Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
Complex Evaluation of Surfactant Protein A and D as Biomarkers for the Severity of COPD
Authors Lv MY ![]() , Qiang LX, Wang BC, Zhang YP, Li ZH, Li XS, Jin LL, Jin SD
, Qiang LX, Wang BC, Zhang YP, Li ZH, Li XS, Jin LL, Jin SD
Received 18 March 2022
Accepted for publication 25 June 2022
Published 2 July 2022 Volume 2022:17 Pages 1537—1552
DOI https://doi.org/10.2147/COPD.S366988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Mei-Yu Lv, Li-Xia Qiang, Bao-Cai Wang, Yue-Peng Zhang, Zhi-Heng Li, Xiang-Shun Li, Ling-Ling Jin, Shou-De Jin
Department of Respiratory Medicine, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, 150001, People’s Republic of China
Correspondence: Shou-De Jin, Department of Respiratory Medicine, The Fourth Affiliated Hospital of Harbin Medical University, No. 37 Yiyuan Street, Nangang District, Harbin, 150001, People’s Republic of China, Tel/Fax +86 0451-85939123, Email [email protected]
Purpose: Pulmonary surfactant proteins A (SP-A) and D (SP-D) are lectins, involved in host defense and regulation of pulmonary inflammatory response. However, studies on the assessment of COPD progress are limited.
Patients and Methods: Pulmonary surfactant proteins were obtained from the COPD mouse model induced by cigarette and lipopolysaccharide, and the specimens of peripheral blood and bronchoalveolar lavage (BALF) in COPD populations. H&E staining and RT-PCR were performed to demonstrate the successfully established of the mouse model. The expression of SP-A and SP-D in mice was detected by Western Blot and immunohistochemistry, while the proteins in human samples were measured by ELISA. Pulmonary function test, inflammatory factors (CRP, WBC, NLR, PCT, EOS, PLT), dyspnea index score (mMRC and CAT), length of hospital stay, incidence of complications and ventilator use were collected to assess airway remodeling and progression of COPD.
Results: COPD model mice with emphysema and airway wall thickening were more prone to have decreased SP-A, SP-D and increased TNF-α, TGF-β, and NF-kb in lung tissue. In humans, SP-A and SP-D decreased in BALF, but increased in serum. The serum SP-A and SP-D were negatively correlated with FVC, FEV1, FEV1/FVC, and positively correlated with CRP, WBC, NLR, mMRC and CAT scores (P < 0.05, respectively). The lower the SP-A and SP-D in BALF, the worse the lung function and the increased probability of complications and ventilator use. Moreover, the same trend emerged in COPD patients grouped according to GOLD severity grade (Gold 1– 2 group vs Gold 3– 4 group). The worse the patient’s condition, the more pronounced the change.
Conclusion: This study suggests that SP-A and SP-D may be related to the progression and prognostic evaluation of COPD in terms of airway remodeling, inflammatory response and clinical symptoms, and emphasizes the necessity of future studies of surfactant protein markers in COPD.
Keywords: COPD, pulmonary surfactant protein A and D, airway remodeling
Introduction
Chronic obstructive pulmonary disease (COPD), which has been considered by many scholars for years as a heterogeneous disease caused by multiple factors, is the third leading cause of death in the world and characterized by chronic airway inflammatory infiltration and destruction of normal alveolar structural integrity.1,2 As the disease progresses, the initiation of inflammatory mechanism and immune system can destroy the structural integrity of the airway and alveolar cells by activating inflammatory cells and promoting the release of a large quantity of cytokines and inflammatory mediators,3–6 which affects the secretion of pulmonary surfactant proteins (SPs).7,8 This not only aggravates the formation of emphysema and the progress of COPD but also promotes the occurrence and mortality of inflammatory response in patients with COPD.
SPs, which are secreted by alveolar cells and account for 8–10% of pulmonary surfactants, can be divided into four types and named A, B, C, and D according to different structures and molecular weights. In particular, pulmonary surfactant protein A (SP-A) and D (SP-D), which act as defensive guards against airway infection, can activate macrophages and phagocytes by virtue of their hydrophilicity, broad specificity and carbohydrate recognition domain that can bind to microorganisms. Therefore, they could exert the functions of antibacterial and elimination of apoptotic cells, and participate in the process of immune function, inflammatory response and lipid metabolism at the same time.9 At present, some studies have confirmed that SP-A and SP-D can establish a relationship with COPD in both animal and human studies, but the specific connection and the significance of diagnosis and treatment to the disease in the future are still not clear.
Mountains of evidence have proved that the pulmonary emphysema model mice had the following characteristics, such as disorder of phospholipid metabolism on the surface of alveoli, a tendency to decrease SP-A and SP-D expression, and infiltration and aggregation of alveolar macrophages in the airways. These suggested that SP-A and SP-D, which are extremely likely, are involved in the pathogenesis of COPD and have some representative significance and potential value in it.10 What is more, some scholars found that SP-D-deficient mice tend to spontaneous emphysema formation due to abnormal changes in alveolar structure, while macrophages from SP-A and SP-D gene-null mice were more likely to express reactive oxygen species highly.11 Meanwhile, in human studies, SP-A and SP-D have been found to be related to smoking and even to gene polymorphism in the pathogenesis of COPD.12–14 However, relevant studies are still imperfect, especially the significance of SP-A or SP-D in evaluating the severity of illness in patients with Acute Exacerbation of COPD (AECOPD). In addition, the characteristics exhibited by SP-A or SP-D in different types of specimens are unclear.
Based on the above, this study focused on evaluating the effects of SP-A and SP-D on COPD progression. We sought to investigate (1) SP-A and SP-D levels in cigarette and lipopolysaccharide-induced model mice by simulating the pathogenesis of COPD in humans, (2) the relationship between SP-A and SP-D expression and COPD progression by linking pulmonary function, inflammatory parameters, and clinical manifestations in clinical studies, and (3) differences in SP-A or SP-D in serum and BALF collected by standardized operation.
Materials and Methods
Animal Experiments
C57BL/6 male mice (8–12 weeks old, weighing 18-25g) were obtained from the Laboratory Animal Center of Peking University, and maintained in SPF condition. Animal experiments were carried out in accordance with the Chinese Association for Laboratory Animal Science Policy and approved by the ethics committee of the Fourth Affiliated Hospital of Harbin Medical University. The mice were randomly divided into 2 groups as follows: 1) control (CTL) (n = 5), smoking-lipopolysaccharide (LPS) (SM) (n = 5) groups. The commercial cigarettes (Huangguoshu brand, Guizhou, China), containing 11 mg tar, 0.8 mg nicotine and 13 mg CO, were used in this study to construct the mouse model.15 All mice in SM group were exposed to smoking (10 cigarettes/day, 30 min/time) in a self-made plexiglass box (50 × 40 × 30 cm) at regular daily intervals except on days 7, 14, and 21 due to intratracheal instillation of LPS (1 mg/kg) without smoking, while the control mice did not receive any treatment. The mice were sacrificed 24 hours after the last exposure to smoke. Lung tissue was collected from two groups at day 30 for subsequent studies.
