Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 19
MerTK Inhibition Aggravates Pancreatic Inflammation and Structural Damage via the NF-κB Pathway in an in vivo Type 2 Diabetic Rat Model
Authors Su X ![]() , Chen W, Fu Y, Lan D, Zhao Y, Yang Q, Nian X
, Chen W, Fu Y, Lan D, Zhao Y, Yang Q, Nian X ![]()
Received 27 January 2026
Accepted for publication 12 June 2026
Published 9 July 2026 Volume 2026:19 597106
DOI https://doi.org/10.2147/DMSO.S597106
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Xiaoyang Su,1 Wenting Chen,1 Yidan Fu,1 Danfeng Lan,2 Yan Zhao,1 Qiuping Yang,1 Xin Nian1
1Department of Endocrinology, The First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, People’s Republic of China; 2Department of Gastroenterology, The First People’s Hospital of Yunnan Province, Kunming, Yunnan, People’s Republic of China
Correspondence: Xin Nian, Email [email protected]
Background: Type 2 diabetes mellitus (T2DM) is characterized by chronic inflammation and progressive pancreatic β-cell dysfunction. The Mer receptor tyrosine kinase (MerTK) regulates immune homeostasis and inflammation, but its role in T2DM-related islet injury remains unclear. This study aimed to investigate the effects of MerTK and its inhibition on pancreatic structure, metabolic parameters, and NF-κB–mediated inflammation in a rat model of T2DM.
Methods: Seventy male Sprague–Dawley rats were randomized into five groups: normal control (CON), CON+MRX-2843 (MerTK inhibitor), T2DM 2-week (T2DM-2; assessed 2 weeks after diabetes induction), T2DM 12-week (T2DM-12; assessed 12 weeks after diabetes induction), and T2DM-12+MRX-2843. T2DM was induced by a high-fat/high-sugar diet plus streptozotocin injection. In the intervention groups, MRX-2843, a selective MerTK inhibitor (65 mg/kg/day) was administered by gavage for two weeks after successful diabetes induction, including after 10 weeks of diabetes exposure in the T2DM-12+MRX-2843 group. Serum MerTK, glucose, lipids, insulin, and pro-inflammatory cytokines (NF-κB, TNF-α, IL-1β) were measured by ELISA, and pancreatic histology was evaluated by HE staining.
Results: Serum MerTK expression increased with diabetes duration, peaking in the T2DM-12 group (47.5% higher than CON). MerTK inhibition significantly reduced MerTK levels by 21.3% versus T2DM-12 but aggravated metabolic dysfunction, with higher fasting glucose (+13.3%), HbA1c (+2.5%), total cholesterol (+16.9%), triglycerides (+32.6%), and insulin (+8.5%) compared to non-inhibited T2DM rats. Pro-inflammatory cytokines were markedly elevated after MRX-2843 treatment, particularly NF-κB (+35.2%), TNF-α (+48.2%), and IL-1β (+70.3%) in the T2DM-12+MRX-2843 group. Histological analysis showed progressive islet atrophy and vacuolation with diabetes, which became more severe after MerTK inhibition, indicating enhanced structural damage.
Conclusion: MerTK expression is dynamically upregulated in T2DM and exerts a compensatory protective effect by limiting NF-κB–driven inflammation and preserving pancreatic integrity. Pharmacological inhibition of MerTK exacerbates cytokine release, metabolic disturbances, and pancreatic injury, suggesting that MerTK may represent a promising candidate for further therapeutic investigation in T2DM.
Keywords: MerTK, type 2 diabetes, NF-κB pathway, pancreatic islets, inflammation
Introduction
Type 2 diabetes mellitus (T2DM) is a major global metabolic disorder driven by chronic, low-grade inflammation and characterized by insulin resistance and progressive pancreatic β-cell dysfunction,1 which leads to chronic hyperglycemia and multiple vascular complications.2–4 Despite the availability of various therapeutic options, T2DM remains a leading cause of morbidity and mortality worldwide.5 This inflammatory state is now recognized as a key driver of disease progression.1
Among inflammatory pathways, nuclear factor-κB (NF-κB) plays a central role by orchestrating innate immune responses and upregulating pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β).6 These cytokines impair β-cell survival, promote apoptosis, and interfere with insulin signaling, thereby aggravating hyperglycemia. Sustained activation of NF-κB–driven inflammatory cascades has been linked to islet inflammation, loss of β-cell mass, and metabolic deterioration, underscoring the importance of identifying upstream regulators of this pathway in T2DM.7–9
The TAM receptor tyrosine kinase family—Tyro3, Axl, and MerTK—plays a pivotal role in maintaining immune homeostasis.10,11 Among them, MerTK is highly expressed in macrophages and dendritic cells, where it mediates efferocytosis, suppresses excessive inflammation, and promotes tissue repair through ligands such as growth arrest–specific protein 6 (Gas6) and Protein S.12,13 MerTK activation triggers anti-inflammatory signaling cascades and facilitates resolution of inflammation, suggesting that it functions as a gatekeeper of immune balance in metabolic and tissue-injury settings.14,15
Previous studies have implicated MerTK in immune-mediated β-cell destruction in type 1 diabetes (T1DM). Notably, alterations in MerTK signaling—particularly the loss of its protective function on mononuclear phagocytes—have been associated with enhanced autoreactive T-cell activation and accelerated islet loss.16 The central role of these autoreactive T cells in driving disease pathogenesis is well-recognized.17 Conversely, MerTK has also been reported to exert protective effects in models of diabetic neuropathy and tissue injury, where it limited NF-κB activation and enhanced tissue repair.18 These findings suggest that MerTK may exert context-dependent effects: pathogenic in autoimmunity, yet protective in chronic inflammatory or metabolic conditions.
Despite these insights, the role of MerTK in T2DM-associated islet inflammation remains poorly understood. It is not known whether MerTK expression changes dynamically with disease progression or whether MerTK activity protects islets against metabolic stress–induced injury. Furthermore, the potential mechanistic link between MerTK signaling and NF-κB–mediated inflammatory responses in the pancreas has not been clarified. Addressing these questions may help determine whether MerTK serves as a compensatory, protective factor in the diabetic islet microenvironment. Furthermore, elucidating MerTK’s role in islet inflammation may not only advance understanding of inflammation-driven β-cell dysfunction but also inform the development of novel immunomodulatory strategies for T2DM.
