Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
LAMC2 Drives Airway Remodeling in COPD via EMT Regulation Through the AKT Pathway
Authors Wang Z ![]() , Shi J, Zhang Y, Luo Y, Rao Y, Qu J, Gai X, Sun Y
, Shi J, Zhang Y, Luo Y, Rao Y, Qu J, Gai X, Sun Y
Received 28 November 2025
Accepted for publication 15 March 2026
Published 27 March 2026 Volume 2026:21 580964
DOI https://doi.org/10.2147/COPD.S580964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
Zihan Wang,* Jun Shi,* Yue Zhang, Ying Luo, Yafei Rao, Jingge Qu, Xiaoyan Gai, Yongchang Sun
Department of Respiratory and Critical Care Medicine, Peking University Third Hospital; Research Center for Chronic Airway Diseases, Peking University Health Science Center, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoyan Gai, Department of Respiratory and Critical Care Medicine, Peking University Third Hospital; Research Center for Chronic Airway Diseases, Peking University Health Science Center, Beijing, People’s Republic of China, Email [email protected] Yongchang Sun, Department of Respiratory and Critical Care Medicine, Peking University Third Hospital; Research Center for Chronic Airway Diseases, Peking University Health Science Center, Beijing, People’s Republic of China, Email [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) is characterized by irreversible airflow limitation, largely driven by airway remodeling. Epithelial-mesenchymal transition (EMT) is a key mechanism underlying this process. Laminin subunit gamma-2 (LAMC2) is implicated in fibrosis and EMT, but its role in COPD-associated airway remodeling remains unclear.
Methods: Differential expression analysis was performed using airway epithelial cell datasets from COPD patients and TGF-β 1-induced EMT models. Findings were validated in COPD patient lung tissues, smoke-exposed mice, and in vitro experiments. In vivo, chronic smoke-exposed mice were pre-treated intratracheally with adeno-associated virus (AAV)-shLAMC2. Functional assays involved siRNA knockdown or plasmid overexpression of LAMC2 in bronchial epithelial cells. RNA sequencing and pathway analyses were conducted to explore underlying mechanisms.
Results: LAMC2 was significantly upregulated in COPD patient and murine airway epithelia. AAV-shLAMC2 administration alleviated airway remodeling and restored epithelial E-cadherin while reducing mesenchymal markers (N-cadherin, fibronectin), indicating attenuation of EMT. In vitro, LAMC2 was upregulated in TGF-β 1-stimulated epithelial cells, and its modulation significantly influenced EMT progression. Transcriptomic analysis suggested that AKT signaling as a potential downstream of LAMC2, supported by functional assays.
Conclusion: LAMC2 is upregulated in COPD airway epithelium and promotes airway remodeling by regulating EMT, potentially through AKT signaling. These findings suggest that targeting LAMC2 may represent a potential strategy for mitigating COPD-associated airway remodeling.
Keywords: COPD, airway remodeling, EMT, LAMC2
Introduction
Chronic obstructive pulmonary disease (COPD) is the fifth most prevalent disease worldwide, affecting nearly 300 million people, and ranks as the third leading cause of death globally, posing a significant burden on healthcare system.1–3 Persistent and irreversible airflow limitation and dyspnea are hallmarks of COPD,4 with progressively worsening airway remodeling serving as a central driver of its pathogenesis and progression.5,6 Current maintenance therapies for stable COPD–primarily inhaled bronchodilators,7 corticosteroids,8 and emerging biologics9,10 –can alleviate symptoms and reduce inflammation, but have limited efficacy in reversing established airway remodeling. Elucidating the mechanisms driving airway remodeling in COPD may provide a theoretical foundation for the development of novel therapeutic strategies aimed at improving impaired lung function, slowing disease progression, and potentially reversing the disease.
Mounting studies have demonstrated that epithelial–mesenchymal transition (EMT)—a process by which adherent epithelial cells acquire mesenchymal characteristics and migratory capacity–is a key pathological mechanism involved in airway remodeling during the progression of COPD.11–15 Airway epithelium from COPD patients exhibits persistent EMT activation, characterized by the downregulation of epithelial markers such as E-cadherin and the upregulation of mesenchymal markers including N-cadherin and fibronectin.16,17 Among various mediators, transforming growth factor-beta 1 (TGF-β1) has been consistently shown to be overexpressed in the airway epithelium of COPD patients and has emerged as a principal regulator of EMT in bronchial epithelial cells.18–20 However, the precise molecular mechanisms by which TGF-β1 regulates EMT during airway remodeling in COPD remain to be fully elucidated.
Laminin γ2 (LAMC2), an essential structural component of the heterotrimeric laminin-332 glycoprotein composed of α3, β3, and γ2 chains, plays critical roles in fundamental biological processes including cell proliferation, adhesion, migration, and EMT.21,22 Previous studies have established its essential involvement in both tissue repair mechanisms and fibrotic progression.23,24 Additionally, LAMC2 expression demonstrates marked upregulation within airway epithelial cells—with pronounced expression in regenerating epithelial populations—in fibrotic pulmonary disorders including idiopathic pulmonary fibrosis (IPF) and cryptogenic organizing pneumonia (COP),25 raising the possibility that it may be involved in fibrotic remodeling processes in the lung. Nevertheless, the role of LAMC2 in COPD, especially in the context of airway remodeling, remains largely unexplored.
In this study, we found that LAMC2 expression is significantly upregulated in airway epithelial cells of COPD patients and experimental models. In vitro assays demonstrated that TGF-β1 induces LAMC2 expression, and that silencing or overexpressing LAMC2 respectively suppresses or promotes the EMT process. Mechanistically, LAMC2 was shown to regulate EMT through activation of the AKT signaling pathway. In vivo, administration of AAV-shLAMC2 significantly mitigated airway epithelial EMT and airway remodeling in a COPD mouse model compared with the control group (AAV-shNC). Collectively, our findings highlight a pivotal role for LAMC2 in the pathogenesis of COPD. The aberrant upregulation of LAMC2 in airway epithelial cells drives airway remodeling through an AKT-dependent EMT mechanism, offering novel mechanistic insights and a potential therapeutic target for COPD.
Materials and Methods
Data Acquisition
The publicly available transcriptomic datasets GSE87292 (30 air-control and 34 COPD-model mouse lung tissues), GSE5058 (12 non-smokers, 12 smokers, and 6 COPD human small airway epithelial samples), GSE104908, and GSE40374 (each with 3 TGFβ1-treated and 3 control bronchial epithelial cell samples) were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). These datasets were generated using the following platforms: GPL6885 (Illumina MouseRef-8 v2.0 microarray), GPL570 (Affymetrix Human Genome U133 Plus 2.0 array), GPL6480 (Agilent Whole Human Genome Microarray 4x44K), and GPL16791 (Illumina HiSeq 2500 RNA-Seq). Raw data were preprocessed in R using appropriate packages: affy for Affymetrix arrays, limma for Illumina arrays and Agilent arrays, and standard RNA-Seq pipelines for sequencing data. Preprocessing included background correction, normalization, and log2 transformation where applicable. For genes represented by multiple probes, the mean probe expression was used to represent the gene-level expression.
Subjects and Specimen Sampling
Lung tissue samples from non-smokers, smokers, and individuals with COPD were obtained from patients undergoing surgical resection for solitary pulmonary tumors at Peking University Third Hospital. To minimize tumor-associated alterations, samples were collected from areas of macroscopically normal lung parenchyma located at the greatest distance from the tumor, as assessed by a pathologist. Non-smokers were defined as individuals with no history of tobacco use and a post-bronchodilator FEV1/FVC ratio ≥ 0.7. Smokers were defined as those with a smoking history of ≥10 pack-years and preserved lung function (post-bronchodilator FEV1/FVC ≥ 0.7). COPD patients were identified based on a smoking history of ≥10 pack-years and a post-bronchodilator FEV1/FVC ratio < 0.7, in accordance with the GOLD diagnostic criteria. All procedures involving human participants were approved by the Ethics Committee of Peking University Third Hospital (approval number: S2018193), and written informed consent was obtained from all participants prior to sample collection. Detailed clinical characteristics of the subjects are provided in Table S1.
