Back to Journals » Journal of Inflammation Research » Volume 14
microRNA-223 Deficiency Exacerbates Acute Inflammatory Response to Monosodium Urate Crystals by Targeting NLRP3
Authors Yang QB, Li LQ, Zhang QB, He YL, Mi QS, Zhou JG
Received 26 February 2021
Accepted for publication 14 April 2021
Published 11 May 2021 Volume 2021:14 Pages 1845—1858
DOI https://doi.org/10.2147/JIR.S307796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Qi-Bin Yang,1,2 Ling-Qin Li,1 Quan-Bo Zhang,2,3 Yong-Long He,1 Qing-Sheng Mi,2 Jing-Guo Zhou4
1Department of Rheumatology and Immunology, Affiliated Hospital of North Sichuan Medical College, Nanchong, 637000, Sichuan Province, People’s Republic of China; 2Henry Ford Immunology Program, Departments of Dermatology and Internal Medicine, Henry Ford Health System, Detroit, MI, 48202, USA; 3Department of Gerontology, Affiliated Hospital of North Sichuan Medical College, Nanchong, 637000, Sichuan Province, People’s Republic of China; 4Department of Rheumatology and Immunology, Clinical Medical College and The First Affiliated Hospital of Chengdu Medical College, Chengdu, 610500, Sichuan Province, People’s Republic of China
Correspondence: Qing-Sheng Mi
Henry Ford Immunology Program, Departments of Dermatology and Internal Medicine, Henry Ford Health System, 1 Ford Place, Detroit, MI, 48202, USA
Tel +1-313-876-1017
Email [email protected]
Jing-Guo Zhou
Department of Rheumatology and Immunology, Clinical Medical College and the First Affiliated Hospital of Chengdu Medical College, Chengdu, 610500, Sichuan Province, People’s Republic of China
Tel +86-28-8301-6078
Email [email protected]
Objective: MicroRNAs were identified as master-switch molecules limiting acute inflammatory response. This study investigated the potential role of microRNA (miR)-223 in the mechanism of gout.
Methods: Wild-type (WT) and miR-223 knock-out (KO) mice were used to evaluate the phenotypes of gout models. Inflammatory cytokines were measured in air pouch and peritoneal cavity lavage fluid. In addition to miR-223 level in gout patients, miR-223 and pro-inflammatory genes were examined in bone marrow-derived macrophages (BMDMs) from mice as well as peripheral blood mononuclear cells from healthy controls (HC) treated with monosodium urate (MSU) crystals in vitro.
Results: MiR-223 was up-regulated in the early phase in BMDMs from WT mice after MSU challenge and decreased rapidly, and this was not observed in miR-223 KO mice in vitro. In addition, miR-223 was required for macrophages homeostasis. In comparison with WT mice in vivo, miR-223 deficiency exacerbated swelling index of MSU-induced inflammation in foot pad and ankle joint models. MiR-223 deficiency also markedly aggravated inflammatory cells infiltration and cytokines release including interleukin (IL)-1β, IL-6 and monocyte chemotactic protein-1 (MCP-1) in the air pouch and peritonitis models. In the in vitro experiments, miR-223 deficiency promoted the inflammatory response by targeting NLR family pyrin domain containing protein 3 (NLRP3). Besides, miR-223 level was down-regulated in gout patients and in HC exposed to MSU in vitro.
Conclusion: MiR-223 was down-regulated in gout patients and miR-223 deficiency exacerbated inflammatory response in diverse murine models, suggesting that up-regulation of miR-223 could be a potential therapeutic strategy for alleviating gouty inflammation.
Keywords: miR-223, gout, animal models, cytokines, monosodium urate
Introduction
Gout is an auto-inflammatory disease caused by the accumulation of monosodium urate (MSU) crystals in joints and connective tissues causing an intense inflammatory response and joint dysfunction. Recurrent attacks and spontaneous remission are typical clinical characteristics of acute gouty arthritis, which are important in the differential diagnosis of gouty arthritis from other types of arthritis. Currently, an acute gout attack is mediated by interleukin (IL)-1β, which is central to the initiation of acute arthritis and is produced due to activation of Toll-like receptor 4 (TLR4)/nuclear factor (NF)-κB and NLR family pyrin domain containing protein 3 (NLRP3) inflammasome signaling pathways by MSU crystals.1–6 However, the key regulatory mechanisms related to gouty inflammation have yet to be clarified.
MicroRNAs are a class of small, endogenous non-coding RNAs of approximately 20 nucleotides (nt) in length that regulate gene expression post-transcriptionally by binding to 3ʹ-untranslated regions (UTRs), coding sequences or 5ʹ-UTRs of target messenger RNAs (mRNAs) and lead to inhibition of translation or mRNA degradation.7,8 MicroRNAs control the expression of genes involved in several biological processes such as apoptosis, proliferation, differentiation and metastasis.7,8 In addition, several microRNAs have been identified to be negative regulators of inflammation.9,10 One recent study has shown that microRNA-223 (miR-223) acts as a negative regulator of NLRP3 inflammasome activity in vitro.11 Given that miR-223 is a negative regulator of NLRP3, and that NLRP3 is important for MSU-induced inflammation,12,13 we hypothesized that the loss of miR-223 may exacerbate the inflammatory response to MSU crystals.
In this study, miR-223 level was analyzed in gout patients and diverse phenotypes of MSU-induced gout murine models, including ankle arthritis, foot pad arthritis, subcutaneous air pouch and peritoneal cavity models, were evaluated in miR-223 knockout (KO) and wild-type (WT) mice in vivo. In addition, the targeting gene of miR-223 for regulating gouty inflammation was confirmed in bone marrow-derived macrophages (BMDMs) in vitro.
Materials and Methods
Patients
A total of 243 male patients with primary gout visited the Department of Rheumatology of the Affiliated Hospital of North Sichuan Medical College, China. These patients were confirmed to have primary gout according to the American College of Rheumatology classification criteria (1977)14 and were divided into acute gout (AG, 140 cases) and intercritical gout (IG, 103 cases) according to their clinical manifestations and C-reactive protein or erythrocyte sedimentation rate. Patients with gout had no history of cancer, hematopathy, nephropathy, infection or other autoimmune diseases, and did not receive any systemic anti-inflammatory treatments or medications to control the production and elimination of uric acid before the blood samples were obtained. Medical history and clinical data were obtained from all patients (Table 1). One hundred age-matched males who underwent regular physical examination during the same periods were enrolled as healthy controls (HC). They have no personal or familial history of gout and any systemic inflammatory diseases. In addition, routine blood counts and laboratory biochemical examinations of conventional parameters were normal. All participants in this study were from the Chinese Han population. The study was approved by the institutional research ethics committee of Affiliated Hospital of North Sichuan Medical College and written informed consent was obtained from each subject. Experiments using human blood were performed in accordance with the Declaration of Helsinki (No:2021ER(A)033).