Based on the above evidence, we hypothesized that MerTK is dynamically upregulated in T2DM as a compensatory protective response to chronic metabolic stress and inflammation, and that pharmacological inhibition of MerTK would exacerbate NF-κB–mediated inflammatory activation, thereby worsening pancreatic islet injury and metabolic dysfunction. Therefore, the present study aimed to elucidate the role of MerTK in T2DM progression by characterizing its dynamic expression, assessing the metabolic and inflammatory consequences of its pharmacological inhibition, and exploring its mechanistic link to NF-κB-mediated islet inflammation.
Materials and Methods
Ethical Approval
All animal procedures were reviewed and approved by the Animal Experiment Ethics Committee of Kunming Medical University (Approval No.: kmmu20221595). This study was conducted in accordance with the ARRIVE guidelines (https://arriveguidelines.org), and we have adhered to the ARRIVE guideline. Experiments were conducted in compliance with national regulations and the International Association of Veterinary Editors’ Consensus Guidelines for Animal Ethics and Welfare. Anesthesia was applied for all surgical procedures, and every effort was made to minimize pain, suffering, and mortality.
Animals and Housing
Seventy male Sprague–Dawley (SD) rats (8 weeks old, 330 ± 20 g) were purchased from Hunan SJA Laboratory Animal Co., Ltd. (license: SCXK (Xiang) 2019–0004). All animals were acclimatized for one week under SPF conditions (temperature 22 ± 2 °C, humidity 55% ± 10%, 12 h light/dark cycle) at the Experimental Animal Department of Kunming Medical University, with free access to food and water.
Experimental Grouping and Diabetes Induction
This study was designed as a randomized, controlled, parallel-group in vivo animal experiment. Rats were randomly assigned into five groups: Normal Control (CON, n=15), Normal Control with MerTK inhibitor (CON+MRX-2843, n=15), Type 2 Diabetes Mellitus 2-week group (T2DM-2, n=10), Type 2 Diabetes Mellitus 12-week group (T2DM-12, n=11), and T2DM-12 with MerTK inhibitor (T2DM-12+MRX-2843, n=12). The control groups received standard chow (Beijing KEAO Xieli Feed Co., Ltd)., while the experimental groups were fed a high-fat, high-sugar diet (Research Diets D12451; 35% carbohydrate calories, 45% fat calories, customized by Jiangsu Xietong Pharmaceutical Bio-engineering Co., Ltd). After six weeks of dietary intervention, experimental groups were injected intraperitoneally with 1% streptozotocin (STZ; Sigma, USA) at 35 mg/kg, while controls received an equal volume of saline. Random tail vein blood glucose was monitored on days 3, 7, and 14 post-injection, and rats with glucose ≥ 300 mg/dL were considered successfully modeled as diabetic.
MerTK Inhibitor Intervention
At 8 weeks after successful modeling, CON and T2DM-12 groups were further randomized to receive either vehicle (0.9% saline, 0.2 mL/day) or the MerTK inhibitor MRX-2843 (65 mg/kg/day, 0.2 mL gavage) for two weeks. The dose of 65 mg/kg/day was selected based on previous preclinical studies demonstrating that this regimen achieves effective MERTK inhibition with acceptable tolerability in murine models.19 During the experiment, all rats were monitored weekly for weight, blood glucose, feeding, and activity. Short-acting insulin (1 U, intraperitoneal) was temporarily administered only as a humane endpoint measure when blood glucose exceeded 504 mg/dL, to prevent life-threatening hyperglycemia-related mortality. This intervention was applied minimally and did not constitute a therapeutic modification of the diabetic model.
Sample Collection and Assays
After a 12-hour fast, rats received general anesthesia via intraperitoneal injection of 0.9% sodium pentobarbital (30 mg/kg), followed by sciatic nerve conduction velocity measurement. Upon completion of the measurement, an additional intraperitoneal dose of sodium pentobarbital (30 mg/kg) was administered to ensure deep anesthesia. Blood was then collected via the abdominal aorta, and complete exsanguination under deep anesthesia was performed for euthanasia. Serum was separated by centrifugation at 2500 rpm for 15 min at 4 °C. Fasting plasma glucose (FPG), triglycerides (TG), total cholesterol (TC), glycated hemoglobin (HbA1c), and insulin (INS) were measured using ELISA kits (BioSharp Inc., China). Pro-inflammatory cytokines (NF-κB p65, TNF-α, IL-1β) were quantified with rat ELISA kits (#E-EL-R0674, BioSharp Inc). The NF-κB p65 ELISA measures total circulating NF-κB p65 protein in serum. Given that NF-κB is primarily an intracellular transcription factor, serum NF-κB p65 levels were interpreted as a surrogate marker of systemic NF-κB–associated inflammatory activity rather than reflecting intracellular activation or nuclear translocation status. Plasma MerTK levels were determined using a sandwich ELISA kit (Thermo Fisher, Item No.: EHMER). The measured serum MerTK represents soluble MerTK (sMerTK), which is generated by proteolytic cleavage of membrane-bound MerTK and released into circulation. Therefore, circulating MerTK levels were interpreted as a surrogate indicator of systemic MerTK shedding and related inflammatory status, rather than direct measurement of cellular receptor expression.
Histological Examination
Pancreatic tissues were harvested after perfusion with phosphate-buffered saline (PBS, ~50 mL) via the left ventricle until the effluent was clear. Tissues were fixed in either 4% paraformaldehyde or 10% neutral buffered formalin, dehydrated, embedded in paraffin, and sectioned. Sections were stained with hematoxylin (10–20 min), differentiated with hydrochloric acid alcohol (5–10 s), counterstained with eosin (3–5 min), and dehydrated in graded alcohols. Slides were mounted with neutral gum and observed under a light microscope. All histological images were captured under identical microscope settings, including magnification, illumination, and camera parameters, to ensure consistency across groups.
