Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
Maternal Nutrition, Toxicants, and Epigenetic Programming of Obesity Across Generations
Authors Alum EU ![]() , Aloh HE, Obasi DC, Okoroh PN, Aniokete UC, Emeruwa AP
, Aloh HE, Obasi DC, Okoroh PN, Aniokete UC, Emeruwa AP
Received 5 November 2025
Accepted for publication 18 December 2025
Published 31 December 2025 Volume 2025:18 Pages 4873—4911
DOI https://doi.org/10.2147/DMSO.S579409
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Donald McClain
Esther Ugo Alum,1,2 Henry Egi Aloh,3 David Chukwu Obasi,4,5 Prince Nkemakolam Okoroh,4,6 Ugonna Cassandra Aniokete,7,8 Akunna Perpetua Emeruwa8,9
1Department of Research and Publications, Kampala International University, Kampala, Uganda; 2Department of Biochemistry, Faculty of Science, Ebonyi State University, Abakaliki, Ebonyi State, Nigeria; 3Health Economics and Policy Research Unit, Directorate of Health Services, Alex Ekwueme Federal University Ndufu-Alike, Ikwo, Ebonyi State, Nigeria; 4Department of Medical Biochemistry, David Umahi Federal University of Health Sciences, Uburu, Ebonyi State, Nigeria; 5International Institute for Toxicology, Environmental & Occupational Health and Safety Research, David Umahi Federal University of Health Sciences, Uburu, Ebonyi State, Nigeria; 6International Institute for Biomedical, Tissue Engineering and Regenerative Medicine Research, David Umahi Federal University of Health Sciences, Uburu, Ebonyi State, Nigeria; 7Department of Medical Laboratory Science, David Umahi Federal University of Health Sciences, Uburu, Ebonyi State, Nigeria; 8International Institute for Infectious Diseases, Biosafety and Biosecurity, David Umahi Federal University of Health Sciences, Uburu, Ebonyi State, Nigeria; 9Department of Microbiology and Parasitology, David Umahi Federal University of Health Sciences, Uburu, Ebonyi State, Nigeria
Correspondence: Esther Ugo Alum, Email [email protected]; [email protected]
Background: The developmental origins of health and disease (DOHaD) framework highlights the importance of the intrauterine environment in shaping lifelong health outcomes. Maternal nutrition, toxic exposures, and epigenetic reprogramming are key factors influencing offspring susceptibility to obesity and cardiometabolic disorders. However, prior reviews have typically addressed nutrition and toxicants separately, limiting insights into their combined effects on the fetal epigenome. This review integrates current evidence on how maternal nutrition and toxicant exposures converge through epigenetic mechanisms to influence obesity risk, while outlining translational opportunities for mitigating intergenerational metabolic disease.
Methods: A narrative review was conducted of studies published from 2000 to 2025, sourced from PubMed, Scopus, and Web of Science, supplemented by manual screening. Search terms included maternal nutrition, environmental toxicants, epigenetic mechanisms, and offspring obesity outcomes. Studies on animal models, human cohorts, and intervention trials were included, focusing on links between maternal exposures, epigenetic changes, and metabolic disease.
Results: Maternal dietary imbalances, such as deficiencies in one-carbon donors or excess caloric intake, cause persistent epigenetic changes on genes regulating adipogenesis and energy homeostasis, increasing offspring obesity risk. Prenatal exposure to environmental toxicants, including endocrine disruptors and heavy metals, amplifies these vulnerabilities by altering DNA methylation, histone modifications, and noncoding RNA networks. Combined nutritional deficits and toxicant exposures, particularly in low- and middle-income countries (LMICs), create a “dual burden” that intensifies epigenetic instability. Nutrients like methyl donors and antioxidants may mitigate toxicant-induced epimutations, offering potential for precision maternal nutrition interventions.
Conclusion: Maternal nutrition and toxicant exposures interact through epigenetic mechanisms to program obesity and related diseases. Addressing these factors through precision nutrition, stricter environmental regulations, and early-life epigenetic biomarkers offers promising prevention strategies. Large, diverse, multi-generational cohorts and multi-omics approaches are needed to strengthen causal inference and inform equitable policies to break the intergenerational cycle of metabolic disease.
Plain Language Summary: This study explores how a mother’s diet and exposure to environmental pollutants during pregnancy can shape her child’s future risk of obesity and related diseases such as diabetes and heart disease. It explains that what happens in the womb can “program” a baby’s metabolism for life through changes in gene activity known as epigenetic modifications which are chemical tags that switch genes on or off without changing DNA.
Poor maternal nutrition, whether from eating too much fat and sugar or lacking key vitamins like folate and B12 can alter these epigenetic marks, increasing the child’s tendency to gain weight. At the same time, exposure to environmental toxicants like plastics (which contains BPA), heavy metals, or pesticides can make these effects worse. When both poor nutrition and toxic exposure occur together, especially in low-income settings, the risk becomes even greater.
The review also highlights that healthy diets rich in folate, antioxidants, and omega-3 fats may help protect against these harmful effects. New technologies that study genes, metabolites, and gut bacteria together (multi-omics) are helping scientists identify early biomarkers of risk.
Ultimately, the study calls for integrated maternal health policies that combine good nutrition with stronger environmental protections to prevent obesity from being passed down across generations.
Keywords: toxic exposures, epigenetics, histone modifications, endocrine disruptors, fetal programming, maternal precision nutrition
Graphical Abstract:

Introduction
Obesity and its associated metabolic disorders represent one of the most pressing global health challenges of the 21st century. The prevalence of obesity has tripled over the past four decades, with current estimates indicating that more than 890 million adults and over 160 million children and adolescent are affected worldwide. Unfortunately, the prevalence of this menace has been predicted to surge by 2050 with the overall number of adults and children with overweight and obesity projected to reach 3.80 billion and 186 million, respectively.1,2 Obesity not only increases the risk of type 2 diabetes, cardiovascular disease, and nonalcoholic fatty liver disease but also contributes to reduced life expectancy and escalating healthcare costs.3,4 Traditional explanations centered on lifestyle and genetic predisposition cannot fully account for the rapid rise in obesity, suggesting that early-life determinants play a pivotal role in shaping lifelong metabolic risk.5
At the same time, global populations, particularly those in low- and middle-income countries face rising exposures to environmental toxicants. Biomonitoring surveys indicate that >90% of pregnant women in the United States have detectable levels of endocrine disruptors such as bisphenol A (BPA) and phthalates in their urine, while similar findings have been reported in European birth cohorts.6 In South Asia, especially Pakistan and Bangladesh, tens of millions of people are chronically exposed to arsenic in drinking water, with exposure rates reaching up to 60 million individuals in Pakistan and 28–46% of the Bangladeshi population exceeding WHO safety limits.7,8 Moreover, global food safety reports estimate that approximately 25% of food crops are contaminated with mycotoxins like aflatoxins, and in some regions detection reveals contamination in up to 60–80% of staple crops, notably maize and groundnuts in sub-Saharan Africa.9,10
The World Health Organization further estimates that environmental pollution accounts for 24% of global deaths and disproportionately affects women and children.11 Together, these statistics reveal a “dual burden” in which widespread maternal exposure to obesogenic toxicants coincides with the escalating prevalence of obesity.
Growing evidence supports the concept that the intrauterine environment exerts a profound influence on long-term health outcomes, as articulated by the developmental origins of health and disease (DOHaD) hypothesis.12 Maternal nutrition, exposure to environmental toxicants, and other gestational factors can induce biological changes in the developing fetus that persist well into adulthood. These developmental “programming” effects create a biological memory of early exposures, predisposing offspring to obesity and metabolic syndrome long before lifestyle factors take effect.13
At the molecular level, epigenetic programming provides a mechanistic framework for understanding how intrauterine exposures shape disease susceptibility without altering the DNA sequence.14 Epigenetics refers to the study of inheritable modifications in gene expression that transpire without changes to the fundamental DNA sequence. These alterations are facilitated by mechanisms including DNA methylation, histone modification, chromatin remodelling, and non-coding RNAs, which govern the activation or repression of genes in response to developmental or environmental stimuli.15,16
Beyond DNA methylation and histone acetylation/methylation, several emerging epigenetic mechanisms may also mediate the impact of maternal nutrition and toxicants on obesity risk. Histone lactylation, derived from glycolytic lactate, links cellular metabolic state directly to chromatin regulation and may be particularly relevant in obesogenic, high-glycolytic intrauterine environments. Altered maternal fuel availability and placental hypoxia could, in principle, shift lactate flux and histone lactylation marks at genes controlling adipogenesis, inflammation, and energy expenditure.17 According to Li et al,18 changes in chromatin accessibility and nucleosome positioning, evaluated by ATAC-seq method, offer a more integrated view of how maternal exposures remodel cis-regulatory landscapes in adipose and hepatic lineages. Additionally, 3D genome architecture including chromatin looping, topologically associated domains, and long-range enhancer–promoter contacts may orchestrate coordinated transcriptional programs in developing metabolic tissues.19 Although human pregnancy data remain sparse, early animal and in vitro work suggest that maternal dietary imbalance and toxicant exposure can rewire higher-order chromatin organization, with potential long-term consequences for obesity-related gene networks. Together, these processes are highly responsive to nutritional inputs and environmental insults during critical windows of development, establishing long-lasting alterations in metabolic pathways such as adipogenesis, insulin sensitivity, and energy homeostasis.20
Despite significant progress, several critical knowledge gaps remain. First, most studies examine maternal nutrition or toxicant exposures in isolation rather than within an integrated maternal “exposome” that reflects real-world co-occurrence of dietary imbalances and multi-toxicant burdens. Second, much of the mechanistic insight derives from animal models, which, while invaluable, may not fully capture the complexity of human pregnancy, placental function, and long-term metabolic outcomes. Third, human evidence largely comes from high-income cohorts, with relatively few data from LMICs where the dual burden of maternal undernutrition and pollution is greatest. Third, longitudinal, multi-omics studies that link specific maternal exposures to epigenetic marks and subsequent obesity phenotypes are still sparse, limiting causal inference and translational application. Observational cohort studies demonstrate associations between maternal exposures and offspring obesity but cannot establish causality. Few randomized controlled trials have tested nutritional or exposure-reduction interventions during pregnancy, and ethical considerations limit experimental manipulations in human populations.21 Lastly, many studies rely on single-exposure assessments, overlooking the real-world complexity of combined nutritional deficiencies, excesses, and multi-toxicant exposures that constitute the maternal exposome.
Therefore, the aim of this narrative review is to fuse current knowledge on how maternal nutrition and toxic exposures individually and synergistically reconfigure the fetal epigenome to amplify intergenerational risk for obesity and related disorders. Specifically, we seek to: (i) summarize epigenetic mechanisms linking maternal macronutrient, micronutrient, and bioactive intake to offspring adiposity and metabolic disease; (ii) examine how major classes of environmental toxicants reconfigure the fetal epigenome to promote obesogenic phenotypes; (iii) evaluate evidence for interactive nutrition–toxicant effects, including microbiome-mediated and sex-specific pathways; and (iv) identify key knowledge gaps and translational opportunities for precision maternal nutrition, exposure-reduction approaches, and policy actions aimed at mitigating intergenerational cycles of obesity.
Methodology
This review adopts a narrative approach, aiming to integrate evidence across disciplines rather than constrain the synthesis to rigid systematic review protocols. Literature was identified through extensive searches of PubMed, Scopus, and Web of Science, complemented by manual screening of reference lists from relevant reviews and primary research papers. Search terms included maternal nutrition, macronutrient and micronutrient imbalances, epigenetics, DNA methylation, histone modifications, non-coding RNAs, obesogens, endocrine disruptors, and offspring obesity. To balance breadth with relevance, we focused on publications in English from 2000 to 2025, ensuring inclusion of both seminal studies and recent advances.
Studies were considered if they examined the relationship between maternal dietary exposures or toxicant burdens during pregnancy and epigenetic outcomes relevant to obesity risk. Evidence was drawn from animal experiments that provide mechanistic insights, observational human cohorts that capture real-world exposures, and randomized controlled trials or intervention studies where available. Reports without direct relevance to epigenetic mechanisms or obesity outcomes were excluded. In synthesizing the literature, emphasis was placed on mapping exposures to epigenetic modifications (DNA methylation, histone changes, chromatin remodeling, or non-coding RNA regulation), identifying the molecular targets most frequently implicated (IGF2, LEP, PPARγ, insulin signaling genes), and highlighting offspring outcomes such as adiposity, insulin sensitivity, and metabolic syndrome. Human cohort studies including famine exposures, prospective birth cohorts, and multi-omics investigations were particularly valued for their translational relevance, while animal models were used to fill mechanistic gaps that cannot be ethically or practically addressed in humans.
Because this is a narrative rather than a systematic review, the synthesis privileges integration and critical interpretation over exhaustive cataloguing. Findings are presented thematically, beginning with the effects of maternal macronutrient and micronutrient status, moving to the impact of environmental toxicants, and finally exploring their synergistic or antagonistic interactions. Throughout, attention is drawn to research gaps such as the underrepresentation of low- and middle-income countries, challenges in distinguishing causality from correlation, and the need for multi-omics approaches that frame opportunities for advancing both science and policy.
Maternal Nutrition and Epigenetic Programming of Obesity
Maternal nutrition during pregnancy is a critical determinant of the intrauterine environment, exerting profound influence on fetal growth trajectories and long-term metabolic health.22 Beyond the direct supply of substrates for fetal development, maternal diet regulates the establishment of epigenetic marks that shape gene expression patterns in tissues central to energy balance, including adipose tissue, liver, pancreas, and hypothalamus. Nutritional perturbations, whether deficiencies or excesses, can induce lasting epigenetic alterations in pathways governing adipogenesis, appetite regulation, and insulin signaling, thereby programming an increased risk of obesity in offspring.23
Macronutrient Imbalances and Epigenetic Regulation
High-Fat Diets
Maternal overnutrition and high-fat intake have been consistently linked to obesity in offspring.24 Mechanistically, excess maternal fat induces DNA methylation changes at adipogenic regulators such as PPARγ and C/EBPα, leading to enhanced adipocyte differentiation and lipid storage.20 Histone acetylation changes in fetal liver further promote lipogenic gene expression, while alterations in hypothalamic DNA methylation disrupt appetite-regulating neurocircuitry.2 Animal studies demonstrate that maternal high-fat feeding enhances activating histone marks (H3K9ac, H3K4me3) in lipogenic gene promoters, establishing a chromatin landscape permissive for obesity-related phenotypes.25,26 Importantly, systematic reviews of human cohorts (eg, ALSPAC and Generation R) confirm that higher maternal dietary fat intake during pregnancy is associated with differential DNA methylation at obesity-related loci, including LEP and IGF2, which in turn correlate with childhood BMI trajectories.27,28 Meta-analyses further suggest that maternal overnutrition during pregnancy increases offspring risk of obesity by 30–40%, partly mediated by epigenetic alterations.29,30
High-Sugar Diets
Gestational exposure to high-sucrose or high-fructose diets promotes epigenetic dysregulation of glucose–insulin signaling pathways.31 In rodent models, maternal high-sugar intake reduces DNA methylation at IGF2 and IRS1, genes central to insulin signaling, resulting in hyperinsulinemia and early-onset adiposity.23,32 Histone deacetylase (HDAC) activity is also altered, shifting chromatin states toward increased glycolytic and lipogenic capacity.2 Human studies support these findings: the Project Viva cohort reported that higher maternal intake of sugar-sweetened beverages was linked to epigenetic modifications in cord blood insulin signaling genes, which predicted higher adiposity in early childhood.33 A meta-analysis review of 2003 mother-offspring pairs from three cohorts concluded that maternal glycemic load during pregnancy is consistently associated with methylation changes in glucose–insulin pathways, though replication across diverse populations is still limited.34
Protein Restriction
Low-protein maternal diets induce a “thrifty phenotype,” whereby offspring develop enhanced metabolic efficiency that predisposes them to obesity in nutrient-rich postnatal environments.35 Mechanistically, protein restriction is associated with hypomethylation of glucocorticoid receptor (NR3C1) and peroxisome proliferator-activated receptor alpha (PPARα) genes, altering stress responses and lipid metabolism.36 Reduced histone acetylation in the pancreas and liver further modifies energy partitioning, enhancing susceptibility to metabolic syndrome.37
The “thrifty phenotype” hypothesis proposes that poor nutrition during fetal development and early life leads to long-term metabolic adaptations that help the individual survive in a nutrient-scarce environment. These adaptations include reduced insulin secretion and sensitivity, and altered energy metabolism. While these changes may be beneficial in conditions of chronic undernutrition, they increase susceptibility to metabolic disorders such as type 2 diabetes, obesity, and cardiovascular disease if the individual later encounters a nutrient-rich environment.38
The “thrifty phenotype” hypothesis, originally derived from low-protein diet models in rodents, has human parallels. Evidence from the Dutch Hunger Winter and Chinese Famine cohorts demonstrates that prenatal protein-energy restriction is associated with hypomethylation of NR3C1 (glucocorticoid receptor) and IGF2, leading to long-term dysregulation of stress responses and metabolic pathways.23 A meta-analysis and systematic analysis combined study confirmed persistent DNA methylation differences at these loci decades after exposure, directly linking undernutrition to increased obesity and cardiometabolic risk in adulthood.39
Micronutrients and One-Carbon Metabolism
Methyl Donors (Folate, Choline, Vitamin B12, Methionine)
One-carbon metabolism provides substrates for DNA and histone methylation, making maternal micronutrient status a cornerstone of epigenetic regulation. Deficiency in folate or vitamin B12 during pregnancy disrupts S-adenosylmethionine (SAM) availability, leading to global hypomethylation and instability in obesity-related genes.40 Classic evidence comes from the Dutch Hunger Winter cohort, where prenatal famine exposure altered DNA methylation at IGF2 decades later, linking nutritional stress to lifelong obesity risk.41 Conversely, optimal methyl donor intake supports stable imprinting and healthy metabolic programming. Human evidence is particularly robust: maternal folate and B12 deficiencies have been associated with altered DNA methylation at IGF2, LEP, and other obesity-related genes in multiple cohorts (ALSPAC, Pune Maternal Nutrition Study).40,42 A systematic review of 46 studies concluded that inadequate maternal folate and B12 significantly increase the risk of obesity and insulin resistance in offspring, highlighting the importance of optimal one-carbon donor status.43
Minerals and Trace Elements
Micronutrients such as zinc and selenium act as cofactors for epigenetic enzymes (eg, DNA methyltransferases, histone demethylases). Deficiency alters enzyme activity, promoting aberrant methylation patterns in metabolic genes. For example, in pregnant mice, zinc deficiency in pregnancy has been associated with altered methylation at leptin (LEP) and adiponectin (ADIPOQ) genes, affecting appetite regulation and adipocyte function.44
Rodent studies implicate zinc and selenium in maintaining DNA methyltransferase and histone demethylase activity. Human reviews note associations between zinc deficiency and altered gene-specific methylation, and state that zinc influences key epigenetic processes.45,46 Selenium status has been inversely associated with offspring insulin resistance, although evidence is less consistent across populations.47
Dietary Bioactive Compounds as Epigenetic Modulators
Polyphenols (Resveratrol, Epigallocatechin Gallate, Curcumin)
Maternal consumption of polyphenol-rich diets exerts protective epigenetic effects against obesogenic programming. Resveratrol, for instance, activates sirtuin 1 (SIRT1), promoting histone deacetylation of pro-inflammatory genes and reducing adipogenesis.2 Epigallocatechin gallate (EGCG) inhibits DNA methyltransferases (DNMTs), modulating methylation at metabolic genes involved in lipid oxidation.48 These effects suggest bioactive polyphenols function as natural “epigenetic regulators” capable of mitigating toxicant- or nutrient-induced epimutations. In animals, polyphenols act as natural epigenetic regulators by modulating sirtuins and DNMT activity. Human intervention studies remain scarce, but small randomized trials indicate that maternal polyphenol-rich diets improve oxidative stress markers and may influence placental methylation patterns.49 A recent systematic review of randomized control trials concluded that evidence in humans is promising but insufficient, calling for larger, well-controlled trials.50
Omega-3 Fatty Acids
Long-chain polyunsaturated fatty acids (PUFAs), especially docosahexaenoic acid (DHA), regulate histone acetylation and microRNA expression in pathways linked to inflammation and adipogenesis. Maternal omega-3 supplementation has been linked in human trials to altered cord blood DNA methylation at inflammatory genes (TNFα, IL6) and regulators of adipogenesis.51 These changes are associated with improved offspring insulin sensitivity and lower risk of childhood obesity. A systematic review of 13 RCTs supports the role of omega-3s in modulating epigenetic markers relevant to metabolic programming.52
Other Bioactives
Rodent studies highlight sulforaphane (from cruciferous vegetables) and butyrate (a microbial metabolite of dietary fiber) as histone deacetylase inhibitors, enhancing protective chromatin remodeling.53,54 In humans, evidence is still emerging, but observational data suggest that maternal high-fiber intake (increasing butyrate production) is associated with beneficial cord blood methylation signatures in metabolic pathways.23,55 These findings point to the possibility of maternal dietary interventions as epigenetic therapeutics in preventing obesity programming.
To integrate mechanistic insights from experimental models with evidence from human populations, Table 1 summarizes maternal nutritional factors, their associated epigenetic mechanisms, key gene targets, and the corresponding obesity-related outcomes observed across animal and human studies.
