Back to Journals » Journal of Pain Research » Volume 18
Monosodium Iodoacetate Induces Knee Osteoarthritis in Rats in a Dose- and Time-Dependent Manner
Authors Song M, Han W, Li J, Wu G, Yu H, Yuan Q, Yu J
Received 14 May 2025
Accepted for publication 20 September 2025
Published 17 October 2025 Volume 2025:18 Pages 5507—5522
DOI https://doi.org/10.2147/JPR.S527135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wendy Imlach
Video abstract of “Mono-iodoacetate in knee osteoarthritis” [527135].
Views: 66
Min Song,1,2,* Weichang Han,1,* Jingyi Li,1,* Gaoyi Wu,2 Haibo Yu,2 Qing Yuan,1 Jiani Yu3,4
1Medical College of Acu-Moxi and Rehabilitation, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 2The Fourth Affiliated Hospital of Guangzhou University of Chinese Medicine; Shenzhen Traditional Chinese Medicine Hospital, Shenzhen, Guangdong, People’s Republic of China; 3The Second Affiliated Hospital of Guangzhou University of Chinese Medicine; Guangdong Provincial Hospital of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 4Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qing Yuan, Email [email protected] Jiani Yu, Email [email protected]
Background: Knee osteoarthritis (KOA) is indeed a major global health issue, characterized as the most common form of osteoarthritis. It affects millions of people worldwide, leading to joint pain, stiffness, and functional limitations. As life expectancy increases and populations age, KOA’s impact on quality of life and public health systems becomes more pronounced. Our objective was to evaluate pain-related behaviors, tibial cartilage pathology, and inflammatory factor expression levels in rats following different doses and administration times of monosodium iodoacetate (MIA) to explore its effects on KOA.
Methods: The rats were allocated into two groups: an experimental group and a control group. In the experimental group, MIA was administered as a single intra-articular injection into the left knee joint to induce KOA. The dosage varied between 0.5 mg and 2.0 mg. Following the injection, assessments were conducted on days 3, 5, 7, 14, 21, and 28 to evaluate knee circumference, pain-related behavior, tibial cartilage pathology, and the expression levels of IL-1β and IL-10.
Results: The knee joint diameters expanded in a dose- and time-dependent manner, paralleled by corresponding reductions in MIA-induced paw withdrawal threshold (PWT) and weight-bearing distribution (WBD), both of which followed similar dose- and time-dependent patterns. Compared with controls, MIA-treated rats exhibited significantly heightened defensive behaviors (eg, paw licking, retraction), confirming successful inflammatory pain induction. Pain intensity analysis revealed lower pain scores in the 0.5 and 1.0 mg MIA groups relative to 1.5 and 2.0 mg groups across the study. Pain severity escalated sharply from day 0 to 7, stabilized until day 14, and peaked again at day 28. Histologically, the 1.5 and 2.0 mg MIA groups demonstrated early-stage KOA pathology (days 5– 20), characterized by chondrocyte cloning and matrix disorganization, followed by mid-stage features (days 21– 27) including localized cartilage defects, surface irregularities, hypocellularity, and vascular infiltration. By day 28, late-stage KOA hallmarks emerged, such as widespread cartilage degeneration, subchondral bone exposure, and peripheral osteophytes. These histological findings aligned with behavioral pain outcomes and ELISA results, collectively illustrating synchronized progression of inflammatory and structural pathology across all assays.
Conclusion: The severity of pain, the extent of articular cartilage degeneration, and the level of inflammatory response following intra-articular MIA injection progressively worsened in a dose- and time-dependent fashion. In the MIA group, early-stage KOA cartilage pathology was observed between days 5 to 20 post-injection. By day 21, cartilage lesions progressed to mid-stage KOA cartilage, and by day 28, they advanced to late-stage KOA cartilage. The pain trajectory corresponded with the pathological features of MIA-induced KOA, offering valuable insights into optimal dosing and timing for targeted KOA pain management interventions.
Keywords: animal models, pain, cartilage, injections, intra-articular, monosodium iodoacetate, osteoarthritis, knee
Introduction
Osteoarthritis (OA), the most prevalent form of arthritis, predominantly affects individuals over 40, with higher incidence in females, commonly affecting the knees, hands, hips, and feet.1 Its pathogenesis stems from an imbalance between joint tissue anabolism and catabolism, driving progressive structural deterioration and chronic mobility limitations, exacerbated by age-dependent progression.2 The global economic burden is staggering, including 100 billion in direct medical costs, with age-standardized prevalence rising 9.3% from 1990 to 2017, a trend fueled by aging populations.3,4 Key risk factors include age, female sex, obesity.5 In knee osteoarthritis (KOA), the most disabling subset of OA, pain, swelling, stiffness, and mobility loss dominate clinical presentations.6–8 While inflammation initiates KOA, pain persists throughout its progression, becoming the primary clinical challenge, yet no pharmacotherapy fully alleviates it, underscoring the urgent need for deeper mechanistic insights to improve therapeutic outcomes.9–12
Animal models are essential in investigating the pathogenesis and potential treatments for KOA. These models include drug induction, joint braking, surgical induction, spontaneous OA, and transgenic animal model.13 Monosodium iodoacetate (MIA), a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) inhibitor, is widely used in rat osteoarthritis (OA) models to disrupt glycolysis and induce chondrocyte apoptosis.14,15 Although MIA doses for KOA models in various studies range from 0.1–4.8 mg,16 we selected a dose range of 0.5–2.0 mg. At these concentrations, KOA progresses in a staged manner17 and reliably induces degenerative changes consistent with human KOA pathology (eg, chondrocyte apoptosis, cartilage erosion,18–20 while avoiding excessive tissue necrosis observed at higher doses. However, the nonlinear relationship between MIA dosage, modeling timeline, and disease severity remains poorly quantified, complicating inter-study comparisons.
Furthermore, MIA exhibits high toxicity, with the potential for lethal systemic effects.21 Hence, it is essential to ascertain the ideal dosage that optimizes pain induction while minimizing the need for high doses. Additionally, the MIA model exhibits a two-phase pain mechanism, characterized by an initial inflammatory phase followed by a non-inflammatory phase.22–24 These distinct pain phases have been reported to respond differently to specific treatments, emphasizing the need to elucidate the onset and end of each phase.25 However, the absence of a clearly defined pain trajectory following MIA injection complicates the timing of interventions for these distinct phases. Therefore, we established MIA-induced KOA models using varied MIA doses and time and chose 2mg/50 μL as the highest dose to meet the actual needs and well simulate the slow pathological process of human knee joint degeneration. Subsequently, evaluating changes in pain intensity, histopathology, and expression levels of inflammatory factors induced by these differing doses at different points in time. Our objective is to furnish dose selection and timing guidance for forthcoming investigations targeting KOA pain.
We established MIA-induced KOA models and selected 2 mg/50 μL as the optimal dose to simulate the slow pathological progression of human knee osteoarthritis. Subsequent experiments evaluated temporal changes in pain behaviors, histopathological alterations, and inflammatory mediator expression under this regimen.
Materials and Methods
Animals
The research protocol was examined and endorsed by the Animal Ethics Committee of Guangzhou University of Chinese Medicine. All aspects of animal handling and experimental protocols complied strictly with the regulations set forth by the Animal Ethics Committee. A total of 126 male Sprague Dawley rats, aged 6 weeks and weighing between 230–250 g, were acquired from the Guangdong Medical Laboratory Animal Center. The rats were kept and cared for at the Experimental Animal Center of the Medical College of Acu-Moxi and Rehabilitation of Guangzhou University of Chinese Medicine.
Preparation of MIA Arthritis Model for Cartilage Evaluation
MIA (Aladdin, Shanghai, China) was diluted in 50 µL of normal saline and injected exclusively into the left knee joint. Doses of 0.5, 1.0, 1.5, and 2.0 mg/50 µL of MIA were delivered (n=30 per dose). Anesthesia was induced using isoflurane (RWD, Jinan, Shandong, China) at 3–4.5 vol %, with oxygen ventilation at 4–5 vol %; rats were then placed on the fixed plate for observation. Continuous anesthesia was carried out under 2–3 vol % isoflurane and 4–5 vol % oxygen. The knee joint was maintained at a 90° angle, and MIA was administered via the patellar tendon. Following the procedure, the animals were returned to their cages and maintained under a 12-hour light/12-hour dark cycle with unrestricted access to food and water. Subsequently, the rats were randomly allocated into six experimental groups: 3-day group (n = 5), 5-day group (n = 5), 7-day group (n = 5), 14-day group (n = 5), 21-day group (n = 5), and 28-day group (n = 5). Additionally, six rats were designated to the blank group, and behavioral tests were performed at the following time points: 0, 3, 5, 7, 14, 21, and 28 days. At each time point, one rat was sacrificed for tissue collection (n = 1 per time point). Rats in the blank group received no intervention, placebo, or sham injections. Following pain behavior assessment, rats were euthanized with intraperitoneal overdose of anesthetic.
