Back to Journals » International Journal of Nanomedicine » Volume 21
Mitochondria-Targeted MPDA Nanosystem Co-Delivering Evodiamine and IR820 for Chemo-Photothermal Therapy of Hepatocellular Carcinoma
Authors Kong S, Lin H, Liu Y, Tang R, Li H, Lin L
Received 9 January 2026
Accepted for publication 20 May 2026
Published 5 June 2026 Volume 2026:21 594951
DOI https://doi.org/10.2147/IJN.S594951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Eng San Thian
Shasha Kong,1– 3,* Hongmei Lin,4,5,* Yuling Liu,1,* Ruying Tang,1 Hui Li,1– 3 Longfei Lin1
1Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China; 2Institute of Traditional Chinese Medicine Health Industry, China Academy of Chinese Medical Sciences, Nanchang, Jiangxi, People’s Republic of China; 3Jiangxi Health Industry Institute of Traditional Chinese Medicine, China Academy of Chinese Medical Sciences, Nanchang, Jiangxi, People’s Republic of China; 4Beijing Research Institute of Chinese Medicine, Beijing University of Chinese Medicine, Beijing, People’s Republic of China; 5National Medical Products Administration Key Laboratory for Research Evaluation of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hui Li; Longfei Lin, Email [email protected]; [email protected]
Introduction: Hepatocellular carcinoma (HCC) is a highly malignant tumor characterized by considerable heterogeneity, aggressive invasiveness, and a high recurrence rate. Monotherapy often yields limited clinical efficacy, and evodiamine (EVO), a natural alkaloid with promising antitumor activity, is hindered by low bioavailability and potential systemic toxicity.
Methods: A mesoporous polydopamine (MPDA)-based theranostic nanosystem was constructed for the co-delivery of EVO and the photothermal agent IR820. To enhance tumor specificity and minimize off-target toxicity, the platform was functionalized with cyclic RGD (cRGD) peptides for active tumor homing and triphenylphosphonium (TPP) for mitochondrial localization. The resulting IR820/EVO@MPDA-TPP/cRGD nanoparticles were characterized for drug loading, photothermal conversion stability, and fluorescence properties, and evaluated for antitumor efficacy and mechanistic actions in vivo.
Results: The nanosystem demonstrated high drug loading efficiencies, excellent photothermal conversion stability, and robust near-infrared fluorescence emission suitable for real-time diagnostic tracing. Upon NIR laser irradiation, IR820/EVO@MPDA-TPP/cRGD exhibited potent synergistic anticancer activity and significantly inhibited tumor growth in vivo. Mechanistically, the combined photothermal and chemotherapeutic effects triggered severe mitochondrial dysfunction, leading to the collapse of mitochondrial membrane potential and subsequent release of pro-apoptotic factors.
Discussion: The dual-targeting strategy effectively shifted cellular homeostasis toward programmed cell death while simultaneously engaging quality control pathways, underscoring a mitochondria-centered mechanism linking photothermal effect and chemotherapy.
Conclusion: IR820/EVO@MPDA-TPP/cRGD represents a dual-targeting theranostic nanoplatform that integrates imaging and chemo-photothermal combination therapy. This strategy offers a promising and clinically relevant approach for advanced HCC. Nanoparticle system: synthesis, targeting and effects like ROS, autophagy, apoptosis.The image shows a nanoparticle delivery system involving multiple stages of synthesis and cellular effects. The synthesis process begins with MPDA, followed by IR820/EVO@MPDA, IR820/EVO@MPDA-cRGD and finally IR820/EVO@MPDA-TPP@cRGD. Components include EVO, IR820, cRGD and TPP. A mouse is shown receiving an injection, with nanoparticles targeting a tumor. Inside the cell, the nanoparticles are exposed to near-infrared light (NIR), leading to decreased mitochondrial membrane potential (Δψ) and ATP levels. This results in increased reactive oxygen species (ROS) production, triggering autophagy marked by LC3-II and apoptosis through cytochrome C and activated caspase pathways. Arrows indicate the flow of processes and interactions within the cell.
Keywords: hepatocellular carcinoma, evodiamine, mesoporous polydopamine, photothermal therapy, drug delivery, mitochondria
Introduction
Hepatocellular carcinoma (HCC) represents a major global health burden with particularly high prevalence in China and other regions of Asia. Clinically, the disease is characterized by insidious onset, aggressive progression, frequent metastasis, and high recurrence rates. The absence of distinct early-stage symptoms often results in delayed diagnosis, with most patients presenting at advanced stages where curative surgical intervention is no longer feasible. Even among surgical candidates, postoperative recurrence remains common.1 Conventional treatment modalities, including radiotherapy and chemotherapy, provide limited efficacy,2 while responses to emerging options such as targeted agents and immunotherapy are often variable and inconsistent. Consequently, a substantial improvement in overall survival rates remains elusive. There is therefore an urgent need to develop novel and effective therapeutic strategies to address these clinical challenges in HCC management.
Photothermal therapy (PTT), a non-invasive treatment modality, utilizes near-infrared (NIR) light-activated photothermal agents to generate localized hyperthermia for selective ablation of tumor tissue.3 This approach has garnered significant attention due to its high spatiotemporal precision, rapid treatment cycles, and minimal invasiveness.3 PTT is frequently combined with chemotherapy, immunotherapy, or photodynamic therapy (PDT), synergistically enhancing tumor eradication and mitigating drug resistance. However, conventional PTT often requires high-temperature thermal effects, which can damage surrounding healthy tissue and impair hepatic function. Recent advances have identified mitochondria, which are central organelles involved in regulating energy metabolism and apoptosis, as promising subcellular targets for phototherapy due to their sensitivity to heat and reactive oxygen species (ROS).4,5 Mitochondria-targeted photothermal strategies facilitate apoptosis under mild thermal conditions while promoting ROS generation, thereby augmenting therapeutic synergy. Despite these advantages, most existing phototherapeutic nanomedicines are limited to cellular-level targeting, lack the capability for precise mitochondrial delivery.
Mesoporous polydopamine (MPDA) nanoparticles are a class of melanin-like polymeric nanomaterials notable for their excellent biocompatibility and richly porous structure. Their high surface area and abundant mesopores confer strong drug adsorption capacity, allowing efficient loading of therapeutic agents. In addition, MPDA exhibits photothermal conversion efficiency, enabling rapid generation of localized hyperthermia under NIR irradiation for effective tumor ablation.6 The surface of MPDA contains diverse functional groups that facilitate conjugation with targeting ligands, such as peptides or antibodies, to enhance tumor-specific accumulation.7 This modifiable surface also enables the integration of other functional components, extending the applicability of MPDA to multimodal therapeutic strategies. Owing to these features, MPDA has emerged as a promising nanocarrier platform, capable of supporting synergistic antitumor therapies, including combinations of PTT with chemotherapy,8 gene therapy,9 immunotherapy,6 and others.
Evodiamine (EVO), a natural alkaloid with diverse antitumor mechanisms, has been shown to induce apoptosis,10,11 suppress metastasis,12 and reverse chemoresistance.13 Notably, EVO sensitizes tumor cells to PTT by inhibiting the expression of heat shock proteins 70 and 90 (HSP70/90),14 thereby enhancing therapeutic efficacy. IR820, a near-infrared heptamethine dye, not only acts synergistically with MPDA to augment the photothermal heating and improve tumor ablation, but also enables real-time fluorescence imaging for noninvasive tracking of nanodrug distribution, assessment of targeting efficiency, and delineation of tumor margins. To achieve organelle-specific delivery, the nanosystem was functionalized with cRGD peptides to promote active tumor targeting and accumulation in hepatocellular carcinoma tissues.15 Furthermore, mitochondrial tropism was enhanced through surface modification with triphenylphosphonium (TPP), enabling its precise localization to mitochondria and facilitating subcellular-specific therapy.
Although mesoporous polydopamine (MPDA)-based photothermal delivery platforms have been extensively documented,16,17 and the respective roles of cRGD in tumor targeting and TPP in mitochondrial tropism are well established, current nanocarrier designs for hepatocellular carcinoma (HCC) largely remain confined to single-level targeting strategies.18–20 Such configurations typically emphasize either enhanced intratumoral accumulation or improved cellular internalization, yet they often fail to adequately address the sequential biological barriers spanning from vascular extravasation to subcellular organelle localization.21 In distinct contrast to recently described single-ligand MPDA systems,22,23 the novelty of the present study resides in the strategic exploitation of the spatial synergistic effect between cRGD and TPP to engineer a “Membrane-to-Mitochondria Sequential Relay Targeting” modality. Specifically, surface conjugation of cRGD facilitates evasion of the first-pass effect to achieve robust intratumoral retention within HepG2 xenografts, while the subsequent exposure of TPP moieties directs the nanoparticles to bypass lysosomal sequestration and anchor precisely at the mitochondria outer membrane, a site exquisitely sensitive to thermal stress. This strategy effectively reduces nonspecific thermal damage to surrounding normal tissues and markedly circumvents the systemic toxicity induced by free EVO. Notably, this design constitutes more than a mere physical admixture of established ligands; rather, it leverages the temporally programmed exposure of cryptic TPP cationic sites, thereby mitigating the non-specific scavenging by the hepatic reticuloendothelial system that frequently compromises the efficacy of conventional TPP-only modifications.
In summary, we developed a dual-ligand functionalized nanosystem, termed IR820/EVO@MPDA-TPP/cRGD, and evaluated its therapeutic efficacy in a HepG2 cell-derived xenograft (HCDX) model. The nanocomposite preserved the fluorescence properties of IR820 while preventing molecular aggregation. Moreover, IR820 and EVO were efficiently encapsulated into the mesoporous channels of MPDA via hydrophobic interactions, achieving high loading capacities of 5.0% and 4.6%, respectively. Guided by the cooperative targeting effect of cRGD and TPP, the nanoparticles demonstrated enhanced tumor-specific accumulation and precise mitochondrial localization. As a chemotherapeutic agent, EVO acted synergistically with IR820/MPDA-mediated photothermal therapy, leading to potent tumor suppression both in vitro and in vivo. Mechanistic studies revealed that the combined chemo-photothermal treatment induced severe mitochondrial dysfunction, apoptosis, autophagy, and activation of programmed cell death pathways. Furthermore, the nanosystem exhibited favorable biosafety at the administered dosage, supporting its potential for further translational development.
Materials and Methods
Materials
Evodiamine (EVO) was obtained from Baoji Yirui Biotechnology Co., Ltd. (Shaanxi, China). Poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) diacrylate (F127), new Indocyanine Green (IR820), dopamine hydrochloride, and 3,3′,5,5′-Tetramethylbenzidine (TMB) were purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). Triphenylphosphine-NH2 (TPP-NH2) and cRGD were provided by Xi’an Ruixi Biotechnology Co., Ltd. (Xian, China).
Preparation of IR820/EVO@MPDA-TPP/cRGD
Synthesis of MPDA
F127 and TMB were added to a mixed solvent (water/ethanol=1:1, v/v) under vigorous stirring for 30 minutes, to form a stable white emulsion. Subsequently, aqueous ammonia and dopamine hydrochloride were added to the reaction mixture, followed by continuous stirring at 25 °C for 2 hours. The resulting suspension was centrifuged at 13,000 rpm for 10 minutes, and the precipitate was thoroughly washed twice with water/ethanol (1:1, v/v) to remove residual surfactants and unreacted precursors. The purified product was redispersed in ultrapure water to yield MPDA nanoparticles.
