Back to Journals » Journal of Blood Medicine » Volume 17
Malignancy-Associated Hemophagocytic Lymphohistiocytosis: An Experience of 15 Years in Polish Pediatric Hematology Centers
Authors Wolowiec M ![]() , Drabko K, Maciejka-Kemblowska L
, Drabko K, Maciejka-Kemblowska L ![]() , Irga-Jaworska N, Smalisz K, Skoczen S, Tomaszewska R, Szczepanski T
, Irga-Jaworska N, Smalisz K, Skoczen S, Tomaszewska R, Szczepanski T ![]() , Babol-Pokora K
, Babol-Pokora K ![]() , Mlynarski W, Popko K, Demkow U, Furmanczyk K
, Mlynarski W, Popko K, Demkow U, Furmanczyk K ![]() , Malinowska I
, Malinowska I
Received 1 October 2025
Accepted for publication 11 April 2026
Published 27 May 2026 Volume 2026:17 571561
DOI https://doi.org/10.2147/JBM.S571561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Magdalena Wolowiec,1 Katarzyna Drabko,2 Lucyna Maciejka-Kemblowska,3 Ninela Irga-Jaworska,3 Katarzyna Smalisz,4 Szymon Skoczen,4 Renata Tomaszewska,5 Tomasz Szczepanski,5 Katarzyna Babol-Pokora,6 Wojciech Mlynarski,6 Katarzyna Popko,7 Urszula Demkow,7 Konrad Furmanczyk,8,9 Iwona Malinowska1
1Department of Oncology, Pediatric Hematology, Clinical Transplantology, Pediatrics and Rare Diseases, Medical University of Warsaw, Warsaw, Poland; 2Department of Pediatric Hematology, Oncology and Transplantology, Medical University of Lublin, Lublin, Poland; 3Department of Pediatrics, Pediatric Hematology and Oncology, Medical University of Gdansk, Gdansk, Poland; 4Department of Pediatric Oncology and Hematology, Institute of Pediatrics, Jagiellonian University Medical College, Krakow, Poland; 5Department of Pediatrics, Pediatric Hematology and Oncology, Silesian Medical University, Zabrze, Poland; 6Department of Pediatrics, Oncology and Hematology, Medical University of Lodz, Lodz, Poland; 7Department of Laboratory Diagnostics and Clinical Immunology of Developmental Age, Medical University of Warsaw, Warsaw, Poland; 8Department of Applied Mathematics, Warsaw University of Life Sciences, Warsaw, Poland; 9Department of the Prevention of Environmental Hazards, Allergology and Immunology, Medical University of Warsaw, Warsaw, Poland
Correspondence: Magdalena Wolowiec, Department of Oncology, Pediatric Hematology, Clinical Transplantology, Pediatrics and Rare Diseases, Medical University of Warsaw, 02-091 Żwirkii Wigury Str 63, Warsaw, Poland, Tel +4822317-94-84, Email [email protected]
Background: M-HLH is a type of HLH that most often occurs in children in the course of hematological malignancies. The diagnosis is based on meeting the criteria of the HLH-2004 Protocol. The disease has a rapid course, and the lack of proper diagnosis and treatment threatens the patient’s life.
Methods: The paper presents the thorough clinical characteristics, course of treatment, and treatment results in 23 patients treated in pediatric hemato-oncology centers in Poland and reported to Polish HLH registry. HLH diagnostic criteria, CNS involvement, coexisting viral infections, genetic test results, treatment modalities, therapeutic responses, relapse incidence, and final outcomes were analyzed. Clinical and laboratory factors were subjected to statistical analysis. Overall survival was estimated using the Kaplan–Meier method for the entire cohort and separately for subgroups defined by type of malignancy and treatment strategy. Separate univariable Cox proportional hazards models were used to explore the association between selected clinical factors and survival.
Results: The median age at diagnosis was 11.7 years, in 8 patients HLH symptoms preceded the cancer diagnosis. Acute leukemia was the factor causing M-HLH in 12 patients, in the remaining patients it was lymphoma. The treatment included tumor-directed therapy with or without HLH-directed therapy. The probability of survival in the study group was 0.539 (95% CI 0.364– 0.798). Remission of HLH was statistically significant (p-value = 0.0007) and reduced the risk of death by 90% (HR=0.10).
Conclusion: Quick and precise diagnosis and implementation of appropriate treatment bringing the patient into HLH remission is extremely important for survival.
Keywords: hemophagocytic lymphohistiocytosis, HLH, malignancy, leukemia, lymphoma, HLH-directed therapy
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome of life-threatening inflammation resulting from an excessive, prolonged, and ineffective activation of the immune cells. The pathogenesis of HLH involves hyperactivation of CD8+ T lymphocytes and stimulation of macrophages leading to a cytokine storm.1–5 HLH presents with fever, severe cytopenia, organomegaly, hyperferritinemia, coagulation defects, impaired liver function and frequent central nervous system (CNS) involvement.2,3,6–8 HLH can be defined as fulfillment of at least five of eight clinical and laboratory criteria.2 HLH is classified into two types: genetic (primary) and acquired (secondary).2,3 Secondary HLH may occur at any age, and is often triggered by infection, rheumatic condition, or malignancy.