Statistical Analysis
Continuous variables were assessed for normality using the Shapiro–Wilk test. In both the three-group comparison (CON, T2DM-2, and T2DM-12) and the five-group comparison following MRX-2843 administration (including CON+MRX-2843 and T2DM-12+MRX-2843), at least one group deviated from a normal distribution. Therefore, continuous variables were summarized as median (interquartile range, IQR).
Between-group comparisons were performed using the Kruskal–Wallis test. When the overall test was statistically significant, post hoc pairwise comparisons were conducted using Dunn’s test, with p-values adjusted for multiple comparisons using the False Discovery Rate (Benjamini–Hochberg method).
For the changes in body weight and blood glucose between 2 and 8 weeks after STZ injection, effect sizes were additionally calculated for the nonparametric paired comparisons. Since the Wilcoxon signed-rank test was used, the effect size was reported as r, calculated as r = z/sqrt(N), where Z denotes the standardized Wilcoxon test statistic and N denotes the number of paired observations. An r value of 0.1 was considered small, 0.3 medium, and 0.5 large.
To further evaluate the robustness of group comparisons among the five groups, post-hoc power analyses were performed for all outcomes after MRX-2843 administration. As no standard approach exists for directly estimating power in the Kruskal–Wallis test, an approximation based on the parametric one-way ANOVA framework was adopted, with effect sizes derived from the observed group differences. Effect sizes of ANOVA were quantified using eta-squared (η2), representing the proportion of total variance explained by group differences. A η2 value of 0.01 was considered small, 0.06 medium, and 0.14 large. Conventionally, a power value of 0.8 or higher is considered indicative of adequate statistical sensitivity.
Analysis of covariance (ANCOVA) was performed to evaluate the association between group and MerTK levels, adjusting for FPG, HbA1c, TG, TC, and INS. Type III sums of squares were used to assess the independent effect of Group. Effect sizes were reported as partial eta-squared (η2).
All statistical analyses were performed using R version 4.5.2 (R Foundation for Statistical Computing, Vienna, Austria). A two-tailed P < 0.05 was considered statistically significant.
Box plots for histopathological quantitative data were generated using GraphPad Prism version 10 (GraphPad Software, USA).
Results
Study Flow, Mortality, and Baseline Comparability
A total of 70 SD rats were initially enrolled and subjected to the experimental protocol (Figure 1). During the model establishment phase, rats in the T2DM-12w group experienced a total of seven deaths under high-fat diet feeding and STZ induction, resulting in 23 animals being successfully included in the final T2DM-12w model group. No mortality occurred in the CON or T2DM-2w groups during this phase.
 |
Figure 1 The experimental flow diagram. |
Subsequently, during the intervention period, the T2DM-12w group was further divided into T2DM-12w + NS (n = 12) and T2DM-12w + MRX-2843 (n = 11). One additional death occurred in the T2DM-12w + MRX-2843 group during drug administration, whereas no mortality was observed in the other four groups. Overall, only one death occurred after model establishment and group allocation.
To ensure comparability among groups, baseline characteristics were evaluated at two different groupings (3 groups or 5 groups). At the completion of model establishment, there were no statistically significant differences among the three primary groups (CON, T2DM-2w, and T2DM-12w) in body weight (H = 4.19, P = 0.123) or blood glucose levels (H = 2.20, P = 0.332). Similarly, in the grouping after intervention (five-group comparison), baseline body weight (H = 4.63, P = 0.328) and blood glucose levels (H = 4.50, P = 0.343) remained comparable across all groups, including the T2DM-12w + MRX-2843 group after accounting for one death.
Taken together, although mortality occurred during model establishment in the T2DM-12w group, the minimal loss after group allocation and the absence of significant baseline differences indicate that group comparability was well maintained throughout the study.
Metabolic Parameters and Animal Health Status
Before the initiation of the high-fat and high-sugar diet, no significant differences in body weight were observed among the groups (P = 0.123, Table 1). Similarly, prior to STZ injection, blood glucose levels remained within the normal range and were comparable across all groups (P = 0.332).
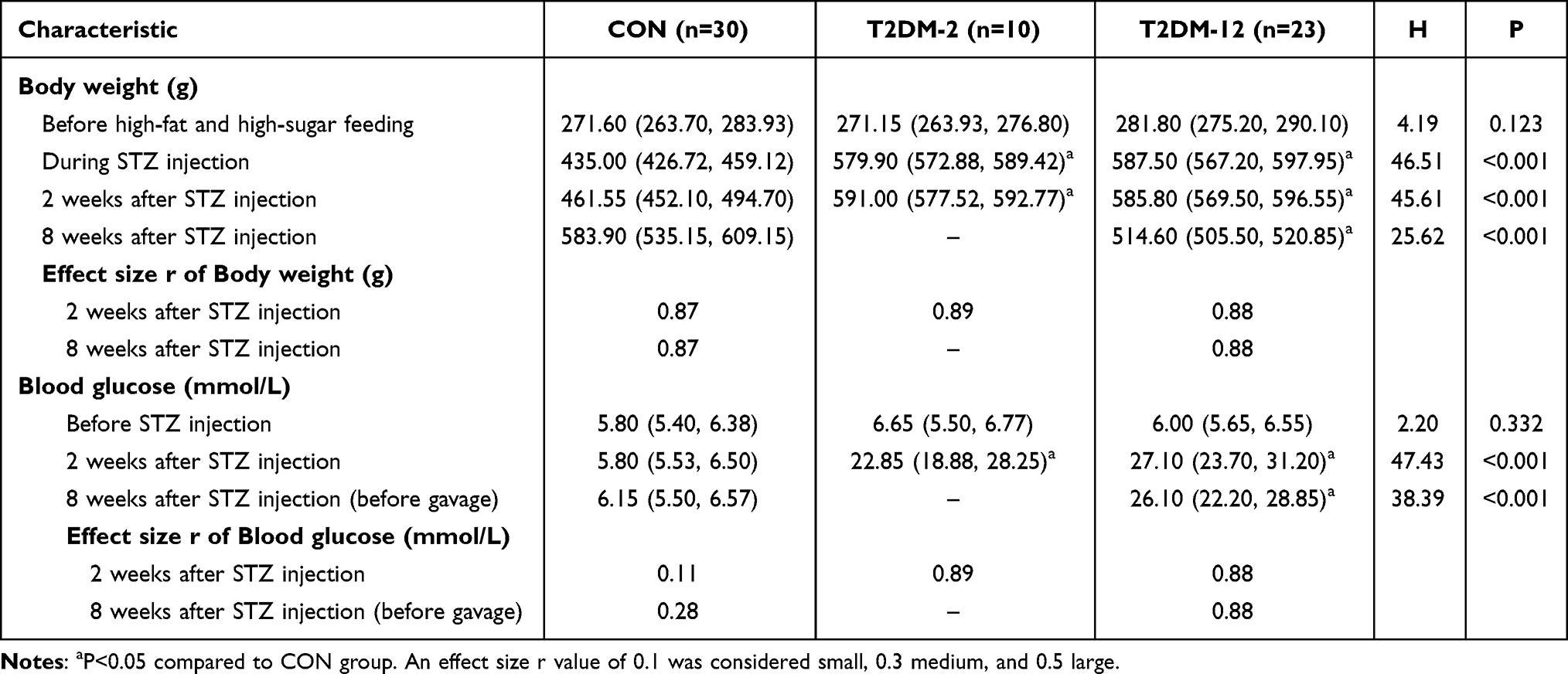 |
Table 1 Changes of Body Weight and Blood Glucose in Each Group |
During the high-fat diet period and at the time of STZ injection, rats in the experimental groups (T2DM-2 and T2DM-12) exhibited significantly higher body weights compared with the CON group (both P < 0.001, Table 1), indicating successful induction of metabolic alterations before diabetes modeling.