Animal Model and Administration of Adenovirus
Six-week-old male C57BL/6J mice (specific pathogen-free, SPF) were purchased from Cyagen Biosciences Inc. (Santa Clara, CA, USA) and housed in individually ventilated cages under controlled conditions (12-h light/dark cycle, 24 ± 2 °C, 50 ± 10% humidity) at the Department of Laboratory Animal Science, Peking University Health Science Center. To establish the cigarette smoke (CS)-induced COPD model, mice were exposed to CS generated from 20 Marlboro cigarettes (tar: 10 mg, nicotine: 0.8 mg, CO: 11 mg per cigarette) twice daily (2 h/session, 4 h apart), 6 days/week for 12 weeks using a whole-body exposure system. Control mice were exposed to filtered air under the same conditions.
The adeno-associated virus (AAV) 6-shCtrl and AAV6-shLAMC2 were synthesized by WZ Biosciences Inc. (Shandong, China). Mice were randomly divided into four groups: Air + AAV-shCtrl, Air + AAV-shLAMC2, CS + AAV-shCtrl, and CS + AAV-shLAMC2. Mice were injected via intratracheal instillation with 5 × 1012 V.G/mouse. Prior to CS exposure, mice were intranasally administered the respective AAV vectors.
All animal experiments were approved by the Institutional Animal Care and Use Committee of Peking University Health Science Center. All procedures involving animals, including anesthesia and euthanasia, were performed in accordance with the AVMA Guidelines for the Euthanasia of Animals (2020). Mice were anesthetized with isoflurane inhalation (3–4% for induction and 1–2% for maintenance) prior to intratracheal instillation and other experimental procedures. At the end of the study, mice were euthanized under deep isoflurane anesthesia followed by cervical dislocation to ensure death.
Cell Culture and Treatment
The 16HBE cells were obtained from Bai Ye Biotechnology Center (Shanghai, China) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Hyclone, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Procell, China) and 1% penicillin-streptomycin (Gibco, USA). All cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. The human TGF-β1 protein (Abbkine, Wuhan, China) was used to stimulate 16HBE cells at various concentrations for 24 hours. To investigate the function and underlying mechanism of LAMC2, 16HBE cells were transfected with either a control vector or a LAMC2 overexpression vector (LST Bio-tech ShanDong co, Ltd), or with a small interfering RNA (siRNA) negative control or LAMC2-targeting siRNA (Generalbiol, Chuzhou, China), using Lipofectamine 3000 (Lipo3000; Livning, China) following the manufacturer’s protocol. The LAMC2 siRNAs sequences are as follows:
siRNA-1: 5’-GCUCACCAAGACUUACACAUU-3’;
siRNA-2: 5’-GCCCUGCAAUUGUAACUCCAA-3’.
Western Blot
Total protein was extracted using RIPA buffer (APPLYGEN, China) supplemented with phosphatase and protease inhibitors (APPLYGEN, China). Equal amounts of protein (20–50 µg) were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, MA, USA). The membranes were blocked with 5% bovine serum albumin for 1 hour at room temperature and then incubated overnight at 4°C with the following primary antibodies: GAPDH (1:5000; Cell Signaling Technology, Danvers, MA, USA), LAMC2 (1:1000; Proteintech, China), E-Cadherin (1:1000; Cell Signaling Technology), N-Cadherin (1:1000; Cell Signaling Technology), Fibronectin (1:1000; HUABIO, China), AKT (1:1000; Cell Signaling Technology), and phospho-AKT (p-AKT, 1:1000; Cell Signaling Technology). After washing with Tris-buffered saline containing 0.1% Tween-20 (TBST), the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (H+L) secondary antibody (1:5000; Abcam, USA) for 1.5 hours at room temperature. Finally, the membranes were washed again with TBST and visualized using an enhanced chemiluminescence (ECL) detection system (Millipore, MA, USA).
Quantitative Real‑time‑polymerase Chain Reaction (qRT‑PCR)
Total RNA was extracted using the RNA Fast 200 Extraction Kit (Fastagen Biotech, China). RNA purity and concentration were assessed using a spectrophotometer, and 1 µg of RNA was used for reverse transcription. Complementary DNA (cDNA) was synthesized using a reverse transcription kit (Vazyme, Nanjing, China). Quantitative real-time PCR (qRT-PCR) was performed using the ChamQ Universal SYBR qPCR Master Mix (Vazyme, China) on a Bio-Rad CFX96 Real-Time PCR Detection System to assess mRNA expression levels. The specific primer sequences used are provided in Table S2.
Wound Healing Assay
16HBE cells subjected to different treatments were seeded into 6-well plates and cultured until they reached approximately 95% confluence. A vertical scratch was carefully made across the cell monolayer using a 200 μL pipette tip. Following the scratch, the cells were gently washed with PBS to remove detached cells and debris, and then maintained in serum-free medium. Images of the scratch area were captured at 0 h and 24 h, and the wound width was measured to evaluate cell migration.
Transwell Assay
Migration assays were conducted using transwell chambers with 8 μm pore filters (Corning, USA). 16HBE cells subjected to different treatments were harvested, washed, and resuspended in serum-free DMEM. A cell suspension containing 5 × 105 cells per well was seeded into the upper chamber, while the lower chamber was filled with DMEM supplemented with 10% fetal bovine serum (FBS) as a chemoattractant. The chambers were incubated at 37 °C for 48 hours to allow cell migration through the membrane. After incubation, the cells that had migrated to the lower surface of the membrane were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and counted under a microscope.
RNA Sequencing and Data Analysis
Total RNA from 16HBE cells in the TGFβ1 + siRNA-NC and TGFβ1 + siRNA-LAMC2 groups was extracted and immediately submitted to Metware Biotechnology (Wuhan, China) for RNA sequencing. Differentially expressed genes (DEGs) were identified and retrieved from Metware’s cloud-based analysis platform (https://cloud.metware.cn). Subsequent enrichment analyses of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were conducted using the DAVID database (https://david.ncifcrf.gov/home.jsp).
Histopathological Evaluation and Immunohistochemistry
Lung tissues were carefully excised and immediately fixed in 4% paraformaldehyde for 24 hours. Following fixation, the tissues were embedded in paraffin and sectioned into 5-μm-thick slices for various staining procedures. For instance, Masson’s trichrome staining was performed to evaluate the degree of airway remodeling, while immunohistochemistry (IHC) staining was conducted to examine the expression of markers related to EMT. Specifically, after deparaffinization and rehydration, the lung tissue sections underwent antigen retrieval using citrate unmasking solution. Endogenous peroxidase activity was then quenched by incubating the sections with 3% hydrogen peroxide. To minimize non-specific binding, the sections were blocked with goat serum for 1 hour at room temperature. Subsequently, the sections were incubated overnight at 4°C with primary antibodies, including anti-LAMC2 (1:200, Proteintech), anti-E-cadherin (1:400, Cell Signaling Technology), anti-N-cadherin (1:100, Cell Signaling Technology), and anti-Fibronectin (1:100, HUABIO). On the following day, the sections were incubated with biotinylated anti-rabbit IgG secondary antibody to bind the primary antibodies. Finally, the chromogenic substrate was applied (ZSGB BIO, China), and color development was monitored to assess staining intensity. For histological scoring, slides were randomly selected and coded so that the investigator performing the scoring was blinded to the experimental groups.
Immunofluorescence (IF) Staining
For both lung tissue sections and cultured cells, samples were incubated with anti-phospho-AKT (p-AKT) primary antibody (1:200, CST) overnight at 4°C. Prior to antibody incubation, tissue sections underwent deparaffinization, antigen retrieval, and blocking, while cells were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and blocked with 5% goat serum. The next day, samples were incubated with a corresponding fluorophore-conjugated secondary antibody (1:400, Jackson ImmunoResearch, USA) for 1 hour at room temperature, followed by nuclear staining with DAPI (1:200) for 5 minutes.
Statistical Analysis
Data following a normal distribution are presented as mean ± standard deviation (SD), while non-normally distributed data are expressed as median with interquartile range (IQR). Comparisons between two groups were conducted using Student’s t-test for normally distributed variables or the Mann–Whitney U-test for non-normally distributed variables. For comparisons involving three or more groups, normally distributed data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison correction, while non-normally distributed data were analyzed using the Kruskal–Wallis test followed by Dunn’s multiple comparison test. All statistical analyses were conducted using GraphPad Prism version 8.0 and SPSS version 25.0. A two-tailed P value < 0.05 was considered statistically significant.