Knee Diameters
To assess joint swelling as an indicator of inflammation, knee diameters were measured. Left knee diameters were measured using a manual caliper at days 3, 5, 7, 14, 21, and 28 post-injection, with a sample size of 5 rats for each time point (Figure 1).
 |
Figure 1 Study schema. |
Assessment of Pain Behavior
Pain behavioral assessments were conducted with n=5 per group on days 3, 5, 7, 14, 21, and 28 post-injection (Figure 1). Rats were given at least 30 minutes to acclimate to the laboratory environment before testing began. Following acclimation, the rats underwent several assessments: mechanical hypersensitivity was measured using the BIO-EVF4-POIGN apparatus (RWD, China) for an automated Von Frey test; heat hypersensitivity was assessed using the Hargreaves’ paw flick test equipment (UGO BASILE SRL, Italy); and weight-bearing on both hind limbs was determined with the BIO-SWB apparatus (RWD, China).
Von Frey thresholds for mechanical hypersensitivity were measured 30 minutes after the rats had acclimated in the test cage. A mechanical von Frey probe was used to apply gradual pressure perpendicularly to the mid-plantar surface of the paw. Upon a response like foot lifting, rapid withdrawal, licking, ceased pressurization and recorded the value displayed on the pain meter panel, which is the paw withdrawal threshold (PWT, g). For each hind paw, three measurements were taken with a 60-second interval between each measurement, and the results were averaged.
To further investigate pain sensitivity in the inflamed paw, we employed the Hargreaves test to assess heat sensitivity. PWT was measured at specified time points to assess pain sensitivity. To assess heat hypersensitivity, a light source was employed to apply radiant heat to the plantar surface of the hind paw. The heat intensity was carefully regulated to prevent tissue injury, with a maximum latency period of 20 seconds. The thermal stimulus persisted until either paw withdrawal occurred or the cutoff time was attained. Behaviors indicative of paw withdrawal, including lifting, licking, or flinching, were recorded. For each hind paw, three measurements were taken at 5-minute intervals, and the mean values were computed.
To assess spontaneous pain following MIA injection, the static weight-bearing in capacitance test was utilized. Rats were positioned in a slanted Plexiglas chamber, with each hind limb placed on a separate force platform. After acclimation, recordings were obtained while the rats were stationary. The downward pressure applied by each hind limb was documented in grams, with three readings acquired and their mean value calculated. To minimize the potential impact of weight variations among animals, the outcomes were normalized and presented as a percentage relative to body weight. The weight-bearing distribution (WBD) on the right hind paw was determined using the following formula: WBD = [(left hind weight)/(left hind weight + right hind weight)] × 100. Assessments were independently performed by three team members.
Histological Examination
The hind limb knee joints were collected at 3, 5, 7, 14, 21, and 28 days post-injection (n = 5 rats per group for each time point) for histopathological analysis (Figure 1). To evaluate the histological characteristics of the proximal femur and tibia, the specimens were initially fixed in 4% paraformaldehyde for 48 hours. This was followed by decalcification in a 20% EDTA solution at 37°C for 25 days. After decalcification, the samples were embedded in paraffin wax. The fixed specimens were then sectioned sagittally into 4 mm slices, dewaxed using xylene, and dehydrated via an alcohol gradient. Subsequently, the sections were stained with hematoxylin and eosin (H&E) as well as safranin O.26 The histological sections were then visualized with a NanoZoomer S60 scanner (Hamamatsu, Japan). The evaluation of degeneration in the medial tibial plateau cartilage was conducted using the OARSI scoring system (ranging from 0 to 25 points)27 and the Mankin grading scale (spanning 0 to 13 points),28 Both methods were tailored specifically for application in rat models. Histological scoring was performed by two independent observers blinded to group allocation, with interobserver reliability assessed via Cohen’s kappa (κ = 0.85).29 The experimental grouping was blinded twice and only unblinded after the completion of statistical analysis to reveal the actual group allocation of the rats. Pathological changes in cartilage were evaluated by quantifying a composite of factors including cartilage structure, chondrocytes, staining, tidemark, calcification zone, and subchondral bone condition. The final histopathological score is calculated by summing the scores of individual components, where higher total scores signify more severe cartilage damage. The Mankin score classifies OA as follows: score 1–5 indicates early OA, 6–9 indicate middle-stage OA, and 10–14 indicate late-stage OA.
Collection of Joint Cavity Fluid Samples
First, a 0.5 mL microsyringe was filled with 0.3 mL of sterile 0.9% sodium chloride solution (physiological saline) and kept on standby. Following euthanasia of the rats, the knee joints were gently flexed and extended to promote synovial fluid mobilization. The joint capsule was then immediately incised at the lateral superolateral aspect of the patellar region, and synovial fluid was carefully withdrawn via the microsyringe. The syringe was gently aspirated to mix the synovial fluid with the saline, ensuring homogenization. A final retrieved volume of at least 0.2 mL was collected. The sample was centrifuged at 3000 rpm for 10 minutes to pellet cellular debris, and the supernatant was carefully aspirated and transferred to cryovials. Samples were immediately stored at −80°C until further analysis.
ELISA
Serum and joint cavity fluid samples were gathered at 3, 5, 7, 14, 21, and 28 days post-procedure (n=5 rats per group for each time point) for cytokine quantification (Figure 1). The expression levels of IL-1β and IL-10 in serum and joint cavity fluid were quantified using antigen-based sandwich ELISA. The Rat IL-1β ELISA Kit (catalog number MM-0047R1) and Rat IL-10 ELISA Kit (catalog number MM-0195R1), both provided by Meimian (Wuhan, Jiangsu, China), were utilized in this study. Following the addition of the stop solution, the optical density (OD) values were determined at a wavelength of 450 nm using Skanlt Software Research Edition (Thermo Fisher Scientific, USA). The levels of IL-1β and IL-10 in serum and joint cavity fluid were determined based on the standard curve and then subjected to statistical analysis.
Statistical Analysis
Three independent observers evaluated the scores, two of whom conducted blinded assessment. The sample size was determined based on a prior power analysis (effect size d = 0.8, power = 0.8, α = 0.05), ensuring sufficient statistical power to detect meaningful differences between groups (n=5 per group at each time point).30 Data were presented as mean ± standard deviation (SD). Statistical evaluations were performed using IBM SPSS software (version 26.0; IBM, Armonk, NY, USA). To examine differences among groups at each time point and within each group, a two-way repeated measures ANOVA was applied. When data satisfied parametric assumptions, Bonferroni’s post hoc test was conducted. For non-parametric datasets, the Mann–Whitney U-test was utilized to compare intergroup differences at each time point. Friedman test with post-hoc Wilcoxon analysis was employed to assess intragroup variations compared to baseline values. A two-way ANOVA was implemented to evaluate knee diameter, pain behavior, Mankin and OARSI scores, as well as IL-1β and IL-10 expression levels across different dosages and time points. The significance level was established at p < 0.05. GraphPad Prism 8 was used for data visualization.
Results
Knee Diameters
The findings revealed that the knee diameters varied in accordance with both dosage and time (Figure 2A). Compared to the control group, MIA injection resulted in an increase in left knee diameters, which became more pronounced as the dose increased (Figure 2A, g). Knee diameter were maximal after 2.0 mg MIA injections and minimal after 0.5 mg MIA injections (Figure 2A, g). In the 0.5mg, 1.0, 1.5, and 2.0 mg MIA groups, knee diameter peaked on day 3, indicating pronounced knee swelling that gradually decreased after day 3 but remained notably elevated compared to the control group throughout the study (Figure 2A, g). By day 7, acute inflammation and joint swelling began to diminish, with no significant differences among MIA concentrations by day 14 (Figure 2A, d, P > 0.05). Thereafter, knee diameters in each group showed a continuous increase from day 14 to day 28.