According to the Histiocyte Society malignancy-associated HLH (M-HLH) can present either at the onset of malignancy or as a result of chemotherapy. Its pathophysiology involves cytokine secretion (including interferon-γ and interleukin-6), persistent antigen stimulation by malignant cells, immunosuppression or infection.9,10 M-HLH is most often linked to hematologic malignancies, primarily lymphomas and less frequently leukemias, with solid tumors being exceedingly rare.11,12 The disease can also occur after hematopoietic cell transplantation, organ transplantation, CAR-T cell therapy or treatment with checkpoint inhibitors.13
Adolescents demonstrate a prevalence from 8.4% to 11.5% for malignancy-triggered HLH, though this may be significantly underestimated.14,15 Hypomorphic mutations in HLH-related genes may lead to delayed onset16–18 and primary immunodeficiencies – such as MAGT1 deficiency or X-linked lymphoproliferative syndromes – can predispose individuals to HLH and related cancers.19–22
Although rare in children, M-HLH is clinically aggressive and has a poor prognosis, with 1-year survival rates below 40% in retrospective studies.13 Despite increasing recognition in Poland, data on pediatric HLH regarding its clinical background and treatment response remain lacking.
To date, no published data have addressed the clinical development and outcome predictors of M-HLH in Polish children. Furthermore, a comprehensive analysis of HLH treatment efficacy and response patterns has not been undertaken.
This multicenter retrospective study, inspired by data from the national HLH treatment coordinating center in Warsaw, was aimed to investigate the clinical course and survival outcomes of Polish children diagnosed with M-HLH.
Materials and Methods
The study utilized the data from Polish Registry of HLH, encompassing all pediatric hematology and oncology centers in Poland. Clinical records of 23 patients under 18 years old diagnosed with M-HLH between April 2008 and December 2024 were retrospectively analyzed. Variables examined included HLH diagnostic criteria, CNS involvement, coexisting viral infections, genetic test results, treatment modalities, therapeutic responses, relapse incidence, and final outcomes. Diagnostic and laboratory evaluations were performed locally, following HLH protocols.
Immunological tests (soluble interleukin 2-receptor concentration, sIL-2R) were conducted at the Department of Laboratory Diagnostics and Clinical Immunology of the Developmental Age of the Medical University of Warsaw. Molecular tests using the Next Generation Sequencing method followed by Sanger direct sequencing were carried out in the Laboratory of Immunopathology and Genetics, Department of Pediatrics, Oncology and Hematology in Łódź, Poland.
The diagnosis was made based on the fulfilled clinical and laboratory criteria according to the HLH-2004 protocol - unremitting fever (>7 days), splenomegaly, cytopenia ≥2 cell lines, hyperferritinemia >500μg/l, hypofibrinogenemia <1.5g/l or hypertriglyceridemia >3mmol/l, elevated sIL-2R >2400U/mL, hemophagocytosis in bone marrow or other tissues, reduced or absent NK cytotoxicity.2
Ethical approval was obtained from the Medical University of Warsaw. Informed consent was secured from parents and patients aged ≥16 years. The study complies with the Declaration of Helsinki.
Statistical Analysis
Categorical variables are presented as counts and percentages, whereas continuous variables are summarized using mean and standard deviation (SD) or median and interquartile range (IQR), depending on distribution.
The starting point of follow-up was defined as the date of HLH diagnosis. Follow-up time was calculated from the date of HLH diagnosis to death or last clinical contact. Median follow-up was estimated using the reverse Kaplan–Meier method.
Overall survival was estimated using the Kaplan–Meier method for the entire cohort and separately for subgroups defined by (1) type of malignancy triggering HLH (leukemia vs. lymphoma) and (2) treatment strategy (tumor-directed therapy alone vs. tumor-directed therapy combined with HLH-directed therapy).
Due to the small sample size and limited number of events, differences between survival curves were assessed using a permutation version of the two-sample Log rank test with 10,000 permutations. This approach was chosen because asymptotic assumptions of the standard Log rank test may not be reliable in small samples, whereas permutation testing provides valid inference without relying on large-sample approximations.
Because of the limited sample size (n = 23) and the risk of model overfitting, a full multivariable Cox proportional hazards model was not performed. Instead, separate univariable Cox proportional hazards models were used to explore the association between selected clinical factors and survival, and hazard ratios (HRs) with 95% confidence intervals (CIs) were reported. The only exception was an exploratory multivariable Cox proportional hazards model including four predefined variables, which was interpreted with caution given the limited sample size.
A significance level of 0.05 was applied. All analyses were performed using R statistical software (version 3.4.3; R Foundation for Statistical Computing, Vienna, Austria, www.r-project.org).
Results
Clinical Course
Participant selection flow is presented in Figure 1 and patients’ baseline characteristics is presented in Table 1.