Synthesis of IR820/EVO@MPDA
IR820 and EVO were dissolved in methanol and mixed with MPDA nanoparticles. The mixture was stirred overnight at 25 °C. The resulting suspension was centrifuged at 13,000 rpm for 10 minutes, and the precipitate was collected. After two washes with water/ethanol (1:1, v/v), the product was redispersed to obtain IR820/EVO@MPDA.
Synthesis of IR820/EVO@MPDA-cRGD
cRGD was dissolved in deionized water and added to the IR820/EVO@MPDA nanoparticle suspension. The mixture was incubated overnight under continuous stirring. The resulting suspension was centrifuged at 13,000 rpm for 10 minutes, and the precipitate was collected. After two washes with water/ethanol (1:1, v/v), the product was redispersed to obtain IR820/EVO@MPDA-cRGD.
Synthesis of IR820/EVO@MPDA-TPP/cRGD
cRGD and TPP-NH2 were dissolved in deionized water and added to the IR820/EVO@MPDA suspension. The mixture was stirred overnight. The final suspension was centrifuged at 13,000 rpm for 10 minutes, and the precipitate was collected. After two washes with water/ethanol (1:1, v/v), the product was redispersed in ultrapure water to obtain IR820/EVO@MPDA-TPP/cRGD.
Characterization
Transmission Electron Microscopy (TEM)
MPDA, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), and IR820/EVO@MPDA-TPP/cRGD (EVO-3) nanoparticles were each dispersed in ultrapure water and subjected to ultrasonic treatment for 3 minutes to ensure homogeneous suspension. A droplet of each dispersion was deposited onto a copper grid and allowed to dry under ambient conditions. Nanoparticle morphology was subsequently examined using transmission electron microscopy (TEM).
Dynamic Light Scattering (DLS)
The hydrodynamic diameter and zeta potential of MPDA, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), and IR820/EVO@MPDA-TPP/cRGD (EVO-3) were measured using a NanoBrook 90Plus PALS dynamic light scattering system. To evaluate colloidal stability, each nanoparticle sample (MPDA, EVO-1, EVO-2, and EVO-3) was dispersed in fetal bovine serum (FBS)-free DMEM and incubated under constant conditions. Particle size was monitored daily over 7 days.
BET Surface Area and Pore Size Analysis
The specific surface area and pore size distribution of MPDA were determined via nitrogen adsorption-desorption isotherms using a Micromeritics ASAP 2460 system (version 3.01, UK). The Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) were applied for data analysis.
Fourier Transform Infrared (FT-IR) Spectroscopy
FT-IR spectroscopy (Thermo Scientific, USA) was employed to acquire near-infrared absorption spectra of MPDA, IR820/EVO@MPDA, IR820/EVO@MPDA-cRGD, and IR820/EVO@MPDA-TPP/cRGD nanoparticles.
Thermogravimetric Analysis (TGA)
Thermal degradation of nanoparticles was evaluated using a PerkinElmer STA 6000 thermal analyzer (USA). Samples were heated in a platinum crucible under a nitrogen atmosphere with a flow rate of 100 mL/min to maintain inert conditions. A constant heating rate of 10 °C/min was applied throughout the measurements.
Photothermal Performance Testing
The photothermal effect of the nanoparticles was assessed by irradiating aqueous dispersions with an 808 nm NIR laser at a power density of 1.0 W/cm2. Temperature changes were monitored in real time using an infrared thermal camera (FLIR C2, USA), with data recorded at 30-second intervals. The photothermal conversion efficiencies of the various nanoformulations were calculated using Roper’s method.
Determination of Drug Loading Capacity (DLC) and Drug Encapsulation Efficiency (DEE)
Appropriate volumes of IR820/EVO@MPDA, IR820/EVO@MPDA-cRGD, and IR820/EVO@MPDA-TPP/cRGD solutions were centrifuged at 13,000 rpm for 5 minutes. The resulting pellets were redispersed in methanol and sonicated at 25 °C for 10 minutes, followed by a second centrifugation under the same conditions. Supernatants were collected for analysis. EVO content was quantified via high-performance liquid chromatography (HPLC), and IR820 content was determined using UV–vis spectroscopy. DLC and DEE were calculated using the following equations:
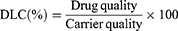
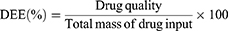
EVO quantification was conducted using a C18 column (250 mm × 4.6 mm; Dalian Elite Analytical Instruments, China) maintained at 25 °C. The mobile phase consisted of methanol (A) and water (B), delivered at 1.0 mL/min under the following elution program: 0–10 minutes, 10% B. Detection was performed at a wavelength of 331 nm.
For IR820, a standard curve was generated using solutions at concentrations of 0.01, 0.02, 0.03, 0.04, and 0.05 mg/mL. UV–Vis spectra were recorded from 300 to 1000 nm (Figure S1). A characteristic absorption peak was observed at 820 nm, and quantitative analysis was based on absorbance at this wavelength.
Drug Release Detection
A total of 8 mg of IR820/EVO@MPDA-TPP/cRGD nanoparticles was dispersed in 4 mL of deionized water and loaded into a dialysis device equipped with a 10 nm polycarbonate membrane. Dialysis was performed against 1 mM PBS (pH 7.2) under continuous stirring. Laser-triggered release was initiated by irradiating the solution with an 808 nm laser (1.0 W/cm 2) for 5 minutes. At predetermined time intervals (0, 1, 2, 4, 6, 24, and 48 hours), samples were collected and replaced with fresh PBS to maintain sink conditions. Each sample was extracted with methanol, centrifuged, and analyzed for EVO content using the HPLC method described in Determination of Drug Loading Capacity (DLC) and Drug Encapsulation Efficiency (DEE). To elucidate the EVO release mechanism from IR820/EVO@MPDA-TPP/cRGD, the cumulative release data were fitted to the Higuchi model and the Ritger-Peppas model.
Cell Culture and Nanoparticle Uptake Analysis
Hepatocellular carcinoma HepG2 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Huh-7 cells were purchased from Xiamen Yimo Biotechnology Co., Ltd (Xiamen, China). The use of these commercial cell lines does not require additional ethical approval as they are obtained from certified repositories. HepG2, and Huh-7 cells were cultured in a high-glucose DMEM supplemented with 10% inactivated FBS and 1% penicillin-streptomycin, and maintained at 37 °C in a 5% CO2 incubator.
To assess cellular uptake, HepG2 cells were seeded in 12-well plates at a density of 1×105 cells per well and allowed to adhere overnight. Cells were treated with EVO, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), or IR820/EVO@MPDA-TPP/cRGD (EVO-3) at a concentration of 20 μg/mL (equivalent EVO concentrations) for 4 hours at 37 °C. After incubation, the medium was removed, and the cells were gently washed three times with ice-cold PBS. Subsequently, 250 μL of RIPA lysis buffer was added for 5 minutes. The lysate was vortexed for 1 minute, mixed with 1 mL of methanol, vortexed again for 3 minutes, and centrifuged at 8000 rpm for 15 minutes. The supernatant was filtered through a 0.45 μm membrane and analyzed via HPLC.
Cell Viability Assay
HepG2 and Huh-7 cells in logarithmic growth phase were seeded into 96-well plates at a density of 8 × 103 cells per well and incubated overnight. Cells were treated with MPDA, EVO, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), or IR820/EVO@MPDA-TPP/cRGD (EVO-3) in culture medium at equivalent EVO concentrations ranging from 2.5 to 160 μg/mL for 24 hours. Cell viability was assessed using the CCK-8 assay by adding 10% (v/v) CCK-8 solution per well. For phototoxicity evaluation, HepG2 cells were incubated with the formulations for 16 hours, irradiated with an 808 nm laser (1.0 W/cm2, 5 min),24 and incubated for an additional 8 hours before viability assessment using the same method.
Evaluation of Mitochondrial Targeting
HepG2 cells were seeded in 35 mm glass-bottom confocal dishes (5 × 104 cells/well) and cultured overnight. Cells were then incubated for 24 hours with IR820, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), or IR820/EVO@MPDA-TPP/cRGD (EVO-3), each at an equivalent IR820 concentration of 10 μg/mL. Following treatment, cells were stained with MitoTracker Green and Hoechst 33258 (Beyotime Biotechnology, China). Imaging was performed using a Leica DMI 600B confocal microscope (Wetzlar, Germany). The following excitation/emission wavelengths were applied: 494/517 nm for green fluorescence (MitoTracker Green), 352/461 nm for blue fluorescence (Hoechst 33258), and 725/850 nm for red fluorescence (IR820).
Observation of Mitochondrial Morphology
Confocal Laser Scanning Microscopy
HepG2 cells were seeded in 35 mm glass-bottom confocal dishes (5 × 104 cells/well) and incubated overnight. Cells were treated with control medium (Con), free EVO, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), or IR820/EVO@MPDA-TPP/cRGD (EVO-3) at an equivalent EVO concentration of 20 μg/mL. After 16 hours of incubation, EVO-2 and EVO-3 groups were exposed to 808 nm laser irradiation (1.0 W/cm2, 5 min) and further incubated for 8 hours. Other groups were cultured for 24 hours. Cells were washed three times with PBS and stained with MitoTracker Red CMXRos and Hoechst 33258 to label mitochondria and nuclei, respectively. Mitochondrial morphology was visualized using a laser scanning confocal microscope.
Biological Transmission Electron Microscopy Imaging
HepG2 cells were seeded in 6-well plates (6 × 105 cells/well) and incubated overnight. Cells were treated with EVO-free medium (Con), EVO, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), or IR820/EVO@MPDA-TPP/cRGD (EVO-3) at an equivalent EVO concentration of 20 μg/mL. Following 16 hours of incubation, EVO-2 and EVO-3 groups were irradiated (808 nm, 1.0 W/cm2, 5 min) and further incubated for 8 hours. Other groups continued incubation for 24 hours. Cells were washed, scraped, centrifuged at 2000 rpm for 5 minutes, and the pellets were fixed in 2.5% glutaraldehyde at 4 °C for 48 hours. Samples were then subjected to gradient dehydration, resin embedding, and ultrathin sectioning. Sections were stained with uranyl acetate and lead citrate before imaging by TEM.
Mitochondrial Membrane Potential (Δψ) Detection
HepG2 cells were seeded in 6-well plates (6 × 105 cells/well) and allowed to adhere overnight. Cells were treated with either nanoparticle-free medium (Con) or medium containing the nanoparticle formulations, all at an equivalent EVO concentration of 20 μg/mL. After 16 hours of incubation, the IR820/EVO@MPDA-cRGD and IR820/EVO@MPDA-TPP/cRGD groups were exposed to 808 nm laser irradiation (1.0 W/cm2) for 5 minutes, followed by an additional 8 hours of culture. All other groups were incubated continuously for 24 hours. Cells were then stained using a JC-1 mitochondrial membrane potential assay kit according to the manufacturer’s protocol and analyzed by flow cytometry.