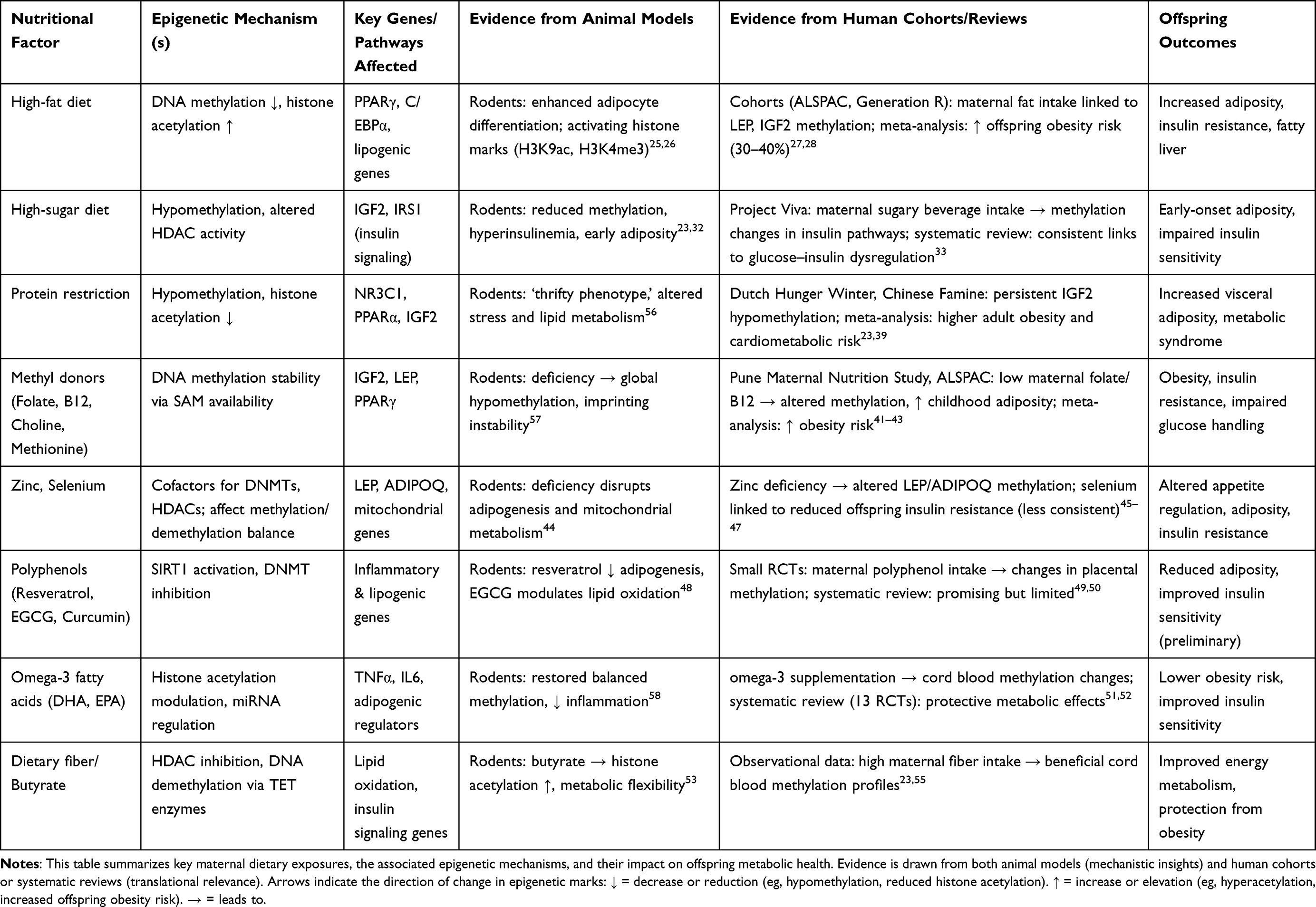 |
Table 1 Maternal Nutritional Factors, Epigenetic Mechanisms, and Offspring Obesity Outcomes |
As summarized in Table 1, maternal nutrition exerts profound epigenetic effects on pathways central to energy balance, adipogenesis, and insulin sensitivity. A consistent pattern emerges: macronutrient excesses (high-fat, high-sugar diets) and deficiencies (protein or one-carbon donor insufficiency) leave persistent epigenetic marks at loci such as IGF2, LEP, and PPARγ, which predict obesity and metabolic dysfunction in offspring. While rodent studies provide mechanistic detail on chromatin remodeling, systematic reviews and cohort data confirm that these associations extend to human populations, particularly in famine cohorts and prospective birth studies. Nonetheless, knowledge gaps remain, especially regarding the long-term persistence of epigenetic marks, the relative contributions of specific micronutrients, and the interactions between maternal diet and other exposures. Future research integrating epigenomics with metabolomics and microbiomics is needed to move from associations toward causal inference, and ultimately to inform precision maternal nutrition strategies.
Maternal Exposure to Environmental Toxicants and Obesity Risk
Beyond nutrition, the intrauterine environment is shaped by maternal exposure to environmental toxicants, many of which function as obesogens, chemicals that perturb developmental pathways to predispose offspring to obesity. These exposures can reconfigure the fetal epigenome through DNA methylation drift, histone modification changes, and altered noncoding RNA expression, thereby disrupting energy metabolism, adipogenesis, and endocrine regulation. The effects are often compounded when toxic exposures coincide with maternal dietary imbalances, amplifying the risk of obesity and related metabolic disorders.59,60 The evidence linking toxicants to obesity risk is growing, but it is important to critically distinguish between correlative associations in human cohorts and causal mechanistic insights from experimental models. Additionally, while numerous studies link toxicants to metabolic dysfunction, translational relevance is strengthened by examining dose–response relationships and identifying exposure thresholds associated with obesity risk.
Endocrine Disruptors (Obesogens)
Bisphenols (BPA and Analogues)
Bisphenol A (BPA), widely used in plastics and food packaging, crosses the placenta and accumulates in fetal tissues. Prenatal BPA exposure has been linked to altered DNA methylation of PPARγ, LEP (leptin), and ADIPOQ (adiponectin), key regulators of adipogenesis.61,62 Histone modifications at estrogen receptor–related genes further enhance lipogenic programming. Emerging evidence suggests that BPA analogues (BPS, BPF), marketed as safer alternatives, may exhibit similar obesogenic epigenetic effects.63 BPA readily crosses the placenta and has been associated in observational studies with altered methylation of PPARγ and LEP, correlating with increased childhood BMI.62 However, most cohort findings are correlative and often rely on single urine measurements, which may not reflect chronic exposure. In contrast, mechanistic animal studies provide stronger evidence, showing that prenatal BPA exposure induces persistent promoter hypomethylation and chromatin remodeling at adipogenic loci.64 Human biomonitoring shows that >90% of pregnant women have detectable urinary BPA.65,66 Multiple birth cohorts report a positive dose–response relationship: higher maternal BPA quartiles correlate with greater offspring BMI scores and altered methylation as well as mesoderm-specific transcript.67 However, most studies use spot urine samples, which may underestimate chronic exposure variability. Animal models show that even low-dose BPA exposure (below current regulatory thresholds of 50 µg/kg/day) induces DNA hypomethylation at adipogenic loci, suggesting that “safe” limits may not adequately protect fetal development.68 BPA analogues (BPS, BPF) demonstrate similar epigenetic effects, but dose–response data remain sparse.63 These findings suggest causality, but the translational gap remains, particularly as human studies often cannot rule out confounding dietary and socioeconomic factors.
Phthalates
Phthalates, commonly found in cosmetics and food contact materials, act as PPARγ agonists, promoting adipocyte differentiation. Epigenetically, maternal phthalate exposure induces hypomethylation at adipogenic and lipid metabolism genes, while altering microRNA networks (such as miR-27 and miR-143) that regulate adipogenesis.69 Human cohort studies (Project Viva, CHAMACOS) have associated prenatal phthalate exposure with increased childhood adiposity, implicating fetal epigenome remodeling as a mechanistic driver, but effect sizes are modest and sometimes sex-specific.23 The strength of evidence here lies in convergence: human cohorts show consistent associations, while animal studies provide plausible molecular mechanisms. Still, a lack of dose–response data in human populations limits causal inference. Epidemiological studies reveal that higher prenatal urinary concentrations of DEHP metabolites are associated with greater childhood adiposity, with some cohorts showing threshold effects above the 75th percentile of exposure.70,71 Mechanistic studies confirm dose-dependent activation of PPARγ and hypomethylation at lipid metabolism genes, but inconsistencies across phthalate subtypes complicate risk assessment.72 Current tolerable daily intakes (TDIs) for phthalates may not account for these epigenetic effects, raising questions about regulatory adequacy.
Heavy Metals
Arsenic
Observational studies link prenatal arsenic exposure with higher obesity and diabetes risk in adulthood, supported by Epigenome-wide association studies (EWAS) findings of persistent methylation changes decades later.73,74 Yet, these human data are largely associative, with limited ability to disentangle arsenic exposure from concurrent nutritional deficiencies (such as low folate). Animal studies provide stronger causal evidence, demonstrating global hypomethylation and specific hypermethylation of GLUT4 and IRS2, leading to impaired insulin signaling. Together, the human and mechanistic evidence is compelling, but further intervention studies (such as folate supplementation trials in exposed populations) are required to establish causality. Dose–response analyses from Bangladeshi and Mexican cohorts demonstrate that higher prenatal arsenic exposure is associated with increased BMI z-scores and insulin resistance in offspring, with risk accelerating at water arsenic levels above 50 µg/L.75 EWAS data reveal progressive global hypomethylation with rising exposure.73 Animal studies corroborate these findings, showing dose-dependent hypermethylation of GLUT4 and IRS2 promoters at environmentally relevant concentrations.76 Notably, the WHO guideline of 10 µg/L may not provide complete protection against epigenetic effects.77
Cadmium and Lead
Both cadmium and lead readily cross the placenta, accumulating in fetal tissues.78 Epidemiological studies suggest that prenatal exposure to cadmium and lead correlates with higher offspring adiposity, but results are inconsistent across cohorts and often confounded by co-exposure to other pollutants.79 Cadmium exposure alters methylation patterns of genes involved in adipogenesis and mitochondrial metabolism, while lead disrupts histone acetylation in neural and metabolic tissues, affecting appetite regulation. These modifications predispose offspring to energy imbalance, early weight gain, and metabolic dysfunction.80 Thus, while biological plausibility exists, human evidence remains heterogeneous, highlighting the need for harmonized exposure measurements and multi-pollutant modeling. Prospective cohorts suggest nonlinear associations, with increased adiposity risk primarily observed in the upper exposure quartiles. For cadmium, cord blood concentrations above 0.44 µg/L have been linked to differential methylation at adipogenic genes.81 Animal studies reveal dose-dependent mitochondrial dysfunction and epigenetic remodeling. Lead exposure shows stronger effects when maternal blood lead levels exceed 5 µg/dL, consistent with public health thresholds, although subtle epigenetic changes occur even below these levels, indicating that no truly safe threshold may exist.82
Food-Borne and Agricultural Toxicants
Aflatoxins
Produced by Aspergillus species in contaminated food, aflatoxins are potent hepatotoxins with emerging roles in metabolic programming. Evidence linking prenatal aflatoxin exposure to obesity risk remains preliminary. While some human studies report associations with impaired growth and liver function, few directly assess obesity outcomes.83 Animal models suggest epigenetic modifications in hepatic metabolic genes, but stronger epidemiological data are needed before causality can be inferred.84
Pesticides and Herbicides
Organochlorine pesticides and glyphosate derivatives have been implicated in metabolic disruption through histone acetylation and microRNA modulation.85 In utero pesticide exposure shifts the chromatin state of adipogenic genes, leading to increased fat deposition.86 Prospective cohorts show inconsistent associations, partly due to variability in exposure measurement. However, dose–response analyses from agricultural communities indicate that higher maternal organophosphate exposure correlates with increased offspring adiposity.87 Animal models show clear dose-dependent histone acetylation changes at adipogenic genes, suggesting plausible thresholds for adverse effects, though human translation remains incomplete. Mechanistic studies, however, demonstrate that pesticides can induce histone acetylation and microRNA modulation at adipogenic genes. The disconnect between correlative human evidence and mechanistic plausibility reflects the challenge of exposure assessment in epidemiological settings, where pesticide mixtures and variable timing of exposure complicate interpretation.
Acrylamide and Other Processed Food Contaminants
Acrylamide, formed during high-temperature cooking, has been shown in animal studies to induce fetal DNA methylation changes in pancreatic and hepatic tissues. These epigenetic disruptions compromise insulin secretion and glucose handling, setting the stage for obesity and diabetes.88 Human data are limited to dietary intake estimates, which are subject to recall bias, and few cohorts have linked acrylamide biomarkers to obesity outcomes. Dietary exposure estimates from the Norwegian Mother and Child Cohort (MoBa) study suggest that higher quartiles of maternal acrylamide intake correlate with lower birth weight and early growth changes, though obesity-specific outcomes remain uncertain.89 Rodent models confirm dose-dependent DNA methylation changes in pancreatic tissue at levels comparable to high human dietary intake.90
Emerging Toxicants: PFAS, Microplastics, and the Expanding Maternal Exposome
In addition to conventional obesogens such as phthalates, and BPA, a growing set of emerging contaminants is now recognized within the maternal exposome. Per- and polyfluoroalkyl substances (PFAS) are environmentally persistent and bioaccumulative compounds can cross the placenta and they have been linked in epidemiological studies to altered birth weight trajectories, adiposity, and dyslipidemia in offspring.91,92 Experimental data suggest that PFAS can modulate nuclear receptor signaling and induce persistent changes in DNA methylation and histone modifications in metabolic tissues.93,94 Additionally, micro- and nanoplastics which are detectable in human placenta and cord blood, represent another rapidly evolving exposure. These particles and their metabolites may induce oxidative stress, low-grade inflammation, and epigenetic reprogramming in placental and fetal cells.95 Thus, incorporating PFAS, microplastics, and other emerging contaminants into DOHaD frameworks is important to embrace the full spectrum of real-world maternal exposures that may converge on shared epigenetic pathways governing energy equilibrium and adiposity.
Translational Evidence from Human Cohorts
Longitudinal studies provide compelling evidence linking maternal toxicant exposure to epigenetic reprogramming and obesity risk. The Dutch Hunger Winter cohort revealed that famine-exposed individuals exhibited altered methylation at IGF2, compounded by exposure to environmental pollutants postnatally.12 Historically, placental epigenomics has moved from candidate loci toward integrative, multi-omics frameworks. Large-scale studies now combine placental DNA methylation, gene expression, and genetic variation to map regulatory circuits linking birthweight loci to placental transcriptional programs, thereby grounding DOHaD concepts in specific molecular pathways.96 Complementary work integrates placental epigenomic profiles with maternal diet, microbiota and metabolite signatures, suggesting that maternal nutritional and microbial milieus can remodel placental chromatin and transcriptomes with consequences for fetal growth and later metabolic risk.97 These placental multi-omics studies accentuate the placenta as a central hub where nutritional and toxicant signals converge on shared regulatory networks, rather than acting solely through cord blood or peripheral tissues.
The emergence of exposome-wide association studies (ExWAS) has been an important methodological development that scientifically test large panels of environmental exposures against obesity-related phenotypes, and it is analogous to genome-wide association studies.98 Early-life exposome projects such as HELIX and related cohorts have applied ExWAS frameworks to prenatal and childhood exposure data, revealing clusters of air pollutants, metals, and lifestyle factors associated with childhood BMI and waist circumference, often with modest but reproducible effect sizes.99–101 Large exposome and omics consortia such as the Pregnancy and Childhood Epigenetics (PACE) consortium and European early-life exposome initiatives (eg, HELIX) now combine high-dimensional exposure assessment with epigenomic, metabolomic, and microbiomic profiling to identify molecular signatures linking complex maternal exposomes to childhood overweight and metabolic traits. These projects illustrate both the promise and the controversy of exposome-wide approaches. PACE consortium pools epigenome-wide methylation data from multiple birth cohorts to perform meta-analyses of maternal characteristics and early-life exposures in relation to offspring CpG methylation and downstream cardiometabolic phenotypes, using harmonized pipelines and causal-inference extensions.102 Furthermore, the Western Australian Pregnancy Cohort (Raine) Study provides multigenerational life-course data that have been used to evaluate how early-life growth, adiposity and cardiometabolic traits track into adolescence and adulthood.103 In the United States, the Environmental influences on Child Health Outcomes (ECHO) Program unites loads of cohorts to evaluate how preconception-to-childhood environments shape obesity and related health outcomes, contributing to unique sample size and exposure diversity.104 While they reveal coherent exposure clusters and candidate molecular pathways, reported effect sizes are often modest, signatures can vary across cohorts and tissues, and results are sensitive to analytical choices in mixture modelling and multiple-testing correction.
Recent birth cohort studies (ALSPAC, Generation R, CHAMACOS) demonstrate associations between prenatal BPA, phthalate, and heavy metal exposures with differential DNA methylation patterns in cord blood, correlating with higher BMI and adiposity in childhood and adolescence.23,105 While DNA methylation remains the most extensively characterized mechanism, these findings likely represent only one layer of a broader epigenetic landscape that also includes histone lactylation, chromatin accessibility, and 3D genome organization, all of which may be sensitive to maternal nutritional and toxicant cues. These findings underscore the clinical relevance of maternal toxicant exposures in shaping the obesity epidemic. However, most of these findings remain associative and are limited by confounding and the difficulty of separating toxicant effects from nutritional or socioeconomic variables. By contrast, animal and in vitro models provide causal evidence of specific epigenetic mechanisms but may not fully capture human pregnancy physiology. Remarkably, not all human studies report positive associations between maternal toxicant exposure and offspring adiposity. Several birth cohorts have observed weak, null, or even inverse associations for specific compounds or time windows, particularly when exposure is assessed using single spot urine samples or limited biomarkers. For instance, while some analyses in ALSPAC, Generation R, and CHAMACOS cohorts link higher prenatal BPA, phthalate, or pesticide levels to increased childhood BMI, other analyses within the same or similar cohorts report no clear dose–response relationship, sex-restricted effects, or outcomes that attenuate after adjustment for socioeconomic and lifestyle confounders.70,106 Inconsistencies are also evident for heavy metals, where associations with adiposity often depend on exposure level, co-exposures, and child age at follow-up. These divergent findings underscore that obesogenic effects of toxicants are not universal; rather, they are contingent on exposure timing, dose, and mixture, underlying nutritional status, epigenetic susceptibility, and residual confounding.
To clarify how different classes of maternal toxicants exert their effects on the fetal epigenome, Table 2 summarizes representative compounds, their primary epigenetic targets, supporting evidence, and the downstream obesity-related outcomes.
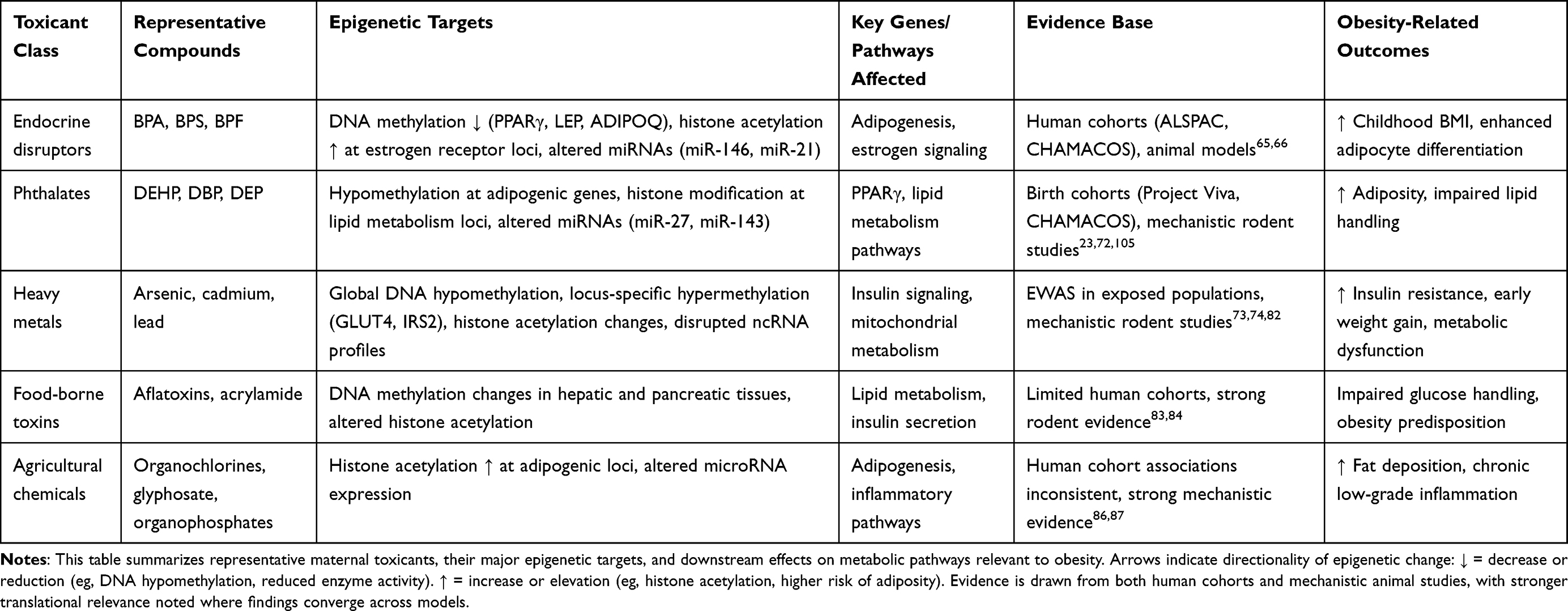 |
Table 2 Maternal Toxicant Classes, Primary Epigenetic Targets, and Offspring Obesity Outcomes |
As shown in Table 2, a consistent theme emerges: endocrine disruptors (BPA, phthalates) and heavy metals (arsenic, cadmium, lead) have the strongest translational evidence, with human cohort associations reinforced by mechanistic studies demonstrating epigenetic reprogramming of adipogenic and insulin signaling pathways. By contrast, food-borne toxins and agricultural chemicals show clear mechanistic plausibility in animal models but remain underexplored in human populations, where exposure assessment and co-exposure confounding limit causal inference. Across toxicant classes, DNA methylation appears to be the most frequently disrupted mechanism, although histone modifications and noncoding RNAs are increasingly recognized as critical mediators. The convergence of multiple epigenetic marks across different compounds suggests shared obesogenic pathways, underscoring the importance of integrating multi-omics approaches in future studies.
Nutrition–Toxicant Interactions: Synergy or Antagonism?
The risk of obesity programmed in utero rarely arises from maternal nutrition or toxic exposures in isolation. Instead, these factors frequently intersect, particularly in vulnerable populations, creating a complex exposome where nutritional status can either exacerbate or buffer toxicant-induced epigenetic disruptions. Comprehending this interplay is critical for elucidating mechanisms of metabolic programming and for designing targeted interventions. More so, human cohort studies increasingly highlight the translational relevance of these interactions.