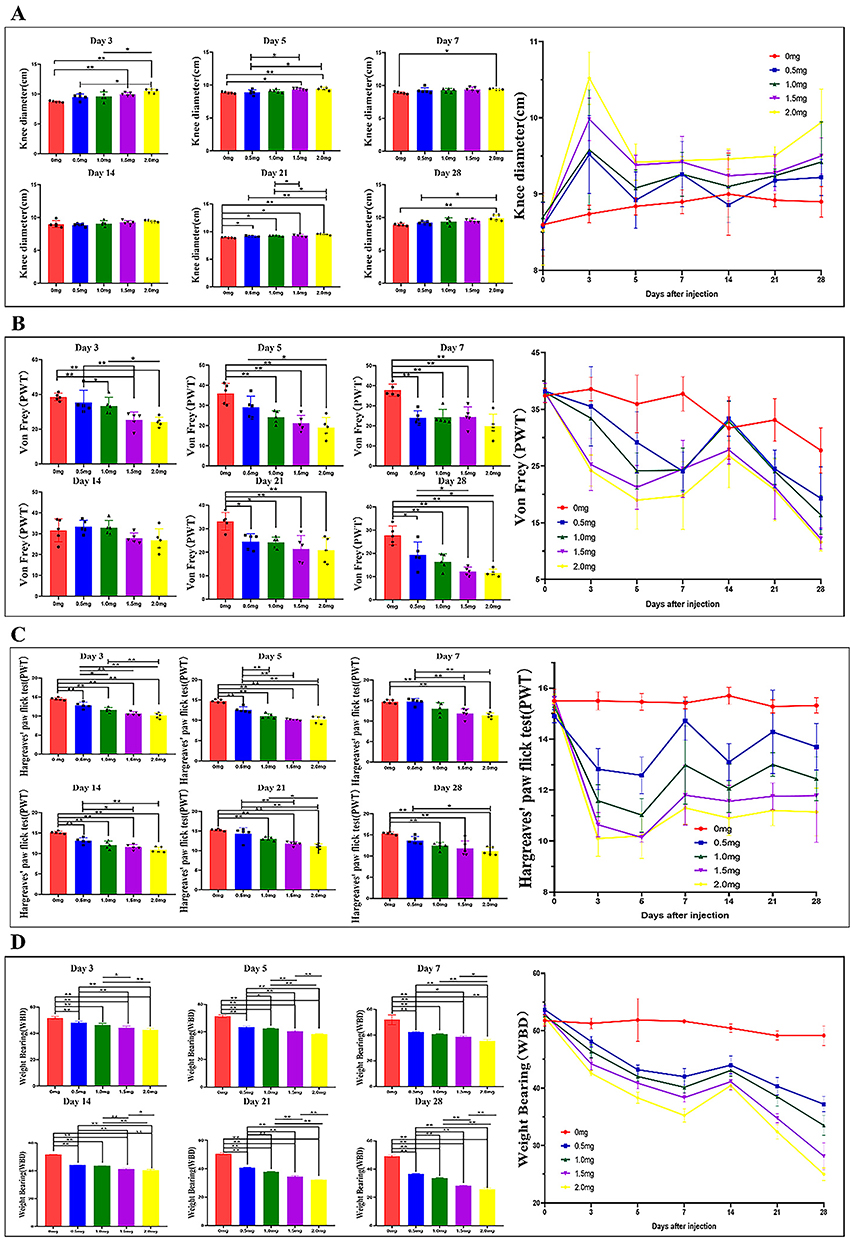 |
Figure 2 Knee diameter-lefts and MIA-induced KOA related pain behaviors induced KOA rat model. (A) a-g Effect of MIA injection on Knee diameter-lefts in rats; (B) a-g Effect of MIA injection on mechanical hypersensitivity in rats; (C) a-g Effect of MIA injection on heat hypersensitivity in rats; (D) a-g Effect of MIA injection on weight-bearing in rats; * P<0.05, ** P<0.001. |
MIA-Induced KOA Related Pain Behaviors
Pain intensity after MIA administration at different doses was assessed using Von Frey, weight-bearing, and Hargreaves tests. In general, pain intensity changes depended on both dose and time (Figure 2). Figure 2B shows all rats in the control group had similar responses in the Von Frey test, demonstrating no tactile allodynia. PWT values of the other four MIA-induced groups gradually decreased with extended modeling time, causing hypersensitivity in the ipsilateral hind paw (Figure 2B, g). Compared to the control group, PWT decreased in MIA groups (Figure 2B a-h, p<0.01). Rats in the 2 mg and 1.5 mg MIA groups exhibited lower PWT compared to those in the 0.5 mg and 1.0 mg MIA groups from day 3 to day 28. This suggests that rats receiving higher doses of MIA had increased sensitization compared to those receiving lower doses (Figure 2B, g). Notably, on day 3 post-induction (Figure 2B, a, p<0.05=, PWT significantly decreased across all groups. However, between day 7 and day 14 (Figure 2B, d, p>0.05), PTW showed an increasing trend before continuously declining. The decline in nociceptive threshold persisted throughout the experiment until day 28 post-induction. The lowest PWT was observed in the 2 mg MIA group on day 28, with an average value of 11.6 ± 1.55 (Figure 2B, f).
In the Hargreaves test, MIA injection significantly reduced the PWT in all model groups (Figure 2C, a-h, p < 0.05). The results indicated that knees injected with MIA exhibited higher thermal hyperalgesia compared to the control group. Notably, MIA injection induced a decrease in PWT, reaching the lowest value of 10.14 ± 0.18 on the acute-phase day 5 (Figure 2C, b). From day 5 onward, the PWT of model groups showed an upward trend but remained below the control group’s levels. By day 7, the trend stabilized, maintaining a chronic pain state for at least 28 days (Figure 2C, g). Additionally, minor fluctuations in PWT values were observed in the 0.5 mg and 1.0 mg groups between days 7–21, but these continued to decline after day 21 until day 28. The lowest PWT values were observed in the 2.0 mg group (Figure 2C, b). These findings were consistent with results from the von Frey test (Figure 2B).
In the weight-bearing test, all groups initially displayed symmetrical support on both hind limbs, with an initial WBD of 52.16 ± 0.73, indicating the absence of joint discomfort (Figure 2D). From day 3, the WBD in the MIA group commenced a decline (Figure 2D, a), suggesting joint discomfort due to symmetrically weight distribution. However, an upward trend in WBD was observed between day 7 and day 14 (Figure 2D, g, c, d), followed by a decline until day 28. At all post-MIA injection time points, rats in the high-dose MIA groups displayed greater weight-bearing deficits than those in low-dose group (Figure 2D, g).
Histological Observations of Tibial Cartilage
The histopathological alterations in MIA-induced KOA were evaluated through hematoxylin and eosin (H&E) staining as well as safranin O fast green staining (Figures 3 and 4). In the control group, the joint surface appeared smooth, and the chondrocytes were orderly organized. Staining of chondrocytes and cartilage matrix appeared normal, with no discernible weakening of staining observed at any time point (Figure 5). However, contrasting histopathological findings were evident in KOA rats, showing a dose- and time-dependent pattern (Figures 3 and 4). By day 5, rats that received injections of 0.5 mg and 1.0 mg of MIA demonstrated minor alterations or a subtle decline in proteoglycan staining, along with a slight reduction in both H&E and safranin O/fast green staining (Figure 4E and F). These alterations became more significant by day 14 (Figure 4M and N). By day 21, evident signs of chondrocyte death and degradation of the extracellular matrix were observed (Figure 3Q and R), together with a significant thinning of the articular cartilage (Figure 4Q and R). By day 28, abnormalities in the superficial layer of the articular cartilage and fissures on the joint surface were observed (Figure 3U and V and 4U and V).