Two weeks after STZ injection, both experimental groups showed marked increases in blood glucose levels compared to the CON group (both P < 0.001), confirming successful establishment of the diabetic model. Body weight remained significantly higher in the experimental groups than in CON at this time point (P < 0.001).
At eight weeks after STZ injection (before gavage intervention), rats in the T2DM-12 group maintained significantly elevated blood glucose levels (P < 0.001), while body weight was significantly reduced compared with the CON group (P < 0.001), suggesting a progression toward a catabolic diabetic state.
Rats in the CON group maintained normal fur coloration and body shape, with no observed abnormalities in food and water intake, behavior, or urination. Rats in the T2DM-2 group showed polyphagia, polydipsia, polyuria, occasional presence of a rotten-apple odor in urine, reduced activity, and lethargy. Rats in the T2DM-12 and T2DM-12+MRX-2843 groups, after 12 weeks of high-fat and high-sugar diet, presented with smaller body size, dry and unkempt fur, bluish skin, cyanosis around the mouth and nose, reduced mobility, and occasional unilateral or bilateral hindlimb claudication.
Serum MerTK Expression
Serum MerTK expression differed significantly among groups (P < 0.001, Table 2) and showed an increasing trend with diabetes duration. Specifically, MerTK levels were significantly higher in both the T2DM-2 and T2DM-12 groups compared with the CON groups (all P < 0.05), with the highest levels observed in the T2DM-12 group.
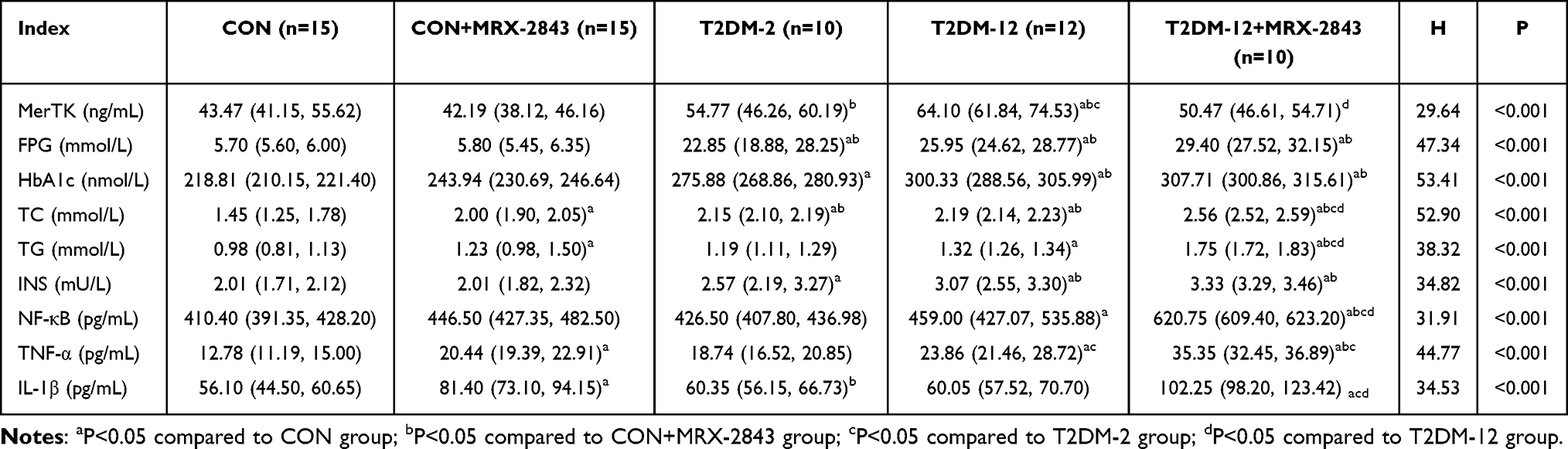 |
Table 2 Changes of MerTK, Blood Glucose, Insulin, and Lipid Levels in Each Group |
Administration of the MerTK inhibitor MRX-2843 significantly reduced MerTK levels. In the T2DM-12+MRX-2843 group, MerTK levels were significantly lower than those in the T2DM-12 group (P < 0.05), approaching levels comparable to the non-diabetic groups. No significant difference was observed between the CON and CON+MRX-2843 groups.