Tissue Collection
After anesthesia, the thoracic cavity of mice was opened to carefully separate the lung tissue and airway, rinsed with 0.9% sodium chloride solution at 4°C, and the left lung was rapidly frozen in liquid nitrogen for RT-PCR and Western blot. The right lungs and airways were fixed in 4% paraformaldehyde for 24 h, and routinely paraffin embedded for HE staining and Immunohistochemistry, as described in previous studies.16,17
H&E and Immunohistochemistry
HE staining was used to observe the destructive changes in airway and lung structure in mice. Three paraffin sections were taken from each lung tissue. Three fields were randomly read from each section. The cross was drawn under a light microscope (100X) to calculate the total length of the cross (L) and the number of alveolar septa with cross shadow (NS). The mean linear intercept (MLI) was calculated using the formula MLI = L/NS and the mean alveolar septal thickness (MAST) was measured. Immunohistochemistry was used to observe the expression of SP-A and SP-D. The IHC Profiler automated analysis software was used in this study to assess the grade of positive staining of immunohistochemical sections, and a score of greater than or equal to 1 was scored as positive. The positive rates of SP-A and SP-D were calculated by taking five areas under a light microscope (400X) for each section and counting the number of alveolar type II cells (ATII) positive for SP-A or SP-D and the total number of ATII.
mRNA Extraction and Real-Time PCR
RNA was extracted by Trizol (Invitrogen) and quantified using a NanoDrop spectrophotometer (NanoDrop Tech, Rockland, DE, USA). cDNA was obtained using ReverTra Ace qPCR RT Kit (TYB-FSQ-101, Toyobo, Shanghai, China) according to the instructions. Quantitative real-time PCR was conducted by using FastStart ™ Universal SYBR ® Green premix (Rox) (Sigma-Aldrich, USA) on a Real-Time PCR machine (Bio-Rad). Gene-specific primers were provided by Thermo Scientific as follows: Mouse TGF-β, forward, 5′-TACAGGGCTTTCGATTCAGC-3’ and reverse, 5′-CGCACACAGCAGTTCTTCTC-3’; Mouse NF-κB, forward, 5′-GCTACACAGAGGCCATTGAA-3’ and reverse, 5′-TCCCGGAGTTCATCTATGTG-3’; Mouse TNF-α, forward, 5′- CCAGACCCTCACACTCAGAT-3’ and reverse, 5′-AACACCCATTCCCTTCACAG-3’; GAPDH, forward, 5′-CACCCACTCCTCCACCTTTG-3’ and reverse, 5′-CCACCACCCTGTTGCTGTAG-3′. GAPDH was treated as an internal control.
Protein Extraction and Western Blot
The protein was extracted by grinding the lung tissue with liquid nitrogen, and the protein concentration was determined using a BCA kit (Beyotime, Shanghai, China). 50 ug protein samples were electrophoresed with SDS-PAGE, transferred to PVDF membranes (Millipore), blocked with 5% skim milk, and finally subjected to immunohybridization (anti-SP-A antibody, ab115791, Abcam; anti-SP-D antibody, ab220422, Abcam; anti-GAPDH antibody, K200057M, Solarbio; HRP conjugated Goat Anti-Mouse IgG, GB23301, Servicebio; HRP conjugated Goat Anti-Rabbit IgG, GB23303, Servicebio) and image acquisition.
Patients
All clinical samples from people without lung disease (Group 1) and with different airway obstructive diseases considered by physicians to be in the acute phase of infection including acute exacerbation of chronic bronchitis (AECB) (Group 2) and AECOPD (Group 3) were collected from April 2019 to January 2022 and approved by the Fourth Affiliated Hospital of Harbin Medical University. The informed consent was obtained from all participants (40–78 years). Groups 1 and 2 served as controls, and people in Group 3 were further divided into Gold 1–2 group (FEV1% pred ≥50%) and Gold 3–4 group (FEV1% pred <50%) based on the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines,18 patients’ clinical symptoms, pulmonary function and other auxiliary examinations.
Exclusion criteria: (1) Patients with tumors, autoimmunity and other respiratory infectious diseases, such as bronchiectasis, bronchial asthma, and tuberculosis, etc. (2) Patients with primary severe cardiovascular and cerebrovascular diseases or severe liver and kidney damage. (3) Patients had received drug therapies, such as antibiotics, glucocorticoids, and so on, before the collection of peripheral blood samples.
Data Source
Peripheral blood laboratory parameters, including white blood cells (WBC), C-reactive protein (CRP), platelets (PLT), procalcitonin (PCT), eosinophil (EOS), neutrophil-to-lymphocyte ratio (NLR), SP-A and SP-D, were collected before patients received any treatment within 24 hours of admission. Additionally, SP-A and SP-D in BALF were obtained by a professional clinician via bronchoscope alveolar lavage when participants’ vital signs were stable. Furthermore, there were other clinical data, including blood gas analysis, clinical symptom assessment questionnaires containing modified Medical Research Council (mMRC) score and COPD assessment test (CAT) score for patients in Group 2 and 3 in medical history, and pulmonary function test (FEV1, FVC, FEV1/FVC) recorded by MedGraphics Profiler spirometer. The use of ventilators, complications, and length of hospital stay were recorded after the patients were discharged, and the time of rehospitalizations was within three years. This work did not intervene in the individualized clinical treatment of patients.
Measurement of Human Surfactant Proteins
3–5 mL of peripheral blood was collected, followed by natural clotting for 20 min at room temperature to separate the serum. The supernatant was obtained by centrifugation at 2500 rpm for 20 min at 4°C using a high-speed refrigerated centrifuge (TGL-16M, China) and stored at –80°C. Correspondingly, the collection of SP-A and SP-D in BALF was accomplished by bronchoalveolar lavage. Under aseptic conditions, a sterilized physiological saline at 37°C was injected into the side of the endoscope with a syringe, and BALF (50 mL × 3) was aspirated from the bronchi (lobar bronchi, segmental bronchi, and subsegmental bronchi) at a negative pressure of 100 mmHg, with a recovery rate over 40%. After filtering out the impurities in the specimen with sterile gauze, the supernatant was obtained by layering at room temperature for 20 min, followed by centrifugation. Centrifugation and storage conditions were the same as above.
SP-A and SP-D in plasma and BALF were detected according to the instructions of the ELISA kit (Shanghai Jianglai Biotechnology Co., Ltd.) and analyzed with a microplate reader at 450 nm (Synergy HTX, BioTek Instruments). The final concentrations of surfactant proteins were calculated based on the standard curve and OD values.
Statistical Analysis
All the data were analyzed using statistical analysis software (IBM SPSS Statistics V25). Quantitative data were normally distributed using one-way ANOVA with Bonferroni’s multiple comparison test or t-test, and non-normally distributed using Kruskal–Wallis test or Mann–Whitney test. Qualitative data were analyzed by chi-square test. Spearman’s rank correlation coefficient (SR) was used to evaluate the correlation between variables. Graphics were drawn using Prism 8 software. P < 0.05 indicated a statistically significant difference.
Results
Evaluation of the Smoking-LPS Induced COPD Model
All mice were included in this analysis. After a 30-day experimental period, mice in the SM group had worse mental status, increased hair loss, decreased food intake, and less weight gain than mice in the CTL group. Especially on day 30, there was a significant difference in the rate of weight growth of two groups of mice (P = 0.041, 95% CI 0.68–25.92; Figure 1A and B).