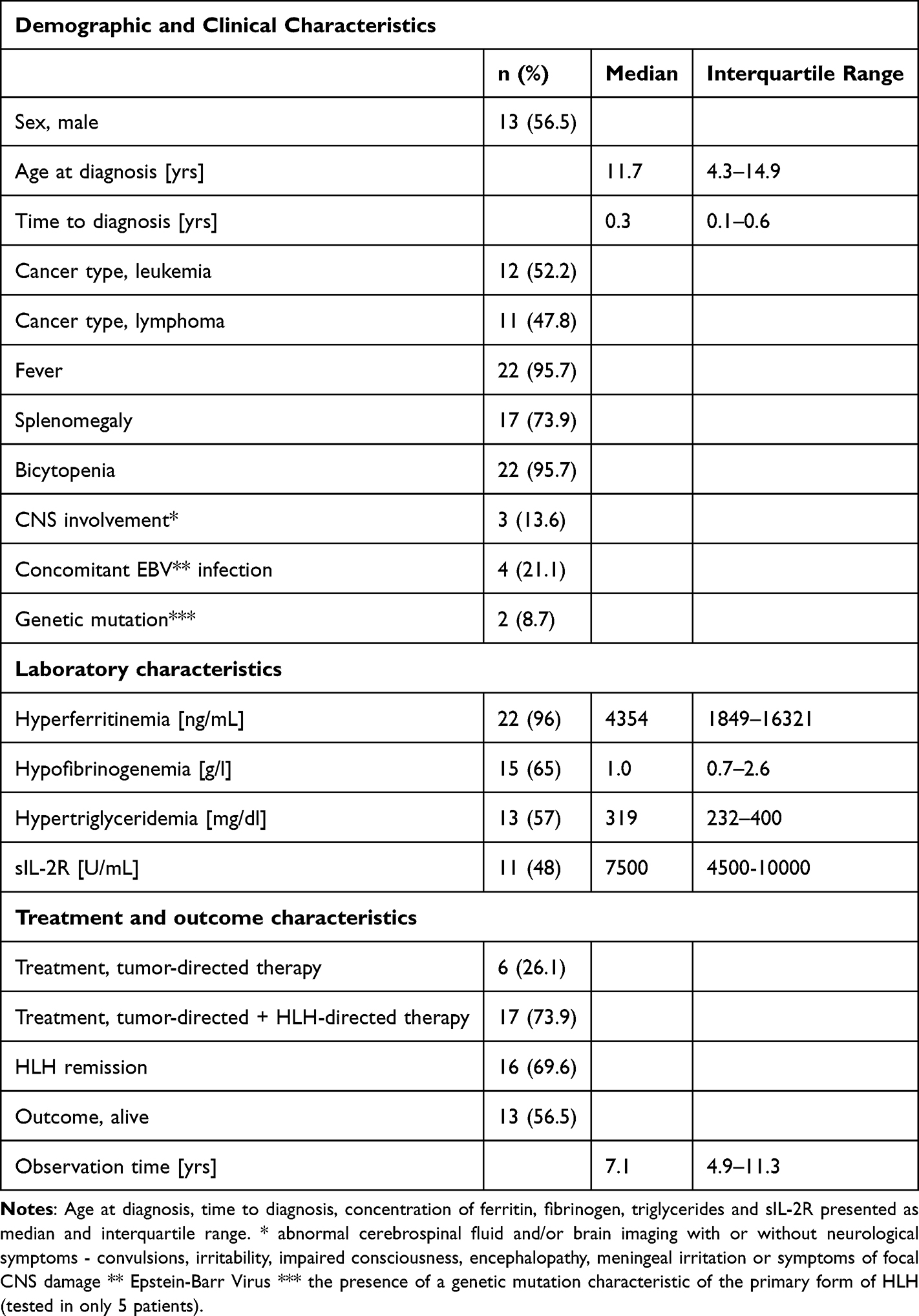 |
Table 1 Baseline Characteristics |
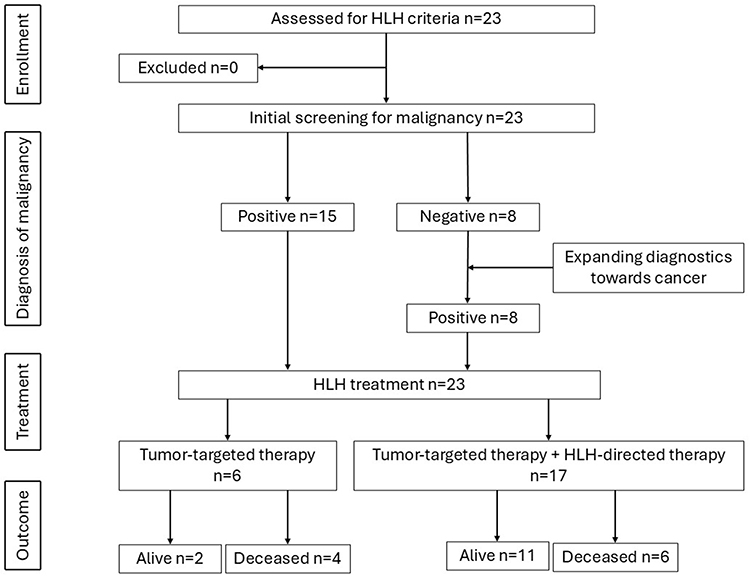 |
Figure 1 Participant selection flow. |
Age at Onset
In the study group, there were 23 patients, including 13 boys and 10 girls. M-HLH was diagnosed in patients from the first days of life to over 17 years, with a median of 11.7 years. HLH diagnosis at birth was made in one infant with congenital myeloid leukemia.