Detection of Intracellular ATP Levels
HepG2 cells were seeded in 6-well plates (6 × 105 cells per/well) and incubated overnight. Cells were then treated with culture medium either without nanoparticles (Con) or with the nanoparticle formulations, each at an equivalent EVO concentration of 20 μg/mL. After 16 hours of incubation, the IR820/EVO@MPDA-cRGD and IR820/EVO@MPDA-TPP/cRGD groups were irradiated with an 808 nm laser (1.0 W/cm2) for 5 minutes and incubated for an additional 8 hours. The remaining groups were incubated continuously for 24 hours. Intracellular ATP levels were measured using a commercial ATP assay kit (Solaibao Technology Co., Ltd., Beijing, China) according to the manufacturer’s instructions.
Detection of Intracellular ROS Levels
HepG2 cells were seeded in 6-well plates at a density of 6×105 cells per well and cultured overnight. After the treatment procedure as described in Detection of Intracellular ATP Levels, cells were harvested and incubated with 10 μM DCFH-DA (Beyotime Biotechnology, China) at 37 °C for 20 minutes. After three washes with PBS, the cells were immediately analyzed by flow cytometry.
Establishment of HepG2 Cell-Derived Xenograft (HCDX) Model
Male BALB/c nude mice (14 ± 2 g) were purchased from SPF (Beijing) Biotechnology Co., Ltd. (license number: SCXK (Beijing) 2024–0001). Mice were housed under specific pathogen-free (SPF) conditions at 25 °C and 60% relative humidity, with a 12-hour light/dark cycle. Five mice were housed per cage, maintained in a laminar flow clean bench. All animal procedures complied with the Regulation on the Administration of Laboratory Animals (2017 Revision) and were approved by the Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences (Ethics Approval No. 2024B353).
After a 3-day acclimatization period, the HCDX model was established. HepG2 cells were harvested and resuspended at a concentration of 4 × 107 cells/mL. The cell suspension was mixed 1:1 (v/v) with matrix gel on ice to maintain homogeneity. A total of 0.2 mL of the mixture was subcutaneously injected into the right flank of each mouse. Mice were monitored daily for health status. By day 10 post-injection, palpable bean-sized tumors had formed at the inoculation site. Tumor length (a) and width (b) were measured every three days, and tumor volume (V) was calculated according to the following equation:

Intracellular Imaging and Biodistribution
When tumor volume reached approximately 200 mm3, the biodistribution of nanoparticles was evaluated. Mice were intravenously injected with IR820, IR820/EVO@MPDA, IR820/EVO@MPDA-cRGD, or IR820/EVO@MPDA-TPP/cRGD (0.1 mL, containing 200 μg IR820). At 0, 24, 48, 72, and 120 hours post-injection, mice were imaged using a small animal live imaging instrument for fluorescence detection (excitation wavelength λex = 710 nm, emission wavelength λem = 820 nm). After the final imaging session, mice were anesthetized and euthanized by decapitation. Major organs (hearts, livers, spleen, lungs, kidneys) and tumors were excised, and ex vivo fluorescence imaging was performed.
Animal Grouping and Administration
When tumor volumes in the axilla of the tumor-bearing mice reached approximately 250 mm3, mice were randomly assigned to nine experimental groups (n = 6 per group): Con, MPDA, EVO, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA + Laser (EVO-1+L), IR820/EVO@MPDA-cRGD (EVO-2), IR820/EVO@MPDA-cRGD + Laser (EVO-2+L), IR820/EVO@MPDA-TPP/cRGD (EVO-3), IR820/EVO@MPDA-TPP/cRGD + Laser (EVO-3+L). Each formulation was administered via tail vein injection every other day for a total of six treatments. The first treatment was initiated on the third day after grouping. On the day of the first injection and for two subsequent days after each injection, laser-treated groups (EVO-1+L, EVO-2+L, EVO-3+L) were irradiated with an 808 nm laser (1 W/cm2) for 10 minutes. Body weight and tumor volume were recorded every two days. Twenty-four hours after the final injection, mice were anesthetized with 5% isoflurane. Approximately 0.7 mL of blood was collected via orbital bleeding and centrifuged at 12,000 rpm for 10 minutes at 4 °C to obtain serum for safety assessment (see Supplementary Material). Mice were then euthanized by cervical dislocation. Tumors were excised, weighed, and photographed. Tumor suppression rate (TSR) was calculated using the equation:

Tumors from three mice per group were fixed in 4% paraformaldehyde for histological and immunohistochemical analysis. Tumors from the remaining mice were stored at −80 °C for Western blot analysis. Major organs (livers, hearts, spleen, lungs, and kidneys) from all mice were collected for histopathological examination.
Hematoxylin and Eosin (H&E) Staining
Tumor tissues were collected from each group. The tissues were dehydrated using a graded ethanol series, cleared in xylene, and embedded in paraffin. Sections were cut using a rotary microtome, deparaffinized in xylene, and stained with H&E. After washing with PBS, the sections were mounted with neutral gum and examined under an optical microscope. Representative fields were selected for morphological analysis.
Immunohistochemistry
Paraffin-embedded tumor tissue sections were deparaffinized in xylene and rehydrated through a graded ethanol series. Antigen retrieval was performed, followed by incubation with primary antibodies against Ki67 and UCP2 at 4 °C overnight. After washing, the sections were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature and developed using 3,3′-diaminobenzidine. Hematoxylin was used to counterstain nuclei. The stained sections were mounted and imaged under an optical microscope. Quantitative analysis was performed using ImageJ software (Media Cybernetics, USA).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling (TUNEL) Assay
Fixed tumor tissues were washed three times with PBS (15 min per wash), then immersed in 30% sucrose solution and incubated overnight at 4 °C with gentle shaking. Tissues were embedded, and cryosections were prepared. Apoptotic cells were detected using the TUNEL assay kit (Wuhan Solarbio Biotechnology Co., Ltd.) according to the manufacturer’s protocol. Fluorescence imaging was performed with a DMI 6000B confocal laser scanning microscope, and apoptosis was quantitatively analyzed from the acquired images.
Western Blotting
Total protein was extracted from tumor tissues and quantified. Equal amounts of protein (30 μg) were separated via SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Membrane were blocked with 5% skim milk for 2 hours at room temperature, then incubated overnight at 4 °C with primary antibodies against BCL-2 (Proteintech, 12789-1-AP), BAX (Proteintech, 50599-2-Ig), cytochrome c (Proteintech, 10993-1-AP), cleaved caspase-3 (Proteintech, 82707–13-RR), ATP5A (Proteintech, 14676-1-AP), UCP2 (Proteintech, 11081-1-AP), PGC-1α (Proteintech, 66369-1-Ig), LC3-II (Affinity, AF5402), PINK1 (Affinity, DF7742), GPx4 (Affinity, DF6701), and GAPDH (Proteintech, 60004-1-Ig) After washing, membranes were incubated with HRP-conjugated secondary antibodies for 2 hours at 25 °C Protein bands were visualized using an enhanced chemiluminescence detection system and quantified using ImageJ software (National Institutes of Health, USA) Original blot images are provided in the Supporting Information (Figures S2–S4).
In vivo Safety Evaluation
The systemic safety of the nanoparticle formulations was assessed by measuring serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (Cr) Elabscience Biotechnology, China) using commercial assay kits (Solabao and Elabscience Biotechnology, China). All procedures followed the manufacturers’ protocols.
Statistical Analysis
All data are expressed as mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism version 6.02. Comparisons between two groups were assessed using an unpaired Student’s t-test. For comparisons among multiple groups, one-way analysis of variance (ANOVA) was used, and when a significant overall effect was detected, group differences were further evaluated using Tukey’s honestly significant difference post hoc test to control for multiple comparisons. A p-value of less than 0.05 was considered statistically significant, with *P < 0.05, **P < 0.01, and ***P < 0.001 indicating increasing levels of significance.
Results
Preparation and Characterization of IR820/EVO@MPDA-TPP/cRGD
Monodisperse MPDA nanoparticles were synthesized in aqueous solution using Pluronic F127 and TMB as soft templates. Dopamine polymerization was catalyzed by ethanol and ammonia, and π–π stacking interactions drove the self-assembly of polydopamine on the TMB template. The template was subsequently removed to yield MPDA nanoparticles.25–27 IR820 and EVO were loaded via mesopore adsorption and π–π interactions between the MPDA framework and the drug molecules. To improve tumor-specific accumulation and enhance antitumor efficacy, the cRGD peptides were conjugated onto the surface of MPDA via Michael addition reaction.28,29 Furthermore, mitochondrial targeting was achieved through surface modification with TPP via Schiff base and Michael addition reactions.30
Morphological and physicochemical characterization of MPDA, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), and IR820/EVO@MPDA-TPP/cRGD (EVO-3) was performed using TEM and DLS. TEM images showed spherical MPDA nanoparticles possessed a well-defined spherical morphology with a clear mesoporous structure (Figure 1A). The average hydrodynamic diameter and zeta potential of MPDA were 209.52 ± 3.9 nm and −19.34 ± 0.85 mV, respectively (Figure 1B). BET analysis confirmed mesoporosity, showing a pore size of 18.89 nm and a surface area of 5.58 m2/g (Figure 1C). After loading EVO and IR820, partial pore blockage was observed in EVO-1 (Figure 1A), accompanied by an increase in diameter (212.11 ± 10.3 nm) and a zeta potential of −17.75 ± 0.61 mV (Figure 1B). Successful drug incorporation was corroborated by FT-IR spectra confirmed successful drug loading, with peaks at 2925.0 cm−1 (EVO)31 and 813.3 cm−1 (IR820)32 (Figure 1D). Quantification via UV–vis analysis showed IR820 encapsulation efficiencies of of 45.0%, 46.7%, and 41.7% for EVO-1, EVO-2, and EVO-3, respectively, with loading capacities of 5.4%, 5.6%, and 5.0%. HPLC revealed EVO encapsulation efficiencies of 35.0%, 34.7%, and 37.3% and loading values of 4.2%, 4.2%, and 4.6%, respectively (Table S1 and Supplementary Material). Functionalization with cRGD further occluded the mesopores, yielding an average diameter of 218.98 ± 14.58 nm and shifted the zeta potential to −20.41 ± 2.06 mV for EVO-2 (Figure 1B). FT-IR analysis showed a shift in the carbonyl stretching vibration (νC=O) from 1512.5 cm−1 to 1515.0 cm−1 (Figure 1D), indicating amide bond formation between cRGD and MPDA. Upon subsequent TPP modification, EVO-3 showed complete pore sealing (Figure 1A), accompanied by an average diameter of 218.53 ± 3.39 nm and a zeta potential of −21.63 ± 1.5 mV (Figure 1B). A pronounced broad absorption band emerged at 1115.9 cm−1 in the FT-IR spectrum (Figure 1D), attributable to the stretching vibration of the C–P bond (νC–P) in TPP,33 confirming successful surface functionalization.