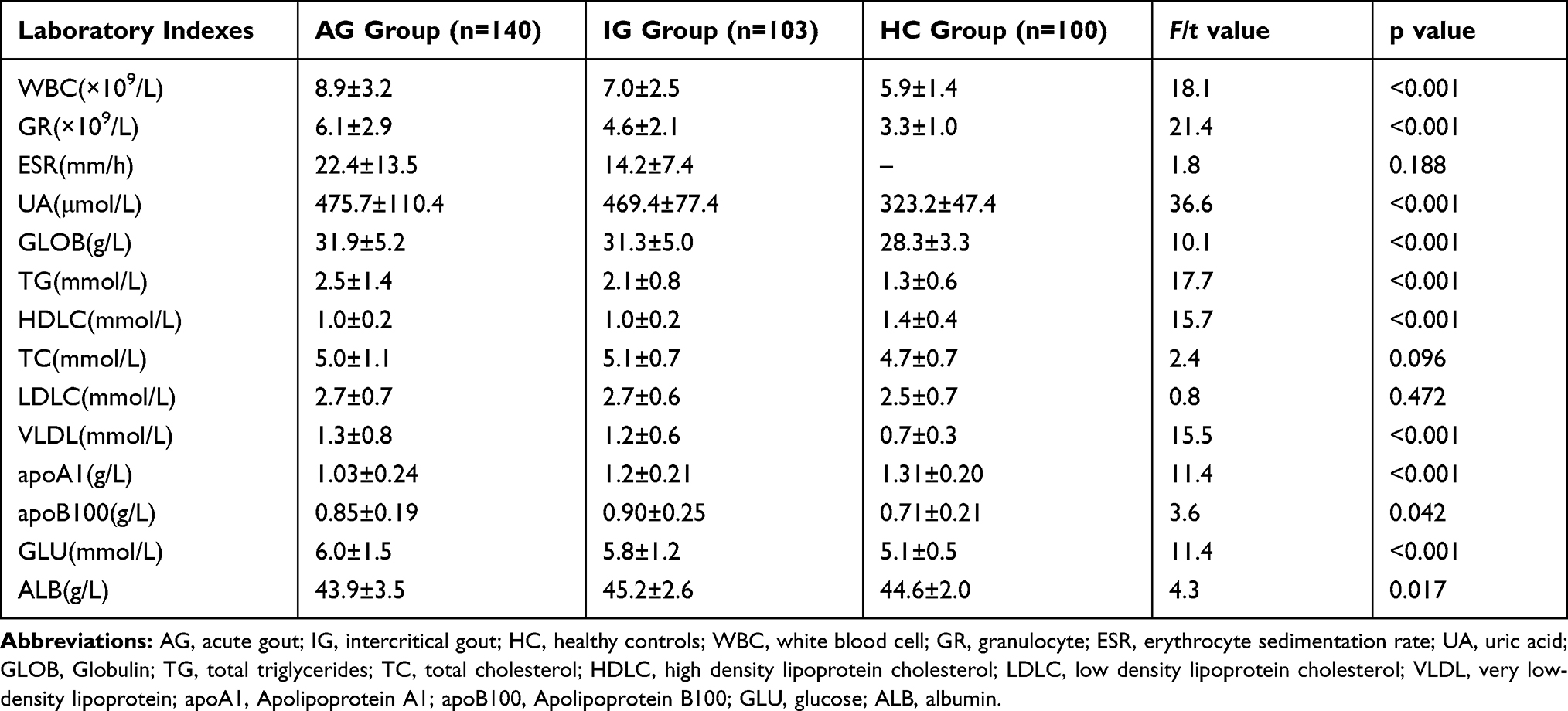 |
Table 1 Comparison of Laboratory Indexes in Patients with Acute Gout, Intercritical Gout and Healthy Controls |
Animals
MiR-223 KO mice were produced refer to the described method15 and kindly provided by the CBR Institute for Biomedical Research, Harvard Medical School, Boston, USA. WT C57BL/6 mice were purchased from the Jackson Laboratory, Bar Harbor, USA. All mice used in these experiments were 8–10 weeks old. All handling of the mice and experimental procedures were developed in accordance with the requirements of the Institutional Animal Care and Use Committee of Henry Ford Health System (Detroit, USA) and also were approved by the Committee.
MSU Crystals Formation
MSU crystals were prepared as described previously.16 Briefly, 1.0 g uric acid (Sigma-Aldrich, USA) was dissolved in 200 mL sterile water containing 6.0 mL of 1 M NaOH. The pH was adjusted to 7.2 and the solution was sterilized by heating at 180°C for 2 h. The precipitate was filtered from the solution, dried under low heat and suspended in phosphate-buffered saline (PBS) at a concentration of 50 mg/mL. All reagents were prepared under pyrogen-free conditions.
BMDMs Culture and MSU Stimulation
BMDMs were cultured in RPMI-1640 (Gibco, Life Technologies, USA) supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL), streptomycin (100 μg/mL) and 30 ng/mL macrophage colony-stimulating factor (#0914245, Peprotech, USA). After 7 days, the cells were harvested with 0.25% trypsin. The phenotypic validation of BMDMs (Figure 1B) was measured by flow cytometry and staining with FITC-conjugated anti-CD11b and PE-conjugated anti-F4/80 antibodies (both diluted 1:200). The BMDMs were double-positive for CD11b and F4/80. The BMDMs were harvested at 30 or 60 minutes after MSU crystal challenge for MSU phagocytosis of macrophages. Phagocytosis of MSU crystals was analyzed by the side scatter (SSC) change in flow cytometry.
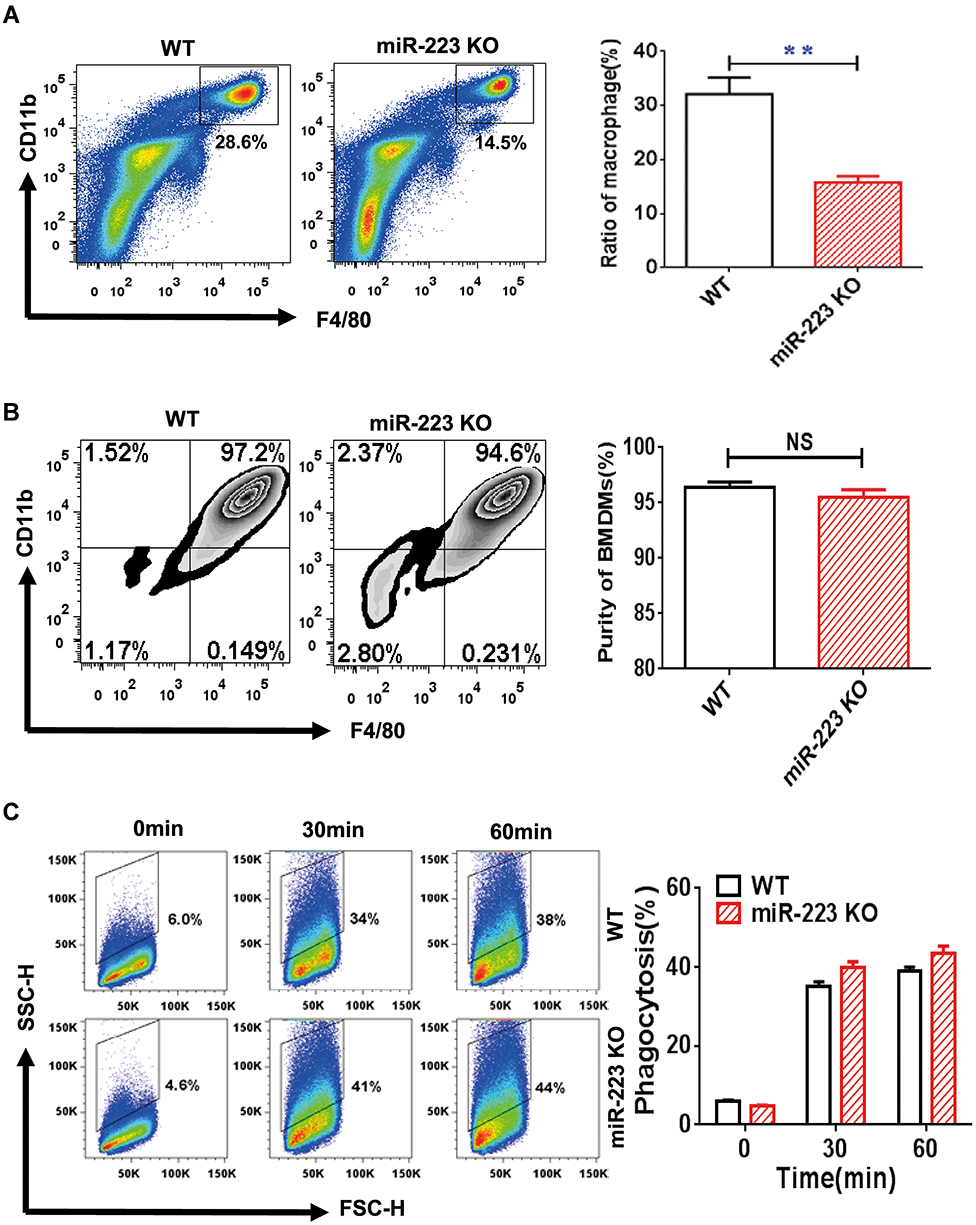 |
Figure 1 miR-223 was required for macrophage homeostasis, but not development and phagocytosis. (A) The cells harvested from peritoneal cavity with 2 mL PBS were stained with anti-F4/80 and anti-CD11b antibodies and analyzed by flow cytometry. (B) The bone marrow cells were cultured and induced by Dulbecco’s modified Eagle’s medium (DMEM) with 30 ng/mL macrophage colony-stimulating factor for 7 days. The DMEM was refreshed every 2 days until the cells were harvested and the purity of the bone marrow-derived macrophages (BMDMs) were then identified with flow cytometry. The BMDMs were represented by positive F4/80 and CD11b. (C) Monosodium urate (MSU) crystal phagocytosis was analyzed by flow cytometry in BMDMs from miR-223 KO and WT mice at different time points after MSU crystal challenge in vitro. n=3–5 for each group and unpaired t-test was used for each group at indicated time points. The results are representative of 3 independent experiments. n=3–5 for each group. **P<0.01. Abbreviation: NS, not significant. |
The harvested BMDMs were divided into 2×106 cells/well in a 6 well/plate containing 1 mL RPMI-1640+10% FBS. According to the experimental protocol for the in vitro experiment,17 the BMDMs were primed with 50ng/mL lipopolysaccharide (LPS) for 4 hours before the BMDMs were stimulated with MSU crystals suspension (MSU 100 μg/mL final concentration) for 4 or 8 h. The cells were harvested for measurement of miRNA, mRNA or protein. In addition, intracellular tumor necrosis factor (TNF)-α production in BMDMs treated with MSU for 2 or 4 h was measured by a FACSAria II (BD Biosciences, USA).