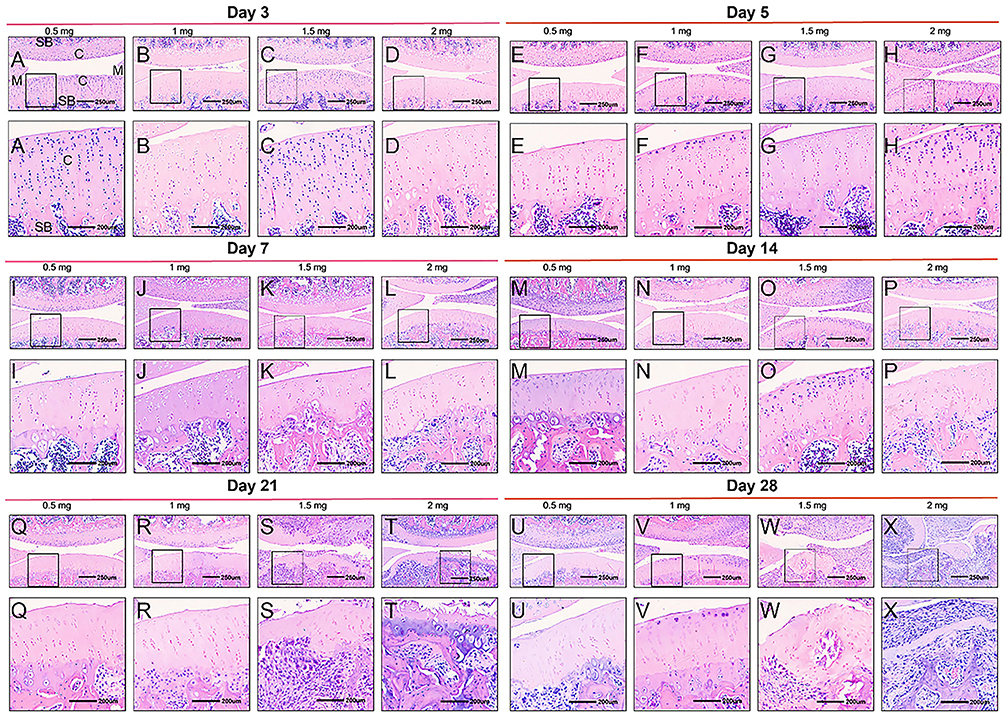 |
Figure 3 H&E staining of knee sections of KOA animals. Rats received an intra-articular injection of 0.5 (A, I, Q, E, M, U), 1 (B, J, R, F, N, V), 1.5 (C, K, S, G, O, W) or 2 mg (D, L, T, H, P, X) of MIA and were sacrificed at 3 (A–D), 5 (E–H), 7 (I–L), 14 (M–P), 21 (Q–T) and 28 days (U–X) post-MIA injection. Each rat knee image is representative of a group of 5 animals. C indicates cartilage; SB, subchondral bone; M, meniscus. Black square boxes: Indicate regions of the cartilage surface localized within the 250 μm scale overall images (top row). The corresponding enlarged views of these boxed areas are shown in the 200 μm scale images directly below in the same column. Scale bars: 250 μm for the top-row overall images and 200 μm for the bottom-row magnified images. |
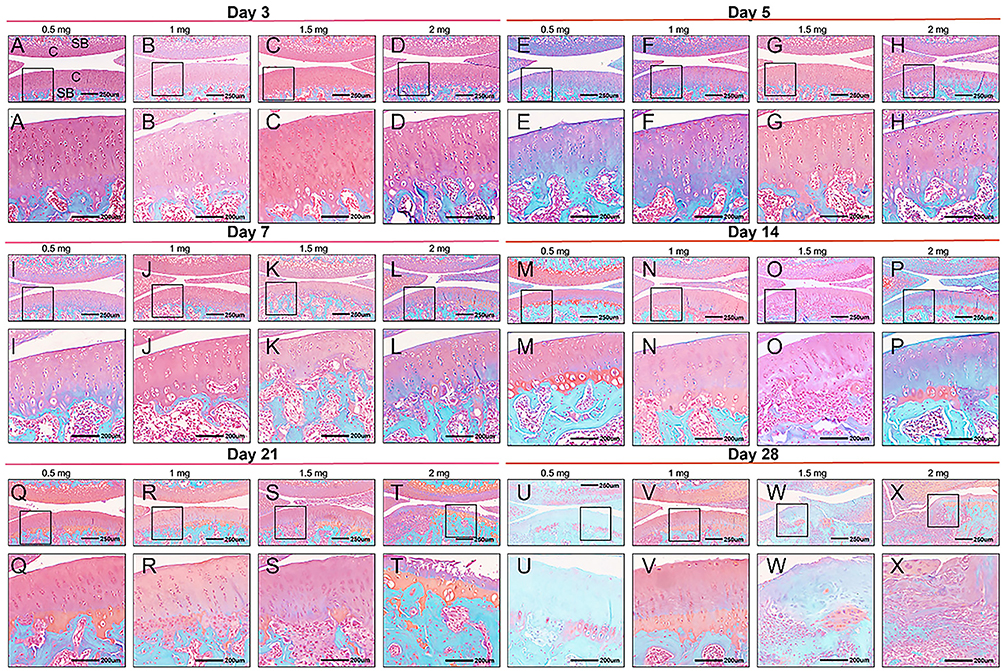 |
Figure 4 Safranin O fast green staining of knee sections of KOA animals. Rats received an intra-articular injection of 0.5 (A, I, Q, E, M, U), 1 (B, J, R, F, N, V), 1.5 (C, K, S, G, O, W) or 2 mg (D, L, T, H, P, X) of MIA and were sacrificed at 3 (A–D), 5 (E–H), 7 (I–L), 14 (M–P), 21 (Q–T) and 28 days (U–X) post-MIA injection. Each rat knee image is representative of a group of 5 animals. C indicates cartilage; SB, subchondral bone; M, meniscus. Black square boxes: Indicate regions of the cartilage surface localized within the 250 μm scale overall images (top row). The corresponding enlarged views of these boxed areas are shown in the 200 μm scale images directly below in the same column. Scale bars: 250 μm for the top-row overall images and 200 μm for the bottom-row magnified images. |
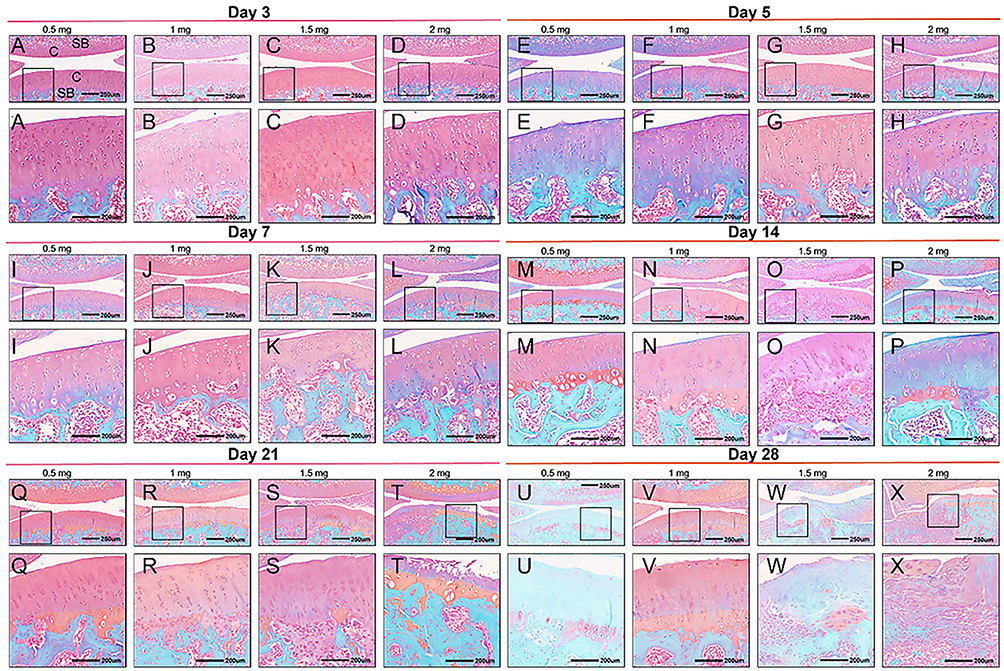 |
Figure 5 H&E (A–F) and Safranin O fast green (G–Q) staining of blank group animals. Rats were sacrificed at 3 (A–D), 5 (E–H), 7 (I–L), 14 (M–P), 21 (Q–T) and 28 days (U–X). C indicates cartilage; SB, subchondral bone; M, meniscus. Black square boxes: Indicate regions of the cartilage surface localized within the 250 μm scale overall images (top row). The corresponding enlarged views of these boxed areas are shown in the 200 μm scale images directly below in the same column. Scale bars: 250 μm for the top-row overall images and 200 μm for the bottom-row magnified images. |
In the groups treated with higher doses of MIA (1.5 and 2.0 mg), evident chondrocyte proliferation and disorganization were observed (Figure 3G and H). On day 5, a more significant decrease in proteoglycan staining within the extracellular matrix of the articular cartilage was observed compared to lower doses (Figure 4G and H). Furthermore, chondrocyte death was evident by day 7 (Figure 3K and L; 4K and L), by day 14, significant loss of extracellular matrix and reduced articular cartilage thickness were observed, along with fissures formation (Figure 3O and P; 4O and P). By day 21, complete erosion of hyaline articular cartilage was apparent, characterized by an irregular hypo-cellular cartilage surface and blood vessel penetration, indicative of mid-stage KOA (Figure 3T and U; 4T and U). By day 28, the structural integrity of the femur and tibia was impaired, revealing the subchondral bone and leading to osteophyte development at the joint’s peripheral edge, characteristic signs of advanced KOA (Figure 3W and X; 4W and X).