Time to Diagnosis
The median time from symptom onset to diagnosis of HLH was 12 days. The diagnosis of HLH was made at the same time as the diagnosis of cancer or its recurrence in 15 patients. Eight patients were initially diagnosed with HLH and treated with the HLH-2004 protocol. However, broader diagnosis aimed at searching for the cause of HLH, led to diagnosis of malignancy. Among them there were 5 patients with lymphoma and 3 patients with acute leukemia, including 2 in the relapsed phase. HLH symptoms preceded cancer diagnosis by 2 days to 3 months, with an average of 43 days.
Cancer Types
In all patients from the study group, the development of M-HLH was caused by proliferative diseases of the hematopoietic system - leukemias and lymphomas, and none of the patients had a solid tumor. Of the 12 patients with acute leukemia, 4 had newly diagnosed acute lymphoblastic leukemia (4/12, 33.3%), 4 had a relapse of acute lymphoblastic leukemia (4/12, 33.3%), and 4 had acute myeloid leukemia (4/12, 33.3%). Of the 11 patients with lymphoma, 4 were diagnosed with anaplastic large cell lymphoma (ALCL) (4/11, 36.4%), 5 with other T-cell lymphoma (5/11,45.5%) – 2 with peripheral T-cell lymphoma (PTCL), 2 with non-Hodgkin lymphoma, 1 with hepatosplenic lymphoma, 1 patient with B-cell lymphoblastic lymphoma (1/11, 9.1%), and 1 with Hodgkin lymphoma (1/11, 9.1%).
Central Nervous System Involvement
In 3 patients CNS involvement was diagnosed, including 1 with neurological symptoms (seizures, disorders of consciousness) and CNS imaging lesions, 1 with neurological symptoms only, and 1 with elevated cytosis in the cerebrospinal fluid. 2 out of 3 patients live without late neurological sequelae, 1 patient died during the course of the disease.
Concomitant Infections
A concomitant symptomatic viral infection was found in 7 (30%) patients from the study group. 4 patients had EBV infection (17.5%), Cytomegalovirus and Human Herpesvirus 6 was identified by PCR in 2 (9%) and 1 (4.5%) patients, respectively.
Molecular Analysis
Molecular tests were performed in 5 patients from the study group. Genetic mutations were found in 2 patients. In a 15-year-old boy diagnosed with PTCL a heterozygous mutation c.[659G>T]; [Arg220Leu] of the STX11 gene was found, and the lesion came from the mother. The boy initially developed symptoms of HLH, and in the course of extended diagnostics, lymphoma was diagnosed. The boy achieved HLH remission, but due to the presence of a pathogenic variant of the HLH gene, he successfully underwent hematopoietic cell transplantation. The second patient, a 17-year-old boy, presented initially non-specific symptoms that met the clinical criteria for HLH. HLH was caused by Hodgkin’s lymphoma. In this boy, a molecular test confirmed a mutation in the MAGT1 gene responsible for X-linked MAGT1 deficiency with increased susceptibility to Epstein–Barr virus (EBV) infection and N-linked glycosylation defect (XMEN syndrome).
Treatment of HLH
In the analyzed group, 17/23 (73.9%) patients received antineoplastic chemotherapy and HLH-targeted therapy, while 6 patients (26%) received only treatment with the antineoplastic regimen appropriate for the underlying disease. Treatment targeting HLH consisted of dexamethasone only in 2 patients, dexamethasone and intravenous immunoglobulins in 3 patients, the complete HLH-2004 protocol in 5 patients, as well as the HLH-2004 protocol in combination with biologic therapy (anakinra, ruxolitinib, tocilizumab) in 7 patients. In terms of biologic therapy, 3 patients received anakinra, 3 patients received tocilizumab, and 1 patient was treated with ruxolitinib.
The clinical course of M-HLH in patients from the study group is presented in Table 2.