Gout Model
Mice were placed under anesthesia [150:10 mg/kg ketamine:xylazine injected intraperitoneally] and were injected with the MSU crystal suspension into the right foot pad (1 mg suspended in 40 μL PBS) or ankle joint (0.5 mg suspended in 20 μL PBS).18 Inflammation parameters were evaluated with an electronic caliper post-MSU crystal injection at different time points (6, 24 and 48 h).
An injection of 5 mL sterile air into the subcutaneous tissue on the back of each mouse was carried out to form an air pouch. On day 7, the MSU crystal suspension (3 mg) was injected into the air pouch.19 In the MSU-induced peritonitis model, MSU crystals (3 mg) were injected into the peritoneal cavity.20 The total number of cells in air pouch lavage fluid (APLF) and peritoneal cavity lavage fluid (PCLF) containing 2 mL PBS were harvested at different time points and counted using a hematocytometer.
Flow Cytometry Analysis
The BMDMs (2×106) treated with MSU for 2 or 4 h were harvested and stained with monoclonal antibody (mAb) F4/80-PE or CD11b-FITC for 30 min at 4°C. The cells were then fixed by intracellular fixation buffer for 30 min at 4°C. After rinsing twice with permeabilization buffer, the cells were dyed with mAb TNF-α-PE-Cy7 for 30 min at 4°C.
APLF or PCLF cells (2×106) were incubated with Fc block (clone 2.4G2) for 15 min and then stained with mAb Ly-6G-PE, F4/80-APC, and CD11b-FITC for 30 min at 4°C. The cells were suspended in running buffer (PBS containing 0.5% FBS and 2 mM EDTA), and then sorted and analyzed on a FACSAria II (BD Biosciences). All antibodies were purchased from eBioscience (USA). Data were acquired using CellQuest software (BD Biosciences) and analyzed by FlowJo software (Tree Star Inc.).
RNA Isolation, cDNA Synthesis and Quantitative PCR (qPCR)
Peripheral blood mononuclear cells (PBMCs) from patients with gout and HC were isolated using Ficoll-Hypaque density gradient centrifugation. Total RNA in human PBMCs and mouse BMDMs was extracted using Trizol reagent (Invitrogen, USA) and reverse-transcribed into cDNA using reverse transcription reagents (Invitrogen, USA) according to the manufacturer’s protocols. Real-time qPCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems, USA). The gene primer sequences (Table 2) were synthesized by Eurofins Genomics (Louisville, USA) or a biological engineering company (Shanghai, China). Gene expression analysis was performed using the 2−ΔΔCT or 2−ΔCT method. The expression of miR-223 (ABI: ID002295, miR-223-3p) in PBMCs, sorted cells and BMDMs were measured using TaqMan MicroRNA Assays (Applied Biosystems, USA) according to the manufacturer’s protocols. The TaqMan MicroRNA Assay for U6 snRNA (ABI: ID001973) was used to normalize the relative abundance of microRNAs.
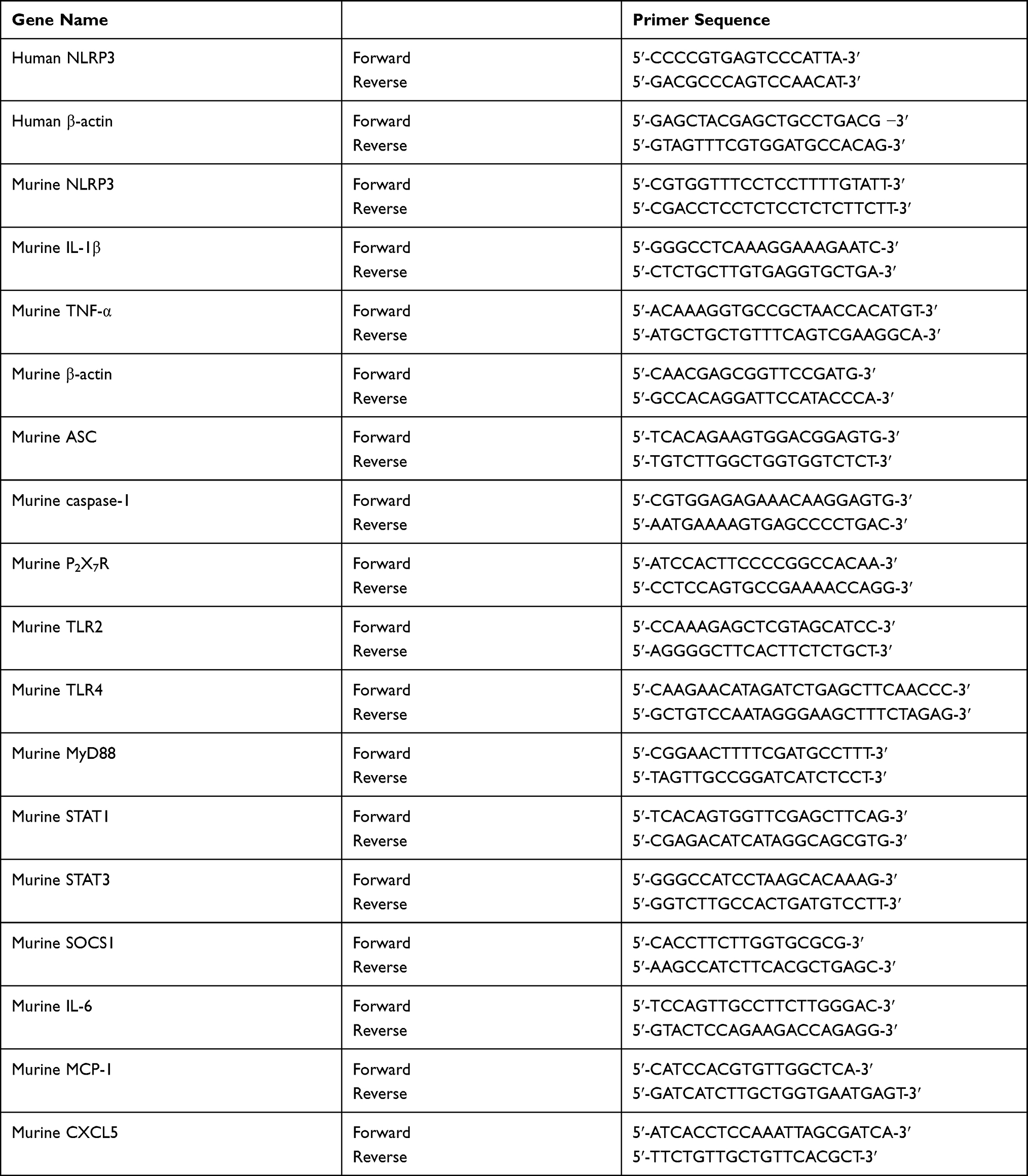 |
Table 2 Sequences of Human and Murine Genes Primers Used in Our Study |
Enzyme-Linked Immunosorbent Assay (ELISA) Analysis
IL-1β (88–7013-88), IL-6 (88–7064-88), and MCP-1 (88–7503-88) in APLF or PCLF were determined using ELISA kits from eBioscience (USA) following the manufacturer’s instructions. The 96-well microplates were read by a VICTOR X3 multilabel plate reader.