Results
Airway Remodeling and LAMC2 Upregulation are Observed in Both COPD Patients and Mouse Models
Masson’s trichrome staining demonstrated significantly increased collagen deposition surrounding the airways in both clinical and experimental contexts: (i) COPD patients exhibited more pronounced collagen deposition relative to both smoking and non-smoking control subjects (Figure 1A and B), and (ii) COPD model mice displayed substantially greater collagen accumulation compared to air-exposed control animals (Figure 1C and D), collectively indicating the presence of significant airway remodeling characteristic of COPD pathogenesis. To investigate LAMC2 expression in the airway epithelium of COPD patients, we analyzed the GSE5058 dataset, which demonstrated significantly elevated LAMC2 levels in COPD patients compared to both smokers and non-smokers (Figure 1E). These findings were further validated by IHC staining (Figure 1F and G). Similarly, transcriptomic analysis of mice lung tissues using the GSE87292 dataset revealed upregulated LAMC2 expression in COPD model mice (Figure 1H), which was corroborated by Western blotting (Figure 1I and J). Furthermore, IHC staining specifically targeting airway epithelium confirmed increased LAMC2 expression in the bronchial epithelial cells of COPD model mice (Figure 1K and L).
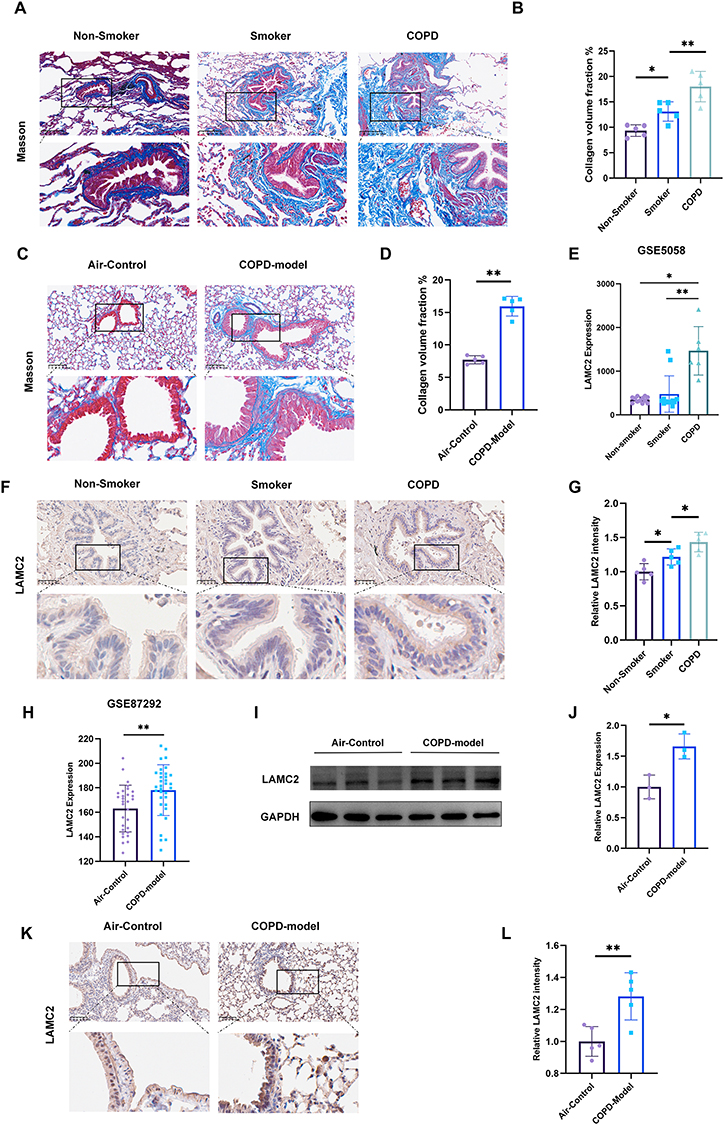 |
Figure 1 Airway remodeling and LAMC2 upregulation are observed in both COPD patients and mouse models. (A) Representative Masson’s trichrome-stained lung sections from non-smokers (n=5), smokers without COPD (n=5), and COPD patients (n=5). Scale bar: 250μm. (B) Quantitative staining of Masson content in non-smoker (n=5), smoker (n=5), and COPD (n=5). (C) Representative Masson’s trichrome staining of lung sections from air-exposed control mice (n=5) and COPD model mice (n=5). Scale bar: 100μm. (D) Quantitative staining of Masson content in air-control (n=5) and COPD model mice (n=5). (E) LAMC2 expression levels in airway epithelial cells from non-smokers (n=12), smokers (n=12), and COPD patients (n=6) based on the GSE5058 dataset. (F) Representative airway LAMC2 immunohistochemical staining on lung sections of non-smoker (n=5), smoker (n=5), and COPD (n=5). Scale bar: 50μm. (G) Quantitative staining of LAMC2 intensity on airway epithelium of non-smoker (n=5), smoker (n=5), and COPD (n=5). (H) LAMC2 expression levels in lung tissues from air-control (n=30) and COPD model mice (n=34) based on the GSE87292 dataset. (I) Western blot and (J) quantitative analysis of LAMC2 protein expression in the lung tissue from mice from different groups (n=3 mice/group). (K) Representative airway LAMC2 immunohistochemical staining on lung sections of air-control (n=5) and COPD model mice (n=5).Scale bar: 100μm. (L) Quantitative staining of LAMC2 intensity on airway epithelium of air-control (n=5) and COPD model mice (n=5). Data presented as the means ± SD. *P < 0.05, **P < 0.01. |
Knockdown of LAMC2 Could Attenuate Airway Remodeling and EMT in a COPD Model
EMT is a key pathological mechanism underlying airway remodeling in COPD.11,12 To investigate whether LAMC2 contributes to airway remodeling in COPD and regulates the EMT process, six-week-old mice were first administered intratracheal instillations of AAV-shCtrl or AAV-shLAMC2, as illustrated in Figure 2A, and Multiple analyses of lung tissues demonstrated that AAV-shLAMC2 effectively reduced LAMC2 expression in the lungs both before and after modeling, indicating that AAV-shLAMC2 can exert a sustained effect in mouse lung tissue (Figure S1A–E). Masson’s trichrome staining revealed that collagen deposition around the airways was significantly reduced in the CS-AAV-shLAMC2 group compared to the CS-AAV-shCtrl group, suggesting that LAMC2 influences extracellular matrix remodeling–such as collagen accumulation–and thereby contributes to airway remodeling (Figure 2B and C). Furthermore, IHC analysis of lung tissues showed that, compared to air control mice, COPD model mice exhibited decreased E-cadherin expression and increased Fibronectin and N-cadherin levels in the airway epithelium. However, following prior intervention with AAV-shLAMC2, these EMT-related markers were significantly altered, with elevated E-cadherin expression and reduced levels of Fibronectin and N-cadherin compared to the COPD control model group (Figure 2D–K). These results were further confirmed by protein analysis of lung tissues (Figure 2J–M).