Dual Burden in Low-Resource Settings
In low- and middle-income countries, pregnant women are often simultaneously exposed to nutritional deficiencies (protein, micronutrients, methyl donors) and high levels of environmental pollutants (air pollution, pesticides, heavy metals). This “dual burden” magnifies epigenetic instability during fetal development.107 Chronic deficiencies in protein, folate, vitamin B12, and micronutrients often coexist with elevated exposures to arsenic-contaminated groundwater, pesticide residues in food, and indoor air pollution from biomass fuels. These overlapping stressors magnify epigenetic instability during fetal development, predisposing offspring to obesity and metabolic disease later in life.108
Human Evidence
In Bangladesh, arsenic exposure combined with low folate status has been associated with global DNA hypomethylation in cord blood and higher childhood adiposity compared with either exposure alone.109
A Chinese famine follow-up study found that individuals prenatally exposed to both famine and industrial pollutants had more pronounced hypomethylation of INSR and IGF2 compared to famine exposure alone, translating into higher rates of adult obesity.110
The INfancia y Medio Ambiente (INMA, Spain) cohorts demonstrated that maternal smoking and cadmium exposure had stronger associations with offspring body weight.111 A similar study reported significant increased anogenital index in male offspring, suggesting altered androgenic signaling.112 According to Chen et al113 in their seven European birth cohorts study, an inflammatory, suboptimal maternal prenatal diet may negatively affect offspring body composition and increase the risk of overweight and obesity, particularly in late childhood. Thus, promoting the use of nutritious, anti-inflammatory maternal prenatal diet may aid in the prevention of childhood obesity. These findings suggest that nutritional deprivation reduces epigenetic resilience, making the developing fetus more vulnerable to obesogen-induced reprogramming.
Mechanistic Evidence
Malnutrition reduces epigenetic resilience by limiting one-carbon metabolites such as folate, methionine, and choline, thereby lowering S-adenosylmethionine (SAM) pools. When coupled with pollutant exposure, DNA methylation fidelity is impaired, leading to stochastic epimutations in metabolic genes (IGF2, PPARγ, LEP).114
Evidence from rat and goat studies reported that protein-energy restriction during gestation alters chromatin remodeling complexes, leaving fetal tissues more vulnerable to obesogen-induced histone modifications.115,116
Pollutant co-exposure (eg, arsenic in drinking water + poor folate intake) further drives global hypomethylation and specific hypermethylation of metabolic genes, compounding obesity risk postnatally.117,118 Thus, in low-resource settings, the interaction of malnutrition and pollutants generates a compounding epigenetic burden, locking offspring into obesogenic trajectories even before birth.
Policy Implications
The dual burden has profound equity dimensions. Nutritional supplementation programs (like folate and iron fortification) are rarely paired with environmental health policies in LMICs, leaving vulnerable populations without integrated protection. Moreover, weak regulatory systems mean that pesticide residues and heavy metals often exceed international safety thresholds, while poor populations are the least able to afford safe food or water alternatives. Without interventions, LMICs risk a “double epidemic” of persistent undernutrition and rising obesity, widening health disparities relative to high-income countries.
Protective Role of Nutrients Against Toxicant-Induced Epimutations
Adequate maternal nutrition can buffer against toxicant-induced epigenetic insults, highlighting a protective antagonism between nutrients and pollutants. In high-income countries, access to balanced diets may mitigate pollutant effects, while in LMICs, nutritional inadequacy limits this resilience. For example, a recent randomized control trials in Bangladesh found that folate supplementation reduced arsenic-induced hypomethylation, yet supplementation coverage remains uneven globally, particularly in rural and low-resource settings.119
Antioxidants
Nutrients such as vitamins C and E, polyphenols, and carotenoids reduce reactive oxygen species (ROS) generated by pollutants. This limits oxidative DNA damage and prevents ROS-driven activation of histone-modifying enzymes (HDACs, HATs) that promote pro-adipogenic chromatin states.120 Polyphenols like resveratrol and EGCG activate SIRT1, a histone deacetylase that suppresses inflammatory and adipogenic gene expression, counteracting BPA- and phthalate-induced chromatin activation.121
Methyl Donors (Folate, Choline, B12, Methionine)
Adequate intake of methyl donors maintains SAM-dependent methylation reactions, stabilizing DNA methylation and preventing pollutant-induced hypomethylation at key metabolic loci. For example, folate supplementation mitigates arsenic-induced global hypomethylation and prevents obesity-related gene dysregulation in animal models.119,122
Trace Elements (Zinc, Selenium)
Zinc as cofactors for epigenetic enzymes (DNMTs, TETs, HDACs), maintaining balanced activity in the presence of toxicant stress.123 Selenium in particular counteracts cadmium-induced epigenetic dysregulation in hepatic metabolic pathways.124 Collectively, these findings suggest that nutritional adequacy functions as an epigenetic shield, buffering fetal development against obesogen-induced programming.
Human Evidence
In Project Viva (USA), higher maternal folate intake attenuated the association between prenatal polyfluoroalkyl substances (PFAS) exposure and adverse birth outcomes, with protective effects persisting into early childhood BMI trajectories.125
Solomon et al23 found that prenatal phthalate exposure altered DNA methylation patterns in Mexican-American neonates’ cord blood, highlighting the impact of environmental exposures on their health and development. This corroborates an earlier research by Huen et al126 concerning Mexican-American infants from the longitudinal birth cohort CHAMACOS. However, experimental and human research indicate that maternal consumption of diets high in antioxidants and polyphenols can affect DNA methylation and other epigenetic mechanisms, implying a possible nutritional route to alleviate environmental impacts.127–129
In prospective cohorts, prenatal mercury exposure correlates with variations in cord-blood DNA methylation; concurrently, omega-3 consumption has been associated with methylation alterations near pro-inflammatory genes (such as TNF and IL6), indicating that nutrition may influence epigenetic inflammatory pathways, although this interaction has not been documented in Generation R.130,131
Together, these findings provide compelling evidence that maternal diet quality can mitigate toxicant-induced epimutations, offering a feasible prevention strategy.
Policy Implications
Embedding environmental exposure reduction into existing maternal nutrition programs could be highly cost-effective. For example, combining folate supplementation with arsenic-free water initiatives in South Asia could address both nutritional and toxicological drivers of epigenetic disruption simultaneously.
More Case Studies in Human Cohorts
Historical famine studies and modern cohort evidence (eg, Dutch Hunger Winter, Chinese Famine, Project Viva, CHAMACOS, and INMA) underscore that maternal nutrition modifies the effects of toxicant exposures on offspring obesity risk. While high-income settings demonstrate protective effects of antioxidants and omega-3s against pollutants, LMIC settings reveal a compounding risk where undernutrition exacerbates pollutant-induced epimutations. The following human cohort studies provide real-world evidence of the nutrition–toxicant interplay in shaping obesity risk through epigenetic pathways:
- Dutch Hunger Winter (1944–1945): Individuals exposed to famine in utero displayed persistent hypomethylation at the IGF2 gene six decades later. Subsequent analyses revealed that those exposed to higher environmental pollutants postnatally had exacerbated obesity risk, highlighting a cumulative effect.23,132
- Chinese Famine (1959–1961): Prenatal undernutrition was associated with altered DNA methylation at INSR (insulin receptor) and higher risk of obesity in adulthood. These effects were stronger in regions with higher environmental pollutant exposure, underscoring the synergistic burden of famine and toxicants.110
- Project Viva (USA): This prospective birth cohort demonstrated that prenatal phthalate and BPA exposure correlated with altered DNA methylation in cord blood, predicting increased adiposity in childhood. Interestingly, maternal diets richer in folate and antioxidants attenuated these associations, supporting a protective nutrient–toxicant antagonism.33,125
- Exposome–Nutrition Interactions in Recent Cohorts: Multi-omics approaches in European and North American cohorts reveal that maternal diet quality modifies the relationship between air pollution exposure and methylation changes at obesity-related loci, further linking nutrition and toxicant exposures in real-world settings.101,133 These findings emphasize the necessity of integrating nutrition and toxicology in exposome research.
Policy Implications
Global maternal health strategies must address the structural inequities that sustain this dual burden. This includes strengthening food safety regulations to reduce pesticide and aflatoxin exposure, scaling up micronutrient supplementation and food fortification, and integrating environmental monitoring into antenatal care. Without coordinated policy action, intergenerational obesity risks will remain disproportionately higher in LMICs, reinforcing cycles of health inequity.
To illustrate how maternal diet can exacerbate or buffer toxicant-induced epigenetic alterations, Table 3 compiles evidence from key human cohorts and famine studies that highlight the interactive effects of nutrition and environmental exposures on obesity risk.
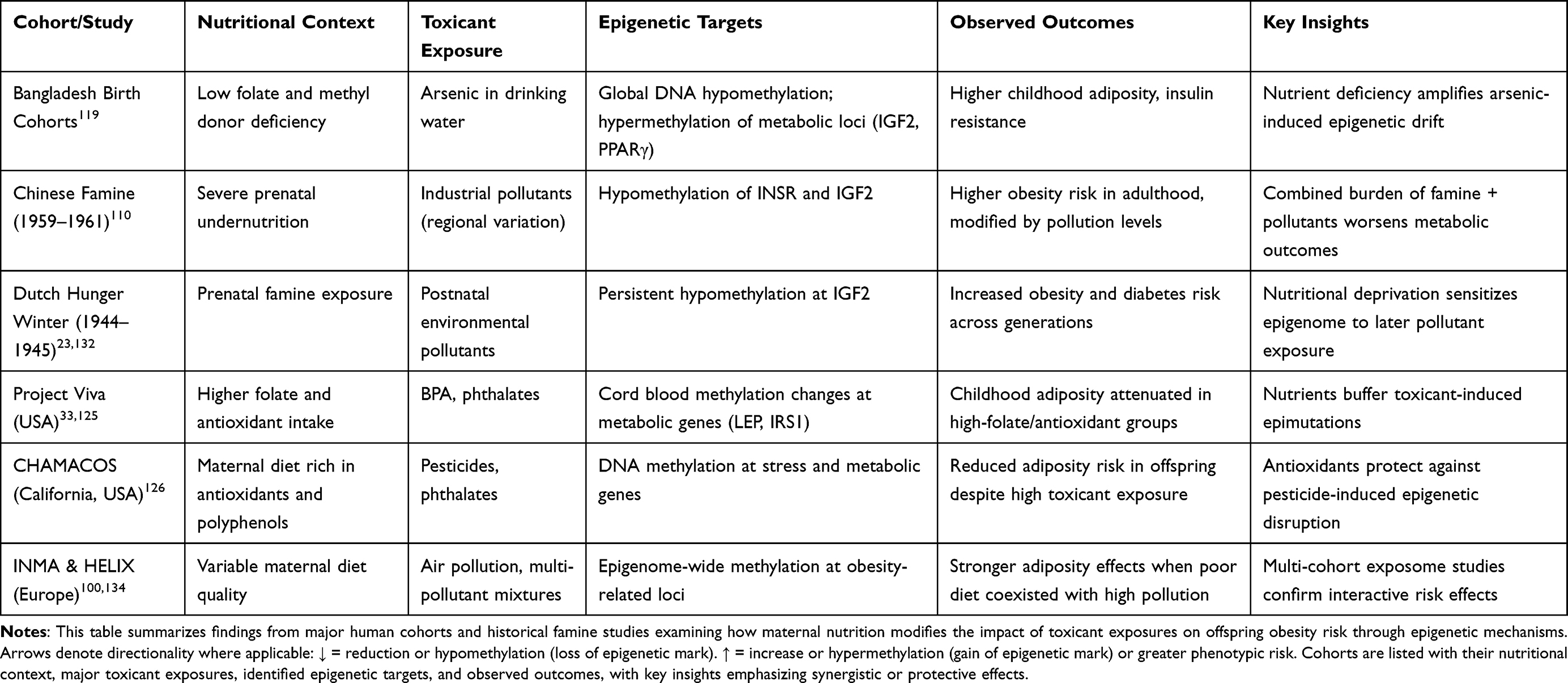 |
Table 3 Human Evidence of Nutrition–Toxicant Interactions in Epigenetic Programming of Obesity |
As summarized in Table 3, evidence from diverse human cohorts demonstrates that maternal nutrition significantly modifies the impact of toxicant exposures on offspring obesity risk through epigenetic mechanisms. Cohorts from famine-exposed populations (Dutch Hunger Winter, Chinese Famine) highlight how undernutrition amplifies pollutant-induced epimutations with long-lasting metabolic consequences, while more studies (Project Viva, CHAMACOS, INMA/HELIX) reveal that adequate folate, antioxidants, and omega-3 fatty acids can attenuate or even neutralize the obesogenic effects of phthalates, pesticides, and heavy metals. These findings underscore the translational importance of maternal diet quality in shaping the fetal exposome, suggesting that nutritional interventions may offer a practical and scalable means of mitigating toxicant-driven obesity programming.
Microbiome–Epigenome Crosstalk in Maternal Programming of Obesity
The maternal gut microbiome is increasingly recognized as a key intermediary linking diet, environmental exposures, and fetal development. Microbial communities metabolize nutrients and xenobiotics into bioactive compounds that cross the placenta or are delivered via breastmilk, directly influencing the fetal and neonatal epigenome. These interactions operate through short-chain fatty acids (SCFAs), bile acids, tryptophan metabolites, and microbial toxins, which serve as substrates or inhibitors of chromatin-modifying enzymes.135 Perturbations in maternal diet or toxicant exposure can shift microbial composition, thereby altering the epigenetic landscape of developing metabolic tissues and programming offspring obesity risk.23 While rodent studies provide direct causal data, human findings are predominantly associative, and distinguishing correlation from causation remains a critical challenge.
Maternal Diet Shapes Gut Microbiota and Epigenetic Signaling
Maternal nutritional status directly modulates the composition and function of the gut microbiota, which in turn generates metabolites with epigenetic activity. For instance, SCFAs produced from dietary fiber fermentation, cross the placenta and act as histone deacetylase (HDAC) inhibitors, enhancing histone acetylation at genes regulating lipid oxidation and insulin sensitivity.136 Butyrate also promotes DNA demethylation by serving as a substrate for α-ketoglutarate–dependent dioxygenases (TET enzymes), facilitating chromatin remodeling toward metabolic flexibility.137
Maternal diets rich in fats alter gut microbiota-derived bile acid pools, which engage nuclear receptors (FXR, TGR5) and modulate histone acetylation in fetal liver. Dysregulated bile acid signaling predisposes offspring to hepatic steatosis and obesity.138 Similarly, indole derivatives produced by microbial metabolism of tryptophan influence aryl hydrocarbon receptor (AhR) activity, which regulates histone acetylation and noncoding RNAs in immune–metabolic pathways.139 Maternal tryptophan availability, therefore, indirectly programs offspring immunity and adiposity via microbiome–epigenome signaling. Thus, maternal diet establishes a microbiota–metabolite–epigenome axis that can either protect against or promote obesity, depending on nutrient quality.
In sum, rodent studies provide strong causal evidence: SCFAs such as butyrate cross the placenta, act as HDAC inhibitors, and directly increase histone acetylation at genes regulating lipid oxidation and insulin sensitivity.140 Supplementation experiments confirm that maternal butyrate administration alters fetal chromatin states and reduces obesity risk.23,141
Longitudinal Human Evidence
Human evidence, however, is largely associative. A prospective cohort study conducted by Fu et al142 aimed to assess the correlation between dietary fibre consumption and short-chain fatty acid-producing bacteria during critical illness, involving 129 patients in the intensive care unit. The main outcome was the relative abundance of SCFA-producing bacteria after 72 hours in the ICU, determined by 16S rRNA gene sequencing of the rectal swab taken at 72 hours. The group with elevated fibre consumption had reduced stomach distension, with no rise in diarrhoea or other negative outcomes.
Emerging evidence challenges the once-held paradigm of a sterile intrauterine environment, underscoring that SCFAs, products of maternal fiber fermentation can reach the fetal compartment and potentially modulate DNA methylation landscapes.143
Epidemiological and mechanistic data suggest that maternal consumption of dietary fiber promotes an increase in SCFA-producing gut microbes, which may in turn influence the fetal epigenome via altered DNA methylation patterns in cord blood.144
The above studies report correlations between maternal fiber intake, increased abundance of SCFA-producing microbes, and differential DNA methylation in cord blood. Yet, direct causal pathways are difficult to establish because SCFA levels are typically inferred from diet or microbiome composition, rather than measured in maternal or cord blood. Thus, while human studies support plausibility, causality remains to be proven.
In analyses within the KOALA Birth Cohort (Netherlands), maternal probiotic use during the last month of pregnancy was included as a confounder when examining how early infant gut microbiota composition related to atopic outcomes.145 Additionally, the INFANTMET study (Ireland) followed maternal–infant dyads and demonstrated that infant gut microbiota development during the first six months of life is strongly influenced by delivery mode and feeding method, with associated changes in urinary metabolite profiles.146 These findings support the hypothesis that maternal diet can shape the maternal–infant microbiome axis, which in turn modulates epigenetic programming of energy balance.
Toxicants Disrupt Microbiota and Epigenetic Pathways
Environmental toxicants disrupt maternal microbial ecology, thereby reprogramming epigenetic signaling in offspring. Mechanistic studies in animals show causality: exposure to BPA, arsenic, or glyphosate reduces microbial diversity and SCFA production, which in turn weakens HDAC inhibition, leading to tighter chromatin states at insulin sensitivity genes. Experimental microbiota transfer studies confirm that maternal toxicant-induced microbiome alterations can be sufficient to reprogram offspring metabolism. Endocrine Disruptors (BPA, Phthalates) reduce microbial diversity and favor pro-inflammatory taxa (like Proteobacteria), leading to increased production of lipopolysaccharide (LPS).147 LPS induces chronic low-grade inflammation and alters histone acetylation in fetal immune cells, predisposing to metabolic dysfunction.148,149
Heavy Metals (Arsenic, Cadmium, Lead) alter gut microbial composition, reducing SCFA producers (eg, Faecalibacterium) while enhancing toxin-producing strains.150 Loss of SCFAs weakens HDAC inhibition, resulting in tighter chromatin states at genes regulating insulin sensitivity.151 Glyphosate and organophosphates disrupt microbial aromatic amino acid metabolism, reducing indole derivatives that regulate AhR-mediated epigenetic signaling. This fosters pro-adipogenic chromatin states in fetal tissues.85
Overall, toxicant-induced microbiome dysbiosis translates into epigenetic silencing of protective metabolic pathways and activation of obesogenic programs, amplifying obesity risk.
Longitudinal Human Evidence
In humans, findings remain associative. For example, the Lifelines NEXT cohort (Netherlands) found that prenatal exposure to air pollution was associated with altered maternal and infant gut microbiota diversity, which correlated with methylation changes at metabolic genes in cord blood.152 However, it cannot definitively prove that microbial shifts mediated the epigenetic changes.
In the CHAMACOS cohort (USA), prenatal exposure to DDT and DDE was associated with increased risk of overweight or obesity by age 9 in male children.153,154 Separately, analyses of prenatal exposure to mixtures of phthalates and phenols revealed epigenetic alterations such as gestational-age acceleration that are linked to increased obesity risk.23,155 Maternal pesticide exposure no doubt is linked to reduced abundance of beneficial SCFA-producing microbes, and these microbial shifts could be associated with epigenetic marks in offspring linked to obesity risk. Nevertheless, confounding by direct toxicant effects cannot be ruled out.
These studies strengthen translational relevance by demonstrating real-world links between environmental toxicants, microbiome shifts, and epigenetic regulation in humans.
Transplacental and Breastmilk-Mediated Microbial–Epigenetic Interactions
Maternal microbiome-derived metabolites reach the fetus not only via the placenta but also through breastmilk, shaping early postnatal epigenetic trajectories. SCFAs, bile acids, and indoles cross the placenta and act directly on fetal hepatocytes, adipocytes, and hypothalamic neurons, establishing chromatin marks that regulate energy balance.156 For example, butyrate-mediated HDAC inhibition in fetal liver promotes histone acetylation at lipid oxidation genes, improving metabolic efficiency.157
Breastmilk contains microbial metabolites (such as SCFAs), immunomodulatory microRNAs, and microbial extracellular vesicles that shape infant gut colonization and epigenetic regulation.158 Epigenetic modifications induced by breastmilk-derived miRNAs (miR-148a, which regulates DNMT1) persist into childhood, influencing adiposity and metabolic health.159 Critically, the prenatal–neonatal transition is a sensitive period in which maternal microbiome-derived signals leave lasting epigenetic imprints. Disruption during this window (eg, maternal antibiotic use, pollutant exposure, or poor diet) has disproportionate long-term effects on offspring obesity risk.160
Longitudinal Human Evidence
While rodent experiments demonstrate causality, human evidence is suggestive but not causal. A study by Fehr et al161 examined mother–infant pairs from the Canadian Healthy Infant Longitudinal Development (CHILD) cohort, showing that bacteria present in breastmilk co-occur with those found in infant stool, suggesting that breastmilk may seed the infant gut microbiome, especially influenced by breastfeeding practices such as exclusivity and pumping.
Similarly, the Finnish HELMi cohort reported that maternal antibiotic use altered breastmilk microbiota and metabolites, which were associated with differences in DNA methylation patterns in infants, correlating with adiposity outcomes at age 3.
Evidence from CHILD birth cohorts indicates that maternal antibiotic exposure and breastfeeding can influence breastmilk and infant microbiota composition,162 while breastfeeding has also been associated with DNA methylation differences in Avon Longitudinal Study of Parents and Children (ALSPAC) cohort in infants.163 Moreover, the result of the Pregnancy and Childhood Epigenetics (PACE) Consortium established an association between pre-pregnancy maternal BMI and methylation in newborn blood DNA which has been linked to later adiposity outcomes.27 The Finnish Health and Early Life Microbiota (HELMi) cohort, with its integrated collection of breastmilk, infant microbiota, and DNA samples, is well positioned to investigate these interrelated mechanisms.164
These longitudinal data confirm that maternal microbiome–epigenome interactions are not transient but persist into early childhood, influencing obesity trajectories. However, causality remains unresolved because breastmilk metabolites coexist with maternal nutrients, hormones, and toxicants, all of which could contribute to epigenetic regulation.