Western Blot Analysis
Proteins in BMDMs or PBMCs were extracted by RIPA lysis buffer (Thermo Scientific) containing protease or phosphatase inhibitors. The proteins (50–70 μg) were separated by 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Bio-Rad). After blocking with 5% non-fat milk (Sigma, USA) for 1 h, the membrane was incubated with primary antibodies [anti-NLRP3 antibody (#15,101), anti-IL-1β antibody (#4283), and anti-β-actin antibody (#3700) (all from Cell Signaling Technology)] at 4°C overnight. Secondary antibodies conjugated to horseradish peroxidase were incubated for 1 h at room temperature. Immunoreactive proteins were visualized using an enhanced chemiluminescence system (Amersham Biosciences), according to the manufacturer’s instructions. Fold changes were assessed using Image J software.
Statistical Analysis
Data were analyzed with Prism 6.0 (GraphPad software). Data are expressed as mean±standard deviation. Differences between experimental groups were tested using the unpaired t-test or a one-way analysis of variance (ANOVA). A p values < 0.05 was considered statistically significant.
Results
MSU Crystals Changed miR-223 Expression in BMDMs from WT Mice
We firstly validated the expression of miR-223 in BMDMs and large peritoneal macrophages (LPMs). As shown in Figure 2A, the levels of miR-223 in BMDMs and LPMs from miR-223 KO mice were significantly lower than those from WT mice. In the in vitro functional experiment, miR-223 was quickly and strongly up-regulated in BMDMs from WT mice after MSU treatment in vitro and then decreased rapidly to an extremely low level. However, the baseline level was maintained in miR-223 KO mice (Figure 2B). These data indicated that miR-223 could be significantly inhibited by MSU crystals although it was promoted at early phase.
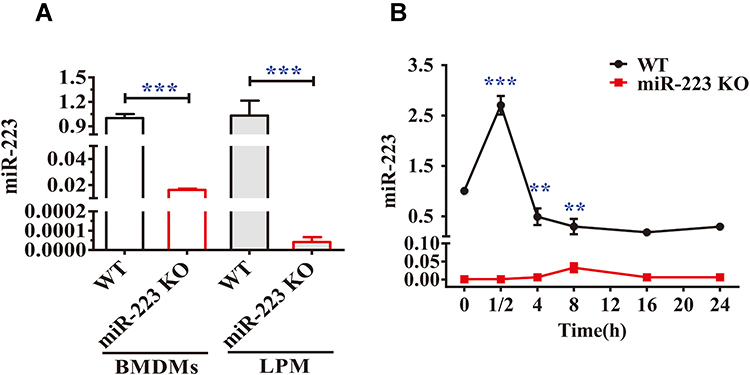 |
Figure 2 miR-223 was validated both in miR-223 knock-out (KO) and wild-type (WT) mice and changed in bone marrow-derived macrophages (BMDMs) from WT mice treated with monosodium urate (MSU) crystals in vitro. (A) Relative expression of miR-223 was measured by TaqMan-PCR in BMDMs and in large peritoneal macrophages (LPMs) from WT and miR-223 KO mice. (B) Relative expression of miR-223 was measured by TaqMan-PCR in BMDMs from WT mice treated with MSU crystals (100 μg/mL) for different time-points up to 24 h. Point 0 represented treatment without MSU crystals. The results are representative of 3 independent experiments. n=3–5 for each group, and the unpaired t-test was used for each group, and the Bonferroni post-test was used for the comparison between baseline and MSU crystals. Compared to the untreated group. **P<0.01, ***P<0.001. |
miR-223 Was Required for Macrophage Homeostasis, but Not Development and Phagocytosis
We examined whether miR-223 deficiency could impair macrophage development and homeostasis using miR-223 KO mice. In comparison with WT mice, the percentage of macrophages was significantly down-regulated in cells profile from the peritoneal cavity of miR-223 KO mice (Figure 1A). The percentage of BMDMs in miR-223 KO mice was not significantly changed compared to WT mice (Figure 1B). It suggested that miR-223 may be required for macrophage homeostasis, but not macrophage development. To further evaluate the potential role of miR-223 in the MSU crystals phagocytosis of macrophages, BMDMs were incubated with MSU crystals. The phagocytosis of macrophages from miR-223 KO mice was comparable to that from WT mice (Figure 1C), suggesting that lack of miR-223 did not block the macrophage phagocytosis.
miR-223 Deficiency Exacerbated MSU-Induced Inflammation in the Foot Pad and Ankle Joint Models
To further address whether miR-223 deficiency affects the phenotype in the MSU-induced gouty inflammatory response in vivo, foot pad and ankle joint models were applied to mimic acute gouty arthritis in humans. Compared with WT mice, significant increases in swelling indices of the foot pad were observed in miR-223 KO mice at 6 h and peaked at 24 h, and were still high at 48 h after administration of MSU crystals although this almost returned to the same level seen at 6 h (Figure 3A). Consistent with the findings with the foot pad, the ankle swelling indices in miR-223 KO mice also markedly increased compared with those in WT mice (Figure 3B). In addition, inflammatory cells were assayed by immunohistochemistry of ankle joint tissue slices. Inflammatory cell infiltration was significantly increased in miR-223 KO mice compared with WT mice (Figure 3C). These data collectively demonstrated that miR-223 deficiency could prominently promote the clinical phenotype of MSU-induced inflammatory response.
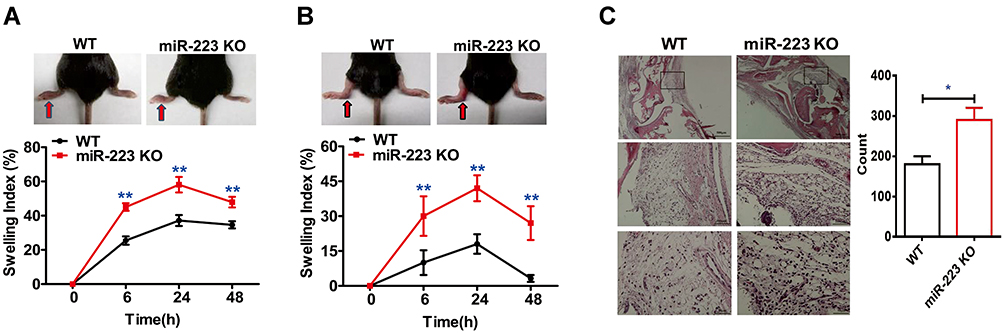 |
Figure 3 miR-223 deficiency exacerbated acute arthritis in response to monosodium urate (MSU) crystals. (A) 1 mg/40 μL MSU suspension was injected into the right foot pad of wild-type (WT) and miR-223 knock-out (KO) mice, and thickness of the foot pad was determined at 0, 6, 24 and 48 h after MSU administration. (B) 0.5 mg/20 μL MSU crystals were injected into the left ankle joints, and the ankles were measured at 0, 6, 24 and 48 h after MSU administration; swelling was expressed as a ratio > 0.15 which indicated inflammation. (C) The ankle joints were harvested 12 h after MSU treatment in 4% formalin and then tissue slices were prepared and stained with hematoxylin and eosin (H&E). Data are expressed as mean ± SEM. The results are representative of 3 independent experiments. n=6–8 for each group and the unpaired t-test was used for each group at the indicated time points. *P<0.05, **P<0.01. |
miR-223 Deficiency Aggravated MSU-Induced Inflammatory Cell Infiltration and Cytokine Release in the Air Pouch and Peritonitis Models
To ascertain the effects of miR-223 deficiency in MSU-induced inflammation in vivo, a murine air pouch model of MSU crystals-induced inflammation was assessed at different time-points. At 12 h after MSU injection, there was a marked increase in the total cell number in subcutaneous APLF (Figure 4A), and the total cell number in APLF in miR-223 KO mice was maintained at a high level at 24 h. Moreover, for neutrophils (Figure 4B), as the major APLF population, both the cell number and the ratio increased at 12 and 24 h in miR-223 KO mice (Figure 4C and D). However, both the cell number and the ratio of macrophages, the second major infiltrating cells, decreased at 6, 12 and 24 h (Figure 4E and F). Furthermore, the levels of IL-1β, IL-6 and MCP-1 in APLF, particularly at the early phase of the inflammatory response, were markedly elevated in miR-223 KO mice compared with WT mice (Figure 4G–I).