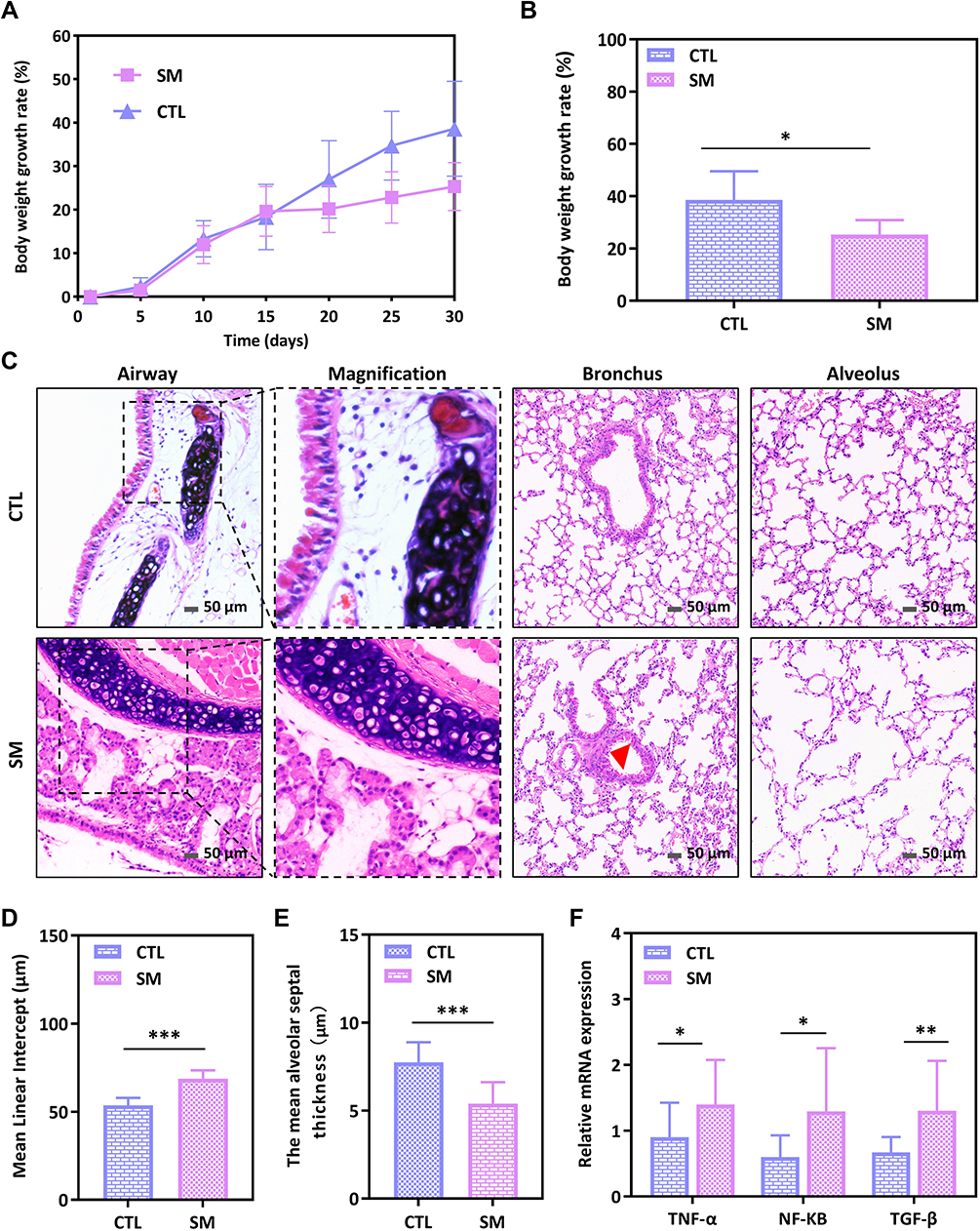 |
Figure 1 Smoking-LPS induced changes in body weight and lung tissue of mice. (A) Weight growth rate over the course of the experiment in two groups. (B) Comparison of weight growth rate of mice on the 30th day. (C) H&E staining showed airway remodeling and destruction of alveolar structure induced by smoking-LPS (200x). Red triangle referred to thickened smooth muscle. Scale bar: 50 μm. (D and E) Comparison of MLI and MAST of two groups. (F) Relative expression of different inflammatory factor mRNA in lung tissues of mice. *P<0.05, **P<0.01, ***P<0.001. |
Representative HE stained images of lung tissue from each group of mice are shown in Figure 1C. In the CTL group, the structure of bronchi, epithelium and alveoli was intact, and the cilia were arranged neatly. Compared with the control group, the SM group had increased airway wall thickness and emphysema-like pathological changes, similar to the population with COPD. The characteristics of airway epithelial cells and cilia shedding, bronchial smooth muscle and collagen fiber proliferation, submucosal glandular hyperplasia, alveolar structure destruction, alveolar wall thinning, smaller MAST, and enlarged MLI (Figure 1D and E) in SM group indicated that the mouse COPD model was successfully established. Moreover, we detected the expression of inflammatory factors in lung tissue by real-time PCR and found that the mRNA expression of TNF-α (P = 0.033), NF-kb (P = 0.017) and TGF-β (P = 0.006) was upregulated in the SM group of mice, suggesting that COPD model mice showed altered pulmonary secretory function (Figure 1F).
SP-A and SP-D Expression in the Mice Lung Tissue
The expression of SP-A and SP-D proteins in lung tissue was evaluated by immunohistochemistry and Western blot, and the result showed that SP-A and SP-D were significantly decreased in the SM group (Figure 2A, C, E and F). The positive rates of SP-A (100% vs 60%, P = 0.017) and SP-D (100% vs 66.67%, P = 0.042) section staining in the CTL group and SM group were calculated to be significantly different by Fisher’s exact method. To more accurately examine the effect on SPs secreted by ATII in the COPD model, we calculated the proportion of SP-A (Figure 2B) or SP-D (Figure 2D) positive pneumocytes II to total II pneumocytes, and the results showed the same trend. The median proportion of SP-A in the CTL and SM groups was 0.78 (0.71, 0.81) and 0.63 (0.58, 0.67), respectively; the median proportion of SP-D in the CTL and SM groups was 0.73 (0.69, 0.77) and 0.65 (0.60, 0.68), respectively, and the difference was statistically significant (Z = −8.023, P < 0.001; Z = −7.613, P < 0.001).
 |
Figure 2 Expression of SP-A and SP-D in lung tissue of mice. (A and C) Representative immunohistochemical images showed reduced SP-A and SP-D expression in the SM group (400x). The black arrow points to the positive expression of pulmonary surfactant protein staining. Scale bar: 20 μm. (B and D) Ratio of SP-A or SP-D positive pneumocytes II to total II pneumocytes decreased in SM group. (E and F) Western blot analysis of lung tissue lysates showed that the relative expression of SP-A and SP-D decreased in COPD model. ***P<0.001. |
Basic Information in Humans
One hundred and twenty-five individuals were eventually incorporated, including 85 cases (COPD, Group 3) serving as the observation group and 40 subjects (Health-control, Group 1; AECB, Group 2) serving as controls. Patients in Group 3 were further divided into two groups based on GOLD guidelines: Gold 1–2 group (FEV1% pred ≥50%, n = 50) and Gold 3–4 group (FEV1% pred <50%, n = 35). All subjects included completed pulmonary function tests. Experimental process and baseline characteristics of participants were presented in Figure 3 and Table 1. There was no significant difference in age, sex, BMI, smoking history and smoking index among the three groups (P > 0.05).