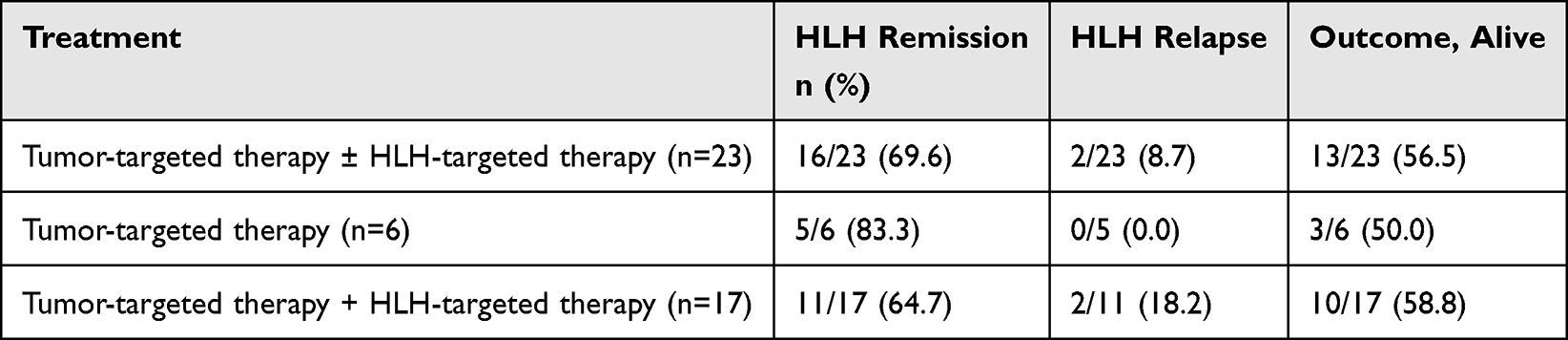 |
Table 2 Clinical Course of M-HLH in the Study Group |
Survival Analysis
The probability of surviving at least 24 months in the study group was 0.539 (95% CI 0.364–0.798) and is shown in Figure 2.
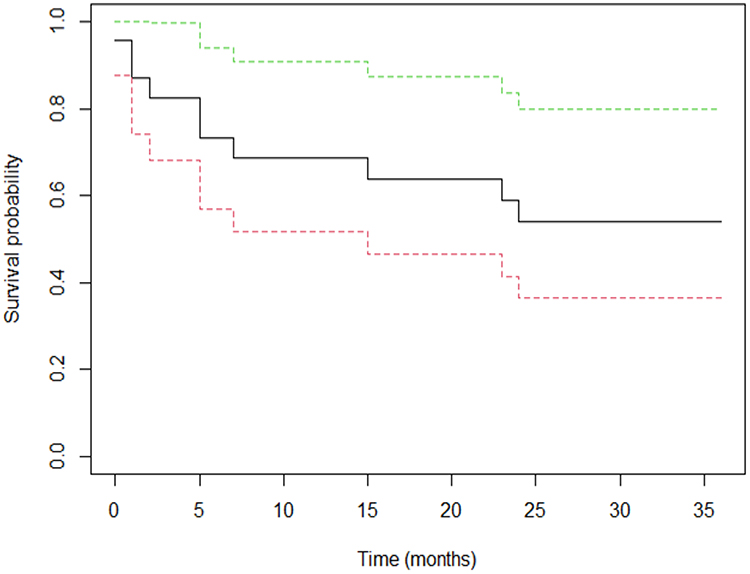 |
Figure 2 Probability of survival in the study group. Kaplan-Meier estimation of overall survival of patients with M-HLH (solid line), 95% CI (dashed lines). |
The probability of survival depending on the underlying disease of M-HLH and treatment regimen is shown in Figure 3.
 |
Figure 3 Probability of survival depending on underlying disease of M-HLH (A) and treatment regimen (B). (A) Probability of survival at least 24 months of patients with leukemia is 0.50 (95% CI 0.284–0.880), at least 24 months of patients with lymphoma is 0.60 (95% CI 0.353–1), p-value=0.7076 for two sample long rank test (no significant differences between the survival curves for both groups). (B) Probability of survival at least 24 months of patients treated only with tumor-directed therapy was 0.50 (95% CI 0.225–1), at least 24 months with tumor directed-therapy + HLH directed therapy was 0.56 (95% CI 0.364–0.871), p-value 0.991 for two sample long rank test (no significant differences between the survival curves for both groups). |
Simple Cox models were made for single survival factors – sex, age at diagnosis, time to diagnosis, underlying malignancy, treatment regimen, CNS involvement, EBV infection. Due to the small amount of data, a combined model was not made for all factors. For the analyzed factors, no relationships affecting the survival time of patients were found. Only achieving remission was statistically significant (p-value = 0.0007). Achieving remission reduces the risk of death by 90% (HR=0.10). The model showed acceptable predictive capabilities (Concordance coefficient = 0.755). Due to the lack of numerical convergence, the multivariate model was created only for 4 variables and it is presented in Figure 4.
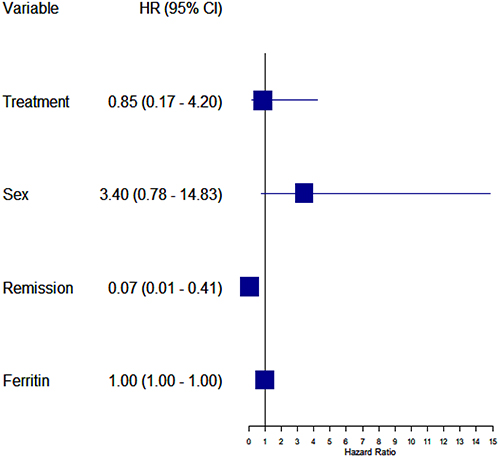 |
Figure 4 Factors influencing survival - multidimensional model. |
Discussion
Although M-HLH is the most common form of HLH in adults, it is a rare condition in children and accounts for 8% of all HLH cases.14 M-HLH can be the first manifestation of cancer, its recurrence, or it can occur at any time during cancer treatment.20,23 In a population-based study conducted in Sweden between 1997 and 2018, M-HLH was diagnosed in 9 children.11 In the study of Chinese team, M-HLH was diagnosed in 27 patients over 10 years.24 Our study included 23 cases of M-HLH for the last 16 years, but due to the fact that this is not a population-based study, these data may be underestimated, which is a limitation of work.