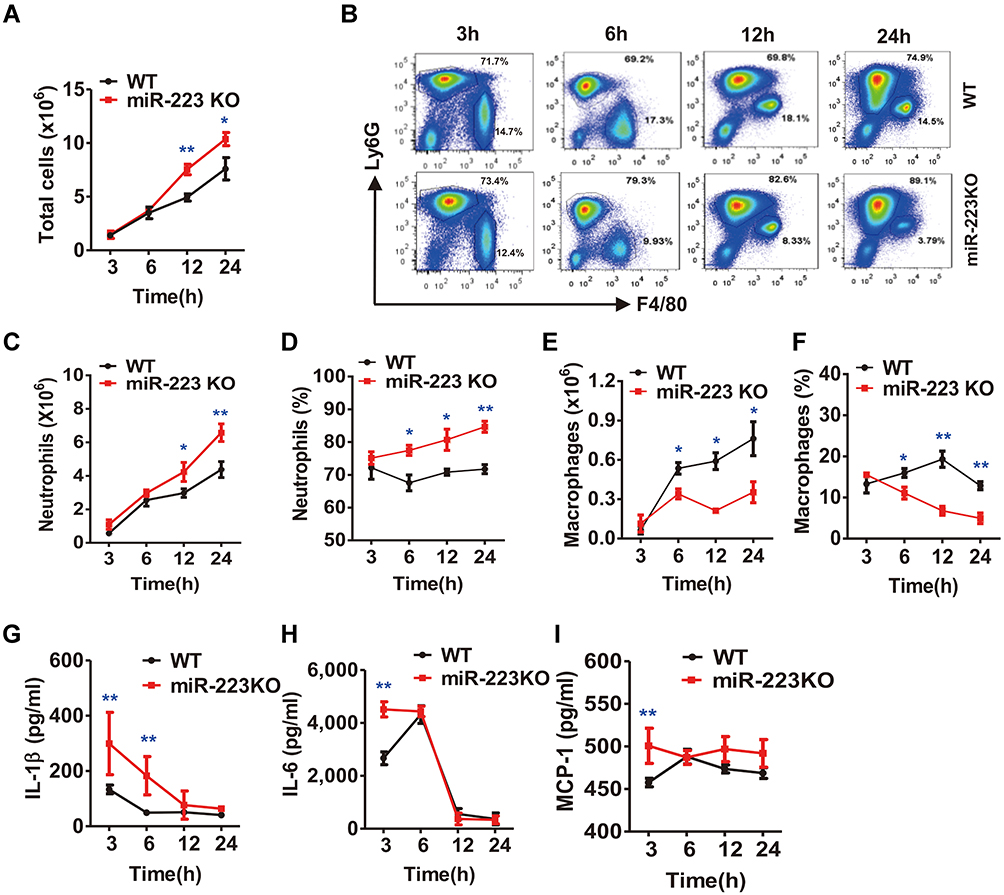 |
Figure 4 miR-223 deficiency enhanced monosodium urate (MSU)-induced inflammatory cell infiltration and cytokine secretion in the air pouch model. (A) Total cell numbers were counted using a hematocytometer. (B) Neutrophils and macrophages were gated by Fluorescence-Activated Cell Sorting (FACS). (C–F) Infiltrated neutrophils (C, D) and macrophages (E, F) were analyzed using FlowJo software. Infiltrated macrophages were represented by F4/80+, while neutrophils were represented by Ly6G+. (G–I) The protein levels of IL-1β (G), IL-6 (H) and MCP-1 (I) in air pouch lavage fluid harvested at 3, 6, 12 and 24 h were measured using ELISA. n=4–6 for each group and the unpaired t-test was used for each group at the indicated time points. *P<0.05, **P<0.01. |
The peritoneal cavity model was used to analyze the cellular phenotype of miR-223 in gout. In comparison with WT mice, it was found that the total cell number in PCLF in miR-223 KO mice increased at 3 and 6 h (Figure 5A). Of note, infiltrating neutrophils (Figure 5B) were not only increased in number but also in ratio in miR-223 KO mice after MSU treatment at 3 and 6 h (Figure 5C and D). However, neither the number nor the ratio of macrophages were significantly different (Figure 5E and F). Moreover, there was a significant elevation of IL-1β, IL-6 and MCP-1 in PCFL from miR-223 KO mice (Figure 5G–I). Our results suggested that deficiency of miR-223 could aggravate infiltration of neutrophils and production of pro-inflammatory cytokines implicated in the process of gouty inflammation.