Transgenerational Epigenetic Inheritance
The concept that maternal nutrition and toxicant exposures can influence not only the immediate offspring but also subsequent generations represents one of the most profound implications of the DOHaD framework. Unlike genetic mutations, which are fixed, epigenetic modifications are dynamic and reversible, yet certain marks established in germ cells can escape epigenetic reprogramming during gametogenesis and early embryogenesis. This raises the possibility of germline epimutations that transmit altered metabolic phenotypes including obesity susceptibility across multiple generations.165
Germline Epimutations Induced by Maternal Diet and Toxins
Nutritional perturbations during pregnancy can induce persistent epimutations in germline DNA of the developing fetus. Maternal overnutrition like high-fat diets during gestation alter DNA methylation and histone modifications in fetal oocytes and spermatogonia, particularly at genes controlling adipogenesis (PPARγ, C/EBPβ). These germline changes are propagated into the next generation, predisposing to increased adiposity even in the absence of continued maternal overnutrition.23 Folate or methyl-donor deficiency reduces SAM availability, leading to global DNA hypomethylation.166 In primordial germ cells where genome-wide demethylation and remethylation occur, this can disrupt the fidelity of imprinting marks.167,168 Indeed, paternal folic acid deficiency in mice has been shown to alter imprinted gene methylation (including H19, Snrpn, Peg3) and increase adverse outcomes in F2 offspring169 while recent work indicates that such methylation changes can persist into F3 progeny.170
Toxicant exposures including obesogens such as bisphenols and phthalates, as well as heavy metals like cadmium, induce heritable epigenetic reprogramming in germline cells. For example, Manikkam et al,171 showed that gestating (F0) rats exposed to a mixture of plastic-derived endocrine disruptors including BPA) during fetal gonadal sex determination produced F3-generation offspring exhibiting increased obesity (adiposity), in conjunction with sperm epimutations involving promoter-region DNA methylation changes. Thus, both diet and toxins act as epigenetic architects of germline memory, encoding metabolic vulnerability that extends beyond direct exposure. These findings support the concept of true epigenetic inheritance beyond direct exposure.
Imprinting Errors and Obesity Inheritance
Genomic imprinting, monoallelic gene expression regulated by parent-of-origin-specific DNA methylation, is particularly vulnerable to nutritional and toxicant perturbations. Disruptions in imprinted loci contribute to obesity and metabolic dysfunction across generations.172,173
Imprinted genes, such as IGF2 and H19, are unusually vulnerable to nutritional and toxicant perturbations because their parent-of-origin–specific methylation states are normally preserved through embryonic reprogramming.174 Altered imprinting at these loci has been linked to obesity risk in both rodent and human studies, supporting their role as potential mediators of multigenerational inheritance. A study by Wu et al175 using mice demonstrated that paternal exposure to a high-fat diet leads to altered methylation of the Igf2/H19 imprinting control region (ICR) in germ cells. This epigenetic change perturbs hepatic glucose metabolism in the offspring, implicating transmission of impaired metabolic health across generations.
Periconceptual exposure of individuals to the Dutch famine was associated with DNA hypomethylation in the differentially methylated region (DMR) that regulates the imprinted gene insulin-like growth factor-2 (IGF2), as assessed in the peripheral blood later in life.176 This hypomethylation has been proposed as a plausible mechanism linking low birth weight to higher risk of later-life metabolic conditions, such as adiposity, insulin resistance, diabetes, and hypertension.177 Animal models further indicate that similar epigenetic changes can persist across multiple generations (for instance, maternal protein-restriction causing promoter hypomethylation and metabolic dysfunction in F1 and F2).178,179
H19 is a long noncoding RNA co-regulated with IGF2 at the imprinted IGF2/H19 locus. H19 exhibits hypermethylation following maternal undernutrition and toxicant exposure, leading to dysregulated growth signaling and enhanced obesity susceptibility in offspring.180 Altered methylation at IGF2/H19 around birth/childhood is associated with greater adiposity/overweight risk in offspring.181,182
Furthermore, altered methylation at imprinted loci such as MEG3, PLAGL1, and PEG3 has been observed in offspring of obese parents, with maternal obesity associated with decreased methylation at MEG3 and increased methylation at PLAGL1, and paternal obesity linked to decreased methylation at PEG3.183 Additionally, epigenetic dysregulation of these loci has been associated with disrupted energy metabolism and transgenerational obesity phenotypes in animal models.184 Imprinting errors are particularly consequential because they bypass the genome-wide epigenetic reprogramming events that typically reset the epigenome during early embryogenesis, ensuring that these obesity-related epimutations persist.
Mechanistic Gaps: How Do Marks Escape Reprogramming?
Despite compelling findings, the mechanisms by which epigenetic marks escape the two major waves of reprogramming in mammals remain poorly understood:
- Germline Reprogramming: During gametogenesis, most DNA methylation marks are erased to reset developmental potential. However, some imprinted regions and transposable element–associated loci resist erasure.185 How diet- or toxin-induced epimutations persist at non-imprinted loci (eg, metabolic genes) is unclear.
- Early Embryonic Reprogramming: After fertilization, the zygote undergoes a second round of genome-wide demethylation, followed by de novo methylation. Certain regions, such as repetitive elements and imprinted loci, retain partial methylation.186 It remains uncertain how environmental exposures introduce “protected” epimutations that can evade this process.
- Histone Modifications and Retained Nucleosomes: In sperm, most histones are replaced by protamines during maturation, but a small fraction of nucleosomes are retained at developmental genes. Animal studies suggest that exposure-induced histone modifications at these sites may be inherited, but direct evidence in humans is lacking.187
- Small Noncoding RNAs: Germline transmission of altered microRNAs and tRNA fragments has been implicated in rodent models of diet- and toxin-induced obesity, but whether these molecules persist long enough to influence human embryogenesis is debated.188
These unresolved mechanisms highlight a major knowledge gap: while rodent studies demonstrate plausible escape routes, the molecular basis for true transgenerational inheritance in humans remains speculative.
Evidence from Rodent and Human Cohorts
Rodent models provide robust mechanistic evidence, while human famine and exposure cohorts demonstrate transgenerational effects in real-world populations. These findings highlight the urgent need for preventive maternal interventions, not only to protect immediate offspring but also to break cycles of obesity inheritance spanning generations.
Rodent Models
Numerous studies confirm true transgenerational inheritance, with obesity phenotypes persisting into the F3 generation despite the absence of continued exposure. These models provide compelling proof-of-principle for germline transmission of epimutations.
- Maternal High-Fat Diets: Multiple studies show that offspring of high-fat-fed animals exhibit obesity and insulin resistance, which persist into F2 and F3 generations despite return to a normal diet. These phenotypes are associated with persistent methylation changes at PPARγ and leptin signaling genes in sperm and oocytes.23,189,190
- Toxicant Models: Prenatal exposure to a mixture of BPA and phthalates induces transgenerational obesity in rodents, with F3-generation animals displaying increased adiposity and associated sperm DNA methylation epimutations, implicating germline epigenetic alterations.171 Additionally, multiple reviews summarize that epigenetic mechanisms, including DNA methylation and histone modifications, underlie the transgenerational inheritance of obesogenic effects following early-life exposure to EDCs like BPA.59 In contrast, while chronic arsenic exposure has been shown to drive transgenerational genotoxicity and global DNA methylation changes into the F3 generation (eg, in rat models), true evidence for F3-generation obesity via hepatic or adipose-specific epigenomic alterations remains to be demonstrated.191,192
Human Evidence
Human evidence is more controversial. Historical famine studies (Dutch Hunger Winter, Chinese Famine) show persistent DNA methylation differences at metabolic loci such as IGF2 in exposed individuals, with some reports suggesting effects in grandchildren. Studies have also identified methylation differences in other metabolic gene loci such as IL10, INSIGF, LEP, ABCA1, MEG3, and GNASAS in individuals exposed to the Dutch famine.41,193 The Överkalix cohort linked paternal prepubertal nutrition to altered health outcomes in grandchildren, suggesting possible germline-mediated effects.194 However, these findings remain associative rather than causal and are subject to major methodological challenges, including small sample sizes, confounding by shared environments, and the inability to disentangle multigenerational exposure (F0, F1, F2) from true transgenerational inheritance (effects in F3 and beyond without direct exposure).
Together, rodent and human evidence underscore that maternal nutrition and toxicant exposures can leave epigenetic “scars” in germline cells, perpetuating obesity risk across generations even in the absence of direct exposure.
Ongoing Controversy and Research Needs
The notion of transgenerational inheritance in humans remains highly debated. Some researchers argue that persistent multigenerational effects may reflect shared social, cultural, and nutritional environments rather than stable germline epimutations. Others point to limited but intriguing evidence from famine and epidemiological cohorts as suggestive of transgenerational epigenetic memory. There is therefore need for cautious framing: while rodent data support causality, human evidence should be described as preliminary, associative, and not yet definitive.
Future research will require large, multi-generational cohorts with detailed exposure, genetic, and epigenetic data, coupled with advanced statistical models that can disentangle shared environment from true inheritance. Until then, the idea of transgenerational epigenetic inheritance in humans should be regarded as a promising but unproven hypothesis.
Sex-Specific Epigenetic Vulnerability
One of the most intriguing and underexplored aspects of developmental programming of obesity is the sex-specific nature of epigenetic responses to maternal nutrition and toxicant exposures. Male and female offspring often exhibit divergent metabolic trajectories following identical in utero exposures, reflecting differences in placental function, sex hormone signaling, and chromatin dynamics. These differences are not merely quantitative but involve qualitatively distinct epigenetic reprogramming events, suggesting that sex-specific epigenetic vulnerability is a critical determinant of obesity risk.
Male vs Female Offspring Differences in Metabolic Programming
Epidemiological and experimental studies consistently demonstrate that males are often more vulnerable to maternal overnutrition, while females may show heightened sensitivity to maternal undernutrition or toxicant exposures.
Earlier findings indicate that maternal high-fat feeding throughout pregnancy and lactation predisposes offspring to molecular insulin resistance and fatty liver in mouse models.195 Male offspring exposed to maternal high-fat diets tend to develop greater adiposity, insulin resistance, and fatty liver compared to female offspring. A 2025 study demonstrated that male offspring of high-fat diet–fed dams exhibited greater weight gain, increased adiposity, and impaired glucose homeostasis compared to female offspring, along with elevated serum insulin, leptin, and cholesterol levels.196 Mechanistically, males exhibit greater DNA hypomethylation at certain liver metabolic gene promoters (for example, Cytochrome P450 2d9), which may contribute to diminished mitochondrial oxidative capabilities.197 Females, by contrast, often maintain metabolic resilience, in part due to activation of oxidative pathways by ERRα, a key regulator of mitochondrial biogenesis and fatty acid metabolism.198
Christians et al199 conducted a systematic review and meta-analysis on prenatal food or protein restriction in rats and mice, looking specifically for sex-dependent effects in offspring physiology. They concluded that majority of studies found no consistent sex-dependent effects.
Rodent offspring exposed to maternal high-fat diet (HFD) often show altered epigenetic regulation in appetite- and metabolism-related genes (like leptin receptor, POMC, NPY, dopamine/opioid genes) in the hypothalamus, which can affect appetite and energy balance.200 In male offspring, maternal HFD has been shown to induce epigenetic modifications in adipose tissue, such as hypomethylation of adipogenic regulators (such as Zfp423) and alterations in PPARγ promoter methylation, contributing to enhanced adipogenesis in offspring.201
Prenatal exposure to endocrine disruptors such as BPA and phthalates often yields sex-specific outcomes: male offspring exhibit pronounced weight gain, while female offspring experience more subtle but enduring alterations in adiposity and inflammatory profiles.202,203 These differences reflect sex-dependent chromatin remodeling at estrogen receptor (ER) and androgen receptor (AR) response elements. Together, these findings emphasize that sex is a biological variable in DOHaD research and must be integrated into epigenetic models of obesity risk.
Human cohort evidence adds important nuance:
- ALSPAC (UK): In ALSPAC, prenatal maternal diets high in fat and sugar were associated with lower IGF2 methylation in male offspring cord blood; this association has been linked to behavioral outcomes, though links to childhood BMI trajectories remain unexplored.204 An epigenome-wide study (NEST), with attempted replication in ALSPAC, identified sex-specific differential DNA methylation associated with maternal pre-pregnancy obesity, which was further linked to offspring BMI and blood pressure in early childhood.205
- Generation R (Netherlands): In the Generation R Study, continued maternal smoking during pregnancy was associated with lower IGF2DMR methylation in newborns, an effect that was more pronounced in girls (β = –1.38) than in boys (β = –0.72).206 Prenatal cadmium exposure has been shown in population studies to produce sex-specific DNA methylation changes, female-predominant hypomethylation and male-predominant hypermethylation.207
- CHAMACOS (USA): Prenatal exposure to certain phthalates and parabens was associated with increased BMI z-score and overweight/obesity status in children at age 5.155 In another study, prenatal phthalate exposure was linked to higher adiposity in boys at age 12, mediated by altered methylation at adipogenic genes, while girls showed weaker associations.153
- Project Viva (USA): In some pregnancy cohorts, sex-stratified analyses suggest that maternal dietary sugar intake may differentially influence offspring epigenetic marks at insulin signaling loci (IRS1), with stronger effects observed in boys than girls.23 Recent human studies corroborate this result. For instance, ex-differentiated DNA methylation patterns in the placenta have also been observed, including male-biased hypermethylation at the ZNF300 locus in first-trimester placenta,23 and widespread sex-dependent methylation regulation across the autosomal genome.208
- HELIX Consortium (Europe): In the HELIX Consortium, a multi-cohort EWAS meta-analysis found that prenatal NO2 exposure was associated with DNA methylation at loci related to mitochondrial metabolism (including LONP1, HIBADH, SLC25A28).209 A related multi-cohort analysis of placentas reported sex-specific DNAm responses to prenatal air-pollution exposure, with several associations stronger in female placentas.210 Another study identified significant sex-specific DNA methylation differences in first-trimester human placentas, notably at the transcription factor ZNF300: hypermethylated in males, with correspondingly lower expression.23
These findings align with rodent studies, showing that males often exhibit greater epigenetic susceptibility to obesogenic diets, while females may display heightened vulnerability to toxicants and stress-related exposures.
Epigenetic Regulation of Sex Hormone Receptors and Adiposity
Sex-specific epigenetic vulnerability arises in part from differential regulation of sex hormone receptors and downstream adipogenic networks.
In females, estrogen receptor α (ERα) promotes lipid oxidation and insulin sensitivity. Dudley et al211 found that maternal high-fat nutrition led to increased methylation of the ERα-1b promoter and reduced ERα expression in the medial preoptic area of female rat offspring. Maternal high-fat diet induces epigenetic changes in metabolic genes (eg, PGC-1α promoter methylation in skeletal muscle, MEF2A promoter hypermethylation in liver), thereby diminishing estrogen’s protective metabolic effects.212 ER-regulated microRNAs are also epigenetically altered in maternal obesogenic environments, modulating adipocyte differentiation.213
In males, androgen receptor (AR) signaling is tightly linked to visceral adiposity and hepatic lipid metabolism.214 Sol et al23 in their Generation R data showed that maternal exposure to endocrine disruptors such as BPA or phthalates may lead to epigenetic changes (especially DNA methylation).23 Mechanistic studies indicate that maternal exposure to endocrine disruptors such as bisphenols or phthalates can perturb AR signaling by altering AR–coregulator interactions, and because AR-driven transcription requires p300/CBP-mediated histone acetylation at AR-binding sites, such disruptions are expected to modify local chromatin acetylation and downstream gene expression at androgen-responsive loci.215,216
Research has shown that obesity in male rats results in altered methylation of epididymal sperm, with differentially methylated regions identified between high-fat diet–fed rats and controls, suggesting potential involvement in obesity transmission across generations.217 Another rodent model highlighted diet-induced alterations in sperm methylome linked to paternal obesity, implicating epigenetic inheritance in offspring’s metabolic health.218 In rats with diet-induced obesity, hypermethylation of the pro-opiomelanocortin (Pomc) gene promoter was observed in sperm and in the hypothalamic arcuate nucleus of their offspring. This epigenetic change correlated with metabolic dysregulation and supports the concept of paternal epigenetic influence on obesity predisposition.219
Although related to environmental toxins rather than obesity, arsenic exposure during gestation in mice induced hypomethylation in F1 sperm, particularly at retrotransposon elements (LINEs and LTRs). These methylation features were seen to re-establish in F2 embryos, suggesting a mechanism for intergenerational epigenetic transmission of disease risk including obesity.220
Importantly, there is sex chromosome–linked epigenetic regulation. Differences in X-chromosome inactivation in females vs Y-linked gene expression in males introduce additional layers of epigenetic complexity.221 For example, a recent study by Lin et al222 demonstrates that KDM6A escapes X-chromosome inactivation, enabling female-biased expression of a histone-demethylase that actively regulates Xist via H3K27 demethylation, promoting effective X–inactivation and potentially endowing females with enhanced chromatin-remodeling adaptability. In sum, histone-demethylase activity confers female-specific flexibility in chromatin remodeling, potentially explaining resilience to certain obesogenic exposures.222
These mechanisms highlight that sex hormone receptor epigenetics acts as a molecular switchboard, modulating how male and female offspring translate maternal exposures into metabolic outcomes. Recognition of these sex-dependent epigenetic landscapes is essential for developing precision maternal interventions and for ensuring that obesity prevention strategies are tailored to both male and female offspring.
Future Directions: Toward Sex-Stratified Interventions and Clinical Monitoring
Recognizing sex-specific vulnerability opens new avenues for precision prevention:
- Sex-Stratified Interventions: Nutritional supplementation interventions could be tailored by sex. For example, ensuring adequate methyl donor and antioxidant intake in pregnancies with male fetuses to buffer obesogenic diet exposures, while prioritizing protein sufficiency and stress-reducing nutrients (like omega-3s) in pregnancies with female fetuses more sensitive to undernutrition and toxicants.223,224 For environmental risk reduction, regulatory policies and clinical guidance may need to account for sex differences, targeting higher-risk exposures (for instance, endocrine disruptors for boys, heavy metals for girls).
- Clinical Monitoring: Development of sex-specific cord blood or placental epigenetic biomarkers stratified could allow early identification of high-risk infants. Again, pediatric follow-up programs may benefit from sex-specific cutoffs and trajectories, recognizing that male and female children exhibit distinct metabolic responses to early-life exposures. Because placental adaptations differ between male and female fetuses, placental epigenetic profiling could inform personalized monitoring strategies during pregnancy.225
- Research Priorities: There is need to expand sex-stratified analyses in ongoing birth cohorts (ALSPAC, Generation R, HELIX) to validate findings across populations. Furthermore, intervention trials that evaluate whether nutritional or exposure-reduction strategies have differential effects in male vs female offspring is necessary. Incorporating sex as a biological variable in epigenome-wide association studies (EWAS) and multi-omics research to avoid obscuring critical differences is essential.
Clinical and Translational Perspectives
The convergence of maternal nutrition, toxic exposures, and epigenetic programming in shaping obesity risk provides not only mechanistic insight but also translational opportunities. Advances in biomarker discovery, nutritional interventions, and exposure-reduction interventions offer realistic near-term opportunities for improving maternal–fetal health, whereas genome- and epigenome-editing technologies are still confined to experimental models offering new avenues for prediction, prevention, and precision healthcare. Integrating these strategies into maternal and public health frameworks has the potential (if supported by robust interventional evidence) to substantially reduce the intergenerational burden of obesity and metabolic disease. Yet, enthusiasm must be tempered by recognition that current evidence is largely observational, effect sizes are modest, and major ethical, safety, and implementation challenges remain. To balance enthusiasm with critical realism, Table 4 contrasts the promises of epigenetic translation in maternal–fetal health with the key pitfalls and challenges that must be addressed before clinical implementation.
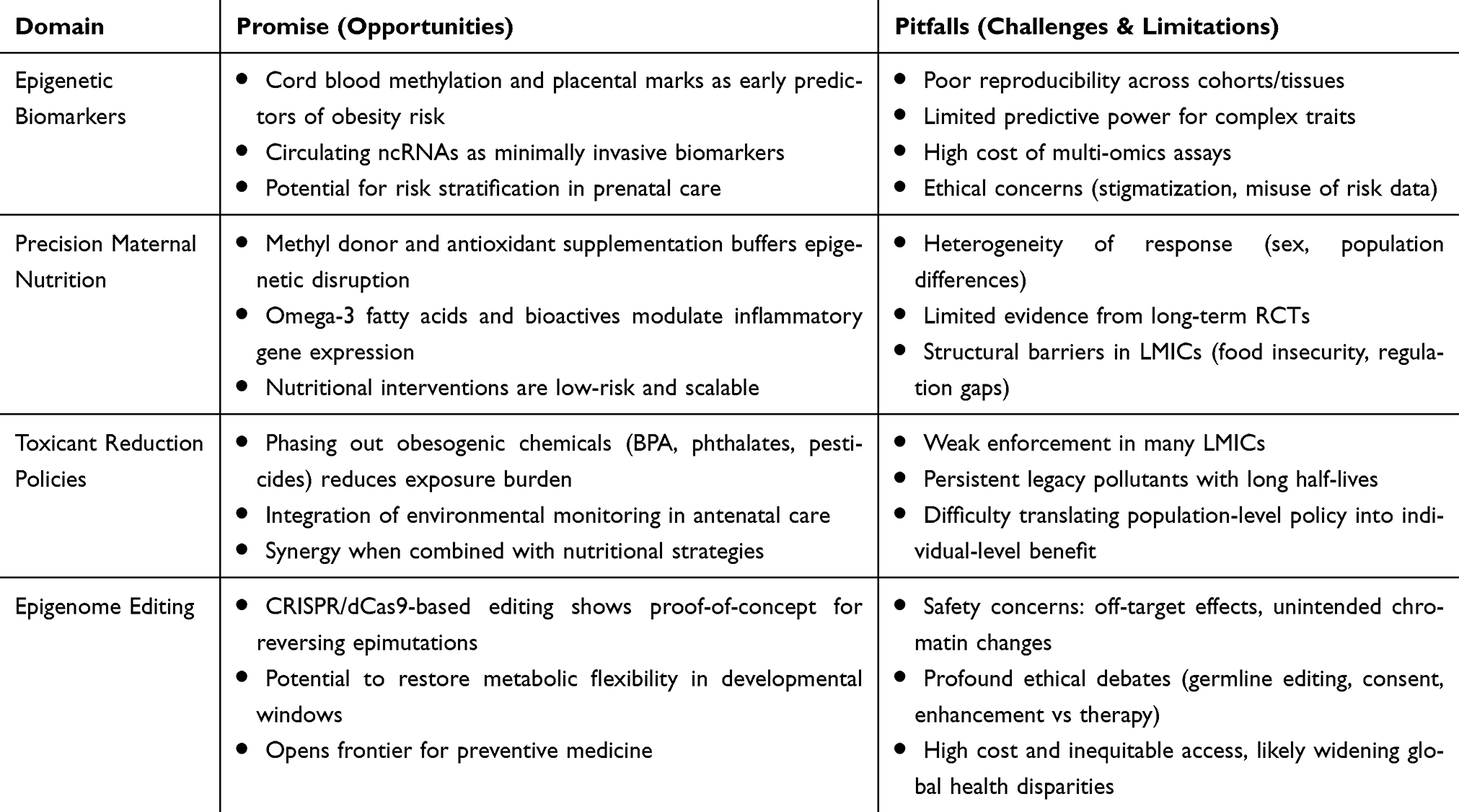 |
Table 4 Promise vs Pitfalls of Epigenetic Translation in Maternal–Fetal Health |
As summarized in Table 4, epigenetic translation offers powerful opportunities for improving maternal–fetal health, yet significant barriers remain. Biomarkers show potential for early risk prediction but face reproducibility and cost challenges. Nutritional interventions are safe and scalable but require sex- and context-specific tailoring, especially in low-resource settings. Policy-driven toxicant reduction has proven effectiveness in high-income countries, yet enforcement gaps leave LMIC populations disproportionately exposed. Epigenome editing represents an exciting frontier, but unresolved safety, ethical, and equity concerns currently preclude clinical use. Addressing these barriers will be critical to move from promising proof-of-concept to equitable and effective implementation in public health and clinical practice.