The histological features of femoral cartilage were assessed using the Mankin score and OARSI score. From day 3 to day 28 post-MIA injection, both the Mankin score and OARSI score in the MIA group demonstrated a significant increase compared to the control group, suggesting progressive pathological changes over time. These increases suggested increased articular cartilage surface damage, decreased chondrocytes, and proliferative clone/osteophyte formation (Figure 6A–M, p < 0.01 vs control group). Rats receiving equivalent MIA dose showed time-dependent increase in Mankin score and OARSI score (Figure 6a and b). In the 0.5 mg and 1.0 mg groups, Mankin score were 1.89 and 2.13 at day 3 day (Figure 6A and G), increasing rapidly after day 14, with a significant increase on day 21 (Figure 6a and b), reaching 8.06 and 8.60 by day 28 (Figure 6F and M). In the 1.5 mg and 2.0 mg groups, the scores were 2.67 and 3.13 on day 3 (Figure 6A and G), respectively, and reached their peak at 8.93 and 9.27 at 4 weeks (Figure 6F and M). In rats that were simultaneously injected with MIA, the increases in Mankin score and OARSI score were dose-dependent. One week after MIA administration, the Mankin score and OARSI score in the 2.0 mg and 1.5 mg MIA groups were significantly higher compared to those in the 0.5 mg and 1.0 mg groups (Figure 6a and b). Consequently, both scores peaked at 4 weeks post-MIA injection in the MIA groups (Figure 6a and b).
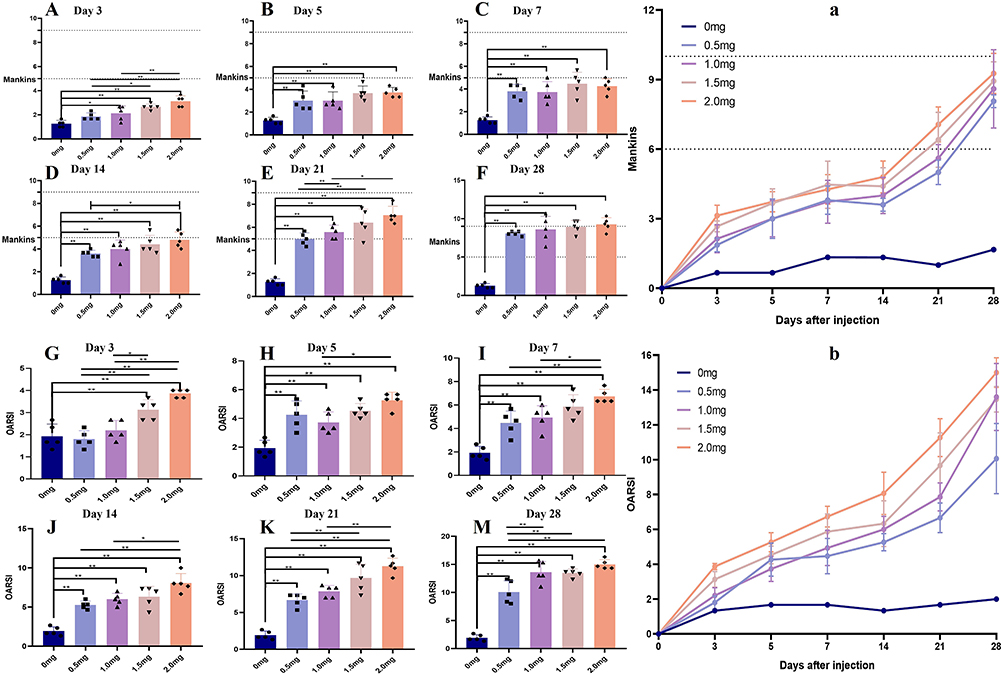 |
Figure 6 Evolution of the Mankins (A–F) and OARSI score (G–M) of blank animals or animals injected with 0.5, 1.0, 1.5 or 2 mg of MIA (KOA; n= 5/group) on various days after the injection until the day of sacrifice (3, 5, 7, 14, 21, 28). At each sacrifice time point 5 animals per group were euthanized. * P < 0.05, ** P < 0.01. |
Upon integrating pathological findings with the Mankin and OARSI scores, in 1.5 mg and 2.0 mg of MIA groups, early-stage KOA cartilage changes were evident at 5 days post-injection, mid-stage changes at 21 days, and late-stage changes emerged at 28 days.
IL-1β and IL-10 Levels
The expression levels of IL-1β and IL-10 in the serum (Figure 7A and C) and joint fluid (Figure 7B and D) were evaluated using ELISA. The results indicated a significant increase in IL-1β levels and a notable decrease in IL-10 levels in both the joint fluid and serum three days after MIA injection compared to the control group (Figure 7A–D). However, the increase in IL-1β levels ceased between days 7 and 14. Thereafter, IL-1β levels continued to rise, reaching their peak by day 28 (Figure 7A and B). Additionally, the levels of IL-1β in both the joint fluid and serum were found to be lower in the low-dose group compared to the high-dose group. In contrast, the trends for IL-10 (Figure 7C and D) were opposite to those of IL-1β. The results suggest that injecting MIA into the knee joint induces an inflammatory response, which is influenced by both the MIA dosage and the time elapsed after injection. Shortly after MIA injection, KOA pain may be triggered by acute inflammation, which later transitions into a chronic inflammatory response that is both spontaneous and induced.
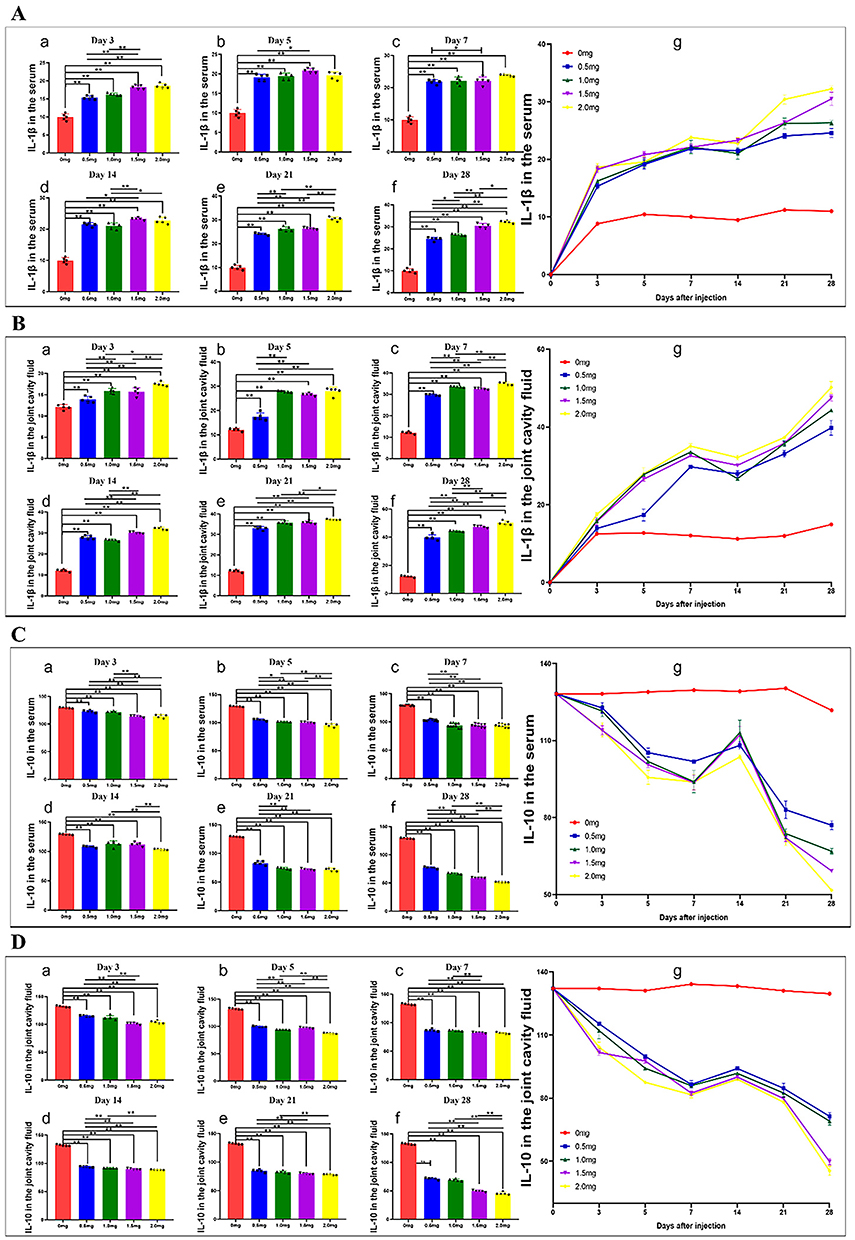 |
Figure 7 The progression of IL-1β in the serum (A) and joint cavity fluid (B); IL-10 expression levels in the serum (C) and joint cavity fluid (D) of animals injected with varying doses of MIA (KOA; n=5/group) or blank animals was monitored on different days post-injection until the day of sacrifice (3, 5, 7, 14, 21, 28). At each time point of sacrifice, 5 animals per group were euthanized. Statistical significance was denoted as * for P < 0.05 and ** for P < 0.01. |
The complete quantitative datasets supporting these findings—including longitudinal knee diameter measurements, ethological pain behavior parameters, histological grading matrices, and cytokine concentration profiles in serum/joint cavity fluid—are systematically organized in Supplementary Table 1 with dedicated worksheets labeled:
Worksheet 1: Knee diameter-lefts
Worksheet 2: Hargreaves’ paw flick test
Worksheet 3: Von Frey test
Worksheet 4: Weight-bearing
Worksheet 5: IL-1B in the serum and joint
Worksheet 6: IL-10 in the serum and joint
Discussion
In general, the progression of pathological changes and pain intensity in KOA is dose- and time-dependent. Over the study period, the high-dose MIA group showed fewer pathological changes and lower pain intensity than the low-dose MIA group. Over the 4-week period following MIA injection, the pain progression exhibited two separate phases: an early phase marked by inflammatory pain (days 0–7), followed by a degenerative phase (days 14–28). These findings are largely consistent with our histological results: pathological changes in KOA were evident from day 5 post-administration, stabilizing between days 5 and 14, and resuming there after until day 28. Inflammatory infiltration, characterized by the influx of neutrophils, lymphocytes, and macrophages, was primarily observed during the first week post-MIA induction.25,30–32 This was succeeded by a transitional phase from day 7 to day 14.33,34 Subsequently, degenerative alterations took precedence, potentially escalating pain intensity and exhibiting neuropathic pain-like traits.22,23,31
At the cellular level, OA is initially characterized by hypercellularity. Chondrocytes in the proliferating state actively release pro-inflammatory cytokines, including IL-1, as well as tissue-degrading enzymes such as matrix metalloproteinases, which in turn worsen their self-destruction.32,33 In the present study, after 5 days post-injection with 1.5–2 mg of MIA, chondrocytes cloning and chondrocytes disorganization, (Figure 3A and B) and enlarged left knee diameter (Figure 2A), and elevated levels of inflammatory factors were indicative of early-stage KOA cartilage pathological changes. The rise in cellular density might possibly result in a decrease in the thickness of the soft articular cartilage and disrupt the boundary between the articular and calcified cartilage. Histologically, this transition appears as a stiff cleft or pannus formation, which ultimately leads to an abnormal joint function and joint instability. This delineates the process through which inflammation culminates in persistent pathological conditions.32,34 Our histological analysis confirmed these findings, revealing local cartilage defects on the knee joint surface in rats by day 21. Additionally, the cartilage surface showed irregularities, hypo-cellularity, and blood vessel penetration. These observations are consistent with the characteristics of the mid-stage of KOA cartilage pathological changes. By day 28, the joint surface showed multiple cartilage defects, subchondral bone exposure, and osteophyte formation at the periphery, indicative of late-stage KOA cartilage pathological changes. All the presented evidence suggests that the MIA model effectively mirrors the cellular-level pathological mechanisms of KOA.