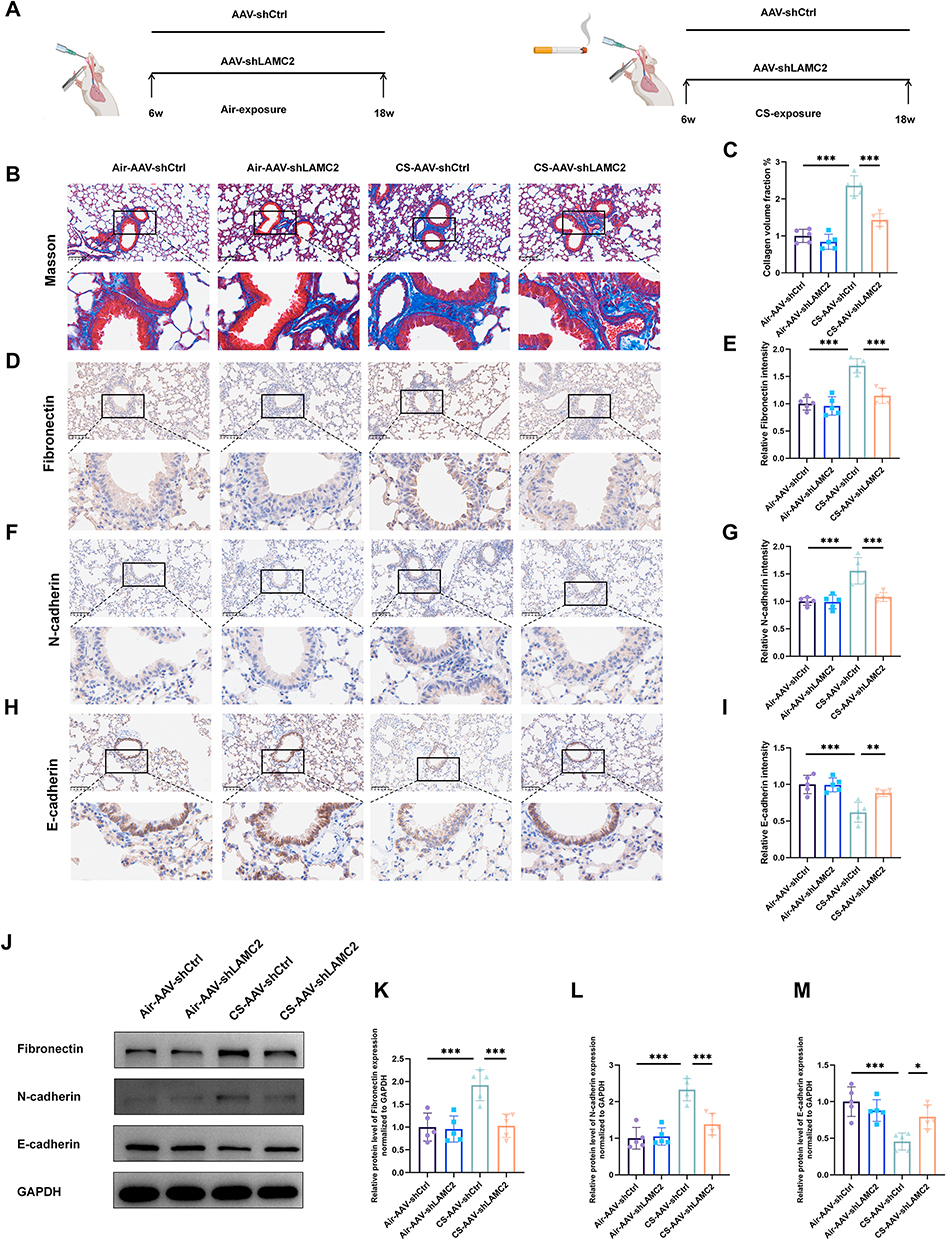 |
Figure 2 Knockdown of LAMC2 attenuates airway remodeling and EMT in a COPD model. (A) Schematic overview of the animal experimental design and grouping. (B) Representative Masson staining on mice lung sections from different groups (n = 5 per group). (C) Quantitative staining of Masson content in different groups (n = 5 per group). (D) Representative airway Fibronectin immunohistochemical staining and (E) quantitative staining of Fibronectin intensity on lung sections of different groups (n = 5 per group). (F) Representative airway N-cadherin immunohistochemical staining and (G) quantitative staining of N-cadherin intensity on lung sections of different groups (n=5 per group). (H) Representative airway E-cadherin immunohistochemical staining and (I) quantitative staining of E-cadherin intensity on lung sections of different groups (n=5 per group). (J–M) Western blot analysis and quantification of EMT-related proteins (Fibronectin, N-cadherin, and E-cadherin) in lung tissues from the different treatment groups. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 100μm. |
LAMC2 Expression is Upregulated in Bronchial Epithelial Cells Following TGF‑β1 Stimulation
TGF‑β1 is markedly elevated in the lung tissue of patients with COPD and serves as a key inducer of EMT, a central process in airway remodeling during COPD progression.12,14,26–28 To explore whether LAMC2 expression is modulated by TGF‑β1 in airway epithelial cells, we first analyzed transcriptomic data from the GSE104908 and GSE40374 datasets, which contain sequencing data comparing airway epithelial cells treated with TGF‑β1 to untreated controls. The results demonstrated that TGF‑β1 stimulation significantly upregulates LAMC2 gene expression (Figure 3A and B). Subsequently, we validated these findings using qPCR and Western blot analyses. To investigate the effects of TGF-β1 on EMT in 16HBE and to determine the optimal stimulating concentration, cells were treated with TGF-β1 at doses of 5, 10, and 20 ng/mL for 24 hours. Compared with the control group, TGF-β1 treatment markedly promoted EMT in airway epithelial cells, as evidenced by significant suppression of the epithelial marker E-cadherin concomitant with pronounced upregulation of the mesenchymal marker N-cadherin and extracellular matrix component fibronectin. Importantly, TGF-β1-mediated signaling robustly enhanced LAMC2 expression, with overall parallel upregulation observed at both mRNA and protein levels (Figure 3C–H).
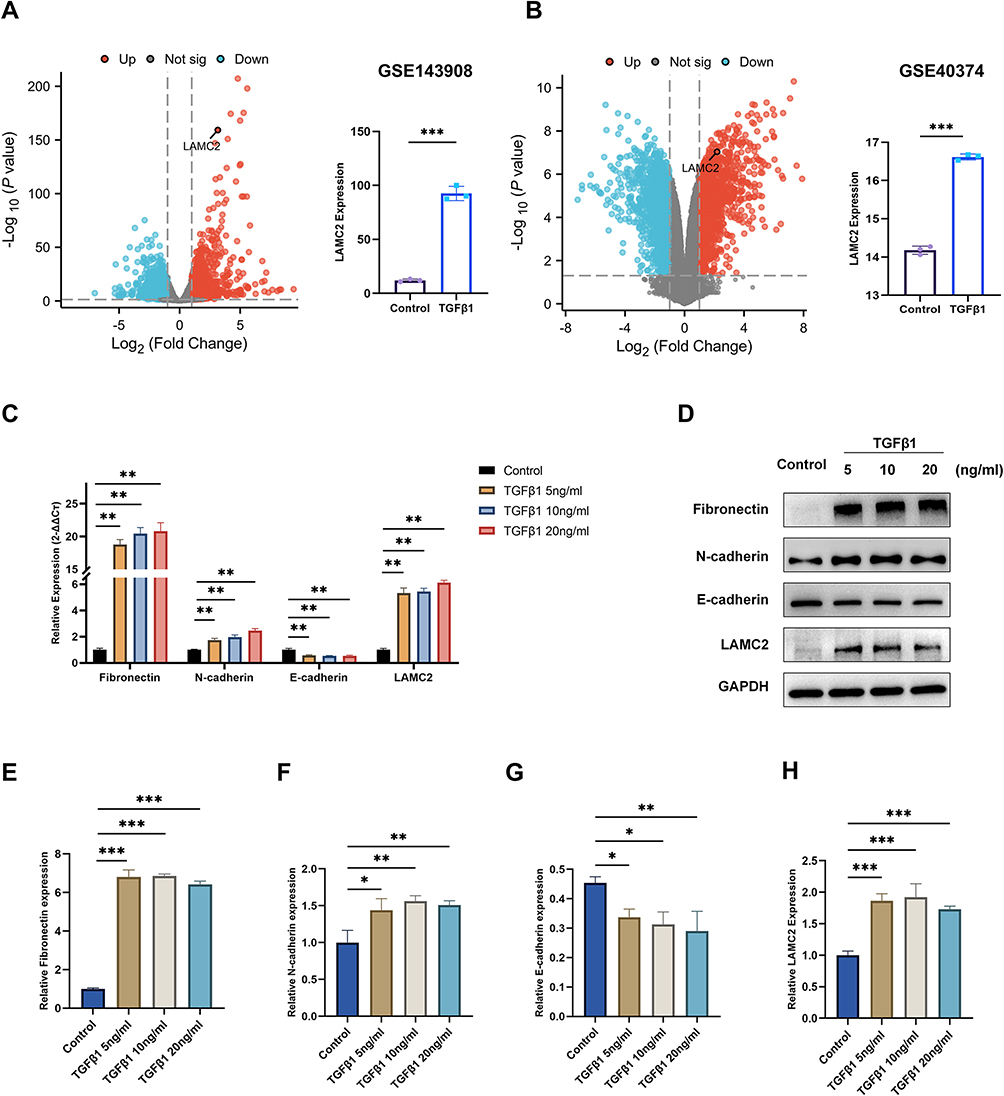 |
Figure 3 LAMC2 expression is upregulated in bronchial epithelial cells following TGF-β1 stimulation. (A and B) Volcano plots illustrating DEGs from (A) GSE104908 and (B) GSE40374 datasets, with corresponding LAMC2 expression levels. Significantly upregulated genes (red) and downregulated genes (blue) are indicated (fold change >1.5, adjusted P < 0.05). (C) The mRNA levels of Fibronectin, N-cadherin, E-cadherin, and LAMC2 after treatment with different concentrations of TGF-β1 for 24 hours. (D) Western blot and (E–H) the relative protein level of Fibronectin, N-cadherin, E-cadherin, and LAMC2 after treatment with different concentrations of TGF-β1 for 24 hours. The data are the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. |
LAMC2 Modulates 16HBE Cell Migration and the Expression of EMT Markers in Response to TGF‑β1 Stimulation
To investigate the role of LAMC2 in EMT, we first knocked down its expression in 16HBE cells using siRNA. Among the siRNA constructs tested, si-LAMC2-1 showed the most efficient silencing and was selected for subsequent experiments (Figure 4A). Based on the comprehensive analysis of TGF-β1 concentration-response results, 10 ng/mL with 24-hour stimulation was chosen as it elicited the most pronounced changes in EMT marker expression, and was therefore used for subsequent experiments. In the presence of TGF-β1, LAMC2 knockdown significantly attenuated the EMT process, as indicated by the upregulation of the epithelial marker E-cadherin and the downregulation of mesenchymal and remodeling markers N-cadherin and fibronectin (Figure 4B–F). Since EMT also confers migratory properties to epithelial cells, we further assessed the impact of LAMC2 knockdown on cell migration. Both wound healing and transwell assays demonstrated that silencing LAMC2 significantly impaired the migratory capacity of 16HBE cells (Figure 4G–I). To further validate the role of LAMC2 in EMT, we overexpressed LAMC2 using a plasmid vector. LAMC2 overexpression markedly increased its expression in 16HBE cells (Figure 5A), and when combined with TGF‑β1 stimulation, it further enhanced EMT progression (Figure 5B–F) and significantly promoted cell migration (Figure 5G–I). Collectively, these findings suggest that LAMC2 plays a critical role in regulating TGF‑β1–induced EMT and cell motility in bronchial epithelial cells.