Epigenetic Biomarkers of Maternal Exposures
Epigenetic modifications serve as molecular archives of intrauterine exposures, making them powerful biomarkers for predicting metabolic risk. The development of epigenetic biomarker panels combining cord blood, placental marks, and circulating ncRNAs could enable stratification of pregnancies at high risk for programming obesity. While promising, these biomarkers require validation across larger and more diverse cohorts before clinical adoption.
- Cord Blood DNA Methylation: Differential methylation at loci such as LEP, IGF2, and PPARγ in cord blood has been associated with maternal high-fat diet, undernutrition, and exposure to BPA and phthalates. Differential methylation at loci such as IGF2 (hypomethylated following prenatal undernutrition during the Dutch Hunger Winter), LEP (induced by maternal high-fat diet in animal models and associated with methylation changes in human cord blood in the context of maternal obesity), and PPARγ (epigenetically altered by maternal protein restriction in animal studies) has been linked to maternal nutrition.226,227 Similarly, exposure to environmental endocrine disruptors like BPA and phthalates during gestation has been associated with DNA methylation changes observed in cord blood.23,228 Epigenome-wide association studies (EWAS) have revealed methylation signatures that correlate with childhood BMI, suggesting cord blood methylation can function as an early-warning system for obesity risk.229
- Placental Epigenome: The placenta is highly sensitive to maternal environment and acts as a critical interface between mother and fetus. Altered methylation at genes involved in nutrient transport, stress responses (NR3C1), and growth regulation (IGF2) reflects both maternal nutritional and toxicant exposures.230 A review by Green and Marsit231 discusses how maternal environmental exposures including both nutritional factors (eg, diet) and toxicants (including heavy metals, endocrine disruptors, smoking) are linked to alterations in gene-specific DNA methylation in fetal tissues. It emphasizes that such epigenetic programming may involve key pathways including nutrient transport, stress response, and growth regulation—highlighting genes like IGF2, NR3C1, and others.231 Placental histone modifications and imprinted gene regulation further predict offspring adiposity, making placental epigenomics a promising diagnostic tool. Placental epigenomic signatures, particularly DNA methylation at specific CpG sites, predict childhood adiposity, suggesting that placental epigenomics is a promising tool for early diagnostic applications.232 Placental histone modifications and imprinted gene regulation also play critical roles in fetal development, underpinning the mechanistic foundation for this predictive potential.233
- Circulating Noncoding RNAs: MicroRNAs (miRNAs) such as miR-21, miR-29, and miR-148a in maternal plasma have been linked to gestational obesity, insulin resistance, and pollutant exposures. Circulating miRNAs including miR-21 and the miR-29 family are altered in the context of maternal metabolic dysregulation such as obesity and insulin resistance.234,235 Additionally, miR-148a-3p has been found to be downregulated in plasma exosomes of women with gestational diabetes mellitus (GDM), linking it to impaired metabolic regulation during pregnancy.236 In parallel, miRNAs such as miR-21 have been shown to respond to environmental pollutant exposures, especially air pollution, acting as epigenetic markers of maternal environmental quality.237 These circulating miRNAs can traffic across the placenta, impact fetal gene expression (as suggested by their presence in fetal and placental tissues and exosomal transfer), and serve as minimally invasive biomarkers that reflect the maternal–fetal environment.
Challenges
- Validation: Epigenetic signatures often lack reproducibility across cohorts and populations due to differences in tissue specificity, measurement techniques, and confounding exposures.
- Predictive value: Many signals explain only a small proportion of obesity variance, limiting their utility as standalone diagnostic tools.
- Feasibility and cost: Multi-omics profiling (EWAS, ncRNA panels) is expensive and not yet feasible for routine clinical screening, particularly in low-resource settings where the dual burden of malnutrition and toxicants is greatest.
- Ethical considerations: The potential use of biomarkers for prenatal risk stratification raises concerns about stigmatization, insurance discrimination, and how to manage high-risk pregnancies when effective interventions remain limited.
Intervention Strategies
Precision Maternal Nutrition
Methyl donor supplementation (folate, choline, B12, methionine) supports one-carbon metabolism and stabilizes DNA methylation patterns during fetal development, reducing the risk of obesogen-induced epimutations. In the Southampton Women’s Survey, poorer maternal and early childhood diet quality across preconception to childhood was associated with higher offspring adiposity at age 6, measured by DXA-assessed fat mass and BMI z-score.238 Periconceptional folic acid exposure and maternal one-carbon status have been linked to altered IGF2 methylation at birth,239 and imbalance of maternal folate and vitamin B12 during pregnancy (high folate with low B12) has been associated with greater childhood adiposity and insulin resistance in the Pune cohort.240 Similarly, supplementation trials in the Pune Rural Intervention in Young Adolescents (PRIYA) study showed that maternal vitamin B12 supplementation during pregnancy improved offspring insulin sensitivity and reduced adiposity at 6 years.241
Randomized trials in Bangladesh show that folic acid supplementation enhances arsenic methylation and lowers blood arsenic.242 Another Bangladeshi birth-cohort data link prenatal arsenic exposure to altered global DNA methylation in cord blood.243 A recent systematic review and meta‐analysis reports that folic acid fortification is projected to be the most effective strategy to ameliorate arsenic exposure and neural tube defects (NTDs) risk in Bangladesh, where arsenic exposures are high.244 Together, these studies illustrate protective nutrient–toxicant antagonism.
The Supplementation with Multiple Micronutrients Intervention Trials (SUMMIT, Indonesia) found that maternal multiple micronutrient supplementation improved infant survival and growth outcomes, with follow-up studies suggesting epigenetic effects on growth-regulatory pathways.245,246 Zinc and selenium supplementation studies, though limited, suggest potential benefits for stabilizing DNA methylation and reducing oxidative stress–related epimutations.247,248
More so, antioxidants (vitamin C, vitamin E, polyphenols) neutralize toxicant-induced ROS, preventing oxidative activation of histone-modifying enzymes. For instance, resveratrol activates SIRT1, counteracting obesogen-driven chromatin activation.249 Furthermore, bioactive supplementation (omega-3 fatty acids, sulforaphane, EGCG) modulate histone acetylation and DNA methylation in metabolic pathways, buffering the fetal epigenome against obesogenic programming.250 The ROLO trial (Ireland) reported that maternal low-glycemic diets during pregnancy reduced offspring birth weight and cord blood insulin resistance markers, with preliminary evidence of DNA methylation changes in metabolic genes.251–253
In the European Nutraceuticals for a Healthier Life (NUHEAL) trial, pregnant women received long-term effects of n-3 (omega-3) LC-PUFA and/or 5-methyltetrahydrofolate report no consistent cognitive benefit, but maternal dietary status may be related to later cognitive function in children.254 However, a follow up study reports that maternal fish oil supplementation during pregnancy could shape resting-state network functioning in their offspring at school age, thus producing long-term effects on children´s cognitive processing.255 Separately, a randomized trial of third-trimester choline supplementation altered methylation of CRH/NR3C1 in placenta and cord and improved early information-processing in infants.256 Another double-blind randomized controlled trial at Oslo University Hospital reports that supplementation of arachidonic acid and docosahexaenoic acid has the potential to improve brain maturation and reduce inflammation related diseases in infants.257 These interventions underscore the potential for targeted maternal dietary strategies to mitigate epigenetic disruptions.
Reducing Toxicant Exposure During Pregnancy
Phasing out obesogenic chemicals such as BPA and certain phthalates from food packaging; stricter regulation of pesticide residues and heavy metals in food and water are correct policy measures. Public health interventions such as education programs promoting safer food storage practices, avoidance of contaminated water sources, and use of toxin-free cosmetics and household products are critical. Incorporating environmental exposure assessments into prenatal care, enabling early risk identification and intervention, are good clinical integration approaches.
Together, precision nutrition and toxicant reduction represent complementary strategies to reshape the maternal exposome toward healthier developmental outcomes.
Challenges
- Heterogeneity of response: Nutritional interventions may have sex-specific or population-specific effects, requiring stratified recommendations rather than one-size-fits-all guidelines.
- Implementation barriers: In many LMICs, structural issues such as food insecurity and weak regulatory oversight limit the impact of individual-level interventions.
- Long-term follow-up: Few trials have tracked whether maternal dietary interventions translate into reduced obesity risk in offspring, making clinical translation premature.
Practical Implications for Clinical Care and Public Health
From a clinical perspective, one of the closely actionable indicators evolving from this study relate to established maternal-health priorities rather than novel strategies. Recent evidence consistently supports optimizing preconception and antenatal nutrition (via sufficient micronutrients intake, evading of extreme energy restriction or highly obesogenic diets), managing maternal BMI and gestational weight gain within guideline ranges, and reducing avoidable exposure to known obesogens (such as tobacco smoke, excessive air pollution, some job-related chemicals) using existing public-health and regulatory frameworks. These recommendations are already endorsed for other maternal–fetal outcomes; the epigenetic and DOHaD data reviewed here mainly strengthen the rationale and help refine sensitive windows and potentially vulnerable subgroups. In contrast, more speculative applications like epigenetic biomarker panels for routine risk prediction or targeted epigenome-modifying interventions remain at a preclinical or proof-of-concept stage and should not yet inform decision-making in clinical setting. Thus, the evidence is promising but unproven, and the experimental strategies may help clinicians, policymakers, and patients navigate the growing but heterogeneous epigenetic literature.
Future of Epigenome Editing and Preventive Medicine
Looking forward, epigenome editing tools and maternal epigenetic counseling hold promise for personalized preventive medicine. Together, these innovations establish the foundation for a translational epigenetic framework to combat intergenerational obesity.
CRISPR/dCas9-Based Epigenome Editing
Emerging technologies such as CRISPR/dCas9 fused to epigenetic modifiers (including DNMT3A, TET1, histone acetyltransferases) allow targeted editing of DNA methylation and histone states without altering DNA sequence. These tools are being applied in experimental DOHaD models to reverse obesogen-induced epimutations.258 For example, targeted demethylation of PPARα in animal models restored metabolic flexibility, suggesting a potential therapeutic role in correcting developmental epimutations.259 Although not yet clinically feasible, this approach represents a proof-of-principle concept that some developmental epimutations are reversible in animal models, but it remains unknown whether similar approaches will be safe, effective, or acceptable in humans.
Maternal Epigenetic Counseling
Epigenetic biomarker screening has been proposed as a route to more personalized risk assessment for pregnant women. By integrating dietary intake, toxicant exposure profiles, and epigenetic readouts (cord blood methylation, placental marks, circulating ncRNAs), clinicians could provide epigenetic counseling tailored recommendations for nutrition, lifestyle, and environmental exposure management to optimize fetal metabolic programming.260 Such approaches would represent a paradigm shift from treating obesity to preventing it before birth, transforming prenatal care into a cornerstone of metabolic disease prevention. At present, however, such approaches remain speculative as limited trials have demonstrated that epigenetic risk stratification and counselling improve obesity outcomes. Moreover, substantial ethical, logistical, and equity concerns would need to be resolved before clinical implementation.
Challenges
- Safety and off-target effects: Epigenome editing is still in its infancy, with significant risk of unintended chromatin changes that could have unpredictable developmental effects.
- Ethical concerns: Editing the fetal or germline epigenome raises profound ethical questions about consent, intergenerational consequences, and the boundary between therapy and enhancement.
- Equity and cost: Cutting-edge technologies are likely to be prohibitively expensive and available only in high-income settings, risking further widening of global health inequities.
AI and Multi-Omics Approaches to Obesity Risk Prediction
Advances in data science are promising in the translation of DOHaD concepts into predictive tools. Recent reviews and proof-of-concept studies show that machine-learning models can leverage routinely collected clinical information and environmental data to predict pediatric obesity risk with higher accuracy than traditional regression alone, and some groups have developed interoperable pipelines designed for integration with electronic health records.261,262 In the multi-omics space, integrative frameworks combining genomics, epigenomics, metabolomics, and microbiomics have identified multi-omics clusters that stratify children into distinct adiposity and metabolic risk profiles and link these to specific prenatal exposures such as maternal BMI and persistent pollutants.263 While these AI-driven, multi-omics risk models may not be yet ready for routine prenatal or pediatric use, they provide cues for how maternal nutrition and toxicant data could eventually be integrated into personalized early-life risk stratification.
Unresolved Debates in Epigenetic Interpretation
Despite rapid progress, several fundamental debates complicate the interpretation of epigenetic data in the context of maternal nutrition, toxicants, and obesity. The first question is whether observed epigenetic changes are causal mediators of disease or primarily biomarkers of underlying exposures and physiological states. Most human epigenome–phenotype associations arise from observational cohort studies that are vulnerable to confounding, reverse causation, cell-type heterogeneity, and technical artefacts. While experimental animal work and in vitro models support the plausibility of causal pathways, in humans epigenetic marks are better regarded as mechanistic candidates and exposure–response indicators rather than established drivers of obesity risk, awaiting stronger evidence from longitudinal designs, and causal inference frameworks. Results from PACE, HELIX, Raine, ECHO and related consortia highlight both the promise and the limitations of current exposome and omics science. Many reported associations between early-life exposures, epigenetic marks, obesity or adiposity are statistically robust at the consortium level yet modest in magnitude, sensitive to analytic decisions (eg, cell-type adjustment, multiple-testing control, mixture modelling strategy), and sometimes heterogeneous across cohorts, tissues and developmental windows.100,102–104 These patterns fuel debate about how strongly such results should be interpreted as causal, which epigenetic or multi-omics markers will ultimately prove reproducible and clinically relevant, and how best to balance discovery-oriented exposome scans with hypothesis-driven mechanistic work.
The second debate relates to how reversible developmentally induced epimutations are, and over what time windows. Many experimental models signify that some marks established during sensitive periods can be diminished or relayed by enhancing maternal diet, normalizing postnatal nutrition, or modifying obesogenic environments, whereas others show relative stability once key developmental programs are set.264–266 Human trials are still scarce and often limited to a few tissues and time points. Moreover, it is unclear which obesity-related epigenetic changes are truly “hard wired”, which are conditionally plastic, and to what extent postnatal interventions can compensate for suboptimal intrauterine environments. This uncertainty cautions against both overly deterministic narratives and overly optimistic assumptions about complete reversibility.
Another debate comprises the difference between intergenerational and transgenerational inheritance. In mammalian systems, in utero exposure of a pregnant female (F0) directly affects not only the fetus (F1) but also the developing germ cells within that fetus (F2). Accordingly, phenotypes observed in F1 and F2 generations may mirror direct exposure rather than true transmission of epigenetic information through the germline.267 Conventionally, evidence for transgenerational inheritance via epigenetic mechanisms would require effects in the F3 generation (for maternal exposures) or the F2 generation (for paternal preconception exposures), after excluding persistent environmental and social transmission. In humans, long generation times, pervasive confounding across generations, and extensive epigenetic reprogramming during gametogenesis and early embryogenesis make such demonstrations extremely challenging.268 As a result, claims of robust transgenerational epigenetic inheritance of obesity in humans should be viewed as intriguing but provisional. Thus, our review focuses primarily on intergenerational effects that are biologically plausible and empirically supported.
Conclusions and Future Directions
Key Insights
Maternal nutrition and environmental toxicant exposures act, both independently and in combination, as epigenetic architects of obesity risk across the life course. Mechanisms include: DNA methylation, histone modifications, and noncoding RNAs. These processes link maternal macronutrient and micronutrient status, one-carbon metabolism, and obesogenic diets to persistent changes in gene networks controlling adipogenesis, insulin signalling, appetite regulation and energy expenditure. Obesogenic toxicants, including endocrine disruptors, heavy metals, food-borne and agricultural contaminants, appear to converge on overlapping pathways, sometimes amplifying the effects of nutritional imbalance. The evidence reviewed here highlights how nutrient excesses, deficiencies, and environmental pollutants converge on the fetal epigenome to establish enduring “metabolic memories” that predispose offspring to obesity and related disorders. Importantly, these effects are not confined to the immediate generation but may propagate across lineages, raising intergenerational stakes for maternal health. Recent studies demonstrate that non-canonical mechanisms such histone lactylation, chromatin accessibility, and 3D genome architecture, as well as placental multi-omics, give the placenta an important location for turning nutritional and toxicant signals into foetal epigenomic states. Nonetheless, results from extensive consortia and exposome-wide studies indicate that effect sizes are typically modest, heterogeneous, and contingent upon context. This suggests that epigenetic marks ought to be regarded as potential mechanistic candidates and exposure-response indicators, rather than conclusive causal determinants.
Outstanding Gaps and Conceptual Challenges
While substantial progress has been made, significant knowledge gaps remain. Much of the mechanistic understanding derives from animal studies, whereas human evidence is primarily associative and limited by confounding, single-exposure assessments. Moreover, human evidence is dominated by observational cohorts from high-income settings, with limited representation of low- and middle-income populations that bear a disproportionate dual burden of undernutrition and environmental pollution. Clarifying causality, integrating multi-omics approaches, and embedding sex-specific analyses are necessary next steps. Beyond issues of causality, the epidemiological literature on toxicants and obesity is characterized by substantial heterogeneity. Effect estimates frequently vary by sex, developmental window, and analytic approach, and many studies report wide confidence intervals or null associations despite plausible mechanistic data. Publication bias toward positive findings and under-reporting of null results likely further distort the apparent strength of evidence. Together, these limitations indicate that current toxicant–obesity associations should be interpreted as suggestive rather than definitive and highlight the need for large, harmonized multi-cohort analyses with repeated exposure measures and robust mixture modelling.
Future Research Agenda
To fully maximize maternal nutrition–toxicant–epigenetic interactions translational potential, it is necessary to transition from association to intervention. This involves integrating mechanistic endpoints into pragmatic trials, evaluating scalable nutrition and exposure-reduction strategies across varied contexts, and perpetually reassessing epigenetic and multi-omics signatures for their robustness and clinical significance. Only via this kind of iterative, cross-disciplinary work can epigenetic insights be used in an accountable manner to break, rather than just describe, cycles of metabolic disease that pass from one generation to the next. Future research should focus on:
- Integrative multi-omics: Combine epigenomics, transcriptomics, metabolomics, microbiomics, and exposomics to improve predictive accuracy and mechanistic understanding.
- Biomarker development: Conduct large, multi-ethnic, longitudinal cohorts to validate epigenetic signatures (DNA methylation, ncRNAs, histone marks) as robust predictors of obesity risk.
- Sex-specific studies: Incorporate sex as a biological variable in cohort analyses and intervention trials to tailor prevention strategies.
- Intervention studies: Test whether targeted nutritional supplementation and exposure reduction strategies can prevent obesogenic epimutations in randomized controlled trials.
- Transgenerational tracking: Design multi-generational human studies to resolve the controversy around true epigenetic inheritance.
- Ethics and equity focus: Establish global guidelines for safe, equitable, and ethical applications of epigenome editing, ensuring benefits are not restricted to high-income settings.
Translational Outlook
Early-life prevention is widely regarded as a critical leverage point for curbing the obesity epidemic. Optimizing maternal nutrition and minimizing toxicant exposure are plausible and increasingly evidence-supported strategies to improve metabolic programming, although definitive proof that they prevent obesity across the life course is still limited. Moving forward, integration of epigenetic risk assessment into prenatal care, coupled with structural policy interventions (nutrient fortification, environmental regulation, public health campaigns), may contribute to interrupting the intergenerational cycle of obesity. This study highlights a set of promising, but yet incompletely validated, opportunities that will require rigorous evaluation in diverse settings. Policymakers, clinicians, and nutrition scientists must converge on strategies grounded in epigenetic evidence to prevent obesity before it takes root in fetal development. By aligning scientific discovery with public health policy and maternal care, it is possible to interrupt the intergenerational cycle of metabolic disease. In doing so, we may redefine obesity prevention, not as an individual lifestyle challenge, but as a societal commitment to safeguarding the epigenetic foundations of health across generations.