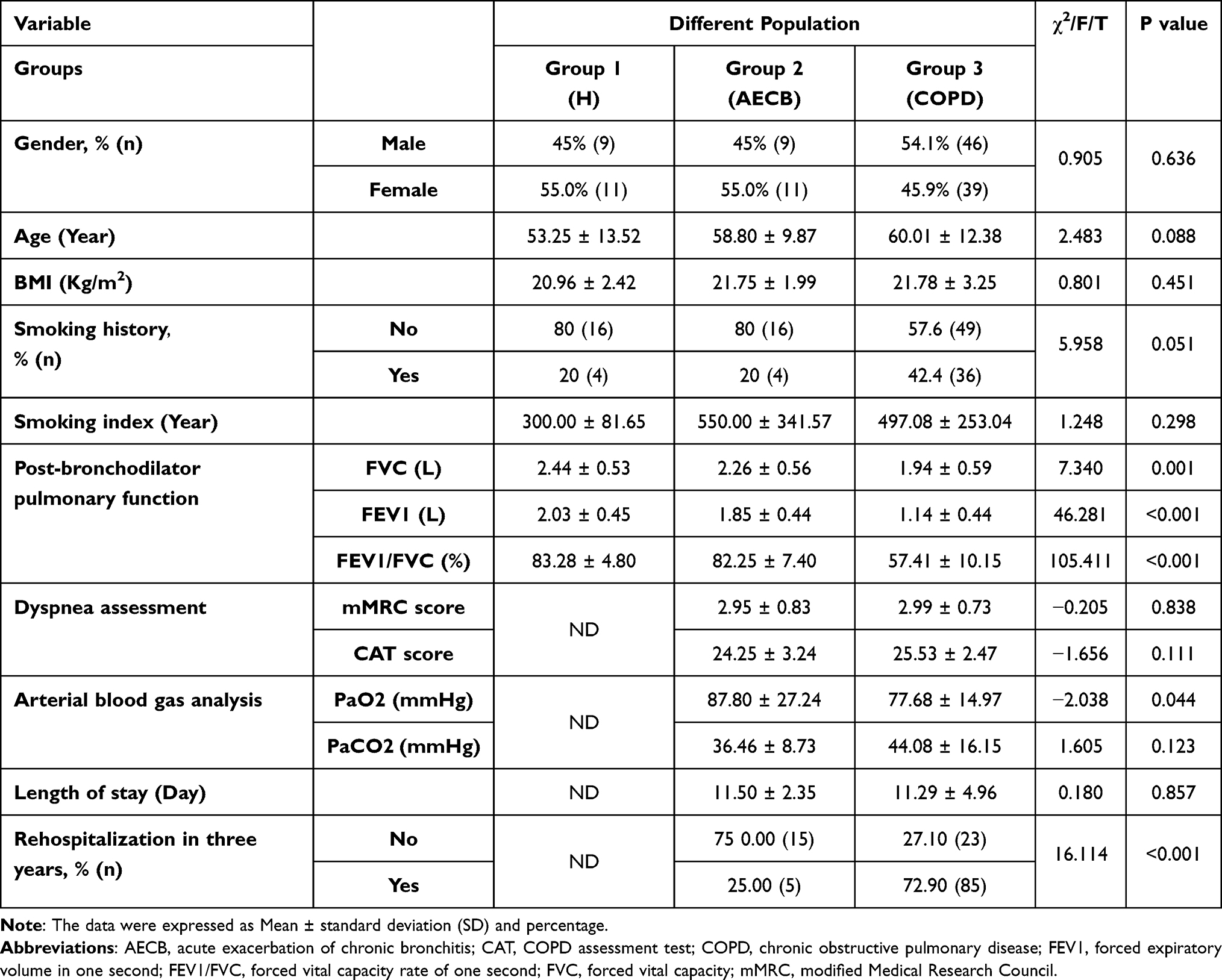 |
Table 1 Baseline Characteristics of the Participants in Three Groups |
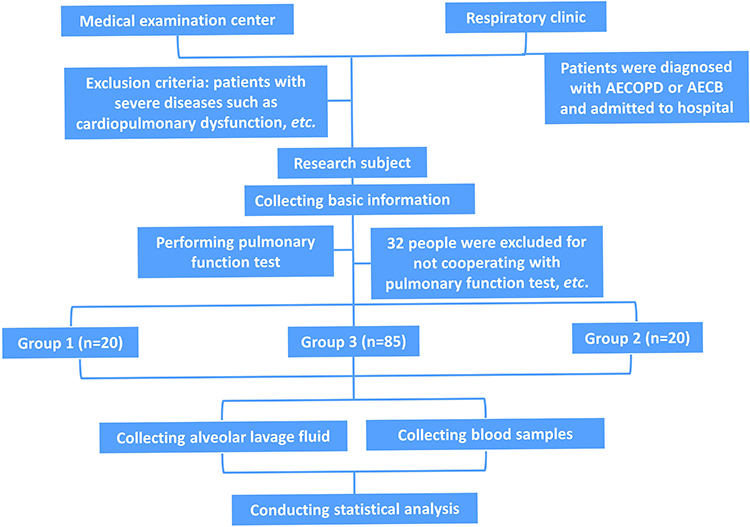 |
Figure 3 Schematic illustration of the clinical experimental process. |
Comparisons of SP-A and SP-D in Different Groups
SP-A and SP-D in serum and BALF were significantly different among the three groups by ANOVA analysis (Table 2). Bonferroni’s multiple comparisons revealed that serum SP-A and SP-D levels were significantly increased in Group 3 compared with Group 1 (P < 0.05); serum SP-A was lower, but serum SP-D was significantly increased in Group 3 compared with Group 2 (P < 0.05). However, the opposite trend was found for these proteins in BALF. SP-A and SP-D in Group 3 were not as high as those in Group 1 and 2, especially SP-D which showed a significant downward trend (P < 0.05; P < 0.05).
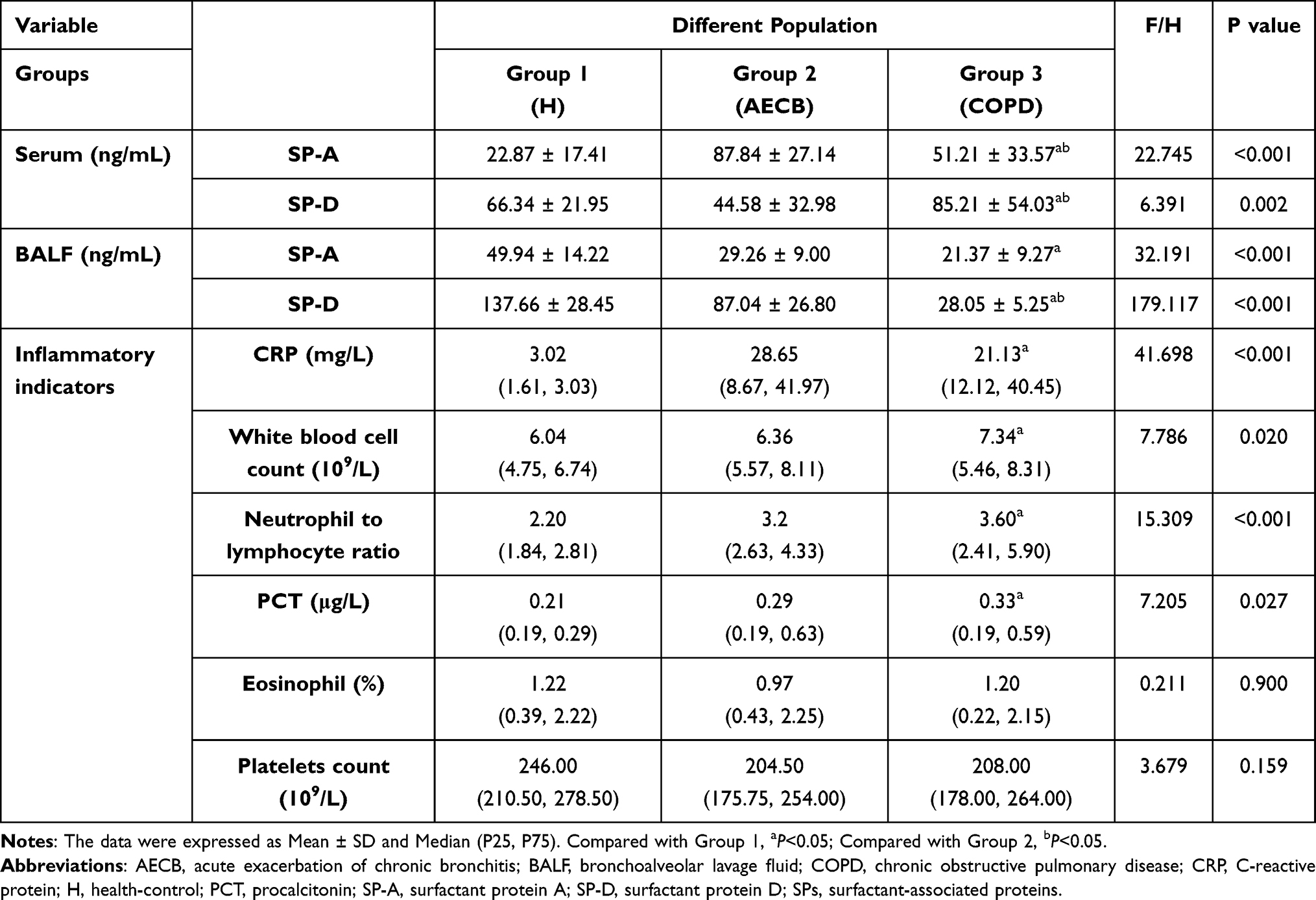 |
Table 2 Laboratory Parameters in COPD Group and Control Groups |
In Group 3, SP-A was found to be lower than SP-D both in serum (P < 0.01) or BALF (P < 0.001). Furthermore, SP-A was higher in serum than in BALF, and a similar trend can be observed in the comparison of SP-D, and the differences were statistically significant (P < 0.001; P < 0.01) (Figure 4A). To investigate the potential relationship between the surfactant proteins and severity of COPD progression, we compared COPD patients in Gold 1–2 group and Gold 3–4 group. The data showed higher SP-A and SP-D in serum but lower SP-A and SP-D in BALF in the Gold 3–4 group compared with Gold 1–2 group, which implies that it was related to the severity and airflow limitation of COPD (P < 0.05, respectively) (Table 3, Figure 4B and C).