After adjusting for metabolic covariates (FPG, HbA1c, TG, TC, and INS), Group remained significantly associated with MERTK levels (F = 4.80, P = 0.002). The effect size was large, with a partial η2 of 0.53, indicating that more than half of the variance in MerTK was explained by group differences. None of the covariates showed significant associations with MERTK (all P > 0.05).
Metabolic Parameters
All metabolic parameters, including fasting plasma glucose (FPG), glycated hemoglobin (HbA1c), total cholesterol (TC), triglycerides (TG), and insulin (INS), differed significantly among groups (all P < 0.001, Table 2).
Compared with the CON groups, both T2DM groups exhibited markedly elevated FPG and HbA1c levels (all P < 0.05), confirming persistent hyperglycemia. Lipid profiles (TC and TG) and insulin levels were also significantly increased in the T2DM groups, indicating pronounced metabolic dysregulation.
Notably, inhibition of MerTK with MRX-2843 further aggravated metabolic abnormalities. The T2DM-12+MRX-2843 group exhibited significantly higher levels of FPG, HbA1c, TC, TG, and INS compared with the T2DM-12 group (all P < 0.05), suggesting that MerTK inhibition exacerbates metabolic impairment in diabetic conditions.
Inflammatory Cytokines
Inflammatory cytokine levels differed significantly among groups (all P < 0.001, Table 2) and increased with diabetes progression. Compared with the CON groups, the T2DM-12 group showed significantly elevated levels of NF-κB, TNF-α, and IL-1β (all P < 0.05), indicating enhanced systemic inflammation associated with prolonged diabetes.
Following MRX-2843 administration, inflammatory responses were further amplified. The T2DM-12+MRX-2843 group exhibited significantly higher levels of NF-κB, TNF-α, and IL-1β compared with the T2DM-12 group as well as all other groups (all P < 0.05), demonstrating that inhibition of MerTK markedly intensifies inflammatory activation.
Effect Size and Post-Hoc Power Analysis
Effect size estimation based on one-way ANOVA revealed large between-group differences across all measured parameters (Table 3). The η2 values ranged from 0.49 to 0.91, all exceeding the threshold for a large effect size (η2 ≥ 0.14), indicating that group differences accounted for a substantial proportion of the variance in each outcome.
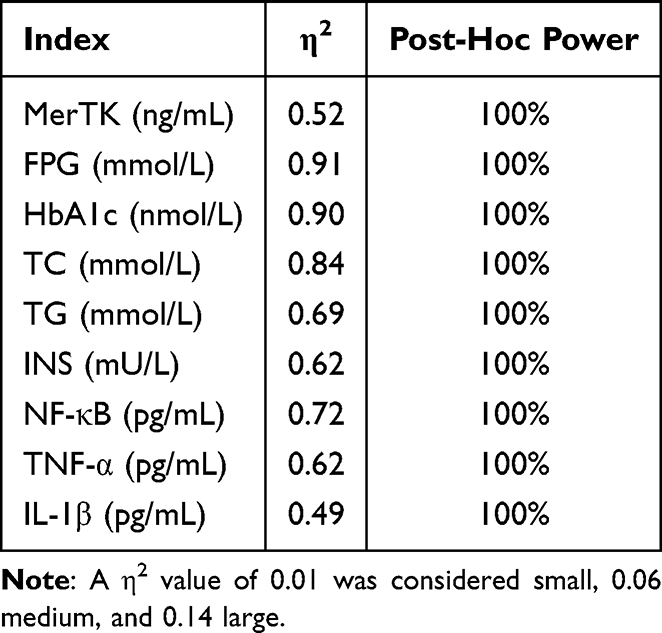 |
Table 3 The Estimated Effect Size and Post-Hoc Power Based on One-Way ANOVA |
Post-hoc power analysis demonstrated that all comparisons achieved a statistical power of 100%, indicating a very low probability of type II error and confirming that the sample size was sufficient to detect the observed group differences.
Pancreatic Histopathology
Histopathological examination revealed distinct morphological changes across groups (Figure 2). The CON group (Figure 2A) displayed normal pancreatic architecture, with round cells, central nuclei, orderly and tightly packed cells, well-defined boundaries, and regular islets with evenly distributed cells. The CON+MRX-2843 group (Figure 2B) showed relatively regular pancreatic islets with clear boundaries. Islets retained round or oval shapes without significant cell loss, and the overall pancreatic structure was largely intact with closely arranged cells.
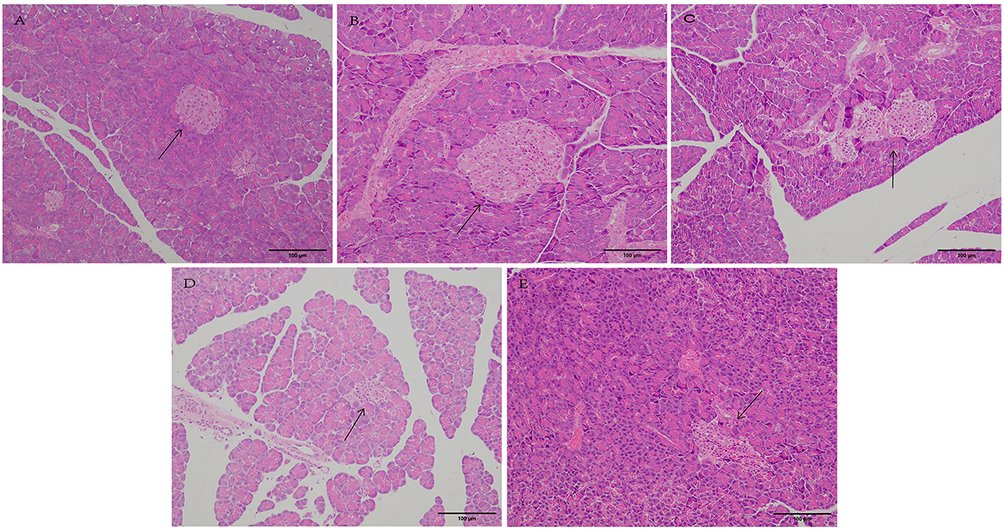 |
Figure 2 Representative photomicrographs of H&E-stained pancreatic sections acquired at 200× magnification. (A) CON group; (B) CON+MRX-2843 group; (C) T2DM-2 group; (D) T2DM-12 group; (E) T2DM-12+MRX-2843 group. Arrows indicate pancreatic islets. Scale bar: 100μm. All images were captured using identical microscope, magnification, illumination, and camera settings across groups. |
The T2DM-2 group (Figure 2C) exhibited irregular islet architecture with poorly defined borders, sparse and unevenly distributed cells, incomplete tissue structure, blurred boundaries, and vacuolar infiltration. The T2DM-12 group (Figure 2D) demonstrated pronounced islet atrophy, sparse and disordered cells, bubble degeneration, unclear boundaries between islets and exocrine tissue, and extensive vacuolar degeneration. The T2DM-12+MRX-2843 group (Figure 2E) showed severe islet atrophy, irregular and disorganized cell morphology, vacuole formation in the interstitium, and overall pancreatic tissue damage.