Strengths and Limitations of the Current Evidence Base
Although this study converges data from animal models, human cohorts, and emerging multi-omics studies, the overall evidence base still has important limitations that temper clinical translation. Most human findings rely on observational designs with modest effect sizes, limited tissue analysis, and potential residual confounding and reverse causation. Epigenetic modifications linked with maternal nutrition or toxicant exposures may act as causal mediators, but they may also function as correlated biomarkers of underlying physiological states. Results are often heterogeneous across cohorts, developmental windows, and null or contradictory findings are imminent. Large consortia and exposome-wide approaches improve power and generalizability but also highlight issues of multiple testing, mixture modelling, and reproducibility. Recognizing these constraints is important. Currently, epigenetic evidence should be regarded as strengthening the biological plausibility of established maternal-health interventions and generating hypotheses for targeted trials, rather than providing definitive, stand-alone grounds for new screening programmes or advanced therapies.
Data Sharing Statement
All data generated during this study are included in the manuscript.
Author Contributions
Conceptualization: Esther Ugo Alum. Methodology: Esther Ugo Alum, Henry Egi Aloh, David Chukwu Obasi, Prince Nkemakolam Okoroh, Ugonna Cassandra Aniokete, Akunna Perpetua Emeruwa. Investigation: Esther Ugo Alum, Henry Egi Aloh, David Chukwu Obasi, Prince Nkemakolam Okoroh, Ugonna Cassandra Aniokete, Akunna Perpetua Emeruwa. Resources: Henry Egi Aloh, David Chukwu Obasi, Prince Nkemakolam Okoroh, Ugonna Cassandra Aniokete, Akunna Perpetua Emeruwa. Supervision: Henry Egi Aloh. Validation: David Chukwu Obasi, Prince Nkemakolam Okoroh. Visualization: Ugonna Cassandra Aniokete, Akunna Perpetua Emeruwa. Writing – original draft: Esther Ugo Alum. Writing – review & editing: Esther Ugo Alum, Henry Egi Aloh, David Chukwu Obasi, Prince Nkemakolam Okoroh, Ugonna Cassandra Aniokete, Akunna Perpetua Emeruwa. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received.
Disclosure
The authors declare no competing interests.
References
1. Kerr JA, Patton GC, Cini KI, et al. Global, regional, and national prevalence of child and adolescent overweight and obesity, 1990–2021, with forecasts to 2050: a forecasting study for the Global Burden of Disease Study 2021. Lancet. 2025;405:785–812. doi:10.1016/S0140-6736(25)00397-6
2. Gujral P, Mahajan V, Lissaman AC, Ponnampalam AP. Histone acetylation and the role of histone deacetylases in normal cyclic endometrium. Reprod Biol Endocrinol. 2020;18:84. doi:10.1186/s12958-020-00637-5
3. Alum EU, Ejemot-Nwadiaro RI, Betiang PA, Basajja M, Uti DE. Obesity and climate change: a two-way street with global health implications. Obesity Med. 2025;56:100623. doi:10.1016/j.obmed.2025.100623
4. Uti DE, Alum EU, Atangwho IJ, Ugwu OP-C, Egbung GE, Aja PM. Lipid-based nano-carriers for the delivery of anti-obesity natural compounds: advances in targeted delivery and precision therapeutics. J Nanobiotechnol. 2025;23:336. doi:10.1186/s12951-025-03412-z
5. Umano GR, Bellone S, Buganza R, et al. Early roots of childhood obesity: risk factors, mechanisms, and prevention strategies. Int J Mol Sci. 2025;26:7388. doi:10.3390/ijms26157388
6. Arbuckle TE, Marro L, Davis K, et al. Exposure to free and conjugated forms of bisphenol A and triclosan among pregnant women in the MIREC Cohort. Environ Health Perspect. 2015;123:277–284. doi:10.1289/ehp.1408187
7. Podgorski J, Berg M. Global threat of arsenic in groundwater. Science. 2020;368:845–850. doi:10.1126/science.aba1510
8. Ahmad SA, Khan MH, Haque M. Arsenic contamination in groundwater in Bangladesh: implications and challenges for healthcare policy. Risk Manag Healthc Policy. 2018;11:251–261. doi:10.2147/RMHP.S153188
9. Eskola M, Kos G, Elliott CT, Hajšlová J, Mayar S, Krska R. Worldwide contamination of food-crops with mycotoxins: validity of the widely cited ‘FAO estimate’ of 25%. Crit Rev Food Sci Nutr. 2020;60:2773–2789. doi:10.1080/10408398.2019.1658570
10. Udomkun P, Wiredu AN, Nagle M, Bandyopadhyay R, Müller J, Vanlauwe B. Mycotoxins in sub-Saharan Africa: present situation, socio-economic impact, awareness, and outlook. Food Control. 2017;72:110–122. doi:10.1016/j.foodcont.2016.07.039
11. SDG Target 3.9 Mortality from environmental pollution. Available from: https://www.who.int/data/gho/data/themes/topics/sdg-target-3_9-mortality-from-environmental-pollution.
12. Lacagnina S. The Developmental Origins of Health and Disease (DOHaD). Am J Lifestyle Med. 2019;14:47–50. doi:10.1177/1559827619879694
13. Alum EU. Metabolic memory in obesity: can early-life interventions reverse lifelong risks? Obesity Med. 2025;55:100610. doi:10.1016/j.obmed.2025.100610
14. Peral-Sanchez I, Hojeij B, Ojeda DA, Steegers-Theunissen RPM, Willaime-Morawek S. Epigenetics in the uterine environment: how maternal diet and ART may influence the epigenome in the offspring with long-term health consequences. Genes. 2021;13:31. doi:10.3390/genes13010031
15. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi:10.1038/nrg.2016.59
16. Alum EU, Ejemot-Nwadiaro RI, Basajja M, Uti DE, Ugwu OP-C, Aja PM. Epitranscriptomic alterations induced by environmental toxins: implications for RNA modifications and disease. Genes Environ. 2025;47:14. doi:10.1186/s41021-025-00337-9
17. Zhang J, Wu D, Zeng F, et al. Lactate metabolic reprogramming and histone lactylation modification in sepsis. Int J Bio Sci. 2025;21:5034–5055. doi:10.7150/ijbs.116088
18. Li S, Zong X, Zhang L, Li L, Wu J. A chromatin accessibility landscape during early adipogenesis of human adipose-derived stem cells. Adipocyte. 2022;11:239–249. doi:10.1080/21623945.2022.2063015
19. Tan B, Hong L, Xiao L, et al. Rewiring of 3D chromatin topology orchestrates transcriptional reprogramming in muscle fiber-type specification and transformation. Nat Commun. 2025;16:5833. doi:10.1038/s41467-025-60866-3
20. Pant R, Firmal P, Shah VK, Alam A, Chattopadhyay S. Epigenetic regulation of adipogenesis in development of metabolic syndrome. Front Cell Dev Biol. 2021;8:619888. doi:10.3389/fcell.2020.619888
21. O’Connor H, Meloncelli N, Wilkinson SA, et al. Effective dietary interventions during pregnancy: a systematic review and meta-analysis of behavior change techniques to promote healthy eating. BMC Pregnancy Childbirth. 2025;25:112. doi:10.1186/s12884-025-07185-z
22. Alum EU, Ugwu OP, Obeagu EI, et al. Nutritional requirements during pregnancy: a comprehensive overview. Int J Innov Appl Res. 2023;11:26–34.
23. Sarubbo F, Esteban S, Miralles A, Moranta D. Effects of resveratrol and other polyphenols on Sirt1: relevance to brain function during aging. Curr Neuropharmacol. 2018;16:126–136. doi:10.2174/1570159X15666170703113212
24. Alum EU, Izah SC, Betiang PA, et al. The ketogenic diet in obesity management: friend or foe? Cell Biochem Biophys. 2025. doi:10.1007/s12013-025-01878-0
25. Glendining KA, Jasoni CL. Maternal high fat diet-induced obesity modifies histone binding and expression of Oxtr in offspring hippocampus in a sex-specific manner. Int J Mol Sci. 2019;20:329. doi:10.3390/ijms20020329
26. Suter MA, Ma J, Vuguin PM, et al. In utero exposure to a maternal high fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol. 2014;210:463.e1–463.e11. doi:10.1016/j.ajog.2014.01.045
27. Sharp GC, Salas LA, Monnereau C, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet. 2017;26:4067–4085. doi:10.1093/hmg/ddx290
28. Alfano R, Robinson O, Handakas E, Nawrot TS, Vineis P, Plusquin M. Perspectives and challenges of epigenetic determinants of childhood obesity: a systematic review. Obes Rev. 2022;23 Suppl 1:e13389. doi:10.1111/obr.13389
29. Reichetzeder C. Overweight and obesity in pregnancy: their impact on epigenetics. Eur J Clin Nutr. 2021;75:1710–1722. doi:10.1038/s41430-021-00905-6
30. Parlee SD, MacDougald OA. Maternal nutrition and risk of obesity in offspring: the Trojan horse of developmental plasticity. Biochim Biophys Acta Mol Basis Dis. 2014;1842:495–506. doi:10.1016/j.bbadis.2013.07.007
31. Alum EU, Obasi DC, Abba JN, Aniokete UC, Okoroh PN, Akwari AA. Evolving paradigms in nutrition therapy for diabetes: from carbohydrate counting to precision diets. Obesity Med. 2025;56:100622. doi:10.1016/j.obmed.2025.100622
32. Hor K, Dearden L, Herzstein E, Ozanne S, Hardingham G, Drake AJ. Maternal high fat and high sugar diet impacts on key DNA methylation enzymes in offspring brain in a sex‐specific manner. J Neuroendocrinol. 2025;37:e70046. doi:10.1111/jne.70046
33. Sen S, Rifas-Shiman SL, Shivappa N, et al. Associations of prenatal and early life dietary inflammatory potential with childhood adiposity and cardiometabolic risk in Project Viva. Pediatr Obes. 2018;13:292–300. doi:10.1111/ijpo.12221
34. Küpers LK, Fernández-Barrés S, Mancano G, et al. Maternal dietary glycemic index and glycemic load in pregnancy and offspring cord blood DNA methylation. Diabetes Care. 2022;45:1822–1832. doi:10.2337/dc21-2662
35. Jahan-Mihan A, Rodriguez J, Christie C, Sadeghi M, Zerbe T. The role of maternal dietary proteins in development of metabolic syndrome in offspring. Nutrients. 2015;7:9185–9217. doi:10.3390/nu7115460
36. Hu P, Li K, Peng X, et al. Nuclear receptor PPARα as a therapeutic target in diseases associated with lipid metabolism disorders. Nutrients. 2023;15:4772. doi:10.3390/nu15224772
37. Kaimala S, Kumar CA, Allouh MZ, Ansari SA, Emerald BS. Epigenetic modifications in pancreas development, diabetes, and therapeutics. Med Res Rev. 2022;42:1343–1371. doi:10.1002/med.21878
38. Adam-Raileanu A, Miron I, Lupu A, et al. Fetal growth restriction and its metabolism-related long-term outcomes—underlying mechanisms and clinical implications. Nutrients. 2025;17:555. doi:10.3390/nu17030555
39. Barouti Z, Heidari-Beni M, Shabanian-Boroujeni A, et al. Effects of DNA methylation on cardiometabolic risk factors: a systematic review and meta-analysis. Arch Public Health. 2022;80:150. doi:10.1186/s13690-022-00907-1
40. van Weelden W, Seed PT, Antoun E, et al. Folate and vitamin B12 status; associations with maternal glucose and neonatal DNA methylation sites related to dysglycaemia, in pregnant women with obesity. J Dev Orig Health Dis. 2022;13:168–176. doi:10.1017/S2040174421000246
41. Shen L, Li C, Wang Z, et al. Early-life exposure to severe famine is associated with higher methylation level in the IGF2 gene and higher total cholesterol in late adulthood: the Genomic Research of the Chinese Famine (GRECF) study. Clin Epigenetics. 2019;11:88. doi:10.1186/s13148-019-0676-3
42. McCullough LE, Miller EE, Mendez MA, Murtha AP, Murphy SK, Hoyo C. Maternal B vitamins: effects on offspring weight and DNA methylation at genomically imprinted domains. Clin Epigenetics. 2016;8:8. doi:10.1186/s13148-016-0174-9
43. Behere RV, Deshmukh AS, Otiv S, Gupte MD, Yajnik CS. Maternal vitamin B12 status during pregnancy and its association with outcomes of pregnancy and health of the offspring: a systematic review and implications for policy in India. Front Endocrinol. 2021;12:619176. doi:10.3389/fendo.2021.619176
44. Ueda H, Nakai T, Konishi T, Tanaka K, Sakazaki F, Min K-S. Effects of zinc deficiency and supplementation on leptin and leptin receptor expression in pregnant mice. Biol Pharm Bull. 2014;37:581–587. doi:10.1248/bpb.b13-00813
45. Mendes Garrido Abregú F, Caniffi C, Arranz CT, Tomat AL. Impact of zinc deficiency during prenatal and/or postnatal life on cardiovascular and metabolic diseases: experimental and clinical evidence. Adv Nutr. 2022;13:833–845. doi:10.1093/advances/nmac012
46. Azimi Z, Isa MR, Khan J, Wang SM, Ismail Z. Association of zinc level with DNA methylation and its consequences: a systematic review. Heliyon. 2022;8:e10815. doi:10.1016/j.heliyon.2022.e10815
47. Rodriguez-Hernandez Z, Bel-Aguilar J, Moreno-Franco B, et al. Differential association of selenium exposure with insulin resistance and β-cell function in middle age and older adults. Nutr Diabetes. 2025;15:5. doi:10.1038/s41387-025-00361-2
48. Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011;82:1807–1821. doi:10.1016/j.bcp.2011.07.093
49. Nacka-Aleksić M, Pirković A, Vilotić A, et al. The role of dietary polyphenols in pregnancy and pregnancy-related disorders. Nutrients. 2022;14:5246. doi:10.3390/nu14245246
50. Hadinata E, Taslim NA, Nurkolis F. Fruit‐ and vegetable‐derived polyphenols improve metabolic and renal outcomes in adults with metabolic syndrome and chronic kidney disease: a systematic review of randomized controlled trials. Nutrición Clínica y Dietética Hospitalaria. 2025;45:1. doi:10.12873/452hadinata
51. Bianchi M, Alisi A, Fabrizi M, et al. Maternal intake of n-3 polyunsaturated fatty acids during pregnancy is associated with differential methylation profiles in cord blood white cells. Front Genet. 2019;10:1050. doi:10.3389/fgene.2019.01050
52. Fazelian S, Moradi F, Agah S, et al. Effect of omega-3 fatty acids supplementation on cardio-metabolic and oxidative stress parameters in patients with chronic kidney disease: a systematic review and meta-analysis. BMC Nephrol. 2021;22:160. doi:10.1186/s12882-021-02351-9
53. Rajendran P, Dashwood W-M, Li L, et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin Epigenetics. 2015;7:102. doi:10.1186/s13148-015-0132-y
54. Ho E, Clarke JD, Dashwood RH. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J Nutr. 2009;139:2393–2396. doi:10.3945/jn.109.113332
55. Mallott EK, Amato KR. Butyrate production pathway abundances are similar in human and nonhuman primate gut microbiomes. Mol Biol Evol. 2021;39:msab279. doi:10.1093/molbev/msab279
56. Williams L, Seki Y, Vuguin PM, Charron MJ. Animal models of in utero exposure to a high fat diet: a review. Biochim Biophys Acta Mol Basis Dis. 2014;1842:507–519. doi:10.1016/j.bbadis.2013.07.006
57. Anckaert E, Romero S, Adriaenssens T, Smitz J. Effects of low methyl donor levels in culture medium during mouse follicle culture on oocyte imprinting establishment1. Biol Reprod. 2010;83:377–386. doi:10.1095/biolreprod.109.082164
58. Wojdasiewicz P, Poniatowski ŁA, Turczyn P, Frasuńska J, Paradowska-Gorycka A, Tarnacka B. Significance of Omega-3 fatty acids in the prophylaxis and treatment after spinal cord injury in rodent models. Mediators Inflamm. 2020;2020:3164260. doi:10.1155/2020/3164260
59. Mohajer N, Joloya EM, Seo J, Shioda T, Blumberg B. Epigenetic transgenerational inheritance of the effects of obesogen exposure. Front Endocrinol. 2021;12:787580. doi:10.3389/fendo.2021.787580
60. Harary D, Akinyemi A, Charron MJ, Fuloria M. Fetal growth and intrauterine epigenetic programming of obesity and cardiometabolic disease. Neoreviews. 2022;23:e363–e372. doi:10.1542/neo.23-6-e363
61. Aja PM, Fasogbon IV, Mbina SA, et al. Bisphenol-A (BPA) exposure as a risk factor for non-communicable diseases. IntechOpen; 2024.
62. Nayan NM, Husin A, Siran R. The risk of prenatal bisphenol A exposure in early life neurodevelopment: insights from epigenetic regulation. Early Hum Dev. 2024;198:106120. doi:10.1016/j.earlhumdev.2024.106120
63. Martínez MÁ, Blanco J, Rovira J, Kumar V, Domingo JL, Schuhmacher M. Bisphenol A analogues (BPS and BPF) present a greater obesogenic capacity in 3T3-L1 cell line. Food Chem Toxicol. 2020;140:111298. doi:10.1016/j.fct.2020.111298
64. Shu L, Meng Q, Diamante G, et al. Prenatal bisphenol A exposure in mice induces multitissue multiomics disruptions linking to cardiometabolic disorders. Endocrinology. 2018;160:409–429. doi:10.1210/en.2018-00817
65. Grohs MN, Reynolds JE, Liu J, et al. the APrON Study Team: prenatal maternal and childhood bisphenol a exposure and brain structure and behavior of young children. Environ Health. 2019;18:85. doi:10.1186/s12940-019-0528-9
66. Callan AC, Hinwood AL, Heffernan A, Eaglesham G, Mueller J, Odland JØ. Urinary bisphenol A concentrations in pregnant women. Int J Hyg Environ Health. 2013;216:641–644. doi:10.1016/j.ijheh.2012.10.002
67. Junge KM, Leppert B, Jahreis S, et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin Clin Epigenet. 2018;10:58. doi:10.1186/s13148-018-0478-z
68. Negri-Cesi P. Bisphenol A interaction with brain development and functions. Dose-Response. 2015;13:1559325815590394. doi:10.1177/1559325815590394
69. Zhong J, Baccarelli AA, Mansur A, et al. Maternal phthalate and personal care products exposure alters extracellular placental miRNA profile in twin pregnancies. Reprod Sci. 2019;26:289–294. doi:10.1177/1933719118770550
70. Harley KG, Berger K, Rauch S, et al. Association of prenatal urinary phthalate metabolite concentrations and childhood BMI and obesity. Pediatr Res. 2017;82:405–415. doi:10.1038/pr.2017.112
71. Sol CM, Delgado G, Kannan K, Jaddoe VWV, Trasande L, Santos S. Fetal exposure to phthalates and body mass index from infancy to adolescence. The Generation R study. Environ Res. 2025;274:121253. doi:10.1016/j.envres.2025.121253
72. Alahmadi H, Martinez S, Farrell R, et al. Mixtures of phthalates disrupt expression of genes related to lipid metabolism and peroxisome proliferator-activated receptor signaling in mouse granulosa cells. Toxicol Sci. 2024;202:69–84. doi:10.1093/toxsci/kfae105
73. Canouil M, Khamis A, Keikkala E, et al. Epigenome-wide association study reveals methylation loci associated with offspring gestational diabetes mellitus exposure and maternal methylome. Diabetes Care. 2021;44:1992–1999. doi:10.2337/dc20-2960
74. Young JL, Cai L, States JC. Impact of prenatal arsenic exposure on chronic adult diseases. System Biol Reprod Med. 2018;64:469–483. doi:10.1080/19396368.2018.1480076
75. Tinkelman NE, Spratlen MJ, Domingo-Relloso A, et al. Associations of maternal arsenic exposure with adult fasting glucose and insulin resistance in the strong heart study and strong heart family study. Environ Int. 2020;137:105531. doi:10.1016/j.envint.2020.105531
76. Pánico P, Velasco M, Salazar AM, et al. Is arsenic exposure a risk factor for metabolic syndrome? A review of the potential mechanisms. Front Endocrinol. 2022;13:878280. doi:10.3389/fendo.2022.878280
