkers for sepsis diagnosis and mechanism via integrated bioinformatics. Sci Rep. 2025;15(1):18575. doi:10.1038/s41598-025-03282-3
15. Liu J, Kuang F, Kang R, Tang D. Alkaliptosis: a new weapon for cancer therapy. Cancer Genet Ther. 2020;27(5):267–269. doi:10.1038/s41417-019-0134-6
16. Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med. 2000;28(4 Suppl):N105–113. doi:10.1097/00003246-200004001-00013
17. Xl L, Gy Z, R G, N C. Ferroptosis in sepsis: the mechanism, the role and the therapeutic potential. Front Immunol. 2022;13:956361. doi:10.3389/fimmu.2022.956361
18. Feng Y, Liu B, Zheng X, et al. The protective role of autophagy in sepsis. Microb Pathogenesis. 2019;131:106–111. doi:10.1016/j.micpath.2019.03.039
19. Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013;126(Pt 9):1905–1912. doi:10.1242/jcs.091181
20. Yan Y, Li B, Gao Q, et al. Intestine-decipher engineered capsules protect against sepsis-induced intestinal injury via broad-spectrum anti-inflammation and parthanatos inhibition. Adv Sci. 2025;12(10):e2412799. doi:10.1002/advs.202412799
21. Xiao Q, Zhong B, Hou Y, et al. Fighting cancer by triggering non-canonical mitochondrial permeability transition-driven necrosis through reactive oxygen species induction. Free Radic Biol Med. 2023;202:35–45. doi:10.1016/j.freeradbiomed.2023.03.020
22. Oikawa D, Gi M, Kosako H, et al. OTUD1 deubiquitinase regulates NF-κB- and KEAP1-mediated inflammatory responses and reactive oxygen species-associated cell death pathways. Cell Death Dis. 2022;13(8):694. doi:10.1038/s41419-022-05145-5
23. Zhang J, Wu Y, Du Y, et al. Cuproptosis-related genes as prognostic biomarkers for sepsis: insights into immune function and personalized immunotherapy. J Inflamm Res. 2024;17:4229–4245. doi:10.2147/jir.S461766
24. Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187(2):235–256. doi:10.1016/j.cell.2023.11.044
25. Cao K, Zhu J, Lu M, et al. Analysis of multiple programmed cell death-related prognostic genes and functional validations of necroptosis-associated genes in oesophageal squamous cell carcinoma. EBioMedicine. 2024;99:104920. doi:10.1016/j.ebiom.2023.104920
26. Lorente L, Martín MM, Ortiz-López R, et al. Parthanatos type programmed cell death and septic patient mortality. Medicina intensiva. 2023;47(12):691–696. doi:10.1016/j.medine.2023.05.007
27. Wu H, Tang T, Deng H, et al. Immune checkpoint molecule Tim-3 promotes NKT cell apoptosis and predicts poorer prognosis in Sepsis. Clin Immunol. 2023;254:109249. doi:10.1016/j.clim.2023.109249
28. Liang S, Xing M, Chen X, et al. Predicting the prognosis in patients with sepsis by a pyroptosis-related gene signature. Front Immunol. 2022;13:1110602. doi:10.3389/fimmu.2022.1110602
29. Song Y, Lin W, Zhu W. Traditional Chinese medicine for treatment of sepsis and related multi-organ injury. Front Pharmacol. 2023;14:1003658. doi:10.3389/fphar.2023.1003658
30. Cai Z, He J, Jiang J, et al. Systematic investigation of the material basis, multiple mechanisms and quality control of Simiao Yong’an decoction combined with antibiotic in the treatment of sepsis. Phytomedicine. 2023;116:154910. doi:10.1016/j.phymed.2023.154910
31. Zhu ZB, Song K, Huang WJ, et al. Si-Miao-Yong-An (SMYA) decoction may protect the renal function through regulating the autophagy-mediated degradation of ubiquitinated protein in an atherosclerosis model. Front Pharmacol. 2020;11:837. doi:10.3389/fphar.2020.00837