M-HLH is diagnosed in children of all age groups. The median age of diagnosis in the study group was 11.7 years, which is consistent with other studies.14,24–26 The risk of developing M-HLH increases with age, due to the higher likelihood of an underlying malignant disease and therefore, in any patient with HLH, cancer should be considered as a potential trigger for HLH.23 All of our patients were diagnosed as malignancy-induced HLH, there were no patients with chemotherapy-induced HLH, but the retrospective nature of the work based on data provided from the centers treating patients poses a risk of inconsistent nomenclature and is an undoubted limitation of the work.
It is often difficult to distinguish the symptoms of HLH from the symptoms of cancer, as both conditions can be accompanied by fever, cytopenia, enlargement of the liver and spleen or neurological symptoms. Due to the severe clinical course of the disease, waiting for the diagnostic criteria to be met may delay the implementation of appropriate treatment.13,27,28 This problem affects not only pediatric patients, but also adults.29 Moreover, the accuracy of the HLH-2004 criteria for the diagnosis of M-HLH is debatable.27,28,30 In adult patients, Zoref-Lorenz et al proposed an optimized HLH inflammatory index for the identification of patients with M-HLH, based on the concentration of soluble IL-2 receptor > 3900U/mL and ferritin concentration > 1000ng/mL This combination of laboratory results is also a significant risk factor for mortality in hematological malignancies.30
In children, M-HLH is most common in the course of T-cell tumors, especially lymphomas. Other proliferative diseases of the hematopoietic system are also described as causes of M-HLH, including acute leukemias and Hodgkin’s lymphoma. Solid tumors are rarely the cause of M-HLH, both in adults and children.14 It is worth noting that M-HLH can also occur after treatment of solid tumors with checkpoint inhibitors.13 In the study group there were 11 patients with M-HLH induced by lymphomas, including 9 in the course of T-cell lymphomas. Contrary to the literature, more patients from the study group had M-HLH in the course of acute leukemia, both lymphoblastic and myeloid leukemia. Cases of M-HLH in the course of treatment of acute leukemias have been described in the literature.31,32 Moreover, acute myeloid leukemia secondary to the treatment of HLH with a high total dose of etoposide was also reported.33
CNS involvement is seen as an unfavorable prognostic factor in HLH.34–36 In the study group, CNS involvement in the form of neurological disorders, abnormalities in brain imaging and cerebrospinal fluid pleocytosis was found only in 3 patients. In the literature, CNS involvement, as well as low platelet count, as a prognostic factor is assessed in the context of all forms of HLH, not only M-HLH.34
EBV infection is an important trigger for HLH, both primary and secondary, including M-HLH.23 Moreover, a high percentage of patients diagnosed with M-HLH are complicated by EBV infection.14 In the study group, EBV infection was found in 4 patients, 2 patients had CMV infection and 1 HHV-6 infection. In the work of researchers from China, EBV infection was described in almost half of the patients studied. According to the M-HLH management guidelines published by Lehmberg et al, infectious agents require rigorous treatment, and patients undergoing immunosuppressive therapy for M-HLH require ongoing infectious screening and antifungal and anti-pneumocystosis prophylaxis.23
Patients with M-HLH may have mutations in genes responsible for the development of primary forms of HLH.14,37,38 Mutations in HLH genes may also increase the risk of developing hematological cancers.14 In the study group, only 5 patients had molecular tests performed. Mutations were detected in 2 of them. A patient with lymphoma and a heterozygous mutation in the STX11 gene, despite a good response to M-HLH treatment, successfully underwent hematopoietic cell transplantation. A patient with a mutation in the MAGT1 gene remains under further close hematological control after completion of M-HLH therapy. According to the literature, patients with mutations predisposing to EBV susceptibility, such as MAGT1, as well as patients with X-linked lymphoproliferative disease (XLP1) should undergo careful screening for lymphoma if they present with HLH.23
Treatment of M-HLH is challenging because it has to balance HLH-directed and tumor-specific therapy.28 On the one hand, it should control hyperinflammation, and on the other hand, anti-cancer therapy should be implemented as soon as possible so as to eliminate the triggering factor of HLH.13 In some patients, the treatment of the cancer that stimulates the development of HLH is sufficient to control the disease. The decision on the optimal treatment method is made on a case-by-case basis.14,23 Moreover, it is important to differentiate between M-HLH, cytokine release syndrome and immune-effector cell-associated HLH-like syndrome to choose the optimal treatment.28 In the therapy of HLH at malignancy presentation, the administration of corticosteroids/anakinra ± etoposide, supported by etoposide, IVIG, ruxolitinib or emapalumab if available, as well as tumor-specific treatment is suggested, while in HLH during cancer treatment, corticosteroids or anakinra, alternatively intravenous immunoglobulins, are proposed.28 In the study group, 6 patients received only tumor-specific treatment to cure the disease that provokes the development of HLH and 17 patients received tumor-specific and HLH-specific treatment, including corticosteroids, IVIG and biologic therapy with anakinra, tocilizumab and ruxolitinib. Less toxic treatment strategies aimed at silencing the stimulated immune system and cytokine storm play an extremely important role in HLH therapy.13,39–41 The probability of survival in the study group was 54% and was comparable to the data from the literature.23–26 In the study group, apart from achieving remission, no statistically significant factors affecting survival were identified. Despite a significant limitation of the study, which is the small size of the examined group resulting from the rare occurrence of the disease in the pediatric population, the study results strongly indicate that rapid implementation of appropriate treatment and bringing the patient into HLH remission is extremely important for survival.