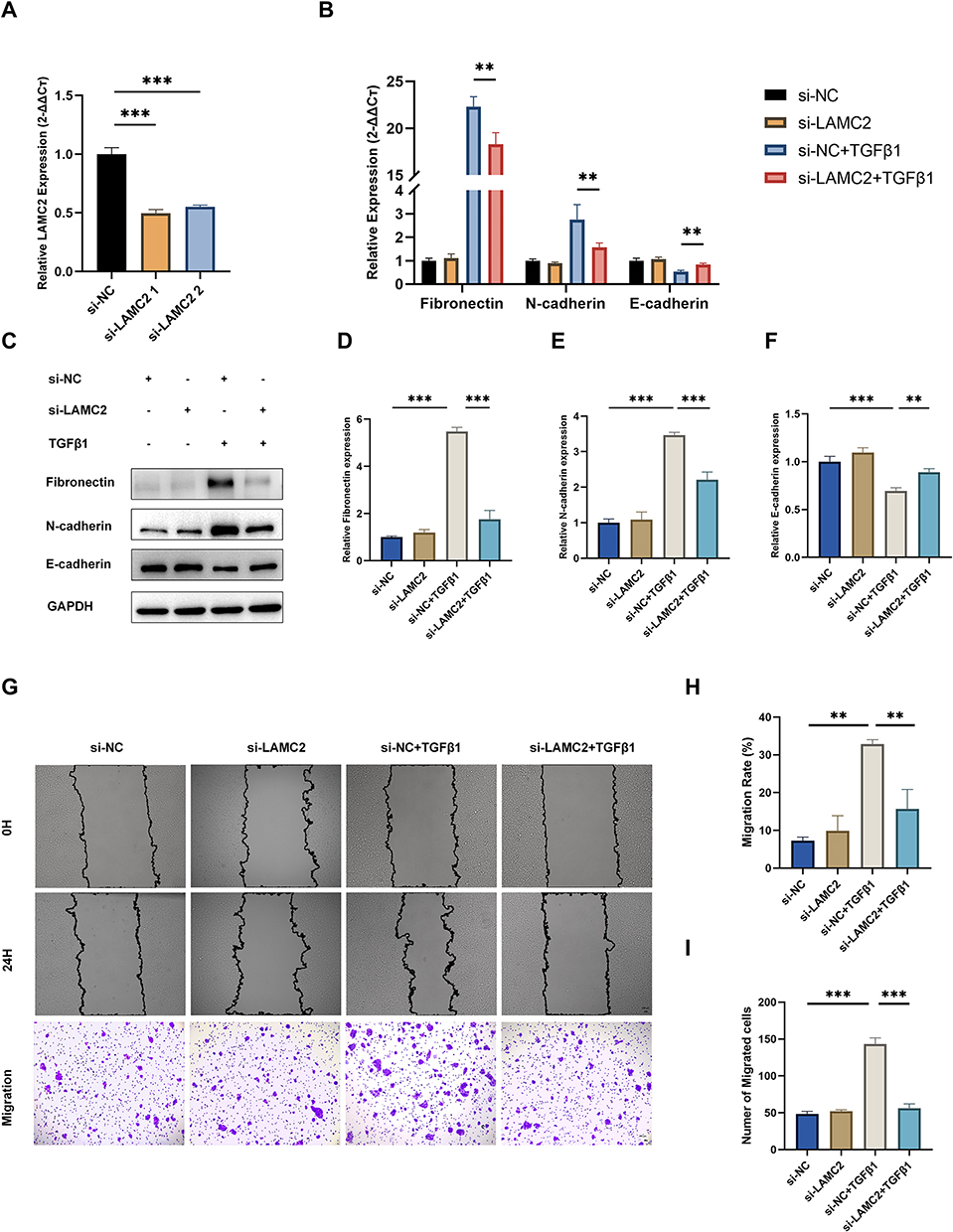 |
Figure 4 Knockdown of LAMC2 reverses TGF-β1-induced EMT marker expression and inhibits 16HBE cell migration. (A) LAMC2 mRNA expression levels following transfection with different siLAMC2 constructs. (B) mRNA expression of Fibronectin, N-cadherin, and E-cadherin after siLAMC2 transfection and subsequent TGF-β1 treatment. (C) Western blot and (D–F) the relative protein level of Fibronectin, N-cadherin, E-cadherin, and LAMC2 after siLAMC2 transfection and TGF-β1 treatment. (G) Representative images of wound healing and transwell migration assays under different treatment conditions. Scale bar: 20 μm. (H) Quantification of migration rate (%) from wound healing assay and (I) Cell migration numbers counted from transwell assays. The data are the mean ± SD. **P < 0.01, ***P < 0.001. |
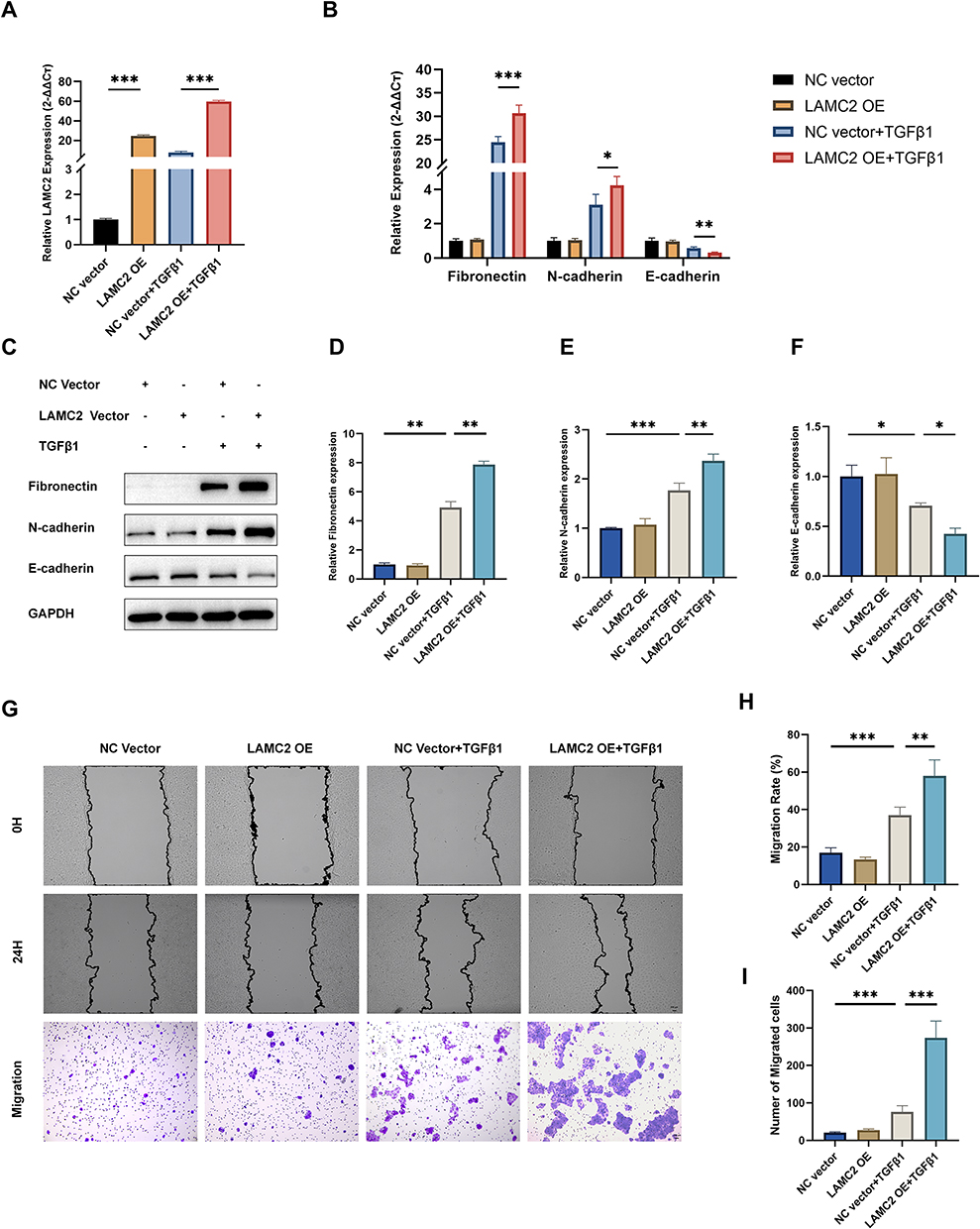 |
Figure 5 Overexpression of LAMC2 promotes TGF-β1-induced EMT and enhances 16HBE cell migration. (A) LAMC2 mRNA expression levels following transfection with LAMC2 plasmid and TGF-β1treatment. (B) mRNA expression of Fibronectin, N-cadherin, and E-cadherin after LAMC2 plasmid vector transfection and subsequent TGF-β1 treatment. (C) Western blot and (D–F) the relative protein level of Fibronectin, N-cadherin, E-cadherin, and LAMC2 after LAMC2 plasmid vector transfection and TGF-β1 treatment. (G) Representative images of wound healing and transwell migration assays under different treatment conditions. Scale bar: 20 μm. (H) Quantification of migration rate (%) from wound healing assay and (I) Cell migration numbers counted from transwell assays. The data are the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. |
LAMC2 Facilitates EMT in 16HBE Cells Through Activation of the AKT Signaling Pathway
To elucidate the precise molecular mechanism by which LAMC2 facilitates EMT in 16HBE cells, we conducted RNA sequencing analysis following TGF-β1 stimulation in cells transfected with either control siRNA (siRNA-NC) or siLAMC2. DEGs showing significant downregulation (fold change ≥ 1.5) upon LAMC2 knockdown were selected for further enrichment analyses. GO enrichment analysis revealed that these differentially expressed genes were predominantly involved in extracellular matrix (ECM) organization and remodeling (Figure 6A). Considering the well-established link between EMT progression and airway remodeling, these results suggest that LAMC2 may contribute to airway remodeling by modulating ECM