 |
Figure 1 (A) Transmission electron microscopy (TEM) images of MPDA, EVO-1, EVO-2, and EVO-3. Representative images are derived from at least three independent experiments. (B) Hydrodynamic size and zeta potential of MPDA, EVO-1, EVO-2, and EVO-3 in aqueous solution. Data represent mean ± SD (n = 3). (C) N2 adsorption-desorption isotherms and corresponding pore size distribution curves of MPDA nanoparticles. (D) Fourier transform infrared (FT-IR) spectra of MPDA, EVO-1, EVO-2, and EVO-3. (E) Stability of MPDA, EVO-1, EVO-2, and EVO-3 nanoparticles suspended in cell culture medium. Data represent mean ± SD (n = 3). (F) Temperature elevation profiles of MPDA, EVO-1, EVO-2, and EVO-3 under continuous 808 nm laser irradiation (1 W/cm2) in vitro. (G) Cumulative EVO release profiles from EVO-3 nanoparticles with or without laser irradiation. Data represent mean ± SD (n = 3). EVO-1, IR820/EVO@MPDA; EVO-2, IR820/EVO@MPDA-cRGD; EVO-3, IR820/EVO@MPDA-TPP/cRGD. |
TGA revealed that the primary weight loss events for the polymer samples occurred within the range of 370–395 °C, indicative of the onset of irreversible chemical degradation. The maximum decomposition temperature was recorded at 372.89 °C for pristine MPDA, while the functionalized nanoparticles EVO-1, EVO-2, and EVO-3 exhibited increased decomposition temperatures of 392.36 °C, 392.99 °C, and 391.07 °C, respectively (Figure S5). This progressive elevation in decomposition temperatures suggests enhanced thermal stability following each modification step. The improvement is attributed to the transformation of the initial loose porous architecture into a highly cross-linked organic–organic composite system, facilitated by physical encapsulation and covalent cross-linking, further corroborating the successful surface modification of MPDA nanoparticles. Additionally, DLS measurements conducted over seven days showed no significant changes in the hydrodynamic size of the nanoparticles (Figure 1E), demonstrating excellent colloidal stability in aqueous media.
The Photothermal Performance of IR820/EVO@MPDA-TPP/cRGD
The photothermal conversion performance of MPDA, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-CRGD (EVO-2), and IR820/EVO@MPDA-TPP/CRGD (EVO-3) nanoparticles was assessed in vitro using an infrared thermal imaging camera and thermocouple measurements. Upon 808 nm laser irradiation at 1 W/cm2, all nanoparticle formulations exhibited a time-dependent increase in temperature. After 10 minutes of irradiation, the temperatures of MPDA, EVO-1, EVO-2, and EVO-3 solutions reached 46.85 °C, 49.9 °C, 43.3 °C, and 47.35 °C, respectively, confirming their photothermal activity. The heating and cooling cycles demonstrated high reproducibility, with certain formulations reaching maximum temperatures above 50 °C under sustained laser exposure (Figure 1F). To quantitatively evaluate the light-to-heat conversion capability, the photothermal conversion efficiency (η) was calculated from the cooling-stage data. The η values of MPDA, EVO-1, EVO-2, and EVO-3 were determined to be 32.73%, 34.76%, 37.20%, and 42.06%, respectively. Notably, EVO-3 exhibited the highest η among the formulations, indicating that co-loading with IR820 and dual-ligand functionalization synergistically enhance the photothermal conversion efficiency. These findings confirm that EVO-3 efficiently converts near-infrared light into heat, indicating its potential for photothermal tumor ablation.
In vitro Release of EVO
The drug release profile of IR820/EVO@MPDA-TPP/cRGD (EVO-3) nanoparticles was evaluated in PBS (pH 7.2) with or without 808 nm laser exposure (1 W/cm2). Under physiological conditions without a laser, EVO exhibited a rapid release within the first 6 hours, followed by a plateau at 24 hours with 73.2% cumulative release. Laser irradiation significantly accelerated the release rate, increasing the cumulative release to 81.0% over the same duration (Figure 1G). This enhancement is likely attributable to the photothermally-induced expansion of the MPDA framework, which facilitates drug diffusion. The elevated temperature also increases EVO’s kinetic energy, promoting its desorption from the carrier by overcoming van der Waals interactions and diffusion energy barriers. Together, these results demonstrate that EVO-3 offers controlled, light-triggered drug release, highlighting its potential for spatially and temporally regulated chemotherapeutic delivery.
To elucidate the underlying release mechanisms, the release data were fitted to the Higuchi and Korsmeyer-Peppas kinetic models (Table S2). Under non-irradiated conditions, the Higuchi model yielded a good fit (R2 = 0.917), suggesting that EVO release is predominantly governed by a diffusion-controlled process through the mesoporous channels of MPDA. The Korsmeyer-Peppas model provided a superior fit (R2 = 0.952), with a release exponent (n) of 0.25, indicative of a Fickian diffusion mechanism typical for drug transport from mesoporous carriers. Upon laser irradiation, while the Higuchi model maintained a reasonable correlation (R2 = 0.895), the Korsmeyer-Peppas model again demonstrated a better fit (R2 = 0.967). Notably, the release exponent decreased to n = 0.20, suggesting that the release mechanism deviates slightly from pure Fickian diffusion toward what may be considered a “Less Fickian” or matrix relaxation-assisted diffusion. This shift is attributed to the photothermally induced expansion of the MPDA framework, which alters the pore architecture and facilitates enhanced drug diffusion. Furthermore, the elevated temperature increases EVO’s kinetic energy, promoting its desorption from the carrier by overcoming van der Waals interactions and diffusion energy barriers. Collectively, these kinetic modeling results demonstrate that EVO-3 offers controlled, Fickian diffusion-dominated drug release under physiological conditions, with laser-triggered framework expansion providing an additional mechanism for accelerated on-demand release, highlighting its potential for spatially and temporally regulated chemotherapeutic delivery.
In vitro Cytotoxicity and Cellular Uptake
The cytotoxic effects of MPDA, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), and IR820/EVO@MPDA-TPP/cRGD (EVO-3) nanoparticles were examined in HepG2 and Huh-7 cells using the CCK-8 assay. As illustrated in Figure 2A and Figure S6, MPDA alone exhibited negligible cytotoxicity after 24 hours of incubation, indicating good biocompatibility. In contrast, EVO-loaded nanoparticles induced concentration-dependent decreases in cell viability across a 2.5 to 160 μg/mL range. The half-maximal inhibitory concentration (IC50) values for HepG2 cells were 29.65 μg/mL for free EVO, 28.87 μg/mL for EVO-1, 23.31 μg/mL for EVO-2, and 17.75 μg/mL for EVO-3 (Figure S7). Near-infrared laser irradiation (808 nm, 1 W/cm2) enhanced anticancer efficacy in all photothermal-treated groups (EVO-1+L, EVO-2+L, and EVO-3+L) (Figure 2B). Importantly, EVO-3+L significantly reduced cell viability compared to non-irradiated EVO-3, supporting the synergistic effect of combined photothermal and chemotherapeutic treatment.
 |
Figure 2 In vitro anti-tumor activity and mitochondrial targeting evaluation. (A) Viability of HepG2 cells after 24 h treatment with varying concentrations of MPDA (AI), free EVO (AII), EVO-1 (AIII), EVO-2 (AIV), and EVO-3 (AV). Data represent mean ± SD (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001 vs Con group. (B) Cell viability of HepG2 cells treated with EVO (BI), EVO-1 (BII), EVO-2 (BIII), and EVO-3 (BIV) with or without laser irradiation (808 nm, 1 W/cm2). Data represent mean ± SD (n = 6). **p < 0.01 vs without laser irradiation group. (C) Mitochondrial targeting observed by confocal microscopy after 24 h treatment. Red: IR820 signal; Blue: Hoechst 33342 (nuclei); Green: MitoTracker Green (mitochondria). Scale bar: 5 μm. Representative images are derived from at least three independent experiments. White arrows indicate the colocalization of nanoparticles and mitochondria. (D) Cellular uptake of EVO in HepG2 cells after 24 h incubation with different nanoparticle formulations. Data represent mean ± SD (n = 3). *p < 0.05, **p < 0.01 vs Con group. EVO-1, IR820/EVO@MPDA; EVO-1+L, IR820/EVO@MPDA+Laser; EVO-2, IR820/EVO@MPDA-cRGD; EVO-2+L, IR820/EVO@MPDA-cRGD+Laser; EVO-3, IR820/EVO@MPDA-TPP/cRGD; EVO-3+L, IR820/EVO@MPDA-TPP/cRGD+Laser. |
To assess the cellular uptake and mitochondrial targeting capabilities of the nanoparticles, HepG2 cells were incubated with different formulations. Confocal microscopy revealed minimal cellular uptake of both free IR820 and IR820/EVO@MPDA (EVO-1), with minimal intracellular localization. IR820/EVO@MPDA-cRGD (EVO-2) primarily accumulated at the cell membrane, with limited internalization and partial mitochondrial association. In contrast, HepG2 cells extensively internalized IR820/EVO@MPDA-TPP/cRGD (EVO-3) nanoparticles, which exhibited prominent perimitochondrial distribution (Figure 2C). Quantitative analysis demonstrated that the colocalization of the IR820 signal with MitoTracker Green was significantly higher in the EVO-3 group than in the EVO-1 and EVO-2 groups (Figure S8), confirming the enhanced mitochondrial targeting efficiency conferred by the dual-ligand (cRGD/TPP) modification. Consistent with imaging observations, HPLC quantification confirmed significantly higher cellular uptake of EVO-2 and EVO-3 compared to free EVO. After 24 hours of incubation, EVO-3 uptake was 1.47-fold higher than that of EVO-1, highlighting the enhanced delivery efficacy (Figure 2D). These results validate that dual modification with cRGD and TPP facilitates efficient cellular internalization and mitochondrial delivery of both EVO and IR820.