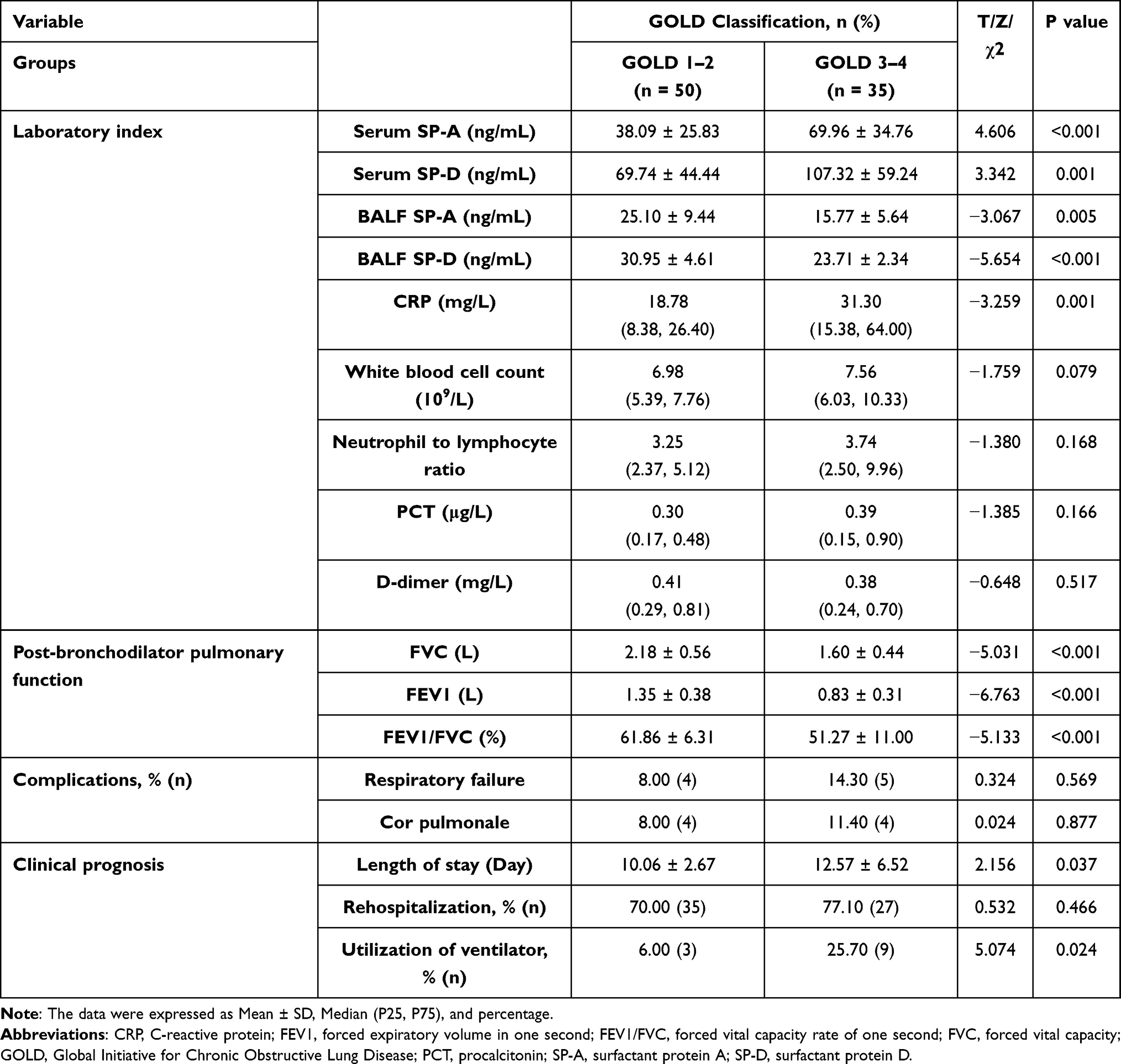 |
Table 3 Indexes of Inflammation and SP-A or SP-D in Different Stages of COPD |
 |
Figure 4 Concentrations of pulmonary surfactant proteins in AECOPD. (A) Comparisons of SP-A and SP-D in serum and BALF. (B and C) Comparisons of SP-A and SP-D in different stages of AECOPD. **P<0.01, ***P<0.001. |
Analysis of SP-A and SP-D to Assess COPD Severity
Our assessment of the ability of pulmonary surfactant protein to reflect the progression and prognosis of COPD was mainly based on inflammatory parameters, pulmonary function parameters, dyspnea score, blood gas analysis, length of hospital stay, hospitalization rate, incidence of complications, and ventilator use.
In terms of peripheral blood infection and inflammation, the indicators in Group 3 and Group 2 were obviously abnormal, while those of Group 1 were within the normal range. After multiple comparisons, the CRP, WBC, NLR, and PCT values in Group 3 were obviously higher than those in Group 1 (P < 0.05, respectively), and not significantly different from those in Group 2 (P > 0.05). There was no significant difference in EOS and PLT among the three groups (P > 0.05). CRP was significantly higher in the Gold 3–4 group than in the Gold 1–2 group (P < 0.05) (Tables 2 and 3).
Pulmonary function parameters, including FVC, FEV1 and FEV1/FVC (%), were significantly different among the three groups, while the degree of airflow limitation was more severe in the population of the GOLD 3–4 group (P < 0.05). Compared with Group 2, Group 3 displayed a lower value of PaO2 in arterial blood gas and a higher rate of rehospitalization within 3 years, suggesting a worse prognosis for COPD patients (P<0.05), although the differences in respiratory symptom severity (mMRC score and CAT score) and length of hospital stay between the two groups were not significant (P > 0.05). In patients with COPD, the ventilator use and length of hospital stay were significantly higher in the GOLD 3–4 group than in the GOLD 1–2 group (P < 0.05), but there was no significant difference in complications and the rate of rehospitalization within 3 years (P > 0.05) (Tables 1–3).
Then, this study analyzed their correlation in COPD (Figure 5A and B, Table 4). The correlation analysis results showed that there existed a positive correlation between serum SP-A or SP-D and numerous inflammatory indicators measured, of which CRP had the largest correlation coefficient (r = 0.621, P < 0.01; r = 0.612, P < 0.01). Similarly, serum SP-A (r = −0.481; r = −0.598; r = −0.545) or SP-D (r = −0.451; r = −0.480; r = −0.296) was negatively correlated with FVC, FEV1 or FEV1/FVC (%), which meant that the higher the level of two proteins, the more severe the patients’ airway limitation. SP-A or SP-D in BALF was positively correlated with FVC or FEV1, verifying the protective effect of the proteins on the lungs. However, no significant correlation was found between SP-A or SP-D in BALF and inflammatory indexes (P > 0.05).