composition through EMT. Moreover, KEGG pathway analysis identified the PI3K-AKT signaling pathway as the most significantly enriched pathway among all differentially expressed genes, indicating that LAMC2 likely exerts its biological effects via activation of the PI3K-AKT axis (Figure 6B). To validate this, we performed both LAMC2 knockdown and overexpression in 16HBE cells, followed by TGF-β1 stimulation. Corresponding changes in phosphorylated AKT (p-AKT) levels were observed, providing direct evidence that LAMC2 regulates EMT through modulation of the AKT signaling pathway (Figure 6C–L). In addition, in vivo experiments also demonstrated that LAMC2 knockdown reduced pulmonary p-AKT levels in the COPD mouse model (Figure S1F and G).
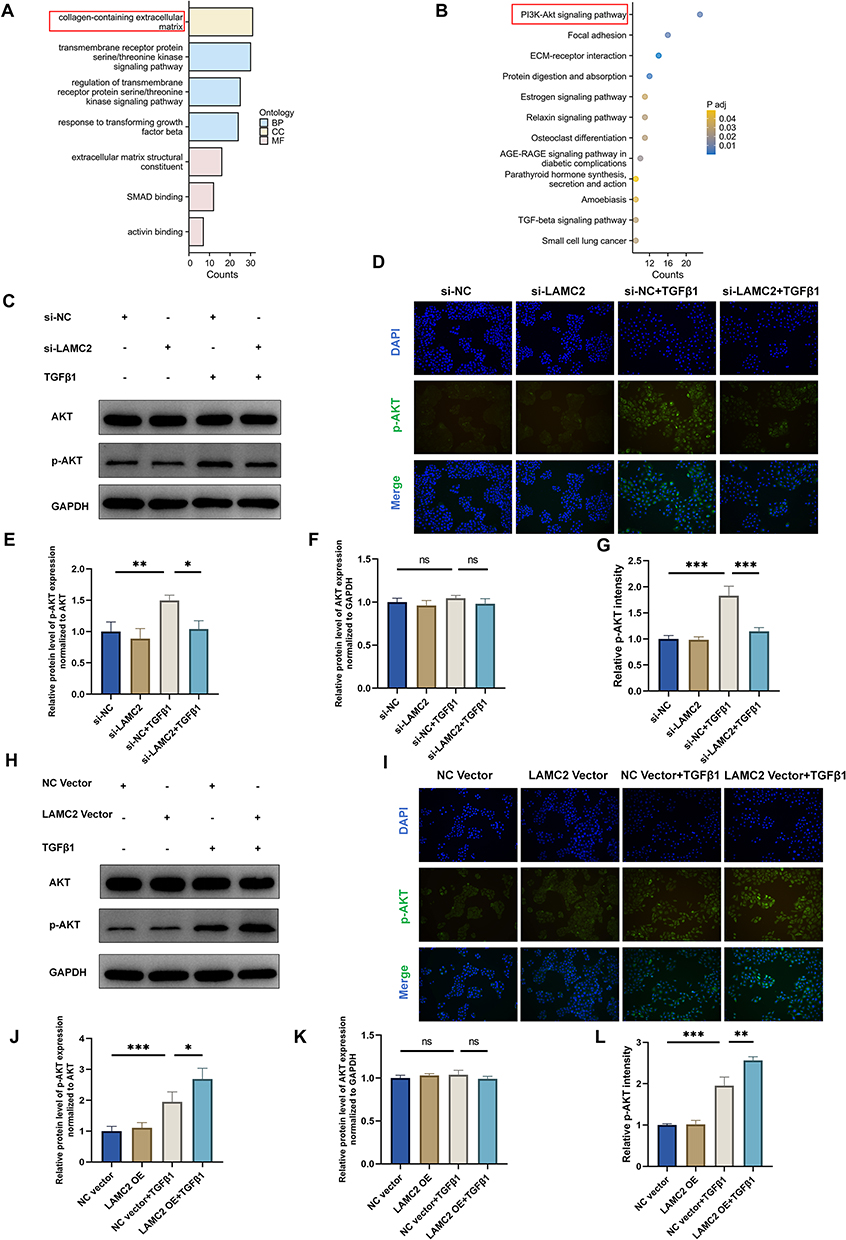 |
Figure 6 LAMC2 facilitates EMT in 16HBE cells through activation of the AKT signaling pathway. (A) GO enrichment analysis of down-regulated DEGs from RNA-seq data, showing the top three most significantly enriched terms in each category: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) (adjusted P < 0.05). Terms are ranked by gene count. (B) KEGG pathway analysis of down-regulated DEGs. Statistically significant pathways (adjusted P < 0.05) are visualized in a dot plot. (C) Western Blot and (D) Immunofluorescence detection, and (E–G) the relative protein level of AKT and p-AKT after si-NC/LAMC2 transfection and TGF-β1 treatment. (H) Western Blot and (I) Immunofluorescence detection, and (J–L) the relative protein level of AKT and p-AKT after LAMC2 plasmid vector transfection and TGF-β1 treatment. Scale bar: 50 μm. The data are the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. |
Discussion
COPD imposes a significant global health burden due to its high morbidity, mortality, and multiple comorbidities.29–33 Irreversible airflow limitation is a hallmark of COPD, with airway remodeling recognized as a key contributor to this pathological feature.34,35 Among the various mechanisms underlying airway remodeling, EMT—a process whereby airway epithelial cells acquire mesenchymal characteristics—plays a pivotal role by driving extracellular matrix (ECM) remodeling and tissue structural changes.36,37 TGF-β1 is a well-established inducer of EMT in bronchial epithelial cells and is known to be upregulated in the airway epithelium of COPD patients.20,38 However, the precise molecular mechanisms through which TGF-β1 promotes EMT in COPD remain incompletely understood.