Conclusions
M-HLH in children, due to its severe clinical course and non-specific symptoms, is difficult to diagnose and treat. The small number of patients and the need for an individual approach to each patient is a limitation in conducting extensive prospective studies. Nevertheless, awareness of the occurrence of M-HLH in children and adults and taking it into account in the differential diagnosis of a severely ill patients is extremely important. Further research on this rare entity, including searching for factors that may influence the course of the disease and treatment outcomes may increase patients’ chances of survival.
Data Sharing Statement
Data are available upon request from the corresponding author.
Ethical Approval
The Ethics Committee of the Medical University of Warsaw, Poland, approved the study (No KB/21/2010). Written, informed consent was obtained from each patient enrolled in the study. Patient data were fully anonymized.
Acknowledgment
Manuscript preparation was supported during Harvard Medical School’s Polish Clinical Scholars Research Training Program, organized by Medical Research Agency, Warsaw, Poland.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. All coauthors approved the final version for publication.
Funding
This work was supported by grant from National Science Centre (No 0989/B/P01/2011/40) received by Iwona Malinowska.
Disclosure
The authors report no conflict of interest.
References
1. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135(16):1332–10. doi:10.1182/blood.2019000936
2. Henter J-I, Horne A, Aricó M. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi:10.1002/pbc.21039
3. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63(1):233–246. doi:10.1146/annurev-med-041610-134208
4. Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66(11):e27929. doi:10.1002/pbc.27929
5. Henter J-I, Longo DL. Hemophagocytic lymphohistiocytosis. N Engl J Med. 2025;392(6):584–598. doi:10.1056/NEJMra2314005
6. Jordan M, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–4052. doi:10.1182/blood-2011-03-278127
7. Madkaikar M, Shabrish S, Desai M. Current updates on classification, diagnosis and treatment of hemophagocytic lymphohistiocytosis (HLH). Indian J Pediatr. 2016;83(5):434–443. doi:10.1007/s12098-016-2037-y
8. Malinowska I, Machaczka M, Popko K, et al. Hemophagocytic syndrome in children and adults. Arch Immunol Ther Exp. 2014;62(5):385–394. doi:10.1007/s00005-014-0274-1
9. Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. doi:10.1182/blood-2016-01-690636
10. Mellgren K, Hedegaard CJ, Schmiegelow K, Müller K. Plasma cytokine profiles at diagnosis in pediatric patients with non-hodgkin lymphoma. J Pediatr Hematol Oncol. 2012;34(4):271–275. doi:10.1097/MPH.0b013e3182431e02
11. Löfstedt A, Jädersten M, Meeths M, Henter J-I. Malignancy-associated hemophagocytic lymphohistiocytosis in Sweden: incidence, clinical characteristics, and survival. Blood. 2024;143(3):233–242. doi:10.1182/blood.2023020715
12. Ishii E, Ohga S, Imashuku S, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86(1):58–65. doi:10.1532/IJH97.07012
13. Setiadi A, Zoref-Lorenz A, Lee CY, et al. Malignancy-associated haemophagocytic lymphohistiocytosis. Lancet Haematol. 2022;9(3):e217–e227. doi:10.1016/S2352-3026(21)00366-5
14. Lehmberg K, Sprekels B, Nichols KE, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170(4):539–549. doi:10.1111/bjh.13462
15. Chinn IK, Eckstein OS, Peckham-Gregory EC, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. 2018;132(1):89–100. doi:10.1182/blood-2017-11-814244
16. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematology. 2013;2013(1):605–611. doi:10.1182/asheducation-2013.1.605
17. Weitzman S. Approach to hemophagocytic syndromes. Hematology. 2011;2011(1):178–183. doi:10.1182/asheducation-2011.1.178
18. Chia J, Yeo KP, Whisstock JC, et al. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci U S A. 2009;106(24):9809–9814. doi:10.1073/pnas.0903815106
19. Li F-Y, Chaigne-Delalande B, Su H, et al. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood. 2014;123(14):2148–2152. doi:10.1182/blood-2013-11-538686
20. Lackner H, Urban C, Sovinz P, et al. Hemophagocytic lymphohistiocytosis as severe adverse event of antineoplastic treatment in children. Haematologica. 2008;93(2):291–294. doi:10.3324/haematol.11704
21. Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 2014;155(1):118–125. doi:10.1016/j.clim.2014.09.008
22. Sieni E, Cetica V, Mastrodicasa E, et al. Familial hemophagocytic lymphohistiocytosis: a model for understanding the human machinery of cellular cytotoxicity. Cell Mol Life Sci CMLS. 2012;69(1):29–40. doi:10.1007/s00018-011-0835-y
23. Lehmberg K, Nichols KE, Henter J-I, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015;100(8):997–1004. doi:10.3324/haematol.2015.123562
24. Huang Z, Jia Y, Zuo Y, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in children: a 10-year experience of a single pediatric hematology center. Hematology. 2020;25(1):389–399. doi:10.1080/16078454.2020.1833505
25. Strenger V, Merth G, Lackner H, et al. Malignancy and chemotherapy induced haemophagocytic lymphohistiocytosis in children and adolescents—a single centre experience of 20 years. Ann Hematol. 2018;97(6):989–998. doi:10.1007/s00277-018-3254-4
26. Celkan T, Berrak S, Kazanci E, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in pediatric cases: a multicenter study from Turkey. Turk J Pediatr. 2009;51(3):207–213.