MIA administration induces articular cartilage deterioration, subchondral bone structure disruption, inflammatory responses, and joint pain.35–37 Histological evidence indicates that even minimal dose of MIA, when administered chronically, can cause structural alterations in joints.35,38 Higher concentrations of MIA are associated with intensified synovial inflammation, greater exposure and distortion of subchondral bone, and heightened pain intensity.39 According to Mohan et al, 10 weeks following the administration of 0.2 mg of MIA,39 cartilage degeneration was observed, and the histological results were consistent with our observations. Compared to traditional surgical models like stabilized cartilage wear damage (SCWD),40 our MIA-induced model avoids invasive procedures while achieving comparable pathology within a shorter timeframe, enhancing translational value. The pathological observations in this study align with those documented in previous studies. We investigated the joint pathological alterations after injecting 0.5, 1.0, 1.5, and 2.0 mg of MIA intra-articularly into the knee joint. Our results demonstrated that the extent of joint injury was relatively restricted at doses of 0.5 mg and 1.0 mg, as opposed to the more pronounced damage observed at 1.5 mg and 2.0 mg. This dose-dependent progression contrasts with the constant severity in collagenase-induced models,40 highlighting the unique advantages of MIA for studying disease progression dynamics.
The dose and volume of MIA used to induce OA exhibit significant variability across studies. Furthermore, MIA exhibits significant toxicity and may pose systemic lethal risks.21 Histological results demonstrate that higher doses result in greater exposure and deformation of subchondral bone, and consequently elevated pain intensity.39 Existing data implies that distinct pathophysiological consequences arise from varying doses of MIA, with the 2 mg dose linked to a significantly greater extent of neuronal injury and/or central sensitization compared to 1 mg MIA. As previous studies have reported, higher concentrations of MIA within the knee joint can cause significant axonal damage to dorsal root ganglia (DRG) cells, which may lead to more intense pain.41 Importantly, considering the high toxicity of MIA and comparable pain intensity (PWT) between the 1.5 mg and 2 mg doses, evaluating joint damage becomes essential. Interestingly, rats receiving 1.5 mg MIA showed slower joint damage progression, making this dose a more suitable model for mimicking the gradual onset of KOA in humans. Therefore, balancing pain-inducing efficacy with overall safety, 1.5 mg of MIA may be a more appropriate option than 2 mg.
Besides articular cartilage deterioration, chronic pain is another significant issue associated with KOA. Pain-related behavioral findings correlated with histological findings in this study. Osteoarthritic pain mainly originates from nociceptive responses to tissue damage at the lesion site. However, persistent joint cavity inflammation can cause subsequent alterations in nociceptive neurons, inducing neuropathic pain.42 In this study, we evaluated PWT and results demonstrated a significant PWT reduction, which closely correlated with increased left knee diameters. This finding is indicative of edema resulting from inflammation, particularly evident during the acute phase of MIA-induced KOA (Figure 2A and B, D). Chronic inflammation triggers nociceptors, leading to excessive excitation of secondary neurons within the spinal cord—a phenomenon referred to as central sensitization. This mechanism is closely associated with chronic neuropathic pain development following KOA.43,44 Moreover, previous research has demonstrated that MIA sensitizes joint nociceptors in a dose-dependent manner, with higher doses exhibiting a stronger association with paw withdrawal behavior.45 Our findings reveal a clear trend: compared with the 0.5 mg and 1.0 mg groups, the 1.5 mg and 2 mg MIA groups showed a significantly greater reduction in PWT and WBD beginning on day 3 after injection. These results align with prior studies.19,46 The hind paw, affected distally by MIA-induced KOA in the knee joint, exhibited increased sensitivity to mechanical stimuli. This heightened response suggests secondary hyperalgesia development, probably due to central sensitization.42 Thus, the noted PWT decrease post-MIA-induced KOA (Figure 2B and D) likely indicates the onset of neuropathic pain, arising from inflammatory pain attributed to central sensitization. Moreover, weight distribution imbalances (Figure 2C) were consistently observed during the onset and progression of primary and secondary hyperalgesia. Regarding pain behavior, the disrupted weight-bearing balance implies spontaneous pain induced by weight-bearing during unrestricted movement, posing a significant challenge for managing chronic KOA.
The onset of KOA is driven by the complex interplay of multiple cytokines, with families such as IL, TNF, and chemokines playing critical roles throughout KOA progression.47 Within the IL family, cytokines exhibit both pro-inflammatory and anti-inflammatory properties. Research has indicated that specific cytokines, including IL-1α, IL-1β, IL-2, IL-6, IL-8, IL-10, and IL-17, play a significant role in the development of KOA. Among these, IL-10 and IL-1β are particularly important in the development of KOA. IL-1β, an inflammatory cytokine synthesized and secreted primarily by activated monocytes-macrophages,48 can stimulate fibroblasts to secrete IL-6 and IL-8, promoting inflammatory responses in the synovial membrane.49 Moreover, IL-1β promotes the production of nitric oxide (NO) and matrix metalloproteinases (MMPs), while also triggering chondrocyte apoptosis. The combined effects of these actions promote the degeneration of articular cartilage.50 Conversely, IL-10 functions as a strong anti-inflammatory and chondroprotective cytokine. It efficiently neutralizes TNF-α-induced expression of IL-6, MMP-1, and MMP-3,51 while enhancing the expression of chondrocyte markers such as Col II, SOX9, and glycosaminoglycans. Additionally, IL-10 inhibits chondrocyte apoptosis, modulates the inflammatory response in chondrocytes, and alleviates joint inflammation.52,53 In this study, the expression of IL-1β increased gradually with escalating MIA dose (0.5 to 2 mg) and advancing KOA over 28 days, while the expression of IL-10 decreased, indicating a dose- and time-dependent inflammatory response to MIA.