In this study, we employed transcriptomic analysis to compare gene expression profiles in airway epithelial cells from COPD patients and healthy controls. Our results revealed a significant upregulation of LAMC2 in both COPD patient samples and experimental models. Meanwhile, LAMC2 expression was markedly increased in response to TGF-β1 stimulation. These bioinformatic findings were validated by a series of in vitro and in vivo experiments. Functionally, silencing of LAMC2 expression attenuated TGF-β1-induced EMT, while overexpression of LAMC2 enhanced this process. These effects were evidenced by changes in epithelial and mesenchymal markers, as well as alterations in cellular migration ability. Collectively, our findings demonstrate that LAMC2 mediates TGF-β1-induced EMT in airway epithelial cells and may serve as a potential therapeutic target for inhibiting airway remodeling in COPD.
LAMC2 is an essential component of the heterotrimeric glycoprotein laminin-332, composed of α3, β3, and γ2 chains. Although no studies have yet specifically investigated the role of LAMC2 in COPD, previous research has demonstrated that its expression is significantly elevated in the airway epithelium of patients with two fibrotic diseases—COP and IPF—particularly in regenerating epithelial cells, compared to healthy controls.25 This suggests that LAMC2 may contribute to the progression of fibrosis through multiple mechanisms during the process of airway epithelial repair. Prior studies have indicated that LAMC2 plays a pivotal role in the EMT. For instance, LAMC2 induces EMT in lung adenocarcinoma cell, thereby promoting cell migration, invasion, and traction force, ultimately leading to distant metastasis.39 Moreover, in pancreatic cancer, LAMC2 also regulates EMT, influencing not only tumor cell migration and invasion but also modulating sensitivity to chemotherapeutic agents.40 EMT is likewise recognized as a key mechanism in COPD-associated airway remodeling, facilitating the transformation of epithelial cells into fibroblast-like cells, which leads to ECM deposition and subsequent airway remodeling.12,14,15 Our study further confirms the functional significance of LAMC2 in a TGF-β1-induced EMT model of airway epithelial cells. Knockdown or overexpression of LAMC2 significantly alters the expression levels of EMT-related markers and affects cell migratory capacity, both of which are closely associated with airway remodeling. Furthermore, we employed AAV-mediated knockdown of LAMC2 in the lungs of mice, which effectively reduced peribronchial collagen deposition and the expression of EMT markers in a COPD mouse model, thereby alleviating airway remodeling. These findings suggest that LAMC2 may play a critical regulatory role in the pathogenesis of airway remodeling in COPD.
Previous studies have demonstrated that LAMC2 contributes to the development and metastasis of pancreatic cancer via the AKT signaling pathway,41 and regulates macrophage apoptosis by modulating p-AKT expression.42 To further investigate the specific mechanism by which LAMC2 participates in EMT in airway epithelial cells, we performed enrichment analysis based on transcriptomic data. Upon TGF-β1 stimulation of 16HBE cells, knockdown of LAMC2 resulted in the downregulation of multiple genes predominantly enriched in the PI3K-AKT signaling pathway, suggesting that LAMC2 may regulate the EMT process through this pathway. In vitro experiments further confirmed that both silencing and overexpression of LAMC2 altered the p-AKT. Notably, inhibition of the AKT pathway in the context of LAMC2 overexpression significantly attenuated the EMT phenotype. These findings indicate that LAMC2 may promote airway remodeling by modulating EMT through the AKT signaling axis. Despite these findings, several challenges remain for the potential clinical translation of targeting LAMC2 in COPD. Achieving cell-type–specific targeting of LAMC2 in airway epithelial cells in humans remains technically challenging. However, emerging therapeutic strategies, including inhaled RNA-based therapeutics, targeted nanoparticle delivery systems, and airway-directed gene therapy approaches, may provide feasible platforms for selectively modulating gene expression in airway epithelial cells. Further studies are warranted to explore the safety, efficacy, and delivery strategies required to translate LAMC2-targeted interventions into clinical applications.
This study has several limitations that should be noted. First, the experiments were conducted using commercially available cell lines, and future studies should incorporate primary human cells for more robust validation. Second, the current investigation into the mechanistic role of LAMC2 in COPD remains preliminary, and more in-depth research is required to elucidate its precise molecular mechanisms.
Conclusions
LAMC2 is significantly upregulated in the airway epithelium of COPD and may contribute to airway remodeling by regulating EMT via the AKT pathway. This mechanistic insight identifies LAMC2 as a novel molecule involved in COPD airway remodeling. Targeting LAMC2-mediated EMT may therefore offer a therapeutic strategy to attenuate airway remodeling and slow COPD progression.
Abbreviations
COPD, Chronic obstructive pulmonary disease; EMT, Epithelial-mesenchymal transition; LAMC2, Laminin subunit gamma-2; TGF-β1, Transforming growth factor-beta 1; GEO, Gene Expression Omnibus; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; AAV, Adeno-associated virus; siRNA, Small interfering RNA; ECM, Extracellular matrix; cDNA, Complementary DNA; qRT-PCR, Quantitative real-time PCR.
Data Sharing Statement
The datasets used and/or analyzed during the present study are available from the corresponding authors Xiaoyan Gai and Yongchang Sun on reasonable request.
Ethical Approval and Consent to Participate
The present study was authorized by the Ethics Committee of Peking University Third Hospital (approval number: S2018193). All animal experiments were approved by the Institutional Animal Care and Use Committee of Peking University Health Science Center (approval number: BCAJ0269). The present study was performed in compliance with the guidelines described in the Declaration of Helsinki.
Consent for Publication
Consent for publication was obtained from all participants included in the present study.
Acknowledgments
The authors extend their gratitude to all participants who contributed to the GEO database, as well as to the researchers responsible for its construction and ongoing maintenance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Our study is supported by the National Natural Science Foundation of China (82370047); National Natural Science Foundation of China Young Scientist Fund (82300055); Beijing Natural Science Foundation Youth Project (7254446).
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. GBD 2021 Causes of Death Collaborators. Global burden of 288 causes of death and life expectancy decomposition in 204 countries and territories and 811 subnational locations, 1990–2021: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. 2024;403(10440):2100–16. doi:10.1016/s0140-6736(24)00367-2
2. Soriano JB, Kendrick PJ, Paulson KR. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8(6):585–596. doi:10.1016/s2213-2600(20)30105-3
3. Global strategy for prevention, diagnosis and management of chronic obstructive pulmonary disease. 2025 report; 2025. Available from: https://goldcopd.org.
4. Global strategy for prevention, diagnosis and management of COPD: 2025 report; 2025. Available from: https://goldcopd.org/2025-gold-report/.