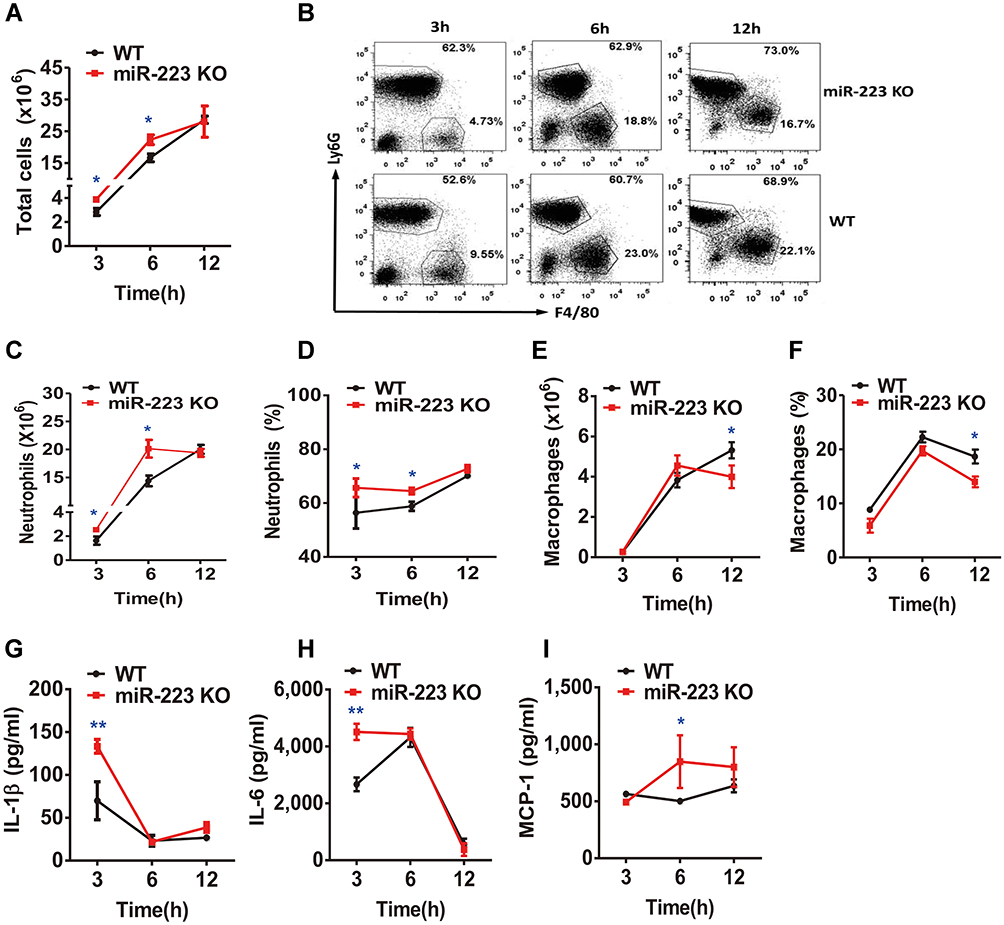 |
Figure 5 miR-223 deficiency aggravated neutrophil infiltration and pro-inflammatory cytokine secretion in monosodium urate (MSU)-induced peritonitis. (A) 3 mg/0.5 mL MSU suspension was injected into the peritoneal cavity, the lavage fluid was harvested at different time points. (A) Total cell numbers were counted using a hematocytometer. (B–F) Neutrophils and macrophages were analyzed by FACS. Macrophages were represented by F4/80+, while neutrophils were represented by Ly6G+. (G–I) IL-1β, IL-6 and MCP-1 levels in peritoneal cavity lavage fluid at the indicated time points were detected using ELISA. The results are representative of 3 independent experiments. n=4–6 for each group and the unpaired t-test was used for each group at the indicated time points. *P<0.05, **P<0.01. |
miR-223 Deficiency Enhanced the MSU-Induced Inflammatory Response by Directly Targeting NLRP3
To further clarify the potential molecular mechanism of miR-223 in the enhancement of MSU-induced gouty inflammation, pro-inflammatory gene expression was evaluated in BMDMs exposed to MSU in vitro. In comparison with WT mice, TNF-α mRNA level was significantly increased in miR-223 KO BMDMs exposed to MSU crystals (Figure 6A). It was further confirmed that the level of TNF-α secretion was markedly elevated in miR-223 KO BMDMs treated with MSU for 2 and 4 h (Figure 6B and C). In addition, the key genes related to the signaling pathways in gouty inflammation were not altered in miR-223 KO mice (Supplementary Figure 1 and 2).
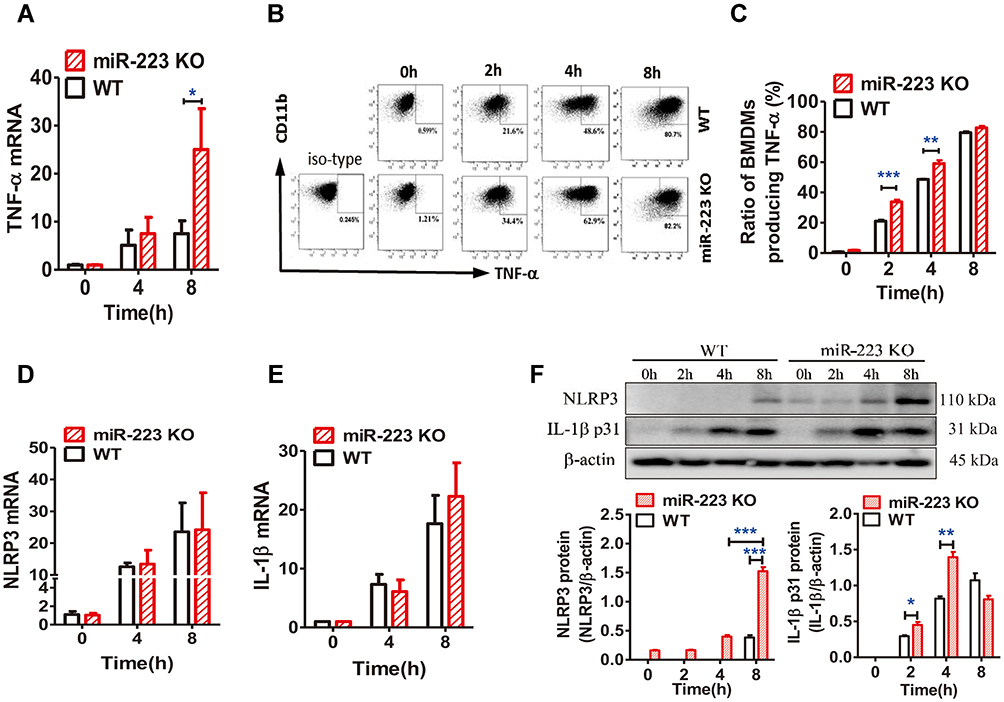 |
Figure 6 miR-223 deficiency promoted the monosodium urate (MSU)-induced inflammatory response by targeting NLRP3. (A) Relative expression of TNF-α mRNA in bone marrow-derived macrophages (BMDMs) treated with MSU (100 μg/mL) at 0, 4 or 8 hours was detected by real-time qPCR. (B and C) BMDMs from WT and miR-223 KO mice were incubated with MSU crystals (100 μg/mL) for 0, 2, 4 or 8h and brefeldin A (1:1000) was added to block cytokine secretion 1 h after MSU crystals administration. The BMDMs were represented by F4/80+CD11b+. (B) TNF-α was detected by FACS. (C) The ratio of BMDMs producing TNF-α was compared between miR-223 KO and WT mice. (D and E) Relative expression of NLRP3 (D) and IL-1β (E) mRNA in BMDMs treated with MSU (100 μg/mL) at 0, 4 or 8 h were detected by real-time qPCR. (F) The protein levels of NLRP3 and IL-1β p31 in the BMDMs from miR-223 KO and WT mice treated with MSU at 0, 2, 4 or 8 h were detected by Western blot and were analyzed using Image J software. The results are representative of 3 independent experiments. n=3–5 for each group, and the unpaired t-test was used for each group at the indicated time points. *P<0.05, **P<0.01, ***P<0.001. |
The mRNA and protein levels of NLRP3, which was one of the miR-223 potential target genes in both humans and mice, were measured in BMDMs treated with MSU in vitro. As expected, NLRP3 mRNA levels were not altered in miR-223 KO BMDMs treated with MSU (Figure 6D), while NLRP3 protein levels increased significantly compared with those in WT mice (Figure 6F), although the NLRP3 protein levels were improved both in miR-223 KO and WT BMDMs after MSU treatment. In addition, IL-1β mRNA expression was also comparable in miR-223 KO and WT mice (Figure 6E), whereas IL-1β protein level in miR-223 KO mice increased significantly compared with WT mice (Figure 6F). Collectively, these data suggested that miR-223 deficiency exacerbated the acute inflammatory response to MSU crystals by targeting NLRP3.
miR-223 Level Was Down-Regulated in PBMCs from Gout Patients
To finally verify miR-223 level in gout patients, the miR-223 level was analyzed in PBMCs from gout patients and healthy controls (HC). Our findings showed that miR-223 was down-regulated in gout patients compared to HC. Furthermore, low miR-223 did not differ between acute gout and intercritical gout (Figure 7A). A functional in vitro study was designed to validate the effects of altered miR-223 level in humans. Consistent with the results observed above, miR-223 level was significantly down-regulated in PBMCs from HC following treatment with MSU crystals to trigger the acute inflammatory response (Figure 7B). In addition, the levels of NLRP3 mRNA in PBMCs, and even IL-1β protein in the supernatant, increased in these cells from HC exposed to MSU in vitro (Figure 7C and D). Thus, it suggested that gout patients with low miR-223 level might have a distinct genetic profile mediated by MSU crystals.