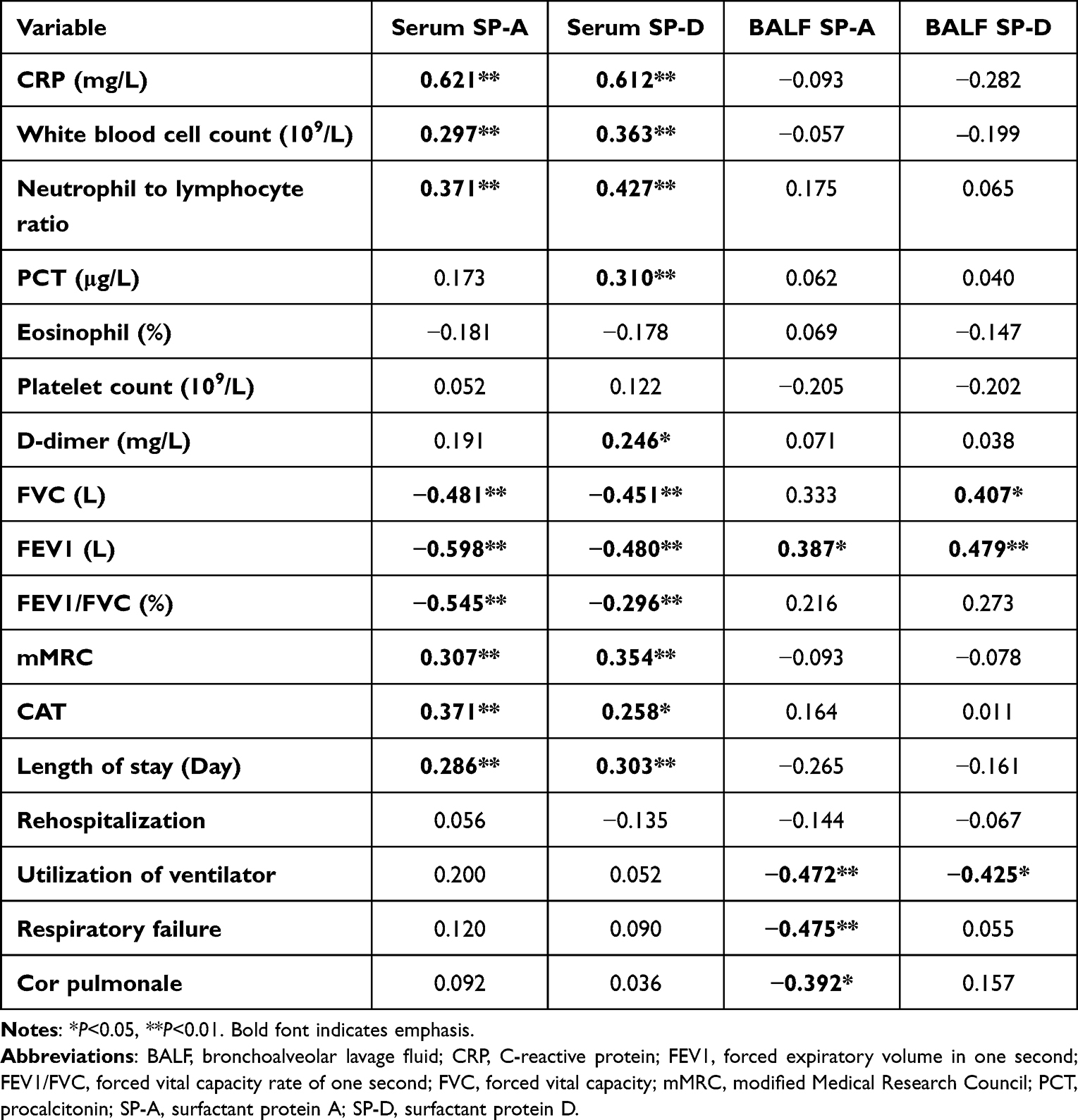 |
Table 4 Correlation Between Clinical Data and SP-A or SP-D in COPD |
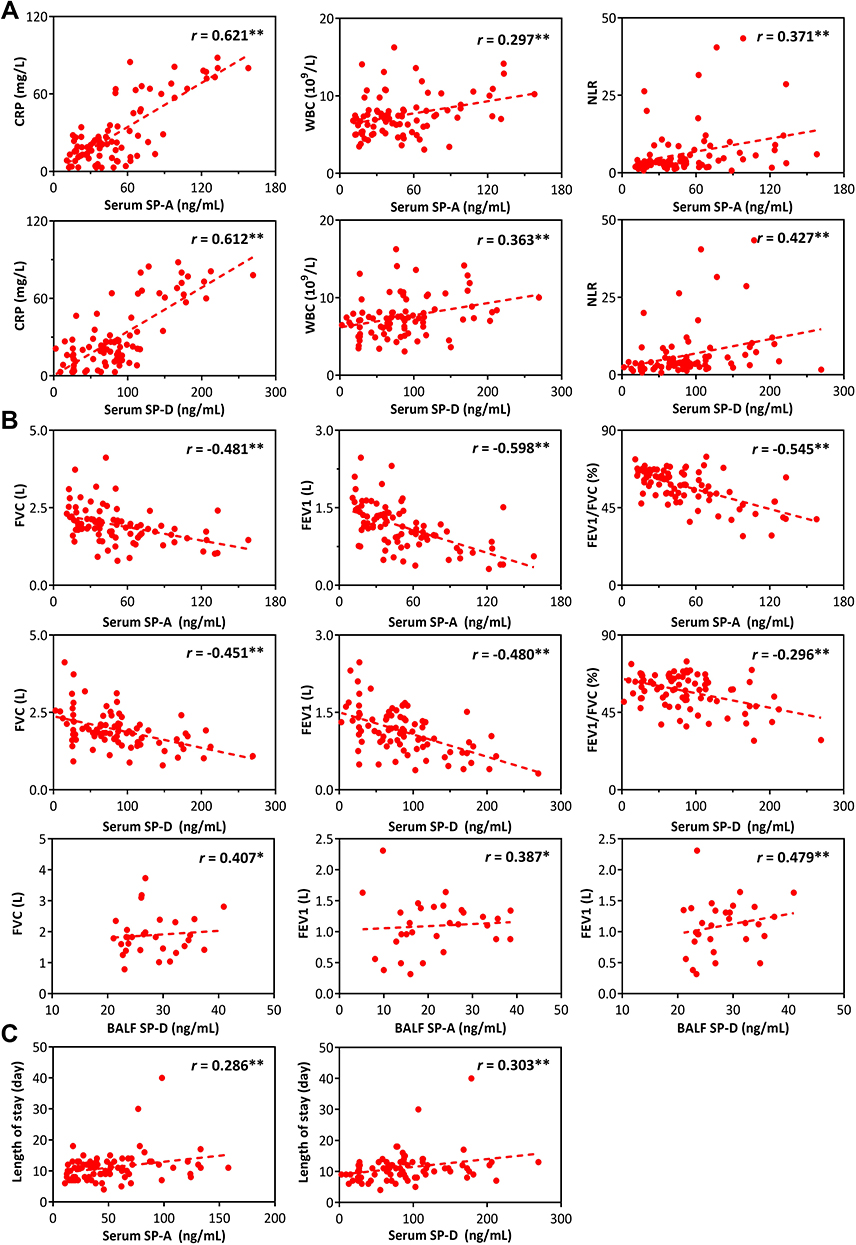 |
Figure 5 Correlation analysis between pulmonary surfactant protein and clinical indexes in COPD. (A) Multiple inflammatory indicators including CRP, WBC, NLR were positively related to serum SP-A and SP-D. (B) A number of pulmonary function parameters were negatively correlated with SP-A and SP-D in serum, but positively correlated with SP-A and SP-D in BALF. (C) The concentration of serum SP-A and SP-D can indicate the length of hospitalization. *P<0.05, **P<0.01. |
In addition, we compared SPs with the dyspnea symptom questionnaire scores (mMRC and CAT scores) and hospitalization data, and found similar trends, ie, the more severe the clinical symptoms, the higher the serum protein level, suggesting that SP-A or SP-D in serum could be used as an index to assess the character of COPD progression (Table 4). In BALF, SP-A or SP-D was negatively correlated with ventilator use and complications, such as cor pulmonale and respiratory failure, suggesting that COPD prognosis could be worse when SPs were lower in BALF (Figure 5C, Table 4).