5. Liu G, Hsu AC, Geirnaert S, et al. Vitronectin regulates lung tissue remodeling and emphysema in chronic obstructive pulmonary disease. Mol Ther. 2025;33(3):917–932. doi:10.1016/j.ymthe.2025.01.032
6. Lu Z, Van Eeckhoutte HP, Liu G, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2021;204(6):667–681. doi:10.1164/rccm.202009-3442OC
7. Oba Y, Pathak M, Maduke T, Fakhouri EW, Dias S. Dual combination therapy versus long-acting bronchodilators alone for chronic obstructive pulmonary disease (COPD): a systematic review and network meta-analysis. Cochrane Database Syst Rev. 2025;4(4):Cd015997. doi:10.1002/14651858.Cd015997
8. Feldman WB, Suissa S, Kesselheim AS, et al. Comparative effectiveness and safety of single inhaler triple therapies for chronic obstructive pulmonary disease: new user cohort study. BMJ. 2024;387:e080409. doi:10.1136/bmj-2024-080409
9. Varricchi G, Poto R. Towards precision medicine in COPD: targeting type 2 cytokines and alarmins. Eur J Intern Med. 2024;125:28–31. doi:10.1016/j.ejim.2024.05.011
10. Calderon AA, Dimond C, Choy DF, et al. Targeting interleukin-33 and thymic stromal lymphopoietin pathways for novel pulmonary therapeutics in asthma and COPD. Eur Respir Rev. 2023;32(167):220144. doi:10.1183/16000617.0144-2022
11. Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014;69(8):760–765. doi:10.1136/thoraxjnl-2013-204608
12. Gohy ST, Hupin C, Fregimilicka C, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J. 2015;45(5):1258–1272. doi:10.1183/09031936.00135814
13. Ding Y, Wang Z, Zhang Z, You R, Wu Y, Bian T. GLUT3-mediated cigarette smoke-induced epithelial-mesenchymal transition in chronic obstructive pulmonary disease through the NF-kB/ZEB1 pathway. Respir Res. 2024;25(1):158. doi:10.1186/s12931-024-02785-3
14. Mahmood MQ, Reid D, Ward C, et al. Transforming growth factor (TGF) β(1) and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology. 2017;22(1):133–140. doi:10.1111/resp.12882
15. Sohal SS, Mahmood MQ, Walters EH. Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): potential target for prevention of airway fibrosis and lung cancer. Clin Transl Med. 2014;3(1):33. doi:10.1186/s40169-014-0033-2
16. Bracken CP, Goodall GJ. The many regulators of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2022;23(2):89–90. doi:10.1038/s41580-021-00442-x
17. Knight DA, Grainge CL, Stick SM, Kicic A, Schuliga M. Epithelial mesenchymal transition in respiratory disease: fact or fiction. Chest. 2020;157(6):1591–1596. doi:10.1016/j.chest.2019.12.014
18. Brake SJ, Lu W, Chia C, et al. Transforming growth factor-β1 and SMAD signalling pathway in the small airways of smokers and patients with COPD: potential role in driving fibrotic type-2 epithelial mesenchymal transition. Front Immunol. 2023;14:1216506. doi:10.3389/fimmu.2023.1216506
19. Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009;10(1):100. doi:10.1186/1465-9921-10-100
20. Takizawa H, Tanaka M, Takami K, et al. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. 2001;163(6):1476–1483. doi:10.1164/ajrccm.163.6.9908135
21. Erice O, Narayanan S, Feliu I, et al. LAMC2 regulates key transcriptional and targetable effectors to support pancreatic cancer growth. Clin Cancer Res. 2023;29(6):1137–1154. doi:10.1158/1078-0432.Ccr-22-0794
22. Wang D, Keyoumu K, Yu R, et al. Extracellular matrix marker LAMC2 targets ZEB1 to promote TNBC malignancy via up-regulating CD44/STAT3 signaling pathway. Mol Med. 2024;30(1):61. doi:10.1186/s10020-024-00827-6
23. Robinson CJ, Thiagarajan L, Maynard R, et al. Release of miR-29 target laminin C2 improves skin repair. Am J Pathol. 2024;194(2):195–208. doi:10.1016/j.ajpath.2023.11.002
24. Feng J, Li Y, Zhang Y, et al. Endothelium-specific deletion of p62 causes organ fibrosis and cardiac dysfunction. J Transl Med. 2024;22(1):161. doi:10.1186/s12967-024-04946-w
25. Lappi-Blanco E, Kaarteenaho-Wiik R, Salo S, et al. Laminin-5 gamma2 chain in cryptogenic organizing pneumonia and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2004;169(1):27–33. doi:10.1164/rccm.200210-1234OC
26. Yang YC, Zhang N, Van Crombruggen K, Hu GH, Hong SL, Bachert C. Transforming growth factor-beta1 in inflammatory airway disease: a key for understanding inflammation and remodeling. Allergy. 2012;67(10):1193–1202. doi:10.1111/j.1398-9995.2012.02880.x
27. Yang Y, Zhang N, Lan F, et al. Transforming growth factor-beta 1 pathways in inflammatory airway diseases. Allergy. 2014;69(6):699–707. doi:10.1111/all.12403
28. Tam A, Leclair P, Li LV, et al. FAM13A as potential therapeutic target in modulating TGF-β-induced airway tissue remodeling in COPD. Am J Physiol Lung Cell Mol Physiol. 2021;321(2):L377–L391. doi:10.1152/ajplung.00477.2020
29. Amegadzie JE, Mehareen J, Khakban A, Joshi P, Carlsten C, Sadatsafavi M. 20-year trends in excess costs of COPD. Eur Respir J. 2025;65(1):2400516. doi:10.1183/13993003.00516-2024
30. Wang Z, Sun Y. Unraveling the causality between chronic obstructive pulmonary disease and its common comorbidities using bidirectional Mendelian randomization. Eur J Med Res. 2024;29(1):143. doi:10.1186/s40001-024-01686-x
31. Wang Z, Zhang J, Cui H, et al. Comparison of PRISm phenotypes on cardiovascular disease risk and spirometry trajectory: a large prospective cohort study. Respir Med. 2025;243:108137. doi:10.1016/j.rmed.2025.108137
32. Wang Z, Shi J, Liang Y, et al. COPD airway epithelial cells-derived extracellular vesicles contribute to endothelial dysfunction and atherosclerosis via the miR-141-3p/PDCD4 axis. J Nanobiotechnology. 2026;24(1). doi:10.1186/s12951-026-04091-0
33. Zeng J, Xie J, Huang S, Cheng J, Zeng Q. Burden of disease analysis of COPD attributable to occupational PGFs in the BRICS countries, 1990–2021. Int J Chron Obstruct Pulmon Dis. 2026;21:1–31. doi:10.2147/COPD.S543554
34. Higham A, Booth S, Dungwa J, Singh D. Histopathology of the small airways: similarities and differences between ageing and COPD. Pulmonology. 2025;31(1):2430032. doi:10.1080/25310429.2024.2430032
35. Burgel PR. The role of small airways in obstructive airway diseases. Eur Respir Rev. 2011;20(119):23–33. doi:10.1183/09059180.00010410
36. Su X, Chen J, Lin X, et al. FERMT3 mediates cigarette smoke-induced epithelial-mesenchymal transition through Wnt/β-catenin signaling. Respir Res. 2021;22(1):286. doi:10.1186/s12931-021-01881-y
37. Zhu J, Wang F, Feng X, Li B, Ma L, Zhang J. Family with sequence similarity 13 member A mediates TGF-β1-induced EMT in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2021;22(1):192. doi:10.1186/s12931-021-01783-z
38. Gohy ST, Detry BR, Lecocq M, et al. Polymeric immunoglobulin receptor down-regulation in chronic obstructive pulmonary disease. Persistence in the cultured epithelium and role of transforming growth factor-β. Am J Respir Crit Care Med. 2014;190(5):509–521. doi:10.1164/rccm.201311-1971OC
39. Moon YW, Rao G, Kim JJ, et al. LAMC2 enhances the metastatic potential of lung adenocarcinoma. Cell Death Differ. 2015;22(8):1341–1352. doi:10.1038/cdd.2014.228
40. Okada Y, Takahashi N, Takayama T, Goel A. LAMC2 promotes cancer progression and gemcitabine resistance through modulation of EMT and ATP-binding cassette transporters in pancreatic ductal adenocarcinoma. Carcinogenesis. 2021;42(4):546–556. doi:10.1093/carcin/bgab011
41. Kirtonia A, Pandey AK, Ramachandran B, et al. Overexpression of laminin-5 gamma-2 promotes tumorigenesis of pancreatic ductal adenocarcinoma through EGFR/ERK1/2/AKT/mTOR cascade. Cell Mol Life Sci. 2022;79(7):362. doi:10.1007/s00018-022-04392-1
42. Jia Y, Zhang X, Zhao C, et al. miR-212-5p regulates PM(2.5)-induced apoptosis by targeting LAMC2 and LAMA3. Int J Mol Sci. 2025;26(4):1761. doi:10.3390/ijms26041761
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Complex Evaluation of Surfactant Protein A and D as Biomarkers for the Severity of COPD
Lv MY, Qiang LX, Wang BC, Zhang YP, Li ZH, Li XS, Jin LL, Jin SD
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:1537-1552
Published Date: 2 July 2022
Mechanistic Regulation of Wnt Pathway-Related Progression of Chronic Obstructive Pulmonary Disease Airway Lesions
Liu M, Huo Y, Cheng Y
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:871-880
Published Date: 15 May 2023
Abhd2, a Candidate Gene Regulating Airway Remodeling in COPD via TGF-β
Lv MY, Jin LL, Sang XQ, Shi WC, Qiang LX, Lin QY, Jin SD
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:33-50
Published Date: 5 January 2024
Role of Digoxin in Preventing Cigarette Smoke-Induced COPD via HIF-1α Inhibition in a Mouse Model
Zhang K, Zhou F, Zhu C, Yuan L, Li D, Wang J, Lu W
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1665-1678
Published Date: 23 May 2025