Moreover, controlling inflammation and alleviating pain are essential for enhancing the quality of life in individuals with KOA. In contrast to previous studies, our research provides a thorough and quantitative assessment of MIA-induced pain at various time points throughout a standardized 4-week observation period. Moreover, we analyzed the dose-response relationship between different MIA doses and pain intensity following intra-articular injection. Such results provide significant guidance for subsequent KOA pain research, assisting in dose determination and intervention scheduling. Despite being conducted on animal models, the detailed pathological profile of MIA-induced KOA presented here could serve as foundational evidence to guide the development of therapeutic strategies for clinical KOA.
Despite its contributions, this study has several limitations. Firstly, OA is a chronic condition, and changes in pathology and pain may extend beyond 28 days. Our analysis of pain trajectory is limited to 28 days post-MIA injection, given the longer-term pain observations are scarce in existing literature. Therefore, future studies should aim to elucidate the pain trajectory beyond 28 days. Secondly, it has been shown that MIA can suppress chondrocyte metabolism, thereby causing rapid and extensive degradation of joint integrity. This includes cartilage loss, osteophyte formation, and eventual cartilage fibrillation, which closely mimics the pathological features of clinical OA.54 However, the duration of joint modeling post-injection is relatively brief, and controlling the dosage of MIA is challenging too high a dose can result in excessive joint damage, while too low a dose may produce negligible degeneration. Moreover, OA induced by MIA does not entirely mirror the complexities of human OA. Clinically, KOA is categorized into primary and secondary KOA, with complex and multifactorial pathogenesis that develops gradually over time. In this study, KOA pathology was modeled using MIA-induced intra-articular inflammation and cartilage destruction, which may differ from KOA resulting from aging, genetic factors, mechanical trauma, metabolic factors, hormonal changes, or surgical modeling techniques. Lastly, as only one rat per time point was used in the control group for pathological staining and ELISA, variations in individual and activity ability may introduce bias to the results.
Conclusions
The administration of MIA via intra-articular injection into rat knee joints elicited KOA alterations in a manner that was dependent on both dose and time. Our findings indicate that early-stage KOA cartilage pathological changes began at 5 days post-injection with 2mg and 1.5mg MIA, advanced to middle-stage changes by 21 days, and reached late-stage changes by 28 days. These observations establish a theoretical foundation for the optimized use of animal KOA models in future research. Furthermore, the selection of 1.5 mg MIA as an optimal dose balances model reproducibility with clinical relevance, reflecting its alignment with human KOA pathophysiology and informing translational therapeutic strategies. The severity of pain following intra-articular MIA injection escalated in a manner dependent on the dose. The evolution of pain adhered to a distinct pattern consistent with the pathological processes associated with MIA-induced pain. This thorough description offers essential guidance for determining suitable dosages and timing interventions aimed at managing KOA pain.
Abbreviations
KOA, knee osteoarthritis; MIA, monosodium iodoacetate; IL-1β, interleukin 1β; IL-10, interleukin 10; PWT, paw withdrawal threshold; WBD, weightbearing distribution; OD, optical density; SD, standard deviation; ANOVA, a two-way analysis of variance; NO, nitric oxide; MMPs, matrix metalloproteinases.
Data Sharing Statement
The datasets used and analyzed in this study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
The study received approval from the Animal Ethics Committee of Guangzhou University of Chinese Medicine (Approval No. 20220526009).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Youth Science Foundation of China (Grant Number: 82104964; 81973950); The start-up research fund at Guangdong Provincial Hospital of Traditional Chinese Medicine (Grant Number: E595). The funding bodies played no part in the study’s design, the collection, analysis, or interpretation of the data, nor in the drafting of the manuscript.
Disclosure
The authors declare that they have no competing interests or conflicts to disclose for this work.
References
1. Courties A, Kouki I, Soliman N, Mathieu S, Sellam J. Osteoarthritis year in review 2024: epidemiology and therapy. Osteoarthritis Cartilage. 2024;32(11):1397–1404. doi:10.1016/j.joca.2024.07.014
2. Cross M, Smith E, Hoy D, et al. The global burden of Hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1323–1330. doi:10.1136/annrheumdis-2013-204763
3. Hawker GA. Osteoarthritis is a serious disease. Clin Exp Rheumatol. 2019;37 Suppl 120(5):3–6.
4. Ouyang Y, Dai M. Global, regional, and national burden of knee osteoarthritis: findings from the global burden of disease study 2021 and projections to 2045. J Orthop Surg Res. 2025;20(1):766. doi:10.1186/s13018-025-06140-0
5. Palazzo C, Nguyen C, Lefevre-Colau MM, Rannou F, Poiraudeau S. Risk factors and burden of osteoarthritis. Ann Phys Rehabil Med. 2016;59(3):134–138. doi:10.1016/j.rehab.2016.01.006
6. Altman R, Asch E, Bloch D, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. diagnostic and therapeutic criteria committee of the American rheumatism association. Arthritis Rheum. 1986;29(8):1039–1049. doi:10.1002/art.1780290816
7. Akdeniz G, Tığlı K, Akıncı NE, et al. Mental imagery enhances pain reduction and visual processing in knee osteoarthritis patients: a comparative study. Pain Res Manag. 2025;2025(1):5576698. doi:10.1155/prm/5576698
8. Sharma L. Osteoarthritis of the Knee. N Engl J Med. 2021;384(1):51–59. doi:10.1056/NEJMcp1903768
9. Guede-Rojas F, Mendoza C, Rodríguez-Lagos L, et al. Effects of non-immersive virtual reality exercise on self-reported pain and mechanical hyperalgesia in older adults with knee and hip osteoarthritis: a secondary analysis of a randomized controlled trial. Medicina. 2025;61(7). doi:10.3390/medicina61071122
10. Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA. 2021;325(6):568–578. doi:10.1001/jama.2020.22171
11. Iordache SA, Cursaru A, Serban B, et al. Unveiling prognostic and diagnostic biomarkers in knee and hip osteoarthritis: a targeted review. Discov Med. 2025;37(197):960–975. doi:10.24976/Discov.Med.202537197.86
12. Cao F, Xu Z, Li XX, et al. Trends and cross-country inequalities in the global burden of osteoarthritis, 1990-2019: a population-based study. Ageing Res Rev. 2024;99:102382. doi:10.1016/j.arr.2024.102382
13. Kuyinu EL, Narayanan G, Nair LS, Laurencin CT. Animal models of osteoarthritis: classification, update, and measurement of outcomes. J Orthop Surg Res. 2016;11:19. doi:10.1186/s13018-016-0346-5
14. Sabri MI, Ochs S. Inhibition of glyceraldehyde-3-phosphate dehydrogenase in mammalian nerve by iodoacetic acid. J Neurochem. 1971;18(8):1509–1514. doi:10.1111/j.1471-4159.1971.tb00013.x
15. Pitcher T, Sousa-Valente J, Malcangio M. The monoiodoacetate model of osteoarthritis pain in the mouse. J Vis Exp. 2016;111:53746.
16. Okun A, Liu P, Davis P, et al. Afferent drive elicits ongoing pain in a model of advanced osteoarthritis. Pain. 2012;153(4):924–933. doi:10.1016/j.pain.2012.01.022
17. Morais SV, Czeczko NG, Malafaia O, et al. Osteoarthritis model induced by intra-articular monosodium iodoacetate in rats knee. Acta Cir Bras. 2016;31(11):765–773. doi:10.1590/s0102-865020160110000010
18. van der Kraan PM, Vitters EL, van de Putte LB, van den Berg WB. Development of osteoarthritic lesions in mice by “metabolic” and “mechanical” alterations in the knee joints. Am J Pathol. 1989;135(6):1001–1014.