Evaluation of Reactive Oxygen Species (ROS) and Mitochondrial Function
To assess oxidative stress induced by the photothermal–chemotherapy combination in HepG2 cells, intracellular ROS levels were measured using the fluorescent probe DCFH-DA (Figure 3A). Experimental results indicated that both free EVO and IR820/EVO@MPDA (EVO-1) induced only mild increases in ROS generation. In contrast, cRGD-modified nanoparticles (IR820/EVO@MPDA-cRGD, EVO-2) significantly enhanced ROS levels, likely due to improved cellular uptake via cRGD targeting. ROS generation was further elevated in the EVO-2 group following laser irradiation, consistent with IR820’s ability to induce ROS upon photothermal activation. Strikingly, IR820/EVO@MPDA-TPP/cRGD (EVO-3) elicited a substantial increase in ROS even in the absence of laser treatment. The highest ROS levels were achieved in the EVO-3 + L group, indicating a synergistic effect between TPP-mediated mitochondrial targeting and IR820-derived photothermal stimulation in aggravating oxidative stress under thermal conditions.
 |
Figure 3 In vitro anti-tumor activity assessment. (A) ROS levels in HepG2 cells after 24 h exposure to different nanoparticle formulations. Data represent mean ± SD (n = 3). (B) Mitochondrial membrane potential changes in HepG2 cells following 24 h treatment with EVO, EVO-1, EVO-2, EVO-2+L, EVO-3, and EVO-3+L. Data represent mean ± SD (n = 3). (C) ATP levels in HepG2 cells after 24 h exposure to different nanoparticle formulations. Data represent mean ± SD (n = 3). (D) Changes in mitochondrial morphology in HepG2 cells following 24 h treatment with different nanoparticles, visualized by confocal microscopy. Scale bar: 5 μm. Representative images are derived from at least three independent experiments. (E) Morphological changes in mitochondria of HepG2 cells after 24 h treatment with different nanoparticles based on biological TEM. Scale bars: 5 μm (overview), 500 nm (zoomed-in). Representative images are derived from at least three independent experiments. Red dashed box indicates the magnified region. EVO-1, IR820/EVO@MPDA; EVO-2, IR820/EVO@MPDA-cRGD; EVO-2+L, IR820/EVO@MPDA-cRGD+Laser; EVO-3, IR820/EVO@MPDA-TPP/cRGD; EVO-3+L, IR820/EVO@MPDA-TPP/cRGD+Laser. *p < 0.05, **p < 0.01, ***p < 0.001 vs Con group, ns indicates no significant difference. |
Mitochondria serve not only as “power plants” or energy suppliers for physiological processes but also as key regulators of apoptosis in response to cellular stress. Both PTT-induced hyperthermia and ROS accumulation can compromise mitochondrial function and trigger apoptotic signaling pathways.34 To evaluate mitochondrial integrity, changes in membrane potential (ΔΨm) were monitored using the JC-1 fluorescent probe (Figure 3B). A decrease in ΔΨm was observed in all nanoparticle-treated groups, with EVO-1 and EVO-2 showing comparable effects. EVO-3, however, induced an approximately 9-fold greater reduction in ΔΨm compared to EVO-2. This indicates that dual-ligand functionalization (cRGD and TPP) markedly enhances mitochondrial targeting and damage efficiency. In particular, free EVO also caused a pronounced decrease in ΔΨm, suggesting its intrinsic ability to disrupt mitochondrial function through chemical mechanisms at the tested dose. Laser irradiation further intensified the loss of ΔΨm, especially in EVO-2 + L and EVO-3 + L groups. Notably, EVO-3 + L resulted in near-complete membrane potential collapse, with a reduction of 92.5%. These findings demonstrate a synergistic effect between PTT and EVO-induced chemical toxicity. The disruption of ΔΨm directly impairs ATP synthesis, and consistent with this mechanism, ATP measurements (Figure 3C) confirmed reduced levels across all treatment groups. The most substantial ATP depletion was detected in the EVO-3 + L group, suggesting effective mitochondrial pathway activation.
To determine whether photothermal and chemical synergy also alter mitochondrial morphology, we performed confocal imaging of stained HepG2 cells with a green fluorescent mitochondrial dye. As shown in Figure 3D, untreated cells displayed healthy, elongated mitochondria with continuous network structures. Free EVO treatment led to slight mitochondrial swelling, indicating mild stress. More pronounced morphological changes were observed in the EVO-1, EVO-2 + L, and EVO-3 + L groups, with mitochondria transitioning from elongated tubules to elliptical or spherical forms. The most severe abnormalities were evident in the EVO-3 + L group, characterized by extensive swelling and fragmentation. TEM imaging (Figure 3E) further revealed intact mitochondrial ultrastructure in control cells, with intact double membranes and well-defined cristae without vacuolation. Free EVO disrupted cristae organization, while increasing treatment intensity caused progressive structural damage. From EVO-1 to EVO-2 + L and EVO-3 + L, mitochondria became shorter and rounder, with cristae fragmentation and eventual disintegration. The EVO-3 + L group displayed the most extensive damage, including complete loss of cristae architecture. Altogether, these findings confirm that combined photothermal and chemical treatment, particularly EVO-3 with laser irradiation, efficiently causes severe mitochondrial dysfunction, in agreement with functional assays.
Biodistributionand Fluorescence Tracing of IR820/EVO@MPDA-TPP/cRGD in vivo
To evaluate in vivo behavior before therapy, the biodistribution of the nanoparticle formulations was studied in an HCDX mouse model. Formulations including IR820, IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), and IR820/EVO@MPDA-TPP/cRGD (EVO-3) were administered via tail vein injection, and fluorescence was tracked using a small animal in vivo imaging system. The intrinsic fluorescence of IR820 enabled real-time monitoring of nanoparticle accumulation and tumor-targeting efficiency. As illustrated in Figure 4A, fluorescence intensity at the tumor site peaked at 24 hours post-injection and gradually declined thereafter. Throughout the observation period, the fluorescence intensities at the tumor site in the EVO-1, EVO-2, and EVO-3 groups were consistently higher than those in the free IR820 group, indicating that the MPDA carrier modification prolonged the systemic circulation and retention of the drug. The EVO-3 group exhibited the strongest fluorescence signal, indicating that the dual-ligand modification further promoted tumor-specific accumulation. At 120 hours post-injection, mice were euthanized, and major organs, along with tumor tissues, were excised for ex vivo fluorescence imaging (Figure 4B). The results revealed predominant drug accumulation in tumor tissues, with minor distribution in the liver and kidneys, likely due to nanoparticle metabolic and clearance pathways. Furthermore, all nanoparticle formulations exhibited significantly higher fluorescence intensities than the free IR820 group, with EVO-3 showing the strongest signal, confirming its enhanced tumor-targeting and accumulation capacity. Collectively, these findings demonstrate that EVO-3 achieves efficient tumor-specific enrichment, supporting its potential as a targeted theranostic platform for hepatocellular carcinoma.
 |
Figure 4 In vivo anti-hepatocellular carcinoma efficacy of different nanoparticle formulations. (A) In vivo fluorescence images at various time points after intravenous injection of IR820, EVO-1, EVO-2, and EVO-3. (B) Ex vivo fluorescence images of major organs and tumors harvested 120 h post-injection. (C) Schematic illustration of the treatment protocol in the PDX model. (D) Representative photographs of excised tumors from each treatment group. (E) Tumor growth curves throughout the treatment period. Data are presented as mean ± SD (n = 6). (F) Tumor weights measured at the endpoint of the study. Data are presented as mean ± SD (n = 6). (G) Body weight changes of mice during the treatment course. Data are presented as mean ± SD (n = 6). EVO-1, IR820/EVO@MPDA; EVO-1+L, IR820/EVO@MPDA+Laser; EVO-2, IR820/EVO@MPDA-cRGD; EVO-2+L, IR820/EVO@MPDA-cRGD+Laser; EVO-3, IR820/EVO@MPDA-TPP/cRGD; EVO-3+L, IR820/EVO@MPDA-TPP/cRGD+Laser. *p < 0.05, **p < 0.01, ***p < 0.001 vs Con group, ns indicates no significant difference. |
Evaluation of Anti-HCC Efficacy in vivo
Building upon the HCDX model, this study further evaluated the in vivo anti-HCC efficacy of IR820/EVO@MPDA-TPP/cRGD (EVO-3) nanoparticles (Figure 4C). As shown in Figure 4D and E, tumors in both the Con (normal saline) and MPDA-only groups grew rapidly, indicating negligible intrinsic antitumor activity of unloaded MPDA nanoparticles. Free EVO exhibited moderate antitumor effects, with a tumor growth inhibition rate of 40.07% (Table S3). In comparison, groups treated with IR820/EVO@MPDA (EVO-1), IR820/EVO@MPDA-cRGD (EVO-2), and IR820/EVO@MPDA-TPP/cRGD (EVO-3) all showed significantly enhanced antitumor efficacy relative to free EVO. These results suggest that both the MPDA carrier and surface functionalization strategies effectively promote cellular uptake and therapeutic performance. Moreover, upon laser irradiation, the EVO-1, EVO-2, and EVO-3 groups exhibited further suppression of tumor growth. Notably, the EVO-3 + L group demonstrated the most potent antitumor effect, achieving a tumor suppression rate (TSR) of 85.23% (Table S3). The average tumor weight in this group was also significantly lower than that in the free EVO and non-irradiated EVO-3 groups (Figure 4F), highlighting the strong synergistic effect between photothermal therapy (PTT) and chemotherapy.
Regarding biosafety, although minor fluctuations in body weight were observed across all groups during the treatment period, no statistically significant differences were detected (Figure 4G). This finding indicates the absence of observable systemic toxicity at the administered dose. Further evaluation of hepatic and renal function using serum biochemistry showed no significant elevation in key biomarkers, including ALT, AST, BUN, and Cr, across any of the nanoparticle-treated groups (Figure 5A–D). These observations suggest that the nanomaterials did not induce acute liver or kidney injury. Importantly, mice treated with free EVO exhibited a marked increase in ALT levels, implying potential drug-induced hepatotoxicity at the equivalent dose. In contrast, all MPDA-based nanoformulations, especially those with ligand modifications, effectively reduced this adverse effect and demonstrated improved biocompatibility and safety.