27. Gurunathan A, Boucher AA, Mark M, et al. Limitations of HLH-2004 criteria in distinguishing malignancy-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2018;65(12):e27400. doi:10.1002/pbc.27400
28. Zoref-Lorenz A, Witzig TE, Cerhan JR, et al. Malignancy-associated HLH: mechanisms, diagnosis, and treatment of a severe hyperinflammatory syndrome. Leuk Lymphoma. 2024; 1–9. doi:10.1080/10428194.2024.2436037
29. Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123(17):3229–3240. doi:10.1002/cncr.30826
30. Zoref-Lorenz A, Murakami J, Hofstetter L, et al. An improved index for diagnosis and mortality prediction in malignancy-associated hemophagocytic lymphohistiocytosis. Blood. 2022;139(7):1098–1110. doi:10.1182/blood.2021012764
31. Lackner H, Seidel MG, Strenger V, et al. Hemophagocytic syndrome in children with acute monoblastic leukemia-another cause of fever of unknown origin. Support Care Cancer. 2013;21(12):3519–3523. doi:10.1007/s00520-013-1937-x
32. Richardson AI, Yap KL, Leuer K, et al. Hemophagocytic lymphohistiocytosis with predominant T-lymphocytes in young child: an unusual presentation of evolving acute myeloid leukemia. J Clin Med. 2025;14(5):1511. doi:10.3390/jcm14051511
33. Imashuku S. Etoposide-related secondary acute myeloid leukemia (t-AML) in hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):121–123. doi:10.1002/pbc.21082
34. Harnchoowong S, Soponkanaporn S, Vilaiyuk S, et al. Central nervous system involvement and thrombocytopenia as predictors of mortality in children with hemophagocytic lymphohistiocytosis. Front Pediatr. 2022;10:941318. doi:10.3389/fped.2022.941318
35. Kim -M-M, Yum M-S, Choi H-W, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol. 2012;47(4):273–280. doi:10.5045/kjh.2012.47.4.273
36. Horne A, Trottestam H, Aricò M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;140(3):327–335. doi:10.1111/j.1365-2141.2007.06922.x
37. Booth C, Gilmour KC, Veys P, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. 2011;117(1):53–62. doi:10.1182/blood-2010-06-284935
38. Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160(3):275–287. doi:10.1111/bjh.12138
39. Bami S, Vagrecha A, Soberman D, et al. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2020;67(11):e28581. doi:10.1002/pbc.28581
40. Keenan C, Nichols KE, Albeituni S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front Immunol. 2021;12:614704. doi:10.3389/fimmu.2021.614704
41. Merrill SA, Spaner C, Chen LYC. Goodbye etoposide? Taking the leap to ruxolitinib in haemophagocytic lymphohistiocytosis. Br J Haematol. 2025;206(1):391–393. doi:10.1111/bjh.19864
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Why and How Should Ethiopia Establish a Stem Cell Transplant Service? A Review Article
Mekonnen S, Farris H
Biologics: Targets and Therapy 2023, 17:33-40
Published Date: 20 March 2023
Analysis of Causes and Risk Factors for Deaths Among Pediatric Oncology Patients: A 20-Year Observation
Mitura-Lesiuk M, Dubaj M, Drabko K, Zawitkowska J
Cancer Management and Research 2026, 18:604728
Published Date: 5 May 2026
The Danish Lymphoid Cancer Research (DALY-CARE): Genetic Cohort Profile
Dietz JBN, Kadlec TF, Rotbain EC, Werling M, Vainer N, Katsimigas A, Ostrowski SR, Sørensen E, Pedersen OBV, Schwinn M, Nissen J, Magnusson MK, Elhussein HH, Agius R, Frederiksen CM, Rostgaard K, Hjalgrim H, Davidsson OB, Kristjánsson RP, Brieghel C, Niemann CU
Clinical Epidemiology 2026, 18:607221
Published Date: 15 July 2026