32. Hou Y, Li J, Ding Y, et al. Luteolin attenuates lipopolysaccharide-induced acute lung injury/acute respiratory distress syndrome by activating alveolar epithelial sodium channels via cGMP/PI3K pathway. J Ethnopharmacol. 2022;282:114654. doi:10.1016/j.jep.2021.114654
33. He F, Gao F, Cai N, et al. Chlorogenic acid enhances alveolar macrophages phagocytosis in acute respiratory distress syndrome by activating G protein-coupled receptor 37 (GPR 37). Phytomedicine. 2022;107:154474. doi:10.1016/j.phymed.2022.154474
34. Tang Z, Zhang Z, Zhao J, et al. Integrated analysis of multiple programmed cell death-related prognostic genes and functional validation of apoptosis-related genes in osteosarcoma. Int J Biol Macromol. 2025;307(Pt 3):142113. doi:10.1016/j.ijbiomac.2025.142113
35. Severino P, Silva E, Baggio-Zappia GL, et al. Gene expression profiling of mononuclear cells from patients with sepsis secondary to community-acquired pneumonia. Genomics Data. 2014;2:332–334. doi:10.1016/j.gdata.2014.10.004
36. Sutherland A, Thomas M, Brandon RA, et al. Development and validation of a novel molecular biomarker diagnostic test for the early detection of sepsis. Crit Care. 2011;15(3):R149. doi:10.1186/cc10274
37. Cazalis M-A, Lepape A, Venet F, et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Experimental. 2014;2(1):20. doi:10.1186/s40635-014-0020-3
38. Wong HR, Cvijanovich N, Lin R, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7(1):34. doi:10.1186/1741-7015-7-34
39. Qiu X, Li J, Bonenfant J, et al. Dynamic changes in human single-cell transcriptional signatures during fatal sepsis. J Leukocyte Biol. 2021;110(6):1253–1268. doi:10.1002/jlb.5ma0721-825r
40. Darden DB, Dong X, Brusko MA, et al. A novel single cell RNA-seq analysis of non-myeloid circulating cells in late sepsis. Front Immunol. 2021;12:696536. doi:10.3389/fimmu.2021.696536
41. Takahama M, Patil A, Richey G, et al. A pairwise cytokine code explains the organism-wide response to sepsis. Nat Immunol. 2024;25(2):226–239. doi:10.1038/s41590-023-01722-8
42. Stanski NL, Wong HR. Prognostic and predictive enrichment in sepsis. Nat Rev Nephrol. 2020;16(1):20–31. doi:10.1038/s41581-019-0199-3
43. Caraballo C, Jaimes F. Organ dysfunction in sepsis: an ominous trajectory from infection to death. Yale J Biol Med. 2019;92(4):629–640.
44. Kuo WK, Hua CC, Yu CC, et al. The cancer control status and APACHE II score are prognostic factors for critically ill patients with cancer and sepsis. J Formosan Med Assoc. 2020;119(1 Pt 2):276–281. doi:10.1016/j.jfma.2019.05.012
45. Neumann F, Lobitz O, Fenk R, et al. The sepsis-related organ failure assessment (SOFA) score is predictive for survival of patients admitted to the intensive care unit following allogeneic blood stem cell transplantation. Ann Hematol. 2008;87(4):299–304. doi:10.1007/s00277-008-0440-9
46. Wang Y, Bian Z. Development of a ferroptosis-related gene prognostic model and molecular subgroups characterization in sepsis. Mol Immunol. 2025;178:1–11. doi:10.1016/j.molimm.2024.12.008
47. Fu T, Zhou J, Yang L, et al. Neutrophil-induced pyroptosis promotes survival in patients with hepatoblastoma. Cancer Immunol Immunother. 2025;74(3):106. doi:10.1007/s00262-024-03922-z
48. Thanarajasingam U, Jensen MA, Dorschner JM, et al. Brief report: a novel ELANE mutation associated with inflammatory arthritis, defective NETosis, and recurrent parvovirus infection. Arthritis Rheumatol. 2017;69(12):2396–2401. doi:10.1002/art.40314