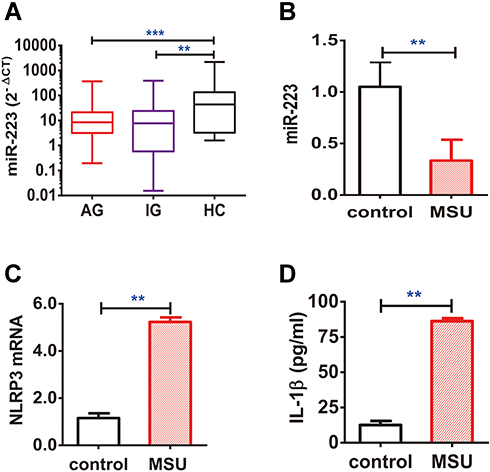 |
Figure 7 miR-223 level was down-regulated in peripheral blood mononuclear cells (PBMCs) from patients with gout. (A) miR-223 level was measured in PBMCs from patients with acute gout (AG, n=80), intercritical gout (IG, n=40) and healthy controls (HC, n=40). (B–D) The PBMCs from HC treated with MSU crystals (100 μg/mL) for 12 h in vitro were harvested for measurement of miR-223 (B), NLRP3 mRNA (C) and IL-1β protein (D) in the supernatant. The miRNA or mRNA level was detected by real-time qPCR, and the protein level was measured using ELISA. The results are representative of 3 independent experiments. n=3 for each group (B–D). One-way analysis of variance or the paired t-test, and the Bonferroni post-test were used for comparisons between the groups. **P<0.01. ***P<0.001. |
Discussion
Although MSU has been identified as a causative agent in gout for more than 100 years, its complicated mechanism was unknown until the discovery of the NLRP3 inflammasome a decade ago.21 Currently, the NLRP3 inflammasome is considered a master switch that modulates gouty inflammatory response. Emerging data have demonstrated that miRNAs negatively regulated gene expression post-transcriptionally by binding to the 3ʹ-UTR of target mRNAs.7,8 Previous in vitro studies reported that miR-223 played a crucial role in modulation of MSU-induced inflammatory response due to negative regulation of NLRP3 inflammasome activity.11,22 However, there is still a lack of direct evidence to further support that miR-223 is indeed involved in the MSU-induced inflammation.
To better understand the biological function of miR-223 involved in gouty inflammation in vitro and in vivo, we confirmed that miR-223 expression was up-regulated in the early phase and then decreased in BMDMs from WT mice after MSU treatment in vitro, but this did not occur in miR-223 KO mice. These results indicated that miR-223 expression was promoted in the early phase of MSU-induced acute gouty inflammation and was suppressed following constant challenge with MSU crystals. However, the exact mechanisms of those change still remained unclear. It could be attribute to the timeliness of miR-223 expression and the several fine-regulatory mechanisms such as regulation of transcript factor,23 circRNA sponge24 and macrophage polarization.10
MiR-223 is involved in determining the identity of cell types in the myeloid compartment, especially the neutrophils and macrophages.10 A previous study25 showed that murine peritoneal cavity harbors a number of immune cells, which is approximately composed of 30% in macrophages, 50% in B cells and 10% in T cells. It has been shown that lack of miR-223 could affect proliferation and differentiation of neutrophils.15 Macrophage, as one of the myeloid cells, plays a critical role in recognition and phagocytosis of MSU crystals and then triggers NLRP3 inflammasome activation, leading to IL-1β release.26–28 To assess if miR-223 affected macrophages development and homeostasis in miR-223 KO mice, we analyzed the macrophages from peritoneal cavity and BMDMs as well. In comparison with WT mice, the percentage of macrophages from miR-223 KO mice was significantly down-regulated, whereas the percentage of BMDMs was similar. Besides, the phagocytosis of macrophages was comparable between miR-223 KO mice and WT mice. Collectively, those data suggested that miR-223 was required for macrophage homeostasis, but not development and phagocytosis.
It is well known that acute arthritis is a clinical manifestation of gout and includes the following clinical features:29 Severe pain, redness, warmth, swelling, and disability. Firstly, we evaluated the clinical phenotype of the foot pad and ankle joint models in miR-223 KO mice in vivo which mimic acute gouty arthritis in humans. Our findings showed that a significant inflammatory response induced by MSU was observed in the foot pad and ankle models in miR-223 KO mice. Neutrophils and macrophages were the major cell populations implicated in the inflammatory response in gout.27,30,31 When the inflammatory response was triggered by MSU at a local site, pro-inflammatory cytokines and chemokines were quickly released to recruit more neutrophil and macrophage/monocyte infiltration at the inflammatory site. According to the literature,32 pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF-α, were elevated within 2 h and peak at 4 h. The air pouch and peritoneal cavity models32,33 were used to identify the cellular phenotype in miR-223 deficient mice. Our analyses revealed that deletion of miR-223 recruited more infiltrating neutrophils to the inflammatory site and produced more cytokines. These data verified that miR-223 deficiency could obviously give rise to enhancement of gouty inflammation in vivo.
The modulatory mechanisms of gout were determined to understand and regulate gouty flares. At the early stage of MSU crystals-triggered inflammatory response, pro-inflammatory mediators from macrophages were activated and released, and then participated in the acute attack process.32,33 It was reported in a previous in vitro study that miR-223 can attenuate IL-1β secretion through inhibition of NLRP3 inflammasome activity,11 suggesting that miR-223 could be a modulator of NLRP3 inflammasome signaling pathway involved in the pathogenesis of gouty inflammation. In our in vitro study, TNF-α mRNA and protein levels from miR-223 KO BMDMs increased markedly compared with WT mice. However, there were no alterations in crucial genes related to the pro-inflammatory cytokines involved in the two signaling pathways of gouty inflammation. Based on the miRBase database and previous studies,11,22 NLRP3, which is one of the potential target genes of miR-223 by binding to the 3ʹ-UTR in both humans and mice, was validated.12,13 We found that NLRP3 protein increased in miR-223 KO compared with WT BMDMs treated with MSU, while NLRP3 mRNA showed comparable changes. Consistent with the two emerging studies that the miR-223 was involved in the regulation of gouty inflammation via targeting NLRP3 in vitro functional experiment,12,13 our findings demonstrated that deletion of miR-223 exacerbated MSU-induced inflammatory response by targeting NLRP3, and alleviating the specific inhibition of NLRP3 inflammasome activity.
In patients with gout, low miR-223 level was observed in PBMCs from those with both acute gout and intercritical gout. Consistent with the observation that miR-223 deficiency was associated with an increase in the MSU-induced inflammation, a reduced miR-223 level in PBMCs from HC was observed following treatment with MSU crystals in vitro, in parallel with increased NLRP3 and IL-1β.
There are some limitations associated with the current study. Firstly, in addition to miR-223 KO mice, we should have determined the phenotype of miR-223 knock-in mice in vivo. Secondly, based on validated targets of miR-223 in literature review described,10 whether miR-223 targeting critical candidate genes in the gouty inflammatory response could target other different genes, such as TLR4,34 should have been explored. Thirdly, more research is necessary to determine the role of miR-223 in the regulatory mechanism of gouty inflammation in gout patients considering that the entire human body is composed of many complicated and sophisticated organs and is also modulated under the accurate surveillance of immune cells.
Conclusion
In this study, we revealed that miR-223 was down-regulated in gout patients and miR-223 deficiency exacerbated inflammatory response in diverse murine models. These findings from humans and mice indicate that miR-223 deficiency exacerbated the acute inflammatory response to MSU crystals by targeting NLRP3 (Figure 8). Therefore, up-regulation of miR-223 may serve as a potential therapeutic strategy for MSU crystal-induced acute gouty inflammation.