Discussion
SP-A and SP-D, as natural barriers for respiratory defense, can be involved in multiple pathogenesis of COPD, such as exerting immunoregulatory and anti-infective effects by virtue of hydrophilicity and special molecular structure. Many studies have shown that long-term repeated stimulation of alveolar epithelial cells by inflammatory factors in COPD affects the secretion of SP-A and SP-D, thereby reducing airway defense and increasing the risk of recurrent infections and disease attacks.19–23 In the prophase animal experiment, domestic and foreign scholars have confirmed that SP-A and SP-D gene deletion do have a significant link with the development of emphysema-like changes.11,24 However, in human studies, the role of SP-A and SP-D for the assessment of COPD is still unknown. To our knowledge, this study is the first to analyze SP-A and SP-D in peripheral blood and BALF from patients diagnosed as COPD, chronic bronchitis and healthy people by combining inflammatory parameters, pulmonary function, and dyspnea index scores to investigate the severity of illness in the process of exacerbation of COPD.
Some interesting findings were listed in this study. First, we demonstrate that cigarette was not the only contributing factor to SPs in COPD. According to WHO statistics, the incidence of COPD can reach more than 50% in smokers,25 and cigarettes are one of the recognized etiological factors. In this study, SP-A and SP-D protein expression decreased in the lung tissue of a cigarette-induced mouse COPD model, but there was no significant difference in smoking metrics on clinical statistics, suggesting that cigarettes were not the only factor affecting SP-A and SP-D. Cigarettes, as an important cause of COPD, can directly destroy the structure of SPs by themselves, and can also enhance the destruction of alveolar epithelial cells by causing the initiation of inflammatory response and oxidative stress, increase the probability of recurrent airway infection, and affect SPs expression, thereby promoting the progression of COPD.
Subsequently, our study confirmed that SP-A and SP-D were associated with infection and inflammation in the period of exacerbation of COPD. In the lung tissue of COPD model mice, SP-A and SP-D expression decreased, accompanied by an increase in TGF-β, TNF-α and NF- kb and airway mucus hypersecretion. Normally, SP-A and SP-D can play an anti-infective role by chemotaxis and binding macrophages for the clearance of microorganisms and tissue necrosis. Abnormal SPs secretion can induce disturbances in the inflammatory system in vivo by affecting the NF-KB signaling pathway and TNF-α release, and increasing the risk of infection. In our clinical studies, a close relationship between SPs and COPD inflammation was demonstrated by comparing the concentrations of SPs in serum or BALF and inflammatory parameters of different populations and performing correlation analysis. Among them, the result that the serum SP-A and SP-D in AECOPD patients were higher than those in healthy individuals was consistent with the research of many scholars at home and abroad.26 The more severe the inflammatory response, the serum SP-A and SP-D levels. However, SP-A and SP-D were markedly reduced in BALF, which was considered to be possibly related to alveolar structural damage caused by inflammatory amplification. The body was susceptible to infection when pulmonary surfactant proteins were low, and lung tissue structure can be destroyed by the pathogen itself or by the inflammatory factors produced. The more severe the inflammatory response of patients with COPD, the greater the damage to the alveolar structure. At the same time, as pulmonary capillary permeability increased, the protective proteins in peripheral blood would increase to further participate in the process of inflammation and exert immune regulation.
Thirdly, SP-A and SP-D expressions in BALF of adults were not static. At present, there are few studies on the measurement of SP-A and SP-D in BALF in lung diseases in humans, especially the expression changes in people of different ages. Generally, it is thought that SP-A levels are consistently higher than SP-D in BALF, as alveolar protein synthesis has been reported to stabilize at gestational age 32, but it remains controversial in children and adults,27 and the measurements we observed in our experiment are the opposite. In this study, we found that the concentration of SP-D in BALF was higher than that of SP-A, which was consistent with the study of Wang et al.28 This may be related to the fact that the synthesis and secretion of SP-D in humans follow SP-A with age as well as airway mucosal damage and secretory dysfunction caused by traction of the alveoli after birth and stimulation of AECII by external harmful substances, such as allergens and smoke, which needs further research. SP-A and SP-D differ in their ability and species to bind pathogens, as do their mechanisms of microbial clearance. It should be noted that SPs in BALF come from the respiratory tract and can be used to reflect the pathological state of alveoli, which is worthy of future attention and exploration.
Fourthly, this study demonstrated that SP-A and SP-D could reflect the degree of airflow limitation in patients with COPD by establishing a link with pulmonary function, which is one of the markers to assess the severity of COPD. Currently, there still remains controversial about the relationship between SPs and the degree of airflow limitation. For example, many scholars at home and abroad have reached different conclusions such as negative or no obvious correlation between serum SP-D and FEV1%pred, which may be caused by various factors such as region, race, DNA, number of participants, and so on.14,29–34 Notably, there were obvious differences in pulmonary function between COPD patients and healthy people. Our findings confirmed that both SP-A and SP-D in serum and BALF were related to pulmonary function, and some of these results were consistent with those of Ju et al.35,36 This conclusion was based on the results of the re-grouping analysis of data from the COPD population according to GOLD guidelines and the correlation analysis between pulmonary function and SPs in the COPD population. Clinically, pulmonary function parameters can provide information on abnormal changes in air and metabolites in the alveoli, especially when the alveolar tension and blood gas barrier regulated by pulmonary surfactant are destroyed, indirectly reflecting airway remodeling. At the same time, we compared patients with COPD and those with chronic bronchitis. The results showed that SP-D was significantly decreased in BALF but significantly increased in serum in hypoxic COPD patients, suggesting that SP-D was more closely related to alveolar structure destruction and COPD pathogenesis. COPD can be understood as an advanced stage of chronic bronchitis progression, characterized by airflow obstruction and alveolar destruction, although the clinical symptoms of the two populations are often very similar. The predominance of SP-D in COPD may be related to the severity and higher sensitivity of patients’ airflow limitation. The above results indicate that SP-A and SP-D in both serum and BALF are useful for clinical assessment of the degree of airflow limitation in COPD and can be used as clinical auxiliary indicators to determine disease progression.
Additionally, a similar pattern can also be obtained when analyzing the relationship between serum SP-A or SP-D and Dyspnea Questionnaire (mMRC and CAT) scores, which are indicators for evaluating the severity of clinical symptoms in COPD patients. In BALF, lower SP-A and SP-D were often associated with high ventilator use and complications, suggesting a poor prognosis for the disease. This further validated our previous conclusions. Our study proposed that SP-A and SP-D can be used as biomarkers to evaluate disease severity and prognosis.
However, there were still some limitations in this work. First, it is clinically a single-center small-sample study with a limited number of samples affected by the outbreak. Second, the collection of SP-A and SP-D in BALF and serum was performed at a single time point due to limitations in patient cooperation and clinical management. In the next step, at the cellular level, we will investigate the relationship between pulmonary surfactant protein and inflammation. At the same time, patient will be followed up to compare SP-A and SP-D in the acute and stable phases of COPD, and carry out a multicenter large-sample study.
In summary, this study demonstrated abnormal SP-A and SP-D expression in COPD at the animal level, and showed in clinical studies that SP-A and SP-D can assess the severity and prognosis of COPD progression, which needs further study. SP-A and SP-D, as regulatory proteins of pulmonary inflammation and immunity, were closely related to the inflammatory response in the process of COPD based on characteristics of certain specificity, and could reflect the degree of airflow limitation and predict the future course of patients. If one can correctly assess the severity and prognosis of the disease in combination with SP-A and SP-D, COPD patients will be able to obtain appropriate airway management, appropriate individualized clinical treatment, and timely preventive measures.
Conclusion
SP-A and SP-D are abnormally expressed in COPD and are able to assess the progression and prognosis of COPD. When SP-A and SP-D are low in the alveoli, the body is prone to infection, airflow limitation, increased probability of complications and ventilator use. Serum SP-A and SP-D can reflect the inflammatory response status and the degree of dyspnea. The more severe the course of the disease, the more significant the changes in pulmonary surfactant protein.
Data Sharing Statement
The data/documents could be obtained from the corresponding author.
Ethics Approval
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study was approved by the institutional ethics committee of the Fourth Affiliated Hospital of Harbin Medical University (2022-SCILLSC-01) and was conducted according to the Declaration of Helsinki and informed consent was given by all patients.