Quantitative analysis of pancreatic histopathology further supported the morphological observations (Figure 3). Significant differences were observed among groups for all evaluated parameters, including islet architectural integrity (H = 30.62, P < 0.001), lobular integrity (H = 32.14, P < 0.001), number of islets per section (H = 30.18, P < 0.001), and islet area (H = 35.63, P < 0.001).
 |
Figure 3 The quantitative analysis of pancreatic histopathology, including (A) islet architectural integrity, (B) lobular integrity, (C) number of islets per section, and (D) islet area. Data are presented as box-plot, the range of the box is from min to max, with the middle line representing the median. Group comparisons were performed using the Kruskal–Wallis test followed by Dunn’s post-hoc test. All parameters showed significant differences among groups (P < 0.001). ns, not significant; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
For islet architectural integrity (Figure 3A), both the CON and CON+MRX-2843 groups exhibited low scores, indicating preserved islet structure, with no significant difference between them. In contrast, integrity scores increased progressively in the T2DM-2 and T2DM-12 groups, reflecting structural disruption, and were further elevated in the T2DM-12+MRX-2843 group, suggesting aggravated islet damage following MerTK inhibition.
A similar trend was observed for lobular integrity (Figure 3B), where no significant difference was detected between CON and CON+MRX-2843 groups, while significantly higher scores were found in diabetic groups, particularly in T2DM-12 and T2DM-12+MRX-2843, indicating worsening lobular disorganization with disease progression and drug intervention.
In contrast, the number of islets per section (Figure 3C) showed a decreasing trend. The CON and CON+MRX-2843 groups had comparable islet counts, whereas significant reductions were observed in T2DM-2 and further in T2DM-12. The lowest counts were observed in the T2DM-12+MRX-2843 group, indicating marked loss of islets.
Similarly, islet area (Figure 3D) progressively declined from CON to diabetic groups. While no significant difference was found between CON and CON+MRX-2843, both T2DM-12 and T2DM-12+MRX-2843 groups exhibited significantly reduced islet area, with the smallest areas observed in the T2DM-12+MRX-2843 group.
Overall, these quantitative findings consistently demonstrate that diabetes induces progressive deterioration in pancreatic islet structure, characterized by increased architectural disruption and reduced islet number and size, and that MerTK inhibition further exacerbates these pathological changes.
Discussion
Principal Findings: MerTK as a Compensatory Protective Factor in T2DM Islets
This study demonstrated that MerTK plays a critical protective role in T2DM. Dynamic analysis revealed that serum MerTK levels progressively increased with diabetes duration, peaking in the T2DM-12 group, suggesting a compensatory response to persistent metabolic stress and inflammation. Pharmacological inhibition of MerTK with MRX-2843 worsened metabolic abnormalities—including elevated FPG, HbA1c, TG, TC, and insulin—while aggravating inflammatory responses and pancreatic structural damage. The observed islet atrophy and vacuolation following MerTK inhibition provide loss-of-function evidence for its essential role. Taken together, our findings indicate that MerTK preserves pancreatic integrity in T2DM, primarily through restraining inflammatory injury.
MerTK in Diabetes: Reconciling Context-Dependent Roles (T1DM vs. T2DM)
These findings should also be interpreted in the broader context of previous research on diabetes subtypes. Our results are consistent with studies showing that MerTK exerts anti-inflammatory and tissue-protective effects in various chronic disease models.20,21 By promoting efferocytosis, limiting pro-inflammatory cytokine release, and supporting tissue repair, MerTK contributes to immune homeostasis.20,21
However, in contrast to our observations, studies in type 1 diabetes (T1DM) suggest that MerTK may facilitate autoimmune-mediated β-cell destruction.22,23 In NOD mice, aberrant MerTK signaling in antigen-presenting cells enhanced autoreactive T-cell activation, accelerating β-cell loss.22 This apparent paradox underscores the context-dependent roles of MerTK in diabetes. In T1DM, where autoimmunity dominates, impaired MerTK function on dendritic cells and macrophages likely promotes antigen presentation and T-cell priming.22,23 In T2DM, however, characterized by metabolic inflammation, MerTK’s beneficial functions—such as enhancing efferocytosis24,25 and dampening NF-κB–driven cytokine release21—are more relevant. Our results provide the first in vivo evidence that MerTK acts as a compensatory protective factor in T2DM-associated islet inflammation, thereby reconciling its divergent roles across diabetes subtypes.
Mechanistic Insights: Linking MerTK to NF-κB–Mediated Pancreatic Inflammation
NF-κB is a central mediator of islet inflammation in T2DM.26 As part of the TAM receptor family, MerTK is activated by ligands such as Gas6 and Protein S to promote efferocytosis and inflammation resolution.21 Consistent with prior reports,27 we observed persistent NF-κB activation and elevated cytokines (TNF-α, IL-1β) during disease progression. Importantly, our study identifies MerTK as an upstream negative regulator of this pathway. MerTK inhibition led to unchecked NF-κB signaling, heightened inflammatory mediator release, and aggravated histopathological injury.