49. Yao L, Xu J, Yang X, Shang Y. Research progress of effect of neutrophil elastase and its inhibitors in sepsis. Zhonghua wei Zhong Bing Ji Jiu Yi xue. 2022;34(11):1209–1212. doi:10.3760/cma.j.cn121430-20220419-00389
50. Tralau T, Meyer-Hoffert U, Schröder JM, Wiedow O. Human leukocyte elastase and cathepsin G are specific inhibitors of C5a-dependent neutrophil enzyme release and chemotaxis. Experimental Dermatol. 2004;13(5):316–325. doi:10.1111/j.0906-6705.2004.00145.x
51. Jamali E, Abbasi M, Tayer AH, et al. The significance of surface neutrophilic MPO expression level in NETosis and NETosis-associated coagulopathies in covid-19 infected patients. Blood Cells Mol Dis. 2022;96:102676. doi:10.1016/j.bcmd.2022.102676
52. Liu L, Shao Y, Zhang Y, et al. Neutrophil-derived heparin binding protein triggers vascular leakage and synergizes with myeloperoxidase at the early stage of severe burns (With video). Burns Trauma. 2021;9:tkab030. doi:10.1093/burnst/tkab030
53. Zhang Q, Ul Ain Q, Schulz C, Pircher J. Role of antimicrobial peptide cathelicidin in thrombosis and thromboinflammation. Front Immunol. 2023;14:1151926. doi:10.3389/fimmu.2023.1151926
54. Yi L, Hu Y, Wu Z, et al. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022;13(7):592. doi:10.1038/s41419-022-05027-w
55. Xing P, Zhou M, Sun J, et al. NAT10-mediated ac(4)C acetylation of TFRC promotes sepsis-induced pulmonary injury through regulating ferroptosis. Mol Med. 2024;30(1):140. doi:10.1186/s10020-024-00912-w
56. Sharma AK, Ismail N. Non-canonical inflammasome pathway: the role of cell death and inflammation in ehrlichiosis. Cells. 2023;12(22):2597. doi:10.3390/cells12222597
57. Jiménez-Sousa M, Medrano LM, Liu P, et al. IL-1B rs16944 polymorphism is related to septic shock and death. Eur J Clin Invest. 2017;47(1):53–62. doi:10.1111/eci.12702
58. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
59. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi:10.1038/nri3552
60. Zheng LY, Duan Y, He PY, et al. Dysregulated dendritic cells in sepsis: functional impairment and regulated cell death. Cell Mol Biol Lett. 2024;29(1):81. doi:10.1186/s11658-024-00602-9
61. Leijte GP, Rimmelé T, Kox M, et al. Monocytic HLA-DR expression kinetics in septic shock patients with different pathogens, sites of infection and adverse outcomes. Crit Care. 2020;24(1):110. doi:10.1186/s13054-020-2830-x
62. Washburn ML, Wang Z, Walton AH, et al. T cell- and monocyte-specific RNA-sequencing analysis in septic and nonseptic critically ill patients and in patients with cancer. J Immunol. 2019;203(7):1897–1908. doi:10.4049/jimmunol.1900560
63. Liu D, Cao S, Zhou Y, Xiong Y. Recent advances in endotoxin tolerance. J Cell Biochem. 2019;120(1):56–70. doi:10.1002/jcb.27547
64. Wang X, Qin W, Xu X, et al. Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc Natl Acad Sci USA. 2017;114(17):4483–4488. doi:10.1073/pnas.1616752114
65. Liu L, Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. 2019;76(11):2031–2042. doi:10.1007/s00018-019-03060-1
66. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–420. doi:10.1038/nri.2017.36
67. Zhang H, Wang Y, Qu M, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med. 2023;13(1):e1170. doi:10.1002/ctm2.1170
68. Davis FM, Schaller MA, Dendekker A, et al. Sepsis induces prolonged epigenetic modifications in bone marrow and peripheral macrophages impairing inflammation and wound healing. Arteriosclerosis Thrombosis Vasc Biol. 2019;39(11):2353–2366. doi:10.1161/atvbaha.119.312754