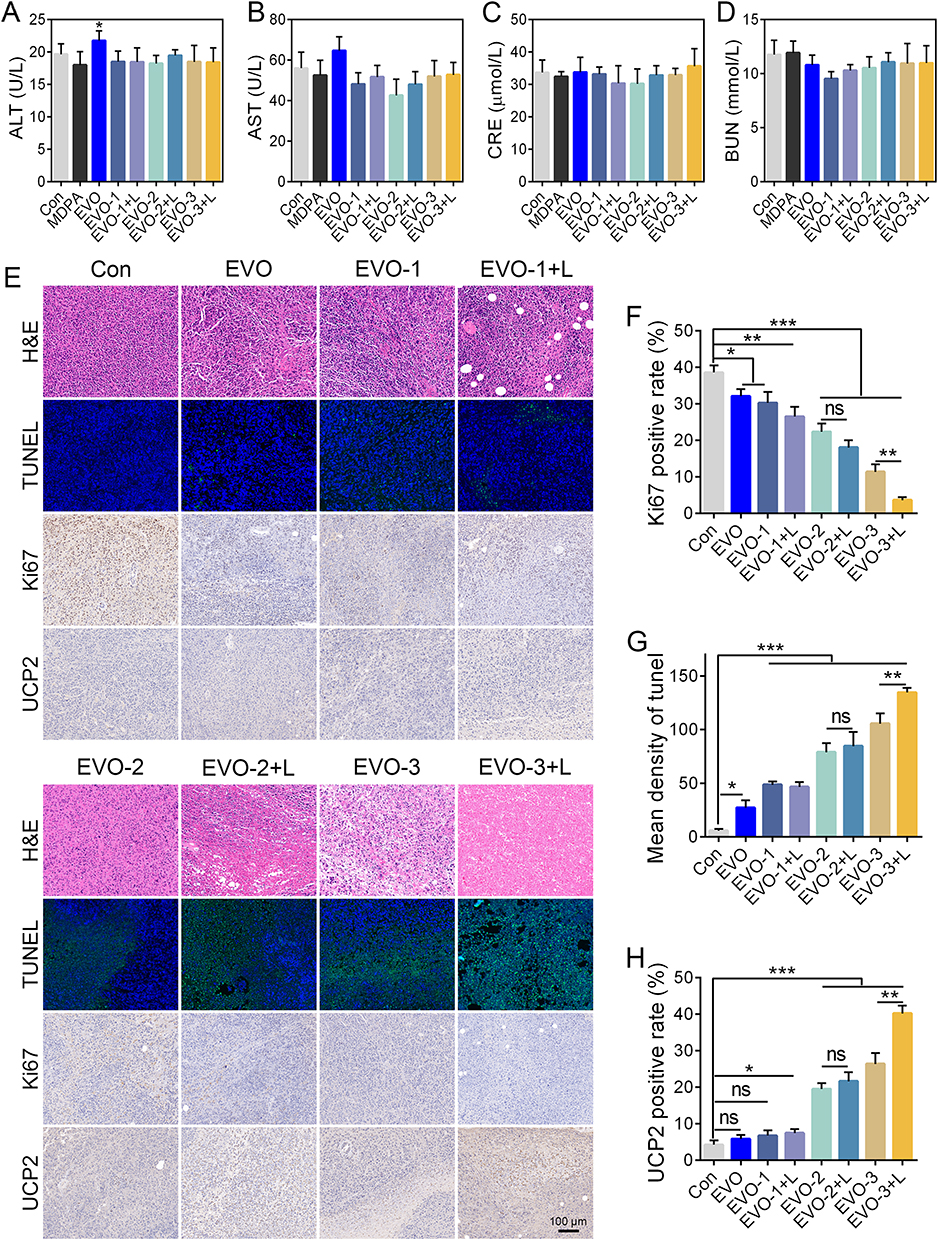 |
Figure 5 Biosafety evaluation and immunohistochemical analysis. Serum levels of alanine aminotransferase (ALT, A), aspartate aminotransferase (AST, B), creatinine (Cr, C), and urea nitrogen (BUN, (D) in mice after treatment. (E) Representative images of H&E, TUNEL, Ki67, and UCP2 staining in tumor tissues following various treatments. Quantitative analysis of Ki67-positive proliferation index (F), TUNEL-positive apoptosis rate (G), and UCP2 expression intensity (H). Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs Con group, ns indicates no significant difference. EVO-1, IR820/EVO@MPDA; EVO-1+L, IR820/EVO@MPDA+Laser; EVO-2, IR820/EVO@MPDA-cRGD; EVO-2+L, IR820/EVO@MPDA-cRGD+Laser; EVO-3, IR820/EVO@MPDA-TPP/cRGD; EVO-3+L, IR820/EVO@MPDA-TPP/cRGD+Laser. |
Histopathological analysis was performed to assess tumor tissue damage in the different treatment groups (Figure 5E). H&E staining revealed drug-induced cytopathological features such as cytoplasmic leakage, nuclear condensation, chromatin aggregation, and membrane rupture. The severity of tissue injury followed the order: EVO < EVO-1 < EVO-1+L < EVO-2 < EVO-2+L < EVO-3 < EVO-3+L. The most extensive tissue damage was observed in the EVO-3+L group. Immunohistochemical analysis of the proliferation marker Ki67 showed an inverse correlation between its expression and antitumor efficacy. All drug-treated groups (EVO, EVO-1, EVO-1+L, EVO-2, EVO-2+L, EVO-3, and EVO-3+L) exhibited a significant reduction in Ki67-positive cells compared to the Con group (Figure 5E and F). The degree of reduction increased progressively, further confirming the effectiveness of the treatments. TUNEL staining (Figure 5E and G) revealed a moderate increase in apoptotic signals (green fluorescence) in the EVO, EVO-1, and EVO-1+L groups relative to the Con group. In comparison, the EVO-2, EVO-2+L, EVO-3, and EVO-3+L groups displayed broader and more intense fluorescent signals. Notably, the EVO-3+L group exhibited a significantly higher fluorescence intensity than its non-irradiated counterpart (EVO-3), highlighting the enhanced efficacy of combined photothermal-chemotherapy in triggering tumor cell apoptosis. Furthermore, immunohistochemistry staining showed markedly elevated expression of UCP2, a marker of mitochondrial dysfunction, in the EVO-2 and EVO-3 groups (Figure 5E and H). Laser irradiation further increased UCP2 expression in the EVO-3 + L group. These findings suggest that TPP-mediated mitochondrial targeting, together with EVO-induced chemical effects, results in profound mitochondrial disruption, thereby improving the overall antitumor response.
Evaluation of Mitochondrial Damage in vivo
Western blot analysis was conducted to assess the expression levels of mitochondrial damage-related proteins in tumor tissues. The results revealed that treatments with IR820/EVO@MPDA-cRGD (EVO-2), EVO-2 + Laser (EVO-2 + L), IR820/EVO@MPDA-TPP/cRGD (EVO-3), and EVO-3 + Laser (EVO-3 + L) significantly reduced ATP5A1 expression, a key protein in energy metabolism, while markedly upregulating UCP2 expression. The most prominent changes were observed in the EVO-3 + L group (Figure 6A and B), indicating that mitochondrial-targeted nanoparticles combined with laser irradiation effectively impair mitochondrial energy metabolism in tumor cells. At the same time, the expression of the antioxidant enzyme GPX4 showed a decreasing trend across EVO-2, EVO-2 + L, EVO-3, and EVO-3 + L groups, with all exhibiting significant differences from the Con group (Figure 6C and D). This suggests that EVO-3 combined with laser exposure leads to elevated oxidative stress. Persistently high oxidative stress levels likely contribute to mitochondrial impairment, as supported by the depletion of PGC-1α protein. Experimental findings confirmed this hypothesis, revealing a decline in PGC-1α expression in all treated groups, with the lowest level in EVO-3 + L (Figure 6A and B). Furthermore, expression of autophagy-related proteins, LC3-II and PINK1, increased to varying extents, with the EVO-3 + L group showing the highest elevation (Figure 6C and D), indicating strong autophagic pathway activation. In terms of apoptosis, upregulation of pro-apoptotic proteins (Bax, Cytochrome C, and Cleaved Caspase-3) and downregulation of the anti-apoptotic protein Bcl-2 were observed, with the EVO-3 + L group showing the most pronounced effects (Figure 6E and F). These results confirm that this treatment strategy activates the mitochondrial apoptosis pathway. In conclusion, EVO-3 + L combination therapy disrupts redox homeostasis, impairs mitochondrial function, and induces both autophagy and apoptosis in hepatocellular carcinoma cells.
 |
Figure 6 Mitochondrial damage-mediated activation of apoptotic and autophagic pathways in tumor cells. (A) Western blot analysis of mitochondrial damage-associated proteins (ATP5A1, UCP2, PGC-1α) in tumor tissues and (B) quantitative densitometry analysis of protein levels. (C) Western blot analysis of oxidative stress marker GPX4 and autophagy-related proteins (LC3-II, PINK1) and (D) quantitative densitometry analysis of protein levels. (E) Western blot analysis of apoptosis-related proteins (Bax, Bcl-2, Cytochrome C, Cleaved Caspase-3) and (F) quantitative densitometry analysis of protein levels. Data are presented as mean ± SD (n = 3). −1, IR820/EVO@MPDA; EVO-1+L, IR820/EVO@MPDA+Laser; EVO-2, IR820/EVO@MPDA-cRGD; EVO-2+L, IR820/EVO@MPDA-cRGD+Laser; EVO-3, IR820/EVO@MPDA-TPP/cRGD; EVO-3+L, IR820/EVO@MPDA-TPP/cRGD+Laser, *p < 0.05, **p < 0.01, ***p < 0.001 vs Con group and ns indicates no significant difference. |
Discussion
Hepatocellular carcinoma (HCC) is recognized as a highly heterogeneous and aggressive cancer with complex pathogenesis and poor prognosis, presenting major clinical challenges.35 The disease is often asymptomatic in early stages, resulting in diagnosis at intermediate or advanced phases when curative interventions like surgical resection or transplantation are no longer viable.2,36 Although targeted therapies and immune checkpoint inhibitors have provided new hope for advanced HCC patients, their effectiveness is hindered by low response rates and the emergence of drug resistance. Moreover, the pronounced heterogeneity of liver cancer and its immunosuppressive tumor microenvironment limit the efficacy of monotherapies.37 As a result, combination therapy strategies based on synergistic mechanisms have become a central focus in liver cancer research. Recently, combinatorial regimens such as targeted therapy with immunotherapy38 or chemotherapy39 integrated with photothermal therapy40 have shown advantages over conventional approaches in clinical settings, offering promising avenues for improving patient outcomes. In this study, we explored a multimodal treatment strategy employing MPDA-based nanoparticles for co-delivery of chemotherapeutic and photothermal agents, achieving enhanced antitumor efficacy against HCC through synergistic chemo-photothermal mechanisms.
The IR820/EVO@MPDA-TPP/cRGD (EVO-3) platform is designed around MPDA nanoparticles, co-loaded with IR820 and the pro-apoptotic natural compound EVO, and functionalized with mitochondrial-targeting TPP41 and tumor-homing cRGD peptides.42 After administration, cRGD facilitates active targeting and tumor accumulation, while TPP ensures mitochondrial and site-specific damage. Confocal imaging confirmed minimal cellular uptake of untargeted IR820/EVO@MPDA (EVO-1), while EVO-3 exhibited extensive internalization and perinuclear mitochondrial accumulation. Quantitative HPLC further validated a 1.47-fold increase in cellular uptake of EVO-3 over EVO-1. In line with in vitro data, in vivo fluorescence imaging showed greater tumor localization of EVO-3 following intravenous injection. EVO-3 treatment also caused more severe mitochondrial impairment than EVO-1, demonstrated by elevated ROS, mitochondrial membrane potential collapse, ATP suppression, and UCP2 overexpression. Elevated UCP2 expression contributes to mitochondrial depolarization, reduces calcium uptake, and worsens dysfunction.43
Upon laser exposure, IR820 absorbs photons, triggering an electron transition to an excited state. Subsequent non-radiative relaxation transfers energy into heat via lattice vibrations, causing localized hyperthermia near the nanocrystals.44 This heat facilitates both tumor ablation and enhanced EVO release and tissue penetration of EVO. These effects improve drug delivery by modifying the tumor microenvironment and disrupting release barriers.45,46 The accelerated EVO release and improved tissue permeability contribute to heightened chemo-efficacy and promote comprehensive tumor eradication. In addition to its role as a highly efficient photothermal converter, IR820 serves as a fluorescent probe for real-time tracking of nanoparticle distribution, enhancing treatment precision. Both in vitro and in vivo experiments showed reduced proliferation and slower tumor growth after photothermal treatment across nanoparticle groups. Notably, EVO-3 + L outperformed IR820/EVO@MPDA + Laser (EVO-1 + L) in antitumor activity, attributed to effective mitochondrial targeting by TPP and tumor homing by cRGD peptides. Without such active targeting, IR820-mediated PTT may suffer from off-target effects, potentially causing severe damage to surrounding healthy tissues.47 Importantly, the IR820-enabled photothermal platform inevitably upregulates the expression of heat shock proteins 70 and 90 (HSP70/90) under thermal stress conditions,48 thereby conferring a thermotolerant phenotype upon hepatocellular carcinoma cells that poses a formidable impediment to effective photothermal ablation. EVO counteracts this adaptive response by suppressing HSP expression while concomitantly promoting mitochondrial-mediated apoptosis,14 thus establishing a synergistic therapeutic modality that is mechanistically distinct from conventional DNA-damaging chemotherapeutics. This mechanistic complementarity provides a cogent explanation for the superior anti-hepatocellular carcinoma efficacy observed in the EVO-3 plus laser irradiation cohort.