Mechanistically, compensatory MerTK upregulation in T2DM likely serves to counterbalance NF-κB activation and preserve immune equilibrium.18 Previous studies have shown that MerTK can induce SOCS1/3 expression28 and promote macrophage polarization toward an anti-inflammatory M2 phenotype,29 both of which may contribute to NF-κB suppression. However, these downstream mechanisms were not directly examined in the present study. Therefore, our proposed MerTK–NF-κB regulatory pathway should be interpreted as a working mechanistic model based on existing literature and our functional observations, rather than definitive mechanistic proof.
Study Limitations and Future Directions
Several limitations should be acknowledged. First, our analyses were limited to serum MerTK and cytokine measurements, which capture systemic changes but not local pancreatic events. Importantly, we did not directly assess pancreatic NF-κB activation, SOCS-related downstream signaling, or macrophage polarization status, which limits mechanistic interpretation of the proposed MerTK-mediated anti-inflammatory pathway. In particular, NF-κB activation was inferred from serum levels of total NF-κB p65 and downstream cytokines, which serve as surrogate markers of systemic inflammation but do not directly reflect NF-κB signaling within pancreatic tissue. Future tissue‑based studies (eg., immunohistochemistry or Western blot) for phosphorylated NF-κB p65, MerTK, and other NF-κB pathway proteins are necessary to confirm the involvement of pancreatic NF-κB in MerTK‑mediated islet protection and to validate the causal signaling pathway proposed in this study. Second, while MRX-2843 inhibition provided functional evidence, additional validation using cell–type–specific MerTK knockout models or MerTK agonists would clarify causal mechanisms.29 Third, the precise cellular sources of MerTK within the pancreas remain uncertain. Although macrophages are likely the major contributors, β cells or stromal cells may also express MerTK. In vitro experiments with macrophage and β-cell lines under metabolic stress could help dissect these contributions. Fourth, we did not perform dynamic functional tests of β‑cell activity, such as intraperitoneal glucose tolerance tests (IPGTT), insulin tolerance tests (ITT), or glucose‑stimulated insulin secretion (GSIS) assays. Therefore, our conclusions regarding β‑cell dysfunction are based on fasting serum insulin levels, pancreatic histology (islet area and architecture), and metabolic parameters (FPG, HbA1c), which provide indirect evidence only. Future studies incorporating these functional readouts are needed to directly assess β‑cell secretory capacity and to determine whether MerTK modulation affects insulin production independently of systemic inflammation. Finally, the therapeutic implications warrant further study. Given MerTK’s protective role, pharmacological activation of this pathway may represent a novel immunometabolic approach to preserve islet function in T2DM.
Conclusion
In summary, this study demonstrates that MerTK expression is dynamically upregulated during T2DM progression as a compensatory protective mechanism. Our findings suggest that MerTK may alleviate pancreatic inflammation and structural damage, potentially involving modulation of NF-κB signaling, thereby contributing to the preservation of islet function. Conversely, MerTK inhibition aggravates metabolic dysfunction, cytokine release, and tissue injury. These findings advance our understanding of β-cell failure in T2DM and support further investigation of MerTK as a potential immunometabolic target. Future studies using gain-of-function approaches or targeted agonists are needed to determine whether modulation of MerTK activity could serve as a viable therapeutic strategy. In addition, future translational and clinical studies are warranted to explore the feasibility of targeting MerTK signaling as a novel therapeutic strategy in patients with type 2 diabetes mellitus.
Data Sharing Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
The study protocol was reviewed and approved by the Animal Experiment Ethics Committee of Kunming Medical University (Approval No. kmmu20221595).
Author Contributions
Xiaoyang Su: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Writing – original draft
Wenting Chen and Yidan Fu: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing
Yan Zhao and Danfeng Lan: Conceptualization, Methodology, Project administration, Supervision, Writing – review & editing
Qiuping Yang and Xin Nian: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the Science and Technology Plan Project of the Yunnan Provincial Department of Science and Technology (202001AY070001-159); The Yunnan Clinical Medical Center for Endocrine and Metabolic Diseases (YWLCYXZXXYS20221005); The Yunnan Clinical Medical Research Center for Endocrine and Metabolic Diseases (2024YNLCYXZX0068); and the “Famous Doctors” Project of the Xingdian Talent Support Program (No. RLMY20220005).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Li H, Meng Y, He S, et al. Chronic inflammation, and insulin resistance. Cells. 2022;11(19):3001. doi:10.3390/cells11193001
2. Witcoski Junior L, de Lima JD, Somensi AG, et al. Metabolic reprogramming of macrophages in the context of type 2 diabetes. Eur J Med Res. 2024;29(1):497. doi:10.1186/s40001-024-02069-y
3. Młynarska E, Czarnik W, Dzieża N, et al. Type 2 diabetes mellitus: new pathogenetic mechanisms, treatment and the most important complications. Int J Mol Sci. 2025;26(3):1094. doi:10.3390/ijms26031094
4. Sun H, Saeedi P, Karuranga S, et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabet Res Clin Pract. 2022;183.
5. Forray A-I, Gavrilaș L, Crăciun A-E, Borzan CM. The global burden of disease: a focus on type ii diabetes. Handb Public Heal Nutr. 2025;1–13.
6. Ma Q, Hao S, Hong W, et al. Versatile function of NF-ĸB in inflammation and cancer. Exp Hematol Oncol. 2024;13(1):1–28. doi:10.1186/s40164-023-00467-2
7. He Y, Zhu W, Qiu Y, Zhou K. Loss of RIP3 alleviates insulin resistance and inflammation in gestational diabetes mellitus mice via TLR4/MyD88/NF-κB signaling pathway. BMC Pregnancy Childbirth. 2025;25(1):163. doi:10.1186/s12884-025-07217-8
8. Nord JA, Makowski SJ, Sidlowski PFW, Bursch KL, Corbett JA, Smith BC. BET bromodomain inhibitors attenuate transcription of a subset of IL-1-induced NF-κB targets that promote inflammation in β-cells. J Biol Chem. 2025;301(7):110358. doi:10.1016/j.jbc.2025.110358
9. Jia R, Liang L, Yin Y, et al. Vitamin D supplementation could enhance the effectiveness of glibenclamide in treating type 2 diabetes by improving the function of pancreatic β-cells through the NF-κB pathway. Biochem Biophys Res Commun. 2024;733.