19. Guingamp C, Gegout-Pottie P, Philippe L, Terlain B, Netter P, Gillet P. Mono-iodoacetate-induced experimental osteoarthritis: a dose-response study of loss of mobility, morphology, and biochemistry. Arthritis Rheum. 1997;40(9):1670–1679. doi:10.1002/art.1780400917
20. Guzman RE, Evans MG, Bove S, Morenko B, Kilgore K. Mono-iodoacetate-induced histologic changes in subchondral bone and articular cartilage of rat femorotibial joints: an animal model of osteoarthritis. Toxicol Pathol. 2003;31(6):619–624. doi:10.1080/01926230390241800
21. Fernihough J, Gentry C, Malcangio M, et al. Pain related behaviour in two models of osteoarthritis in the rat knee. Pain. 2004;112(1–2):83–93. doi:10.1016/j.pain.2004.08.004
22. Ivanavicius SP, Ball AD, Heapy CG, Westwood RF, Murray F, Read SJ. Structural pathology in a rodent model of osteoarthritis is associated with neuropathic pain: increased expression of ATF-3 and pharmacological characterisation. Pain. 2007;128(3):272–282. doi:10.1016/j.pain.2006.12.022
23. Rashid MH, Theberge Y, Elmes SJ, Perkins MN, McIntosh F. Pharmacological validation of early and late phase of rat mono-iodoacetate model using the Tekscan system. Eur J Pain. 2013;17(2):210–222. doi:10.1002/j.1532-2149.2012.00176.x
24. Hoshino T, Tsuji K, Onuma H, et al. Persistent synovial inflammation plays important roles in persistent pain development in the rat knee before cartilage degradation reaches the subchondral bone. BMC Musculoskelet Disord. 2018;19(1):291. doi:10.1186/s12891-018-2221-5
25. Sasaki Y, Kijima K, Yoshioka K. Validity evaluation of a rat model of monoiodoacetate-induced osteoarthritis with clinically effective drugs. BMC Musculoskelet Disord. 2024;25(1):975. doi:10.1186/s12891-024-08083-9
26. Shi X, Yu W, Wang T, et al. Electroacupuncture alleviates cartilage degradation: improvement in cartilage biomechanics via pain relief and potentiation of muscle function in a rabbit model of knee osteoarthritis. Biomed Pharmacother. 2020;123:109724. doi:10.1016/j.biopha.2019.109724
27. Pritzker KP, Gay S, Jimenez SA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14(1):13–29. doi:10.1016/j.joca.2005.07.014
28. Mankin HJ, Dorfman H, Lippiello L, Zarins A. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am Vol. 1971;53(3):523–537.
29. McHugh ML. Interrater reliability: the kappa statistic. Biochem Med. 2012;22(3):276–282. doi:10.11613/BM.2012.031
30. Raman IM. Power analysis. Elife. 2019;8. doi:10.7554/eLife.52232
31. Ikeuchi M, Izumi M, Aso K, Sugimura N, Kato T, Tani T. Effects of intra-articular hyaluronic acid injection on immunohistochemical characterization of joint afferents in a rat model of knee osteoarthritis. Eur J Pain. 2015;19(3):334–340. doi:10.1002/ejp.551
32. Luyten FP, Lories RJ, Verschueren P, de Vlam K, Westhovens R. Contemporary concepts of inflammation, damage and repair in rheumatic diseases. Best Pract Res Clin Rheumatol. 2006;20(5):829–848. doi:10.1016/j.berh.2006.06.009
33. Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1192(1):230–237. doi:10.1111/j.1749-6632.2009.05240.x
34. Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6(11):625–635. doi:10.1038/nrrheum.2010.159
35. Schuelert N, McDougall JJ. Grading of monosodium iodoacetate-induced osteoarthritis reveals a concentration-dependent sensitization of nociceptors in the knee joint of the rat. Neurosci Lett. 2009;465(2):184–188. doi:10.1016/j.neulet.2009.08.063
36. Combe R, Bramwell S, Field MJ. The monosodium iodoacetate model of osteoarthritis: a model of chronic nociceptive pain in rats? Neurosci Lett. 2004;370(2–3):236–240. doi:10.1016/j.neulet.2004.08.023
37. Chien TY, Huang SK, Lee CJ, Tsai PW, Wang CC. Antinociceptive and anti-inflammatory effects of zerumbone against mono-iodoacetate-induced arthritis. Int J Mol Sci. 2016;17(2):249. doi:10.3390/ijms17020249
38. Udo M, Muneta T, Tsuji K, et al. Monoiodoacetic acid induces arthritis and synovitis in rats in a dose- and time-dependent manner: proposed model-specific scoring systems. Osteoarthritis Cartilage. 2016;24(7):1284–1291. doi:10.1016/j.joca.2016.02.005
39. Izumi M, Ikeuchi M, Ji Q, Tani T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J Biomed Sci. 2012;19(1):77. doi:10.1186/1423-0127-19-77
40. Song X, Liu Y, Chen S, et al. Knee osteoarthritis: a review of animal models and intervention of traditional Chinese medicine. Animal Model Exp Med. 2024;7(2):114–126. doi:10.1002/ame2.12389
41. Thakur M, Rahman W, Hobbs C, Dickenson AH, Bennett DL. Characterisation of a peripheral neuropathic component of the rat monoiodoacetate model of osteoarthritis. PLoS One. 2012;7(3):e33730. doi:10.1371/journal.pone.0033730
42. Kim Y, Kim EH, Lee KS, et al. The effects of intra-articular resiniferatoxin on monosodium iodoacetate-induced osteoarthritic pain in rats. Korean J Physiol Pharmacol. 2016;20(1):129–136. doi:10.4196/kjpp.2016.20.1.129
43. De Roover A, Escribano-Núñez A, Monteagudo S, Lories R. Fundamentals of osteoarthritis: inflammatory mediators in osteoarthritis. Osteoarthritis Cartilage. 2023;31(10):1303–1311. doi:10.1016/j.joca.2023.06.005
44. Nees TA, Rosshirt N, Reiner T, Schiltenwolf M, Moradi B. Inflammation and osteoarthritis-related pain. Schmerz. 2019;33(1):4–12. doi:10.1007/s00482-018-0346-y
45. Nwosu LN, Mapp PI, Chapman V, Walsh DA. Relationship between structural pathology and pain behaviour in a model of osteoarthritis (OA). Osteoarthritis Cartilage. 2016;24(11):1910–1917. doi:10.1016/j.joca.2016.06.012
46. Bove SE, Calcaterra SL, Brooker RM, et al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage. 2003;11(11):821–830. doi:10.1016/S1063-4584(03)00163-8
47. Li L, Li Z, Li Y, Hu X, Zhang Y, Fan P. Profiling of inflammatory mediators in the synovial fluid related to pain in knee osteoarthritis. BMC Musculoskelet Disord. 2020;21(1):99. doi:10.1186/s12891-020-3120-0
48. Jrad AIS, Trad M, Bzeih W, El Hasbani G, Uthman I. Role of pro-inflammatory interleukins in osteoarthritis: a narrative review. Connect Tissue Res. 2023;64(3):238–247. doi:10.1080/03008207.2022.2157270
49. Sun G, Ba CL, Gao R, Liu W, Ji Q. Association of IL-6, IL-8, MMP-13 gene polymorphisms with knee osteoarthritis susceptibility in the Chinese Han population. Biosci Rep. 2019;39(2). doi:10.1042/BSR20181346
50. Arora D, Taneja Y, Sharma A, Dhingra A, Guarve K. Role of apoptosis in the pathogenesis of osteoarthritis: an explicative review. Curr Rheumatol Rev. 2024;20(1):2–13. doi:10.2174/1573397119666230904150741
51. Liu W, Guo NY, Wang JQ, Xu BB. Osteoarthritis: mechanisms and therapeutic advances. MedComm. 2025;6(8):e70290. doi:10.1002/mco2.70290
52. Mrosewski I, Jork N, Gorte K, et al. Regulation of osteoarthritis-associated key mediators by TNFα and IL-10: effects of IL-10 overexpression in human synovial fibroblasts and a synovial cell line. Cell Tissue Res. 2014;357(1):207–223. doi:10.1007/s00441-014-1868-y
53. Yu LK, Zhang J, Sun ZY, Ruan CL, Li H, Ruan XJ. Coculture with interleukin-10 overexpressed chondrocytes: a cell therapy model to ameliorate the post-traumatic osteoarthritis development. J Biol Regul Homeost Agents. 2021;35(2):593–603. doi:10.23812/21-40-A
54. Maleeva EE, Palikova YA, Palikov VA, et al. Potentiating TRPA1 by sea anemone peptide ms 9a-1 reduces pain and inflammation in a model of osteoarthritis. Mar Drugs. 2023;21(12):617. doi:10.3390/md21120617
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Therapeutic Effect of Acupotomy at Sanheyang for Cartilage Collagen Damage in Moderate Knee Osteoarthritis: A Rabbit Model
Li Y, Hou Y, Sun J, Wei J, Chai Y, Guo M, Wang R
Journal of Inflammation Research 2023, 16:2241-2254
Published Date: 24 May 2023
Effect of Acupotomy Modulation of NLRP3/Caspase-1/GSDMD Pathway on Chondrocyte Pyroptosis in Knee Osteoarthritis
Wang Y, Chen Y, Yang Y, Zhou Y, Yang Y, Zhu J
Journal of Pain Research 2025, 18:4935-4945
Published Date: 23 September 2025
Acupotomy Ameliorates Articular Cartilage Fibrosis in a Rabbit Model of Knee Osteoarthritis via the TGF-β1/Smad Pathway
Xing L, Zhu W, Chen X, Che R, Wu J, Guo Y, Liang C
Orthopedic Research and Reviews 2026, 18:597057
Published Date: 7 May 2026