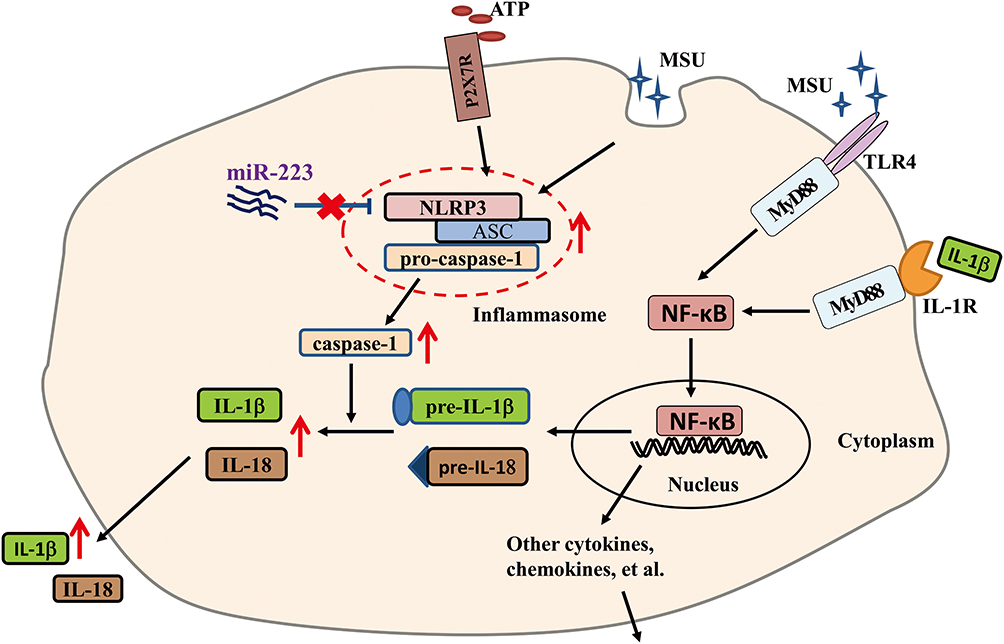 |
Figure 8 The schematic diagram shows that miR-223 deficiency exacerbates acute inflammatory response to monosodium urate crystals by targeting NLRP3. |
Acknowledgments
This work was supported by National Natural Science Foundation of China (81670801, 81800716), Sichuan Science and Technology Program (2017JY0037, 2018JY0498), Henry Ford Health System Research Grants for Immunology Program (T71016), Sichuan Medical Association (S16027), Sichuan Provincial Human Resources and Social Security Department (No.74).
Disclosure
The authors have declared no conflict of interest.
References
1. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi:10.1038/nature04516
2. Chen CJ, Shi Y, Hearn A, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116(8):2262–2271. doi:10.1172/JCI28075
3. Qing YF, Zhang QB, Yang QB, Xie WG, Zhou JG. Altered expression of NLRP3 inflammasome in peripheral blood from gout patients might be associated with gouty arthritis. Gout Hyperuricemia. 2014;1(1):25–32.
4. Qing YF, Zhang QB, Zhou JG, Jiang L. Changes in toll-like receptor (TLR)4-NFkappaB-IL1beta signaling in male gout patients might be involved in the pathogenesis of primary gouty arthritis. Rheumatol Int. 2014;34(2):213–220. doi:10.1007/s00296-013-2856-3
5. Kingsbury SR, Conaghan PG, McDermott MF. The role of the NLRP3 inflammasome in gout. J Inflamm Res. 2011;4:39–49. doi:10.2147/JIR.S11330
6. Martin WJ, Harper JL. Innate inflammation and resolution in acute gout. Immunol Cell Biol. 2010;88(1):15–19. doi:10.1038/icb.2009.89
7. Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi:10.1038/nature02871
8. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi:10.1016/S0092-8674(04)00045-5
9. He X, Jing Z, Cheng G. MicroRNAs: new regulators of toll-like receptor signalling pathways. Biomed Res Int. 2014;2014:945169. doi:10.1155/2014/945169
10. Haneklaus M, Gerlic M, O’Neill LA, Masters SL. miR-223: infection, inflammation and cancer. J Intern Med. 2013;274(3):215–226. doi:10.1111/joim.12099
11. Haneklaus M, Gerlic M, Kurowska-Stolarska M, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J Immunol. 2012;189(8):3795–3799. doi:10.4049/jimmunol.1200312
12. Wang X, Chi J, Dong B, et al. MiR-223-3p and miR-22-3p inhibit monosodium urate-induced gouty inflammation by targeting NLRP3. Int J Rheum Dis. 2021;24(4):599–607. doi:10.1111/1756-185X.14089
13. Tian J, Zhou D, Xiang L, et al. MiR-223-3p inhibits inflammation and pyroptosis in monosodium urate-induced rats and fibroblast-like synoviocytes by targeting NLRP3. Clin Exp Immunol. 2021. doi:10.1111/cei.13587
14. Wallace SL, Robinson H, Masi AT, Decker JL, McCarty DJ, Yu TF. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20(3):895–900. doi:10.1002/art.1780200320
15. Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451(7182):1125–1129. doi:10.1038/nature06607
16. Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum. 2002;46(10):2765–2775. doi:10.1002/art.10526
17. Vyleta ML, Wong J, Magun BE. Suppression of ribosomal function triggers innate immune signaling through activation of the NLRP3 inflammasome. PLoS One. 2012;7(5):e36044. doi:10.1371/journal.pone.0036044
18. Torres R, Macdonald L, Croll SD, et al. Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis. Ann Rheum Dis. 2009;68(10):1602–1608. doi:10.1136/ard.2009.109355
19. Ryckman C, McColl SR, Vandal K, et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum. 2003;48(8):2310–2320. doi:10.1002/art.11079
20. Jin HM, Kim T-J, Choi J-H, et al. MicroRNA-155 as a proinflammatory regulator via SHIP-1 down-regulation in acute gouty arthritis. Arthritis Res Ther. 2014;16(2):R88. doi:10.1186/ar4531
21. McGettrick AF, O’Neill LA. NLRP3 and IL-1beta in macrophages as critical regulators of metabolic diseases. Diabetes Obes Metab. 2013;15(Suppl 3):19–25. doi:10.1111/dom.12169
22. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol. 2012;189(8):4175–4181. doi:10.4049/jimmunol.1201516
23. Zhao G, Jiang K, Yang Y, et al. The potential therapeutic role of miR-223 in bovine endometritis by targeting the NLRP3 inflammasome. Front Immunol. 2018;9:1916. doi:10.3389/fimmu.2018.01916
24. Wang K, Long B, Liu F, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37(33):2602–2611. doi:10.1093/eurheartj/ehv713
25. Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp. 2010;35.
26. Zheng SC, Zhu XX, Xue Y, et al. Role of the NLRP3 inflammasome in the transient release of IL-1beta induced by monosodium urate crystals in human fibroblast-like synoviocytes. J Inflamm. 2015;12:30. doi:10.1186/s12950-015-0070-7
27. Mankan AK, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur J Immunol. 2012;42(3):710–715. doi:10.1002/eji.201141921
28. Yang QB, He YL, Zhang QB, Mi QS, Zhou JG. Downregulation of transcription factor T-bet as a protective strategy in monosodium urate-induced gouty infLammation. Front Immunol. 2019;10:1199. doi:10.3389/fimmu.2019.01199
29. Perez-Ruiz F, Castillo E, Chinchilla SP, Herrero-Beites AM. Clinical manifestations and diagnosis of gout. Rheum Dis Clin North Am. 2014;40(2):193–206. doi:10.1016/j.rdc.2014.01.003
30. Amaral FA, Costa VV, Tavares LD, et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 2012;64(2):474–484. doi:10.1002/art.33355
31. Mitroulis I, Kambas K, Ritis K. Neutrophils, IL-1beta, and gout: is there a link? Semin Immunopathol. 2013;35(4):501–512. doi:10.1007/s00281-013-0361-0
32. Martin WJ, Walton M, Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 2009;60(1):281–289. doi:10.1002/art.24185
33. Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005;52(9):2936–2946. doi:10.1002/art.21238
34. Wang J, Bai X, Song Q, et al. miR-223 inhibits lipid deposition and inflammation by suppressing toll-like receptor 4 signaling in macrophages. Int J Mol Sci. 2015;16(10):24965–24982. doi:10.3390/ijms161024965
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.