10. Ban J, Qian J, Zhang C, Li J. Recent advances in TAM mechanisms in lung diseases. J Transl Med. 2025;23(1):1–18. doi:10.1186/s12967-025-06398-2
11. Wang KH, Ding DC. Dual targeting of TAM receptors Tyro3, Axl, and MerTK: role in tumors and the tumor immune microenvironment. Tzu-Chi Med J. 2020;33(3):250. doi:10.4103/tcmj.tcmj_129_20
12. Wanke F, Gutbier S, Rümmelin A, et al. Ligand-dependent kinase activity of MERTK drives efferocytosis in human iPSC-derived macrophages. Cell Death Dis. 2021;12(6):538. doi:10.1038/s41419-021-03770-0
13. Fabiano MP, Adamczyk AM, Gololobova OA, et al. Plasma extracellular vesicle surface-located GAS6/PROS1 and CD39/CD73 attenuate inflammation. Cell Rep. 2025;44(8):116096. doi:10.1016/j.celrep.2025.116096
14. Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal. 2018;11(549). doi:10.1126/scisignal.aar3721
15. Pastore M, Grimaudo S, Pipitone RM, et al. Role of myeloid-epithelial-reproductive tyrosine kinase and macrophage polarization in the progression of atherosclerotic lesions associated with nonalcoholic fatty liver disease. Front Pharmacol. 2019;10:452744. doi:10.3389/fphar.2019.00604
16. Lindsay RS, Whitesell JC, Dew KE, et al. MERTK on mononuclear phagocytes regulates T cell antigen recognition at autoimmune and tumor sites. J Exp Med. 2021;218(10). doi:10.1084/jem.20200464
17. Yue M, He X, Min X, et al. The role of islet autoantigen-specific T cells in the onset and treatment of type 1 diabetes mellitus. Front Immunol. 2024;15.
18. Su X, Chen W, Fu Y, et al. Protective role of mertk in diabetic peripheral neuropathy via inhibition of the nf-κb signaling pathway. Exp Clin Endocrinol Diabetes. 2024;132(7):396–406. doi:10.1055/a-2301-3970
19. Summers RJ, Jain J, Vasileiadi E, et al. Therapeutic Targeting of MERTK and BCL-2 in T-Cell and Early T-Precursor Acute Lymphoblastic Leukemia. Cancers. 2022;14(24):6142. doi:10.3390/cancers14246142
20. Li B, Zhang X, Liu S, et al. AXL and MERTK facilitate tissue repair in severe acute pancreatitis via a CCR5-dependent neutrophil and macrophage crosstalk. Cell Commun Signal. 2025;23(1):1–16. doi:10.1186/s12964-025-02412-8
21. Cai B, Thorp EB, Doran AC, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A. 2016;113(23):6526–6531. doi:10.1073/pnas.1524292113
22. Wallet MA, Sen P, Flores RR, et al. MerTK is required for apoptotic cell–induced T cell tolerance. J Exp Med. 2008;205(1):219. doi:10.1084/jem.20062293
23. Wallet MA, Flores RR, Wang Y, et al. MerTK regulates thymic selection of autoreactive T cells. Proc Natl Acad Sci U S A. 2009;106(12):4810–4815. doi:10.1073/pnas.0900683106
24. Wu Y, Zhou H, Liu H, et al. Pitavastatin-loaded procyanidins self-assembled nanoparticles alleviate advanced atherosclerosis via modulating macrophage efferocytosis and cholesterol efflux. Acta Pharm Sin B. 2025;15(6):3305–3320. doi:10.1016/j.apsb.2024.08.006
25. Xu Y, Chen J, Liu Y, et al. The macrophage-derived motor protein KIF13B enhances MERTK-mediated efferocytosis and prevents atherosclerosis in mice. Eur Heart J. 2025.
26. Liu Y, Tian Y, Dai X, et al. Lycopene ameliorates islet function and down-regulates the TLR4/MyD88/NF-κB pathway in diabetic mice and Min6 cells. Food Funct. 2023;14(11):5090–5104. doi:10.1039/D3FO00559C
27. Al Madhoun A, Haddad D, Kochumon S, et al. TNF-α/NF-κB mediated upregulation of Dectin-1 in hyperglycemic obesity: implications for metabolic inflammation and diabetes. J Transl Med. 2025;23(1):1–14. doi:10.1186/s12967-025-06303-x
28. Ruan S, Jia R, Hu L, et al. Ozone promotes macrophage efferocytosis and alleviates neuropathic pain by activating the AMPK/Gas6-MerTK/SOCS3 signaling pathway. Front Immunol. 2024;15:151455771. doi:10.3389/fimmu.2024.1455771
29. Chen F, Li Y, Zhao L, et al. Anti-inflammatory effects of MerTK by inducing M2 macrophage polarization via PI3K/Akt/GSK-3β pathway in gout. Int Immunopharmacol. 2024;142(Pt A):112942. doi:10.1016/j.intimp.2024.112942
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Potential Role and Expression Level of Urinary CXCL8 in Different Stages of Incipient Diabetic Nephropathy with Undiminished Creatinine Clearance: A Pilot Study
He Y, Li H, Wang R, Ma N, Liu L, Shi R, Zhang B, Lin N, Tian Y
Diabetes, Metabolic Syndrome and Obesity 2023, 16:1783-1790
Published Date: 17 June 2023
Exploring the Correlation Between the Systemic Immune Inflammation Index (SII), Systemic Inflammatory Response Index (SIRI), and Type 2 Diabetic Retinopathy

Wang S, Pan X, Jia B, Chen S
Diabetes, Metabolic Syndrome and Obesity 2023, 16:3827-3836
Published Date: 24 November 2023
The Inflammation Effect in the Association Between Bilirubin and Chronic Kidney Disease in Patients with Type 2 Diabetes: A Retrospective Cohort Study
Chen Y, Wang S, Chen X, Sun B, Cheng Y, Li X, Chen L
Journal of Inflammation Research 2026, 19:578233
Published Date: 19 January 2026