77. Arsenic. Available from: https://www.who.int/news-room/fact-sheets/detail/arsenic.
78. Alum EU. Highlights of heavy metals: molecular toxicity mechanisms, exposure dynamics, and environmental presence. IAA JAS. 2023;10:8–19. doi:10.59298/IAAJAS/2023/4.2.3222
79. Moynihan M, Telléz-Rojo MM, Colacino J, et al. Prenatal cadmium exposure is negatively associated with adiposity in girls not boys during adolescence. Front Public Health. 2019;7:61. doi:10.3389/fpubh.2019.00061
80. Park SS, Skaar DA, Jirtle RL, Hoyo C. Epigenetics, obesity and early-life cadmium or lead exposure. Epigenomics. 2017;9:57–75. doi:10.2217/epi-2016-0047
81. Sanders AP, Smeester L, Rojas D, et al. Cadmium exposure and the epigenome. Epigenetics. 2014;9:212–221. doi:10.4161/epi.26798
82. Wani AL, Ara A, Usmani JA. Lead toxicity: a review. Interdiscip Toxicol. 2015;8:55–64. doi:10.1515/intox-2015-0009
83. Hassen JY, Debella A, Eyeberu A, Mussa I. Level of exposure to aflatoxins during pregnancy and its association with adverse birth outcomes in Africa: a meta-analysis. Int Health. 2024;16:577–591. doi:10.1093/inthealth/ihae015
84. Kumar P, Mahato DK, Kamle M, Mohanta TK, Kang SG. Aflatoxins: a global concern for food safety, human health and their management. Front Microbiol. 2016;7:2170. doi:10.3389/fmicb.2016.02170
85. Rossetti MF, Canesini G, Lorenz V, Milesi MM, Varayoud J, Ramos JG. Epigenetic changes associated with exposure to glyphosate-based herbicides in mammals. Front Endocrinol. 2021;12:671991. doi:10.3389/fendo.2021.671991
86. Janesick AS, Shioda T, Blumberg B. Transgenerational inheritance of prenatal obesogen exposure. Mol Cell Endocrinol. 2014;398:31–35. doi:10.1016/j.mce.2014.09.002
87. Etzel TM, Engel SM, Quirós-Alcalá L, et al. Prenatal maternal organophosphorus pesticide exposures, paraoxonase 1, and childhood adiposity in the Mount Sinai Children’s Environmental Health Study. Environ Int. 2020;142:105858. doi:10.1016/j.envint.2020.105858
88. Marković Filipović J, Karan J, Ivelja I, Matavulj M, Stošić M. Acrylamide and potential risk of diabetes mellitus: effects on human population, glucose metabolism and beta-cell toxicity. Int J Mol Sci. 2022;23:6112. doi:10.3390/ijms23116112
89. Kadawathagedara M, Botton J, de Lauzon-Guillain B, et al. Dietary acrylamide intake during pregnancy and postnatal growth and obesity: results from the Norwegian Mother and Child Cohort Study (MoBa). Environ Int. 2018;113:325–334. doi:10.1016/j.envint.2018.01.004
90. Zhang Y, Wang Q, Li Y, Cheng J, Chen X, Zhang Y. Comprehensive profile of DNA adducts as both tissue and urinary biomarkers of exposure to acrylamide and chemo-preventive effect of catechins in rats. Chemosphere. 2022;286:131852. doi:10.1016/j.chemosphere.2021.131852
91. Lee YJ, Jung HW, Kim HY, Choi Y-J, Lee YA. Early-life exposure to per- and poly-fluorinated alkyl substances and growth, adiposity, and puberty in children: a systematic review. Front Endocrinol. 2021;12:683297. doi:10.3389/fendo.2021.683297
92. Niemiec SS, Kechris K, Pattee J, et al. Prenatal exposures to per- and polyfluoroalkyl substances and epigenetic aging in umbilical cord blood: the Healthy Start study. Environ Res. 2023;231:116215. doi:10.1016/j.envres.2023.116215
93. Tsai W-J, Hsieh W-S, Chen P-C, Liu C-Y. Prenatal perfluoroalkyl substance exposure in association with global histone post-translational methylation in 2-year-old children. Toxics. 2024;12:876. doi:10.3390/toxics12120876
94. Goodrich JM, Calkins MM, Caban-Martinez AJ, et al. Per- and polyfluoroalkyl substances, epigenetic age and DNA methylation: a cross-sectional study of firefighters. Epigenomics. 2021;13:1619–1639. doi:10.2217/epi-2021-0225
95. Alum EU, Uti DE. Modern perspectives on chelation therapy: optimizing biochemical approaches to heavy metal detoxification. Toxicol Environ Health Sci. 2025;1–4. doi:10.1007/s13530-025-00281-9
96. Tekola-Ayele F, Zeng X, Chatterjee S, et al. Placental multi-omics integration identifies candidate functional genes for birthweight. Nat Commun. 2022;13:2384. doi:10.1038/s41467-022-30007-1
97. Basak S, Mallick R, Navya Sree B, Duttaroy AK. Placental epigenome impacts fetal development: effects of maternal nutrients and gut microbiota. Nutrients. 2024;16:1860. doi:10.3390/nu16121860
98. Chung MK, House JS, Akhtari FS, et al. Members of the Exposomics Consortium: decoding the exposome: data science methodologies and implications in exposome-wide association studies (ExWASs). exposome. 2024;4:osae001. doi:10.1093/exposome/osae001
99. Amine I, Guillien A, Philippat C, et al. Environmental exposures in early-life and general health in childhood. Environ Health. 2023;22:53. doi:10.1186/s12940-023-01001-x
100. Maitre L, De Bont J, Casas M, et al. Human Early Life Exposome (HELIX) study: a European population-based exposome cohort. BMJ Open. 2018;8:e021311. doi:10.1136/bmjopen-2017-021311
101. Wang C, Reimann B, Nawrot TS, et al. Meet-in-the-middle meets multi-omics identifying molecular signatures of environmental drivers of childhood overweight. Environ Int. 2025;202:109630. doi:10.1016/j.envint.2025.109630
102. Felix JF, Joubert BR, Baccarelli AA, et al. Cohort Profile: pregnancy And Childhood Epigenetics (PACE) Consortium. Int J Epidemiol. 2018;47:22–23u. doi:10.1093/ije/dyx190
103. Dontje ML, Eastwood P, Straker L. Western Australian pregnancy cohort (Raine) Study: generation 1. BMJ Open. 2019;9:e026276. doi:10.1136/bmjopen-2018-026276
104. Environmental influences on Child Health Outcomes (ECHO) Program. National Institutes of Health (NIH). Available from: https://www.nih.gov/echo.
105. Khodasevich D, Holland N, Harley KG, Eskenazi B, Barcellos LF, Cardenas A. Prenatal exposure to environmental phenols and phthalates and altered patterns of DNA methylation in childhood. Environ Int. 2024;190:108862. doi:10.1016/j.envint.2024.108862
106. Lichtveld K, Thomas K, Tulve NS. Chemical and non-chemical stressors affecting childhood obesity: a systematic scoping review. J Expo Sci Environ Epidemiol. 2018;28:1–12. doi:10.1038/jes.2017.18
107. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31:363–373. doi:10.1016/j.reprotox.2010.12.055
108. Saxena R, Bozack AK, Gamble MV. Nutritional influences on one-carbon metabolism: effects on arsenic methylation and toxicity. Annu Rev Nutr. 2018;38:401–429. doi:10.1146/annurev-nutr-082117-051757
109. Niedzwiecki MM, Hall MN, Liu X, et al. A dose–response study of arsenic exposure and global methylation of peripheral blood mononuclear cell DNA in bangladeshi adults. Environ Health Perspect. 2013;121:1306–1312. doi:10.1289/ehp.1206421
110. Wang Z, Song J, Li C, et al. DNA methylation of the INSR gene as a mediator of the association between prenatal exposure to famine and adulthood waist circumference. Sci Rep. 2020;10:12212. doi:10.1038/s41598-020-69120-w
111. Iñiguez C, Ballester F, Costa O, et al. INMA Study Investigators: maternal smoking during pregnancy and fetal biometry: the INMA Mother and Child Cohort Study. Am J Epidemiol. 2013;178:1067–1075. doi:10.1093/aje/kwt085
112. García-Villarino M, Fernández-Iglesias R, Riaño-Galán I, et al. Prenatal exposure to cigarette smoke and anogenital distance at 4 years in the INMA-Asturias Cohort. Int J Environ Res Public Health. 2021;18:4774. doi:10.3390/ijerph18094774
113. Chen L-W, Aubert AM, Shivappa N, et al. Maternal dietary quality, inflammatory potential and childhood adiposity: an individual participant data pooled analysis of seven European cohorts in the ALPHABET consortium. BMC Med. 2021;19:33. doi:10.1186/s12916-021-01908-7
114. Serefidou M, Venkatasubramani AV, Imhof A. The impact of one carbon metabolism on histone methylation. Front Genet. 2019;10:764. doi:10.3389/fgene.2019.00764
115. He ZX, Wu DQ, Sun ZH, et al. Protein or energy restriction during late gestation alters fetal growth and visceral organ mass: an evidence of intrauterine programming in goats. Anim Reprod Sci. 2013;137:177–182. doi:10.1016/j.anireprosci.2013.01.005
116. Zheng S, Rollet M, Pan Y-X. Protein restriction during gestation alters histone modifications at the glucose transporter 4 (GLUT4) promoter region and induces GLUT4 expression in skeletal muscle of female rat offspring. J Nutr Biochem. 2012;23:1064–1071. doi:10.1016/j.jnutbio.2011.05.013
117. Lu G, Xu H, Chang D, et al. Arsenic exposure is associated with DNA hypermethylation of the tumor suppressor gene p16. J Occup Med Toxicol. 2014;9:42. doi:10.1186/s12995-014-0042-5
118. Sijko M, Kozłowska L. Influence of dietary compounds on arsenic metabolism and toxicity. Part II—Human Studies. Toxics. 2021;9:259. doi:10.3390/toxics9100259
119. Martinez-Morata I, Parvez F, Wu H, et al. Influence of folic acid and vitamin B12 supplementation on arsenic methylation: a double-blinded, placebo-controlled trial in Bangladeshi children. Environ Int. 2024;187:108715. doi:10.1016/j.envint.2024.108715
120. Alum EU, Aja W, Ugwu OPC, Obeagu EI, Okon MB. Assessment of vitamin composition of ethanol leaf and seed extracts of datura stramonium. Avicenna J Med Biochem. 2023;11:92–97. doi:10.34172/ajmb.2023.2421
121. Flori L, Piragine E, Spezzini J, Citi V, Calderone V, Martelli A. Influence of polyphenols on adipose tissue: sirtuins as pivotal players in the browning process. Int J Mol Sci. 2023;24:9276. doi:10.3390/ijms24119276
122. Wu L, Wu X, Zhang X, et al. Folic acid alleviates gestational arsenic exposure-induced spatial learning and memory impairment in mice offspring via consuming SAM-mediated DNA hypomethylation in the developing brain. Toxicol Lett. 2025;411:61–71. doi:10.1016/j.toxlet.2025.07.1406
123. Brito S, Lee M-G, Bin B-H, Lee J-S. Zinc and its transporters in epigenetics. Mol Cells. 2020;43:323–330. doi:10.14348/molcells.2020.0026
124. Cao Z, Yang F, Lin Y, et al. Selenium antagonizes cadmium-induced inflammation and oxidative stress via suppressing the interplay between NLRP3 inflammasome and HMGB1/NF-κB pathway in duck hepatocytes. Int J Mol Sci. 2022;23:6252. doi:10.3390/ijms23116252
125. Zhang Y, Mustieles V, Sun Q, et al. Association of early pregnancy perfluoroalkyl and polyfluoroalkyl substance exposure with birth outcomes. JAMA Network Open. 2023;6:e2314934. doi:10.1001/jamanetworkopen.2023.14934
126. Huen K, Calafat AM, Bradman A, Yousefi P, Eskenazi B, Holland N. Maternal phthalate exposure during pregnancy is associated with DNA methylation of LINE-1 and Alu repetitive elements in Mexican-American children. Environ Res. 2016;148:55–62. doi:10.1016/j.envres.2016.03.025
127. Milagro FI, Mansego ML, De Miguel C, Martínez JA. Dietary factors, epigenetic modifications and obesity outcomes: progresses and perspectives. Mol Aspects Med. 2013;34:782–812. doi:10.1016/j.mam.2012.06.010
128. Lorente-Cebrián S, Costa AGV, Castillo-Rivas JA, et al. Phenolic compounds and epigenetic mechanisms regulating gene expression: effects on human health. J Physiol Biochem. 2025. doi:10.1007/s13105-025-01105-7
129. Marzag H, Warnault P, Bougrin K, Martinet N, Benhida R. Chapter 7 - natural polyphenols as potent inhibitors of DNA methyltransferases. In: Rahman AU, editor. Studies in Natural Products Chemistry. Elsevier; 2014:195–223.
130. Frankhouser DE, Steck S, Sovic MG, et al. Dietary omega-3 fatty acid intake impacts peripheral blood DNA methylation -anti-inflammatory effects and individual variability in a pilot study. J Nutr Biochem. 2022;99:108839. doi:10.1016/j.jnutbio.2021.108839
131. Lozano M, Yousefi P, Broberg K, et al. DNA methylation changes associated with prenatal mercury exposure: a meta-analysis of prospective cohort studies from PACE consortium. Environ Res. 2022;204:112093. doi:10.1016/j.envres.2021.112093
132. Cheng M, Conley D, Kuipers T, et al. Accelerated biological aging six decades after prenatal famine exposure. Proc Natl Acad Sci U S A. 2024;121:e2319179121. doi:10.1073/pnas.2319179121
133. Margetaki K, Bempi V, Michalaki E, et al. Prenatal air pollution exposure and childhood obesity: effect modification by maternal fruits and vegetables intake. Int J Hyg Environ Health. 2024;256:114314. doi:10.1016/j.ijheh.2023.114314
134. Rammah A, Whitworth KW, Amos CI, et al. Air pollution, residential greenness and metabolic dysfunction during early pregnancy in the INfancia y Medio Ambiente (INMA) Cohort. Int J Environ Res Public Health. 2021;18:9354. doi:10.3390/ijerph18179354
135. Husso A, Pessa-Morikawa T, Koistinen VM, et al. Impacts of maternal microbiota and microbial metabolites on fetal intestine, brain, and placenta. BMC Biol. 2023;21:207. doi:10.1186/s12915-023-01709-9
136. Ziętek M, Celewicz Z, Szczuko M. Short-chain fatty acids, maternal microbiota and metabolism in pregnancy. Nutrients. 2021;13:1244. doi:10.3390/nu13041244
137. Ren Y, Huang P, Zhang L, et al. Dual regulation mechanism of obesity: DNA methylation and intestinal flora. Biomedicines. 2024;12:1633. doi:10.3390/biomedicines12081633
138. Wei M, Huang F, Zhao L, et al. A dysregulated bile acid-gut microbiota axis contributes to obesity susceptibility. EBioMedicine. 2020;55:102766. doi:10.1016/j.ebiom.2020.102766
139. Coretti L, Buommino E, Lembo F. The aryl hydrocarbon receptor pathway: a linking bridge between the gut microbiome and neurodegenerative diseases. Front Cell Neurosci. 2024;18:1433747. doi:10.3389/fncel.2024.1433747
140. Terova G, Díaz N, Rimoldi S, Ceccotti C, Gliozheni E, Piferrer F. Effects of sodium butyrate treatment on histone modifications and the expression of genes related to epigenetic regulatory mechanisms and immune response in european sea bass (Dicentrarchus Labrax) fed a plant-based diet. PLoS One. 2016;11:e0160332. doi:10.1371/journal.pone.0160332
141. Huang Y-J, Wang P-M, Tang K-S, Chen C-J, Huang Y-H, Tiao -M-M. Butyrate ameliorates maternal high-fat diet-induced fetal liver cellular apoptosis. PLoS One. 2022;17:e0270657. doi:10.1371/journal.pone.0270657
142. Fu Y, Moscoso DI, Porter J, et al. The relationship between dietary fiber intake and short chain fatty acid-producing bacteria during critical illness: a prospective cohort study. JPEN J Parenter Enteral Nutr. 2020;44:463–471. doi:10.1002/jpen.1682
143. Qin X, Zhang M, Chen S, Tang Y, Cui J, Ding G. Short-chain fatty acids in fetal development and metabolism. Trends Mol Med. 2025;31:625–639. doi:10.1016/j.molmed.2024.11.014
144. Gray LEK, O’Hely M, Ranganathan S, Sly PD, Vuillermin P. The maternal diet, gut bacteria, and bacterial metabolites during pregnancy influence offspring asthma. Front Immunol. 2017;8:365. doi:10.3389/fimmu.2017.00365
145. Penders J, Thijs C, van den Brandt PA, et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut. 2007;56:661–667. doi:10.1136/gut.2006.100164
146. Hill CJ, Lynch DB, Murphy K, et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome. 2017;5:4. doi:10.1186/s40168-016-0213-y
147. Maradonna F, Carnevali O. Lipid metabolism alteration by endocrine disruptors in animal models: an overview. Front Endocrinol. 2018;9:654. doi:10.3389/fendo.2018.00654
148. Bao M, Hofsink N, Plösch T. LPS versus Poly I: C model: comparison of long-term effects of bacterial and viral maternal immune activation on the offspring. Am J Physiol Regul Integr Comp Physiol. 2022;322:R99–R111. doi:10.1152/ajpregu.00087.2021
149. Akhter N, Kochumon S, Hasan A, et al. IFN-γ and LPS induce synergistic expression of CCL2 in monocytic cells via H3K27 acetylation. J Inflamm Res. 2022;15:4291–4302. doi:10.2147/JIR.S368352
150. Ghosh S, Nukavarapu SP, Jala VR. Effects of heavy metals on gut barrier integrity and gut microbiota. Microbiota Host. 2024;2:e230015. doi:10.1530/MAH-23-0015
151. He J, Zhang P, Shen L, et al. Short-chain fatty acids and their association with signalling pathways in inflammation, glucose and lipid metabolism. Int J Mol Sci. 2020;21:6356. doi:10.3390/ijms21176356
152. Warmink-Perdijk WDB, Peters LL, Tigchelaar EF, et al. Lifelines NEXT: a prospective birth cohort adding the next generation to the three-generation Lifelines cohort study. Eur J Epidemiol. 2020;35:157–168. doi:10.1007/s10654-020-00614-7
153. Warner M, Ye M, Harley K, Kogut K, Bradman A, Eskenazi B. Prenatal DDT exposure and child adiposity at age 12: the CHAMACOS study. Environ Res. 2017;159:606–612. doi:10.1016/j.envres.2017.08.050
154. Warner M, Aguilar Schall R, Harley KG, Bradman A, Barr D, Eskenazi B. In utero DDT and DDE exposure and obesity status of 7-year-old Mexican-American children in the CHAMACOS cohort. Environ Health Perspect. 2013;121:631–636. doi:10.1289/ehp.1205656
155. Berger K, Hyland C, Ames JL, et al. Prenatal exposure to mixtures of phthalates, parabens, and other phenols and obesity in five-year-olds in the CHAMACOS Cohort. Int J Environ Res Public Health. 2021;18:1796. doi:10.3390/ijerph18041796
156. Masse KE, Lu VB. Short-chain fatty acids, secondary bile acids and indoles: gut microbial metabolites with effects on enteroendocrine cell function and their potential as therapies for metabolic disease. Front Endocrinol. 2023;14:1169624. doi:10.3389/fendo.2023.1169624
157. Rios-Morales M, Vieira-Lara MA, Homan E, et al. Butyrate oxidation attenuates the butyrate-induced improvement of insulin sensitivity in myotubes. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166476. doi:10.1016/j.bbadis.2022.166476
158. Le Doare K, Holder B, Bassett A, Pannaraj PS. Mother’s milk: a purposeful contribution to the development of the infant microbiota and immunity. Front Immunol. 2018;9:361. doi:10.3389/fimmu.2018.00361
159. Hatmal MM, Al-Hatamleh MAI, Olaimat AN, et al. Immunomodulatory properties of human breast milk: microRNA contents and potential epigenetic effects. Biomedicines. 2022;10:1219. doi:10.3390/biomedicines10061219
160. Mirpuri J. Evidence for maternal diet-mediated effects on the offspring microbiome and immunity: implications for public health initiatives. Pediatr Res. 2021;89:301–306. doi:10.1038/s41390-020-01121-x
161. Fehr K, Moossavi S, Sbihi H, et al. Breastmilk feeding practices are associated with the co-occurrence of bacteria in mothers’ milk and the infant gut: the CHILD Cohort Study. Cell Host Microbe. 2020;28:285–297.e4. doi:10.1016/j.chom.2020.06.009
162. Azad MB, Konya T, Persaud RR, et al. CHILD Study Investigators: impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. BJOG. 2016;123:983–993. doi:10.1111/1471-0528.13601
163. Hartwig FP, Davey Smith G, Simpkin AJ, Victora CG, Relton CL, Caramaschi D. Association between Breastfeeding and DNA Methylation over the Life Course: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Nutrients. 2020;12:3309. doi:10.3390/nu12113309
164. Korpela K, Dikareva E, Hanski E, Kolho K-L, de Vos WM, Salonen A. Cohort profile: finnish Health and Early Life Microbiota (HELMi) longitudinal birth cohort. BMJ Open. 2019;9:e028500. doi:10.1136/bmjopen-2018-028500
165. Ben Maamar M, Nilsson EE, Skinner MK. Epigenetic transgenerational inheritance, gametogenesis and germline development†. Biol Reprod. 2021;105:570–592. doi:10.1093/biolre/ioab085
166. Araki R, Nishida S, Nakajima Y, et al. Low folate induces abnormal neuronal maturation and DNA hypomethylation of neuronal differentiation-related genes in cultured mouse neural stem and progenitor cells. Heliyon. 2021;7:e08071. doi:10.1016/j.heliyon.2021.e08071
167. Seisenberger S, Andrews S, Krueger F, et al. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–862. doi:10.1016/j.molcel.2012.11.001
168. Seisenberger S, Peat JR, Reik W. Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr Opin Cell Biol. 2013;25:281–288. doi:10.1016/j.ceb.2013.02.013
169. Ly L, Chan D, Aarabi M, et al. Intergenerational impact of paternal lifetime exposures to both folic acid deficiency and supplementation on reproductive outcomes and imprinted gene methylation. Mol Hum Reprod. 2017;23:461–477. doi:10.1093/molehr/gax029
170. Madrid A, Koueik J, Papale LA, et al. Folate-mediated transgenerational inheritance of sperm DNA methylation patterns correlate with spinal axon regeneration. Epigenetics. 2024;19:2380930. doi:10.1080/15592294.2024.2380930
171. Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS One. 2013;8:e55387. doi:10.1371/journal.pone.0055387
172. Cuellar Partida G, Laurin C, Ring SM, et al. Genome-wide survey of parent-of-origin effects on DNA methylation identifies candidate imprinted loci in humans. Hum Mol Genet. 2018;27:2927–2939. doi:10.1093/hmg/ddy206
173. da Rocha ST, Gendrel A-V. The influence of DNA methylation on monoallelic expression. Essays Biochem. 2019;63:663–676. doi:10.1042/EBC20190034