69. Biswas N, Bahr A, Howard J, et al. Survivors of polymicrobial sepsis are refractory to G-CSF-induced emergency myelopoiesis and hematopoietic stem and progenitor cell mobilization. Stem Cell Reports. 2024;19(5):639–653. doi:10.1016/j.stemcr.2024.03.007
70. Da Rosa PC, Bertomeu JB, Royes LFF, Osiecki R. The physical exercise-induced oxidative/inflammatory response in peripheral blood mononuclear cells: signaling cellular energetic stress situations. Life Sci. 2023;321:121440. doi:10.1016/j.lfs.2023.121440
71. Mehdi SF, Pusapati S, Khenhrani RR, et al. Oxytocin and related peptide hormones: candidate anti-inflammatory therapy in early stages of sepsis. Front Immunol. 2022;13:864007. doi:10.3389/fimmu.2022.864007
72. Oh SA, Seol SI, Davaanyam D, et al. Platelet-derived HMGB1 induces NETosis, exacerbating brain damage in the photothrombotic stroke model. Mol Med. 2025;31(1):46. doi:10.1186/s10020-025-01107-7
73. Wang F, Deng D, Zhou Y, Yang R. A case report on the treatment of total placenta increta after transvaginal delivery with integrated traditional Chinese and western medicine. Ann Palliative Med. 2022;11(1):384–393. doi:10.21037/apm-21-3794
74. Zeng Y, Ma Y, Zeng Y, et al. Research progress on modified Simiao Yongan Decoction in treatment of sepsis and its application and development ideas and methods. Chin Traditional Herbal Drugs. 2025;56(1):330–339. doi:10.7501/j.issn.0253-2670.2025.01.031
75. Rosa SI, Rios-Santos F, Balogun SO, Martins DT. Vitexin reduces neutrophil migration to inflammatory focus by down-regulating pro-inflammatory mediators via inhibition of p38, ERK1/2 and JNK pathway. Phytomedicine. 2016;23(1):9–17. doi:10.1016/j.phymed.2015.11.003
76. Mir SM, Ravuri HG, Pradhan RK, et al. Ferulic acid protects lipopolysaccharide-induced acute kidney injury by suppressing inflammatory events and upregulating antioxidant defenses in Balb/c mice. Biomed Pharmacother. 2018;100:304–315. doi:10.1016/j.biopha.2018.01.169
77. Tang X, Liu J, Yao S, et al. Ferulic acid alleviates alveolar epithelial barrier dysfunction in sepsis-induced acute lung injury by activating the Nrf2/HO-1 pathway and inhibiting ferroptosis. Pharm Biol. 2022;60(1):2286–2294. doi:10.1080/13880209.2022.2147549
78. Xu G, Fu S, Zhan X, et al. Echinatin effectively protects against NLRP3 inflammasome-driven diseases by targeting HSP90. JCI Insight. 2021;6(2). doi:10.1172/jci.insight.134601
79. Zhang B, Zeng M, Wang Y, et al. Oleic acid alleviates LPS-induced acute kidney injury by restraining inflammation and oxidative stress via the Ras/MAPKs/PPAR-γ signaling pathway. Phytomedicine. 2022;94:153818. doi:10.1016/j.phymed.2021.153818
80. Li S, Yan G, Chen Z. Influence of “Jiawei Simiao Yong’an decoction” on coagulation dysfunction of patients with sepsis. Acta Universitatis Traditionis Medicalis Sinensis Pharmacologiaeoue Shanghai. 2016;30(1):4. doi:10.16306/i.1008-861x.2016.01.007
81. Zhang S, Shao Y, Jin R, Ma B. Combining network pharmacology, molecular docking, molecular dynamics simulation, and experimental validation to uncover the efficacy and mechanisms of Si-Miao-Yong-An decoction in diabetic wound healing. J Inflamm Res. 2025;18:4087–4101. doi:10.2147/jir.S506739
82. Sun Z, Hu Y, Qu J, et al. Identification of apoptosis-immune-related gene signature and construction of diagnostic model for sepsis based on single-cell sequencing and bulk transcriptome analysis. Front Genetics. 2024;15:1389630. doi:10.3389/fgene.2024.1389630