Besides direct thermal damage, PTT not only mediates direct tumor ablation via thermal effects but also effectively generates substantial oxidative stress within tumor cells. Sustained oxidative stress further promotes mitochondrial lipid peroxidation and leads to damage to mtDNA, thereby worsening the collapse of mitochondrial membrane potential (ΔΨm) and intensifying mitochondrial dysfunction. These effects were particularly evident in the EVO-3 + L group, which showed elevated ROS production, upregulation of GPX4 expression, significant loss of ΔΨm, and depletion of PGC-1α protein levels. Damaged mitochondria activate the caspase cascade49 by releasing pro-apoptotic factors such as cytochrome c, initiating programmed cell apoptosis. Simultaneously, the severe mitochondrial injury triggered by the combined chemo-photothermal treatment led to a marked activation of the PINK1/Parkin-mediated mitophagy pathway,50,51 as evidenced by the substantial upregulation of LC3-II and PINK1. Under physiological or mild stress conditions, mitophagy functions as a protective quality-control mechanism to remove damaged mitochondria and restore cellular homeostasis. However, in the context of the intense oxidative stress and irreversible mitochondrial dysfunction induced by EVO-3 + Laser, this adaptive response was overwhelmed. The hyperactivated mitophagy, together with the release of cytochrome c and activation of caspase cascades, ultimately contributed to programmed cell death rather than cytoprotection. This shift from a pro-survival to a pro-death role is consistent with the robust tumor growth inhibition observed both in vitro and in vivo, indicating that in this therapeutic setting, autophagy cooperates with apoptosis rather than opposing it. Notably, the profound oxidative stress and extensive mitochondrial damage induced by the combined chemo-photothermal treatment may foster immunogenic cell death (ICD). ICD is characterized by the spatiotemporally coordinated emission of damage-associated molecular patterns, including surface-exposed calreticulin, extracellular release of high-mobility group box 1 (HMGB1), and ATP secretion, which collectively promote dendritic cell maturation and the priming of T-cell-mediated antitumor immunity.52,53 In our study, IR820/EVO@MPDA-TPP/cRGD plus laser treatment triggered robust ROS production, near-complete collapse of the mitochondrial membrane potential, and cytochrome c release, all hallmarks closely associated with ICD. Furthermore, observed ATP depletion and UCP2 upregulation suggest metabolic alterations that could favor immunostimulatory ATP release. Although dedicated immunological endpoints were not assessed, these findings raise the possibility that our mitochondria-targeted chemo-photothermal strategy not only directly kills tumor cells but may also convert dying tumor cells into an in-situ vaccine, thereby priming systemic antitumor immunity against residual or metastatic lesions. Future studies incorporating immune cell profiling, cytokine analysis, and direct detection of ICD hallmarks (eg., surface calreticulin, HMGB1 secretion) are warranted to validate this immunogenic potential and to explore IR820/EVO@MPDA-TPP/cRGD as a component of combined photothermal-immunotherapy. In conclusion, the combined approach of PTT and EVO-induced chemotherapy offers a promising strategy for effective elimination of HCC and provides a novel avenue for improving clinical outcomes in HCC patients.
Despite the promising results obtained in this study, several limitations should be acknowledged. Absence of photothermal data for free IR820 alone, which would be essential to quantitatively confirm the synergistic effect. Control groups treated with blank MPDA-TPP or MPDA-cRGD nanocarriers (without EVO/IR820) were not included; such controls would allow the contributions of each ligand to tumor accumulation and mitochondrial targeting to be dissected independently of the therapeutic payload. In vivo antitumor evaluation was conducted solely in a subcutaneous HepG2 xenograft model, which does not fully recapitulate the complex tumor microenvironment, stromal interactions, and immune landscape of clinically relevant orthotopic or metastatic HCC. Consequently, the therapeutic efficacy and biodistribution profile of IR820/EVO@MPDA-TPP/cRGD in more physiologically representative models remain to be established. Furthermore, while the nanosystem demonstrated favorable short-term biosafety based on serum biochemistry and histological analysis, long-term toxicity assessments, including chronic organ damage, immunogenicity, and reproductive toxicity, were not performed within the scope of this study. Comprehensive long-term safety profiling is warranted before clinical translation can be considered. Future studies will focus on incorporating these blank nanocarrier controls to clearly delineate the roles of cRGD and TPP, validating the antitumor efficacy and targeting specificity of this nanosystem in orthotopic and patient-derived xenograft (PDX) models of HCC, as well as conducting systematic long-term toxicological evaluations to rigorously assess its translational potential. These efforts will be critical to advancing IR820/EVO@MPDA-TPP/cRGD from preclinical proof-of-concept toward potential clinical application.
Conclusion
In summary, this study successfully developed a mitochondria-targeted co-delivery system named IR820/EVO@MPDA-TPP/cRGD, based on MPDA nanoparticles, for synergistic chemo-photothermal treatment of HCC. The nanoplatform exhibits favorable biocompatibility, efficient tumor-homing capability through cRGD targeting, and accurate mitochondrial localization enabled by TPP modification. Utilizing the dual function of IR820 as both a photothermal agent and a fluorescence tracer, the system supports real-time tracking and effective photothermal conversion under near-infrared light. Experimental findings revealed markedly enhanced cellular uptake and tumor-specific accumulation of the nanoparticles, resulting in strong inhibition of tumor progression in vitro and in vivo models. Importantly, the dual impact of EVO-induced mitochondrial disruption and IR820-triggered hyperthermia produced a synergistic increase in organelle damage, initiating pronounced apoptosis and autophagy and promoting tumor cell death. These results demonstrate that IR820/EVO@MPDA-TPP/cRGD represents a potential theranostic candidate for image-guided HCC therapy; however, its translational prospect must be further validated. Future studies should incorporate orthotopic HCC models, immune-competent animal studies, comprehensive pharmacokinetic analyses, and long-term toxicity and survival assessments to thoroughly evaluate its clinical applicability.
Abbreviations
ALT, alanine aminotransferase; ANOVA, one-way analysis of variance; AST, aspartate aminotransferase; ATP, adenosine triphosphate; BET, Brunauer–Emmett–Teller; BJH, Barrett–Joyner–Halenda; BUN, blood urea nitrogen; Cr, creatinine; cRGD, cyclic arginine-glycine-aspartic acid; DEE, drug encapsulation efficiency; DLC, drug loading capacity; DLS, dynamic light scattering; EVO, evodiamine; F127, Poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) diacrylate; FBS, fetal bovine serum; FT-IR, Fourier transform infrared spectroscopy; HCC, hepatocellular carcinoma; HCDX, HepG2 cell-derived xenograft; H&E, hematoxylin and eosin; HMGB1, high-mobility group box 1; HPLC, high-performance liquid chromatography; HRP, horseradish peroxidase; ICD, immunogenic cell death; MPDA, mesoporous polydopamine; NIR, near-infrared; PVDF, polyvinylidene difluoride; ROS, reactive oxygen species; PDT, photodynamic therapy; PTT, photothermal therapy; SD, standard deviation; SPF, specific pathogen-free; TEM, transmission electron microscopy; TGA, thermogravimetric analysis; TMB, 3,3’,5,5’-Tetramethylbenzidine; TPP, triphenylphosphonium; TSR, tumor suppression rate; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Author Contributions
Shasha Kong: Conceptualization, Data curation, Validation, Writing-original draft. Yuling Liu: Conceptualization, Supervision. Ruying Tang: Visualization, Supervision. Hui Li: Conceptualization, Supervision. Hongmei Lin: Conceptualization, Formal analysis, Writing-review & editing. Longfei Lin: Conceptualization, Project administration, Writing-review & editing. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Beijing Nova Program (20230484300), Fundamental Research Funds for the Central Public Welfare Research Institutes (ZZ13-YQ-059), and China Postdoctoral Science Foundation (2021M692732).
Disclosure
The authors declare no conflict of interest.