174. Rancourt RC, Harris HR, Barault L, Michels KB. The prevalence of loss of imprinting of H19 and IGF2 at birth. FASEB J. 2013;27:3335–3343. doi:10.1096/fj.12-225284
175. Wu H-Y, Cheng Y, Jin L-Y, et al. Paternal obesity impairs hepatic gluconeogenesis of offspring by altering Igf2/H19 DNA methylation. Mol Cell Endocrinol. 2021;529:111264. doi:10.1016/j.mce.2021.111264
176. Tobi EW, Goeman JJ, Monajemi R, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. doi:10.1038/ncomms6592
177. Lee H-S, Barraza-Villarreal A, Biessy C, et al. Dietary supplementation with polyunsaturated fatty acid during pregnancy modulates DNA methylation at IGF2/H19 imprinted genes and growth of infants. Physiol Genomics. 2014;46:851–857. doi:10.1152/physiolgenomics.00061.2014
178. Sales VM, Ferguson-Smith AC, Patti M-E. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab. 2017;25:559–571. doi:10.1016/j.cmet.2017.02.016
179. Kaspar D, Hastreiter S, Irmler M, Hrabé de Angelis M, Beckers J. Nutrition and its role in epigenetic inheritance of obesity and diabetes across generations. Mamm Genome. 2020;31:119–133. doi:10.1007/s00335-020-09839-z
180. Soubry A, Schildkraut JM, Murtha A, et al. Paternal obesity is associated with IGF2hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 2013;11:29. doi:10.1186/1741-7015-11-29
181. Perkins E, Murphy SK, Murtha AP, et al. Insulin-like growth factor 2/H19 methylation at birth and risk of overweight and obesity in children. J Pediatr. 2012;161:31–39. doi:10.1016/j.jpeds.2012.01.015
182. Huang R-C, Galati JC, Burrows S, et al. DNA methylation of the IGF2/H19 imprinting control region and adiposity distribution in young adults. Clin Epigenetics. 2012;4:21. doi:10.1186/1868-7083-4-21
183. Soubry A, Murphy SK, Wang F, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes. 2015;39:650–657. doi:10.1038/ijo.2013.193
184. Villanueva-Hayes C, Millership SJ. Imprinted genes impact upon beta cell function in the current (and Potentially Next) Generation. Front Endocrinol. 2021;12:660532. doi:10.3389/fendo.2021.660532
185. Braz CU, Passamonti MM, Khatib H. Characterization of genomic regions escaping epigenetic reprogramming in sheep. Environ Epigenet. 2023;10:dvad010. doi:10.1093/eep/dvad010
186. Anvar Z, Chakchouk I, Demond H, Sharif M, Kelsey G, Van den Veyver IB. DNA methylation dynamics in the female germline and maternal-effect mutations that disrupt genomic imprinting. Genes. 2021;12:1214. doi:10.3390/genes12081214
187. Torres-Flores U, Hernández-Hernández A. The interplay between replacement and retention of histones in the sperm genome. Front Genet. 2020;11:780. doi:10.3389/fgene.2020.00780
188. Fullston T, Ohlsson-Teague EMC, Print CG, Sandeman LY, Lane M. Sperm microRNA content is altered in a mouse model of male obesity, but the same suite of microRNAs are not altered in offspring’s sperm. PLoS One. 2016;11:e0166076. doi:10.1371/journal.pone.0166076
189. Schellong K, Melchior K, Ziska T, Rancourt RC, Henrich W, Plagemann A. Maternal but Not Paternal High-Fat Diet (HFD) exposure at conception predisposes for ‘diabesity’ in offspring generations. Int J Environ Res Public Health. 2020;17:4229. doi:10.3390/ijerph17124229
190. Chao S, Lu J, Li L-J, et al. Maternal obesity may disrupt offspring metabolism by inducing oocyte genome hyper-methylation via increased DNMTs. elife. 2024;13:RP97507. doi:10.7554/eLife.97507
191. Nava-Rivera LE, Betancourt-Martínez ND, Lozoya-Martínez R, et al. Transgenerational effects in DNA methylation, genotoxicity and reproductive phenotype by chronic arsenic exposure. Sci Rep. 2021;11:8276. doi:10.1038/s41598-021-87677-y
192. Ceja-Galicia ZA, Daniel A, Salazar AM, Pánico P, Ostrosky-Wegman P, Díaz-Villaseñor A. Effects of arsenic on adipocyte metabolism: is arsenic an obesogen? Mol Cell Endocrinol. 2017;452:25–32. doi:10.1016/j.mce.2017.05.008
193. Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi:10.1073/pnas.0806560105
194. Vågerö D, Pinger PR, Aronsson V, van den Berg GJ. Paternal grandfather’s access to food predicts all-cause and cancer mortality in grandsons. Nat Commun. 2018;9:5124. doi:10.1038/s41467-018-07617-9
195. Fante T, Simino LAP, Fontana MF, et al. Maternal high-fat diet consumption programs male offspring to mitigate complications in liver regeneration. J Dev Orig Health Dis. 2022;13:575–582. doi:10.1017/S2040174421000659
196. Paz HA, Buddha L, Lam T, et al. Maternal high-fat diet-induced obesity in offspring: unraveling adipose tissue dysfunction mediated by increased heat shock proteins. Int J Biochem Cell Biol. 2025;186:106812. doi:10.1016/j.biocel.2025.106812
197. Ito S, Hirabayashi K, Moriishi K, et al. Novel sex-dependent differentially methylated regions are demethylated in adult male mouse livers. Biochem Biophys Res Commun. 2015;462:332–338. doi:10.1016/j.bbrc.2015.04.137
198. LaBarge S, McDonald M, Smith-Powell L, Auwerx J, Huss JM. Estrogen-related receptor-α (ERRα) deficiency in skeletal muscle impairs regeneration in response to injury. FASEB J. 2014;28:1082–1097. doi:10.1096/fj.13-229211
199. Christians JK, Shergill HK, Albert AYK. Sex-dependent effects of prenatal food and protein restriction on offspring physiology in rats and mice: systematic review and meta-analyses. Biol Sex Differ. 2021;12:21. doi:10.1186/s13293-021-00365-4
200. Harmancıoğlu B, Kabaran S. Maternal high fat diets: impacts on offspring obesity and epigenetic hypothalamic programming. Front Genet. 2023;14:1158089. doi:10.3389/fgene.2023.1158089
201. Christoforou ER, Sferruzzi-Perri AN. Molecular mechanisms governing offspring metabolic programming in rodent models of in utero stress. Cell Mol Life Sci. 2020;77:4861–4898. doi:10.1007/s00018-020-03566-z
202. Filardi T, Panimolle F, Lenzi A, Morano S. Bisphenol A and phthalates in diet: an emerging link with pregnancy complications. Nutrients. 2020;12:525. doi:10.3390/nu12020525
203. Yue H, Zhu H, Wu X, et al. Maternal bisphenol A (BPA) exposure induces placental dysfunction and health risk in adult female offspring: insights from a mouse model. Sci Total Environ. 2025;958:177714. doi:10.1016/j.scitotenv.2024.177714
204. House JS, Mendez M, Maguire RL, et al. Periconceptional maternal mediterranean diet is associated with favorable offspring behaviors and altered CpG methylation of imprinted genes. Front Cell Dev Biol. 2018;6:107. doi:10.3389/fcell.2018.00107
205. Martin CL, Jima D, Sharp GC, et al. Maternal pre-pregnancy obesity, offspring cord blood DNA methylation, and offspring cardiometabolic health in early childhood: an epigenome-wide association study. Epigenetics. 2019;14:325–340. doi:10.1080/15592294.2019.1581594
206. Bouwland-Both MI, van Mil NH, Tolhoek CP, et al. Prenatal parental tobacco smoking, gene specific DNA methylation, and newborns size: the Generation R study. Clin Epigenetics. 2015;7:83. doi:10.1186/s13148-015-0115-z
207. Young JL, Cai L. Implications for prenatal cadmium exposure and adverse health outcomes in adulthood. Toxicol Appl Pharmacol. 2020;403:115161. doi:10.1016/j.taap.2020.115161
208. Tekola-Ayele F, Biedrzycki RJ, Habtewold TD, et al. Sex-differentiated placental methylation and gene expression regulation has implications for neonatal traits and adult diseases. Nat Commun. 2025;16:4004. doi:10.1038/s41467-025-58128-3
209. Gruzieva O, Xu C-J, Breton CV, et al. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Perspect. 2017;125:104–110. doi:10.1289/EHP36
210. Broséus L, Guilbert A, Hough I, et al. Placental DNA methylation signatures of prenatal air pollution exposure and potential effects on birth outcomes: an analysis of three prospective cohorts. Lancet Planet Health. 2024;8:e297–e308. doi:10.1016/S2542-5196(24)00045-7
211. Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS One. 2011;6:e21662. doi:10.1371/journal.pone.0021662
212. Zhang L, Zou W, Zhang S, et al. Maternal high-fat diet orchestrates offspring hepatic cholesterol metabolism via MEF2A hypermethylation-mediated CYP7A1 suppression. Cell Mol Biol Lett. 2024;29:154. doi:10.1186/s11658-024-00673-8
213. Kasiappan R, Rajarajan D. Role of MicroRNA regulation in obesity-associated breast cancer: nutritional perspectives. Adv Nutr. 2017;8:868–888. doi:10.3945/an.117.015800
214. Rubinow KB, Wang S, den Hartigh LJ, et al. Hematopoietic androgen receptor deficiency promotes visceral fat deposition in male mice without impairing glucose homeostasis. Andrology. 2015;3:787–796. doi:10.1111/andr.12055
215. Teng C, Goodwin B, Shockley K, et al. Bisphenol A affects androgen receptor function via multiple mechanisms. Chem Biol Interact. 2013;203:556–564. doi:10.1016/j.cbi.2013.03.013
216. Perera L, Li Y, Coons LA, et al. Binding of bisphenol A, bisphenol AF, and bisphenol S on the androgen receptor: coregulator recruitment and stimulation of potential interaction sites. Toxicol in vitro. 2017;44:287–302. doi:10.1016/j.tiv.2017.07.020
217. Le Blévec E, Muroňová J, Ray PF, Arnoult C. Paternal epigenetics: mammalian sperm provide much more than DNA at fertilization. Mol Cell Endocrinol. 2020;518:110964. doi:10.1016/j.mce.2020.110964
218. Bodden C, Hannan AJ, Reichelt AC. Diet-induced modification of the sperm epigenome programs metabolism and behavior. Trends Endocrinol Metab. 2020;31:131–149. doi:10.1016/j.tem.2019.10.005
219. Haberman M, Menashe T, Cohen N, et al. Paternal high-fat diet affects weight and DNA methylation of their offspring. Sci Rep. 2024;14:19874. doi:10.1038/s41598-024-70438-y
220. Nohara K, Nakabayashi K, Okamura K, Suzuki T, Suzuki S, Hata K. Gestational arsenic exposure induces site-specific DNA hypomethylation in active retrotransposon subfamilies in offspring sperm in mice. Epigene Chromat. 2020;13:53. doi:10.1186/s13072-020-00375-3
221. Cabrera Zapata LE, Garcia-Segura LM, Cambiasso MJ, Arevalo MA. Genetics and epigenetics of the X and Y chromosomes in the sexual differentiation of the brain. Int J Mol Sci. 2022;23:12288. doi:10.3390/ijms232012288
222. Lin J, Zhang J, Ma L, et al. KDM6A facilitates Xist upregulation at the onset of X inactivation. Biol Sex Differ. 2025;16:1. doi:10.1186/s13293-024-00683-3
223. Diniz MS, Magalhães CC, Tocantins C, Grilo LF, Teixeira J, Pereira SP. Nurturing through nutrition: exploring the role of antioxidants in maternal diet during pregnancy to mitigate developmental programming of chronic diseases. Nutrients. 2023;15:4623. doi:10.3390/nu15214623
224. Jiao F, Yan X, Yu Y, et al. Protective effects of maternal methyl donor supplementation on adult offspring of high fat diet-fed dams. J Nutr Biochem. 2016;34:42–51. doi:10.1016/j.jnutbio.2016.04.005
225. Tekola-Ayele F, Workalemahu T, Gorfu G, et al. Sex differences in the associations of placental epigenetic aging with fetal growth. Aging. 2019;11:5412–5432. doi:10.18632/aging.102124
226. Kadakia R, Zheng Y, Zhang Z, Zhang W, Hou L, Josefson JL. Maternal pre-pregnancy BMI downregulates neonatal cord blood LEP methylation. Pediatr Obes. 2017;12 Suppl 1:57–64. doi:10.1111/ijpo.12204
227. Zhu Z, Cao F, Li X. Epigenetic Programming and Fetal Metabolic Programming. Front Endocrinol. 2019;10:764. doi:10.3389/fendo.2019.00764
228. Miura R, Araki A, Minatoya M, et al. An epigenome-wide analysis of cord blood DNA methylation reveals sex-specific effect of exposure to bisphenol A. Sci Rep. 2019;9:12369. doi:10.1038/s41598-019-48916-5
229. Alfano R, Zugna D, Barros H, et al. Cord blood epigenome-wide meta-analysis in six European-based child cohorts identifies signatures linked to rapid weight growth. BMC Med. 2023;21:17. doi:10.1186/s12916-022-02685-7
230. Mulligan C, D’Errico N, Stees J, Hughes D. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7:853–857. doi:10.4161/epi.21180
231. Green BB, Marsit CJ. Select prenatal environmental exposures and subsequent alterations of gene-specific and repetitive element DNA methylation in fetal tissues. Curr Environ Health Rep. 2015;2:126–136. doi:10.1007/s40572-015-0045-0
232. Gagné-Ouellet V, Breton E, Thibeault K, et al. Placental epigenome-wide association study identified loci associated with childhood adiposity at 3 years of age. Int J Mol Sci. 2020;21:7201. doi:10.3390/ijms21197201
233. Nelissen ECM, van Montfoort APA, Dumoulin JCM, Evers JLH. Epigenetics and the placenta. Hum Reprod Update. 2011;17:397–417. doi:10.1093/humupd/dmq052
234. Iacomino G, Siani A. Role of microRNAs in obesity and obesity-related diseases. Genes Nutr. 2017;12:23. doi:10.1186/s12263-017-0577-z
235. Dalgaard LT, Sørensen AE, Hardikar AA, Joglekar MV. The microRNA-29 family: role in metabolism and metabolic disease. Am J Physiol Cell Physiol. 2022;323:C367–C377. doi:10.1152/ajpcell.00051.2022
236. Ye Z, Wang S, Huang X, et al. Plasma exosomal miRNAs associated with metabolism as early predictor of gestational diabetes mellitus. Diabetes. 2022;71:2272–2283. doi:10.2337/db21-0909
237. Addo KA, Palakodety N, Hartwell HJ, Tingare A, Fry RC. Placental microRNAs: responders to environmental chemicals and mediators of pathophysiology of the human placenta. Toxicol Rep. 2020;7:1046–1056. doi:10.1016/j.toxrep.2020.08.002
238. Dalrymple KV, Vogel C, Godfrey KM, et al. Longitudinal dietary trajectories from preconception to mid-childhood in women and children in the Southampton Women’s Survey and their relation to offspring adiposity: a group-based trajectory modelling approach. Int J Obes. 2022;46:758–766. doi:10.1038/s41366-021-01047-2
239. Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, et al. Periconceptional maternal folic acid use of 400 µg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One. 2009;4:e7845. doi:10.1371/journal.pone.0007845
240. Yajnik CS, Deshpande SS, Jackson AA, et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: the Pune Maternal Nutrition Study. Diabetologia. 2008;51:29–38. doi:10.1007/s00125-007-0793-y
241. Kumaran K, Yajnik P, Lubree H, et al. The Pune Rural Intervention in Young Adolescents (PRIYA) study: design and methods of a randomised controlled trial. BMC Nutr. 2017;3:41. doi:10.1186/s40795-017-0143-5
242. Gamble MV, Liu X, Slavkovich V, et al. Folic acid supplementation lowers blood arsenic. Am J Clin Nutr. 2007;86:1202–1209. doi:10.1093/ajcn/86.4.1202
243. Pilsner JR, Hall MN, Liu X, et al. Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS One. 2012;7:e37147. doi:10.1371/journal.pone.0037147
244. Wei C, Choma EF, Wang X, et al. Comparing folic acid interventions and arsenic reduction strategies for neural tube defect prevention in bangladesh: a systematic review and decision analysis. Birth Defects Res. 2025;117:e2494. doi:10.1002/bdr2.2494
245. Prado EL, Ullman MT, Muadz H, Alcock KJ, Shankar AH, Group SS. The effect of maternal multiple micronutrient supplementation on cognition and mood during pregnancy and postpartum in indonesia: a randomized trial. PLoS One. 2012;7:e32519. doi:10.1371/journal.pone.0032519
246. Prado EL, Sebayang SK, Apriatni M, et al. Maternal multiple micronutrient supplementation and other biomedical and socioenvironmental influences on children’s cognition at age 9-12 years in Indonesia: follow-up of the SUMMIT randomised trial. Lancet Glob Health. 2017;5:e217–e228. doi:10.1016/S2214-109X(16)30354-0
247. Yildiz A, Kaya Y, Tanriverdi O. Effect of the interaction between selenium and zinc on DNA repair in association with cancer prevention. J Cancer Prev. 2019;24:146–154. doi:10.15430/JCP.2019.24.3.146
248. Mocchegiani E, Malavolta M. Role of zinc and selenium in oxidative stress and immunosenescence: implications for healthy aging and longevity. In: Handbook of Immunosenescence. Springer; 2019:2539–2573. doi:10.1007/978-3-319-99375-1_66
249. Fischer-Posovszky P, Kukulus V, Tews D, et al. Resveratrol regulates human adipocyte number and function in a Sirt1-dependent manner123. Am J Clin Nutr. 2010;92:5–15. doi:10.3945/ajcn.2009.28435
250. Chung M-Y, Kim BH. Fatty acids and epigenetics in health and diseases. Food Sci Biotechnol. 2024;33:3153–3166. doi:10.1007/s10068-024-01664-3
251. Walsh J, Mahony R, Foley M, McAuliffe F. 7: ROLO study: a randomized control trial of low glycemic index diet to prevent macrosomia in euglycemic women. Am J Clin Exp Obstet Gynecol. 2012;206:S4. doi:10.1016/j.ajog.2011.10.033
252. Walsh JM, Mahony RM, Culliton M, Foley ME, McAuliffe FM. Impact of a low glycemic index diet in pregnancy on markers of maternal and fetal metabolism and inflammation. Reprod Sci. 2014;21:1378–1381. doi:10.1177/1933719114525275
253. Geraghty AA, Sexton-Oates A, O’Brien EC, et al. A low glycaemic index diet in pregnancy induces DNA methylation variation in blood of newborns: results from the ROLO randomised controlled trial. Nutrients. 2018;10:455. doi:10.3390/nu10040455
254. Campoy C, Escolano-Margarit MV, Ramos R, et al. Effects of prenatal fish-oil and 5-methyltetrahydrofolate supplementation on cognitive development of children at 6.5 y of age12345. Am J Clin Nutr. 2011;94:S1880–S1888. doi:10.3945/ajcn.110.001107
255. Azaryah H, Verdejo-Román J, Martin-Pérez C, et al. Effects of maternal fish oil and/or 5-methyl-tetrahydrofolate supplementation during pregnancy on offspring brain resting-state at 10 years old: a follow-up study from the NUHEAL Randomized Controlled Trial. Nutrients. 2020;12:2701. doi:10.3390/nu12092701
256. Jiang X, Yan J, West AA, et al. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012;26:3563–3574. doi:10.1096/fj.12-207894
257. Wendel K, Pfeiffer HCV, Fugelseth DM, et al; the ImNuT Collaboration Group. Effects of nutrition therapy on growth, inflammation and metabolism in immature infants: a study protocol of a double-blind randomized controlled trial (ImNuT). BMC Pediatric. 2021;21:19. doi:10.1186/s12887-020-02425-x
258. Cai R, Lv R, Shi X, Yang G, Jin J. CRISPR/dCas9 tools: epigenetic mechanism and application in gene transcriptional regulation. Int J Mol Sci. 2023;24:14865. doi:10.3390/ijms241914865
259. Ehara T, Kamei Y, Yuan X, et al. Ligand-activated PPARα-dependent DNA demethylation regulates the fatty acid β-oxidation genes in the postnatal liver. Diabetes. 2015;64:775–784. doi:10.2337/db14-0158
260. Ahmad F, Uzair SA, Lakshmanan AP, et al. Placental and cord blood DNA methylation changes associated with gestational diabetes mellitus in a marginalized population: the untold role of saturated fats. Mol Nutr Food Res. 2025;69:e70058. doi:10.1002/mnfr.70058
261. Huang L, Huhulea EN, Abraham E, et al. The role of artificial intelligence in obesity risk prediction and management: approaches, insights, and recommendations. Medicina. 2025;61:358. doi:10.3390/medicina61020358
262. Alum EU. Circadian nutrition and obesity: timing as a nutritional strategy. J Health Popul Nutr. 2025;44:367. doi:10.1186/s41043-025-01102-y
263. Saikia AP, Kalita A. Artificial Intelligence and multi-omics integration in obesity: a review of computational models for predicting metabolic comorbidities. Clin Transl Metab. 2025;23:7. doi:10.1007/s12018-025-09310-0
264. Sutton EF, Gilmore LA, Dunger DB, et al. Developmental programming: state-of-the-science and future directions-summary from a pennington biomedical symposium. Obesity. 2016;24:1018–1026. doi:10.1002/oby.21487
265. Tain Y-L, Hsu C-N. Interplay between maternal nutrition and epigenetic programming on offspring hypertension. J Nutr Biochem. 2024;127:109604. doi:10.1016/j.jnutbio.2024.109604
266. Marousez L, Lesage J, Eberlé D. Epigenetics: linking early postnatal nutrition to obesity programming? Nutrients. 2019;11:2966. doi:10.3390/nu11122966
267. Breton CV, Landon R, Kahn LG, et al. Exploring the evidence for epigenetic regulation of environmental influences on child health across generations. Commun Biol. 2021;4:769. doi:10.1038/s42003-021-02316-6
268. Marcho C, Oluwayiose OA, Pilsner JR. The preconception environment and sperm epigenetics. Andrology. 2020;8:924–942. doi:10.1111/andr.12753
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Advances in the Epigenetic Mechanisms of Diabetic Nephropathy Pathogenesis
Zhang C, Xue S, Ren P, Han S, Zhou Y, Si Y, Han X, Zhang X, Zhang Y, Chen N, He H, Feng R, Shang L
Diabetes, Metabolic Syndrome and Obesity 2025, 18:2629-2639
Published Date: 30 July 2025
The Bidirectional Role of Obesity and Aging in the Pathogenesis of Osteoarthritis: Molecular Mechanisms, Epigenetic Insights, and Therapeutic Implications
Lin C, Yang J, Su H, Zhang X, Wang B
Journal of Inflammation Research 2025, 18:12637-12676
Published Date: 12 September 2025
The Role of DNA Methylation in Osteosarcoma Pathogenesis and Therapy
Zhou X, Li B, Tao P
OncoTargets and Therapy 2026, 19:581744
Published Date: 23 February 2026