83. Zhao T, Guo Y, Li J. Identification and experimental validation of cuproptosis regulatory program in a sepsis immune microenvironment through a combination of single-cell and bulk RNA sequencing. Front Immunol. 2024;15:1336839. doi:10.3389/fimmu.2024.1336839
84. She H, Tan L, Yang R, et al. Identification of featured necroptosis-related genes and imbalanced immune infiltration in sepsis via machine learning. Front Genetics. 2023;14:1158029. doi:10.3389/fgene.2023.1158029
85. Yang Z, Kao X, Huang N, et al. Identification and analysis of panoptosis-related genes in sepsis-induced lung injury by bioinformatics and experimental verification. J Inflamm Res. 2024;17:1941–1956. doi:10.2147/jir.S452608
86. Saleem S. Apoptosis, autophagy, necrosis and their multi galore crosstalk in neurodegeneration. Neuroscience. 2021;469:162–174. doi:10.1016/j.neuroscience.2021.06.023
87. Chen Z, Zhang R, Zhao Z, et al. Multiple cell-death patterns predict the prognosis and drug sensitivity of melanoma patients. Front Pharmacol. 2024;15:1295687. doi:10.3389/fphar.2024.1295687
88. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27(9):673–684. doi:10.1016/j.tcb.2017.05.005
89. Adkar SS, Leeper NJ. Efferocytosis in atherosclerosis. Nature reviews. Cardiology. 2024;21(11):762–779. doi:10.1038/s41569-024-01037-7
90. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharmaceutica Sinica B. 2021;11(9):2768–2782. doi:10.1016/j.apsb.2021.02.006
91. Butt AT, Thomas MS. Iron acquisition mechanisms and their role in the virulence of burkholderia species. Front Cell Infect Microbiol. 2017;7:460. doi:10.3389/fcimb.2017.00460
92. Feng J, Wang ZX, Bin JL, et al. Pharmacological approaches for targeting lysosomes to induce ferroptotic cell death in cancer. Cancer Lett. 2024;587:216728. doi:10.1016/j.canlet.2024.216728
93. Mousset A, Lecorgne E, Bourget I, et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell. 2023;41(4):757–775.e710. doi:10.1016/j.ccell.2023.03.008
94. Zambrano F, Uribe P, Schulz M, et al. Antioxidants as modulators of netosis: mechanisms, evidence, and therapeutic potential. Int J Mol Sci. 2025;26(11):5272. doi:10.3390/ijms26115272
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Machine Learning Predictive Model for Septic Shock in Acute Pancreatitis with Sepsis
Xia Y, Long H, Lai Q, Zhou Y
Journal of Inflammation Research 2024, 17:1443-1452
Published Date: 5 March 2024
Identification and Analysis of PANoptosis-Related Genes in Sepsis-Induced Lung Injury by Bioinformatics and Experimental Verification
Yang Z, Kao X, Huang N, Yuan K, Chen J, He M
Journal of Inflammation Research 2024, 17:1941-1956
Published Date: 28 March 2024
Acute Kidney Injury Prediction Model Using Cystatin-C, Beta-2 Microglobulin, and Neutrophil Gelatinase-Associated Lipocalin Biomarker in Sepsis Patients
Susianti H, Asmoro AA, Sujarwoto, Jaya W, Sutanto H, Kusdijanto AY, Kuwoyo KP, Hananto K, Khrisna MB
International Journal of Nephrology and Renovascular Disease 2024, 17:105-112
Published Date: 28 March 2024
Cuproptosis-Related Biomarkers and Characterization of Immune Infiltration in Sepsis
Wang Y, Qiu X, Liu J, Liu X, Pan J, Cai J, Liu X, Qu S
Journal of Inflammation Research 2024, 17:2459-2478
Published Date: 22 April 2024
Comprehensive Analysis of Programmed Cell Death-Related Genes in Diagnosis and Synovitis During Osteoarthritis Development: Based on Bulk and Single-Cell RNA Sequencing Data
Zhou J, Jiao S, Huang J, Dai T, Xu Y, Xia D, Feng Z, Chen J, Li Z, Hu L, Meng Q
Journal of Inflammation Research 2025, 18:751-778
Published Date: 17 January 2025