References
1. Roayaie S, Obeidat K, Sposito C, et al. Resection of hepatocellular cancer ≤2 cm: results from two Western centers. Hepatology. 2013;57(4):1426–24. doi:10.1002/hep.25832
2. Liver. EAftSot. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182–236. doi:10.1016/j.jhep.2018.03.019
3. Liu Y, Bhattarai P, Dai Z, Chen X. Photothermal therapy and photoacoustic imaging via nanotheranostics in fighting cancer. Chem Soc Rev. 2019;48:2053–2108. doi:10.1039/c8cs00618k
4. Haddad S, Abánades Lázaro I, Fantham M, et al. Design of a functionalized metal-organic framework system for enhanced targeted delivery to mitochondria. J Am Chem Soc. 2020;142:6661–6674. doi:10.1021/jacs.0c00188
5. Cao Z, Liu X, Zhang W, et al. Biomimetic macrophage membrane-camouflaged nanoparticles induce ferroptosis by promoting mitochondrial damage in glioblastoma. ACS Nano. 2023;17:23746–23760. doi:10.1021/acsnano.3c07555
6. Li M, Guo R, Wei J, et al. Polydopamine-based nanoplatform for photothermal ablation with long-term immune activation against melanoma and its recurrence. Acta Biomater. 2021;136:546–557. doi:10.1016/j.actbio.2021.09.014
7. Zmerli I, Michel J-P, Makky A. Multifunctional polydopamine-based nanoparticles: synthesis, physico-chemical properties and applications for bimodal photothermal/photodynamic therapy of cancer. Multifunct Mater. 2021;4:022001. doi:10.1088/2399-7532/abf0fa
8. Wang L, He Y, He T, et al. Lymph node-targeted immune-activation mediated by imiquimod-loaded mesoporous polydopamine based-nanocarriers. Biomaterials. 2020;255:120208. doi:10.1016/j.biomaterials.2020.120208
9. Zhang P, Li X, Xu Q, Wang Y, Ji J. Polydopamine nanoparticles with different sizes for NIR-promoted gene delivery and synergistic photothermal therapy. Colloids Surf B. 2021;208:112125. doi:10.1016/j.colsurfb.2021.112125
10. Guo -X-X, Li X-P, Zhou P, et al. Evodiamine induces apoptosis in SMMC-7721 and HepG2 cells by suppressing NOD1 signal pathway. Int J Mol Sci. 2018;19(11):3419. doi:10.3390/ijms19113419
11. Zhang T, Qu S, Shi Q, He D, Jin X. Evodiamine induces apoptosis and enhances TRAIL-induced apoptosis in human bladder cancer cells through mTOR/S6K1-mediated downregulation of Mcl-1. Int J Mol Sci. 2014;15(2):3154–3171. doi:10.3390/ijms15023154
12. Peng X, Zhang Q, Zeng Y, Li J, Wang L, Ai P. Evodiamine inhibits the migration and invasion of nasopharyngeal carcinoma cells in vitro via repressing MMP-2 expression. Cancer Chemother Pharmacol. 2015;76:1173–1184. doi:10.1007/s00280-015-2902-9
13. Khan M, Bi Y, Qazi JI, Fan L, Gao H. Evodiamine sensitizes U87 glioblastoma cells to TRAIL via the death receptor pathway. Molecular Med Report. 2015;11:257–262. doi:10.3892/mmr.2014.2705
14. Hyun SY, Le HT, Min HY, et al. Evodiamine inhibits both stem cell and non-stem-cell populations in human cancer cells by targeting heat shock protein 70. Theranostics. 2021;11:2932–2952. doi:10.7150/thno.49876
15. De Lorenzi F, Rizzo LY, Daware R, et al. Profiling target engagement and cellular uptake of cRGD-decorated clinical-stage core-crosslinked polymeric micelles. Drug Delivery Transl Res. 2023;13:1195–1211. doi:10.1007/s13346-022-01204-8
16. Zhang S, Xu H, Xiang F, et al. A multifunctional injectable MPDA@MTIC-Co hydrogel platform for synergistic chemotherapy-photothermal therapy of postoperative glioblastoma. Bioact Mater. 2026;60:387–400. doi:10.1016/j.bioactmat.2026.01.034
17. Wang H, Wu Y, Zou H, et al. Photothermal therapy with Ag nanoparticles in mesoporous polydopamine for enhanced antibacterial activity. ACS Appl Nano Mater. 2023;6:4834–4843. doi:10.1021/acsanm.3c00509
18. Hou C, Ma N, Shen Z, et al. A GSH-Responsive Nanoprodrug System Based on Self-Assembly of Lactose Modified Camptothecin for Targeted Drug Delivery and Combination Chemotherapy. Int J Nanomed. 2020;15:10417–10424. doi:10.2147/ijn.S276470
19. Yang K, Yang K, Chao S, Wen J, Pei Y, Pei Z. A supramolecular hybrid material constructed from pillar[6]arene-based host-guest complexation and ZIF-8 for targeted drug delivery. Chem Commun. 2018;54:9817–9820. doi:10.1039/c8cc05665j
20. Wang Y, Jin M, Chen Z, et al. Tumor microenvironment responsive supramolecular glyco-nanovesicles based on diselenium-bridged pillar[5]arene dimer for targeted chemotherapy. Chem Commun. 2020;56:10642–10645. doi:10.1039/d0cc04149a
21. Basety S, Gudepu R, Velidandi A. Smart nanoformulations for oncology: a review on overcoming biological barriers with active targeting, stimuli-responsive, and controlled release for effective drug delivery. Pharmaceutics. 2026;18:196. doi:10.3390/pharmaceutics18020196
22. Zhao J, Dai D, Zhou L, et al. A tumor microenvironment responsive mesoporous polydopamine theranostic probe embedded with Gd/I-doped carbon nanodots for CT/MR/FL imaging and chemo/photothermal synergistic therapy. Carbon. 2024;224:119065. doi:10.1016/j.carbon.2024.119065
23. Wang K, Zhu K, Yuan Z, et al. Cardiomyocyte mitochondria targeted biomimetic nanozyme system with calcium ion chelating agent to treat myocardial ischemia–reperfusion injury model in mice. Chem Eng J. 2025;517:164177. doi:10.1016/j.cej.2025.164177
24. Wang T, Song Y, Zhang W, et al. Stable Gold Nanorods Conjugated Liposomal Podophyllotoxin Nanocomposites for Synergistic Chemo-Photothermal Cancer Therapy. J Biomed Nanotechnol. 2017;13:1435–1445. doi:10.1166/jbn.2017.2439
25. Chen T, Zhuang B, Huang Y, et al. Inhaled curcumin mesoporous polydopamine nanoparticles against radiation pneumonitis. Acta pharmaceutica Sinica B. 2022;12:2522–2532. doi:10.1016/j.apsb.2021.10.027
26. Peng L, Hung CT, Wang S, et al. Versatile Nanoemulsion Assembly Approach to Synthesize Functional Mesoporous Carbon Nanospheres with Tunable Pore Sizes and Architectures. J Am Chem Soc. 2019;141:7073–7080. doi:10.1021/jacs.9b02091
27. Chen F, Xing Y, Wang Z, Zheng X, Zhang J, Cai K. Nanoscale Polydopamine (PDA) Meets π-π Interactions: an Interface-Directed Coassembly Approach for Mesoporous Nanoparticles. Langmuir. 2016;32:12119–12128. doi:10.1021/acs.langmuir.6b03294
28. Wu M, Zhong C, Zhang Q, et al. pH-responsive delivery vehicle based on RGD-modified polydopamine-paclitaxel-loaded poly (3-hydroxybutyrate-co-3-hydroxyvalerate) nanoparticles for targeted therapy in hepatocellular carcinoma. J Nanobiotechnol. 2021;19:39. doi:10.1186/s12951-021-00783-x
29. Wang J, Lin C, Li X, et al. Polydopamine nanoparticles loaded with sodium ferulate for targeted therapy of myocardial infarction in endothelial cells. Int J Pharm. 2025;671:125187. doi:10.1016/j.ijpharm.2025.125187
30. Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-Inspired surface chemistry for multifunctional coatings. Science. 2007;318:426–430. doi:10.1126/science.1147241
31. Solanki R, Rajput PK, Jodha B, Yadav UCS, Patel S. Enhancing apoptosis-mediated anticancer activity of evodiamine through protein-based nanoparticles in breast cancer cells. Sci Rep. 2024;14:2595. doi:10.1038/s41598-024-51970-3
32. Feng X, Cao Y, Zhuang P, et al. Rational synthesis of IR820-albumin complex for NIR-II fluorescence imaging-guided surgical treatment of tumors and gastrointestinal obstruction. RSC Adv. 2022;12:12136–12144. doi:10.1039/d2ra00449f
33. de Carvalho FG, Magalhães TC, Teixeira NM, et al. Synthesis and characterization of TPP/chitosan nanoparticles: colloidal mechanism of reaction and antifungal effect on C. albicans biofilm formation. Mater Sci Eng C. 2019;104:109885. doi:10.1016/j.msec.2019.109885
34. Chen D, Zhang J, Tang Y, et al. A tumor-mitochondria dual targeted aza-BODIPY-based nanotheranostic agent for multimodal imaging-guided phototherapy. J Mat Chem B. 2018;6:4522–4530. doi:10.1039/C8TB01347K
35. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nature Reviews Disease Primers. 2021;7:6. doi:10.1038/s41572-020-00240-3
36. Stefanini B, Tonnini M, Serio I, Renzulli M, Tovoli F. Surveillance for hepatocellular carcinoma: current status and future perspectives for improvement. Expert Rev Anticancer Ther. 2022;22:371–381. doi:10.1080/14737140.2022.2052276
37. Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi:10.1158/0008-5472.Can-09-1089
38. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New Engl J Med. 2020;382:1894–1905. doi:10.1056/NEJMoa1915745
39. Peng Y, Nie J, Cheng W, et al. A multifunctional nanoplatform for cancer chemo-photothermal synergistic therapy and overcoming multidrug resistance. Biomater Sci. 2018;6:1084–1098. doi:10.1039/c7bm01206c
40. Ma K, Shi J, Pei Y, Pei Z. A carrier-free supramolecular nanoprodrug based on lactose-functionalized dimeric camptothecin via self-assembly in water for targeted and fluorescence imaging-guided chemo-photodynamic therapy. J Colloid Interface Sci. 2022;609:353–363. doi:10.1016/j.jcis.2021.12.002
41. Zielonka J, Joseph J, Sikora A, et al. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem Rev. 2017;117:10043–10120. doi:10.1021/acs.chemrev.7b00042
42. Wu Y, Zeng C, Lv J, et al. Tumor-Targeted cRGD-Coated Liposomes Encapsulating Optimized Synergistic Cepharanthine and IR783 for Chemotherapy and Photothermal Therapy. Int J Nanomed. 2024;19:6145–6160. doi:10.2147/ijn.S457008
43. Bao W, Liu M, Meng J, et al. MOFs-based nanoagent enables dual mitochondrial damage in synergistic antitumor therapy via oxidative stress and calcium overload. Nat Commun. 2021;12:6399. doi:10.1038/s41467-021-26655-4
44. Fernandez-Fernandez A, Manchanda R, Lei T, et al. Comparative study of the optical and heat generation properties of IR820 and indocyanine green. Molecular Imaging. 2012;11:99–113. doi:10.2310/7290.2011.00031
45. Tang Y, Wang G. NIR light-responsive nanocarriers for controlled release. J Photochem Photobiol C Photochem Rev. 2021;47:100420. doi:10.1016/j.jphotochemrev.2021.100420
46. Chen Q, Hu Q, Dukhovlinova E, et al. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv Mater. 2019;31:e1900192. doi:10.1002/adma.201900192
47. Yoshida S, Kornek M, Ikenaga N, et al. Sublethal heat treatment promotes epithelial-mesenchymal transition and enhances the malignant potential of hepatocellular carcinoma. Hepatology. 2013;58:1667–1680. doi:10.1002/hep.26526
48. Liu W, Di J, Ma Y, et al. Mitochondria-Mediated HSP Inhibition Strategy for Enhanced Low-Temperature Photothermal Therapy. ACS Appl Mater Interfaces. 2023;15:26252–26262. doi:10.1021/acsami.3c00870
49. Shi Y. Mechanisms of Caspase Activation and Inhibition during Apoptosis. Molecular Cell. 2002;9:459–470. doi:10.1016/S1097-2765(02)00482-3
50. Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi:10.1371/journal.pbio.1000298
51. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi:10.1083/jcb.200809125
52. Fucikova J, Kepp O, Kasikova L, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013. doi:10.1038/s41419-020-03221-2
53. Galluzzi L, Vitale I, Warren S, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immuno Ther Cancer. 2020;8:e000337. doi:10.1136/jitc-2019-000337
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Surface Engineering of HEK293 Cell-Derived Extracellular Vesicles for Improved Pharmacokinetic Profile and Targeted Delivery of IL-12 for the Treatment of Hepatocellular Carcinoma
Zhang J, Song H, Dong Y, Li G, Li J, Cai Q, Yuan S, Wang Y, Song H
International Journal of Nanomedicine 2023, 18:209-223
Published Date: 13 January 2023
Traditional Chinese Medicine Monomers and Their Derivatives as a Promising Therapeutic Tool for Hepatocellular Carcinoma by Activation of Mitophagy
Zhu J, Yin S, Luo S, Yu F, Sun K
Drug Design, Development and Therapy 2025, 19:7069-7087
Published Date: 19 August 2025
Melanin@PLGA/Nuciferine Nanoparticles for Enhanced Photothermal Therapy of Hepatocellular Carcinoma by Promoting Autophagy

Mao Y, Du X, Wang W, Dong T, Zhu M, Niu J, Li M, Jiang J, Han L, Yang X
International Journal of Nanomedicine 2025, 20:11081-11097
Published Date: 10 September 2025
Nanocomposites to Overcoming Sorafenib Resistance in Hepatocellular Carcinoma Therapy
Liu J, Zhou S, Wei Y, Du J, Jia F, Dong W, Cao Y, Shi Z, Mu H, Chen L, Yu S
International Journal of Nanomedicine 2025, 20:15723-15767
Published Date: 26 December 2025
Research Progress on Nano-Drug Delivery Systems for Photothermal Cancer Therapy
Li T, Xiao D, Yin Y, Hu L
International Journal of Nanomedicine 2026, 21:567551
Published Date: 30 January 2026