Informed Consent
The information involved in the article has obtained all the patient’s oral informed consent.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81670028) and the Graduate Practice Innovation Project Fund of Harbin Medical University (2018182).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1(1):15076. doi:10.1038/nrdp.2015.76
2. Borg M, Thastrup T, Larsen KL, Overgaard K, Hilberg O, Løkke A. Free diving-inspired breathing techniques for COPD patients: a pilot study. Chron Respir Dis. 2021;18:14799731211038673. doi:10.1177/14799731211038673
3. Hogea SP, Tudorache E, Fildan AP, Fira-Mladinescu O, Marc M, Oancea C. Risk factors of chronic obstructive pulmonary disease exacerbations. Clin Respir J. 2020;14(3):183–197. doi:10.1111/crj.13129
4. Dima E, Kyriakoudi A, Kaponi M, et al. The lung microbiome dynamics between stability and exacerbation in chronic obstructive pulmonary disease (COPD): current perspectives. Respir Med. 2019;157:1–6. doi:10.1016/j.rmed.2019.08.012
5. Cameron SJ, Lewis KE, Huws SA, et al. Metagenomic sequencing of the chronic obstructive pulmonary disease upper bronchial tract microbiome reveals functional changes associated with disease severity. PLoS One. 2016;11(2):e0149095. doi:10.1371/journal.pone.0149095
6. Barker BL, Haldar K, Patel H, et al. Association between pathogens detected using quantitative polymerase chain reaction with airway inflammation in COPD at stable state and exacerbations. Chest. 2015;147(1):46–55. doi:10.1378/chest.14-0764
7. Mantovani A. Wandering pathways in the regulation of innate immunity and inflammation. J Autoimmun. 2017;85:1–5. doi:10.1016/j.jaut.2017.10.007
8. Tasena H, Boudewijn IM, Faiz A, et al. MiR-31-5p: a shared regulator of chronic mucus hypersecretion in asthma and chronic obstructive pulmonary disease. Allergy. 2020;75(3):703–706. doi:10.1111/all.14060
9. Barrow AD, Palarasah Y, Bugatti M, et al. OSCAR is a receptor for surfactant protein D that activates TNF-α release from human CCR2+ inflammatory monocytes. J Immunol. 2015;194:3317–3326. doi:10.4049/jimmunol.1402289
10. Jin S, Zhao G, Li Z, et al. Age-related pulmonary emphysema in mice lacking alpha/beta hydrolase domain containing 2 gene. Biochem Biophys Res Commun. 2009;380(2):419–424. doi:10.1016/j.bbrc.2009.01.098
11. Yoshida M, Whitsett JA. Alveolar macrophages and emphysema in surfactant protein-D-deficient mice. Respirology. 2006;11 Suppl(s1):S37–40. doi:10.1111/j.1440-1843.2006.00806.x
12. Winkler C, Atochina-Vasserman EN, Holz O, et al. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir Res. 2011;12(1):29. doi:10.1186/1465-9921-12-29
13. Li Y, Cho MH, Zhou X. What do polymorphisms tell us about the mechanisms of COPD? Clin Sci (Lond). 2017;131(24):2847–2863. doi:10.1042/CS20160718
14. Obeidat M, Li X, Burgess S, et al. Surfactant protein D is a causal risk factor for COPD: results of Mendelian randomisation. Eur Respir J. 2017;50:1700657.
15. Cavarra E, Bartalesi B, Lucattelli M, et al. Effects of cigarette smoke in mice with different levels of α 1 -proteinase inhibitor and sensitivity to oxidants. Am J Respir Crit Care Med. 2001;164:886–890. doi:10.1164/ajrccm.164.5.2010032
16. Huh JW, Kim SY, Lee JH, et al. Bone marrow cells repair cigarette smoke-induced emphysema in rats. Am J Physiol Lung Cell Mol Physiol. 2011;301(3):L255–66. doi:10.1152/ajplung.00253.2010
17. Lee J-H, Lee DS, Kim E-K, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172(8):987–993. doi:10.1164/rccm.200501-041OC
18. Mirza S, Clay RD, Koslow MA, Scanlon PD. COPD guidelines: a review of the 2018 GOLD report. Mayo Clin Proc. 2018;93(10):1488–1502. doi:10.1016/j.mayocp.2018.05.026
19. Ravi AK, Khurana S, Lemon J, et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir Res. 2014;15(1):103. doi:10.1186/s12931-014-0103-4
20. Kanai K, Koarai A, Shishikura Y, et al. Cigarette smoke augments MUC5AC production via the TLR3-EGFR pathway in airway epithelial cells. Respir Investig. 2015;53(4):137–148. doi:10.1016/j.resinv.2015.01.007
21. Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25(2):250–258. doi:10.1183/09031936.05.00023704
22. Feghali-Bostwick CA, Gadgil AS, Otterbein LE, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(2):156–163. doi:10.1164/rccm.200701-014OC
23. Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. doi:10.1038/nm1238
24. Liu L, Li X, Yuan R, et al. Associations of ABHD2 genetic variations with risks for chronic obstructive pulmonary disease in a Chinese Han population. PLoS One. 2015;10(4):e0123929. doi:10.1371/journal.pone.0123929
25. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi:10.1371/journal.pmed.0030442
26. Vlachaki EM, Koutsopoulos AV, Tzanakis N, et al. Altered surfactant protein-A expression in type II pneumocytes in COPD. Chest. 2010;137(1):37–45. doi:10.1378/chest.09-1029
27. Emmanouil P, Loukides S, Kostikas K, et al. Sputum and BAL Clara cell secretory protein and surfactant protein D levels in asthma. Allergy. 2015;70(6):711–714. doi:10.1111/all.12603
28. Wang LL, Zheng SY, Ren L, et al. Levels of surfactant proteins A and D in bronchoalveolar lavage fluid of children with pneumonia and their relationships with clinical characteristics. Chinese Journal of Contemporary Pediatrics. 2016;18(5):386–390. Chinese. doi:10.7499/j.issn.1008-8830.2016.05.002
29. Koehorst-Ter Huurne K, Groothuis-Oudshoorn CG, vanderValk PD, Movig KL, van der Palen J, Brusse-Keizer M. Association between poor therapy adherence to inhaled corticosteroids and tiotropium and morbidity and mortality in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:1683–1690. doi:10.2147/COPD.S161374
30. Au LH, Chan HS. Severity of airflow limitation, co-morbidities and management of chronic obstructive pulmonary disease patients acutely admitted to hospital. Hong Kong Med J. 2013;19(6):498–503. doi:10.12809/hkmj133909
31. Ozyurek BA, Ulasli SS, Bozbas SS, Bayraktar N, Akcay S. Value of serum and induced sputum surfactant protein-D in chronic obstructive pulmonary disease. Multidiscip Respir Med. 2013;8(1):36. doi:10.1186/2049-6958-8-36
32. Akiki Z, Fakih D, Jounblat R, et al. Surfactant protein D, a clinical biomarker for chronic obstructive pulmonary disease with excellent discriminant values. Exp Ther Med. 2016;11(3):723–730. doi:10.3892/etm.2016.2986
33. Zien Alaabden A, Mohammad Y, Fahoum S. The role of serum surfactant protein D as a biomarker of exacerbation of chronic obstructive pulmonary disease. Qatar Med J. 2015;2015(2):18. doi:10.5339/qmj.2015.18
34. Tantawy AA, Adly AA, Ebeid FSE, et al. Surfactant protein D as a marker for pulmonary complications in pediatric patients with sickle cell disease: relation to lung function tests. Pediatr Pulmonol. 2019;54(5):610–619. doi:10.1002/ppul.24257
35. Ju CR, Liu W, Chen RC. Serum surfactant protein D: biomarker of chronic obstructive pulmonary disease. Dis Markers. 2012;32(5):281–287. doi:10.1155/2012/509063
36. Shakoori TA, Sin DD, Ghafoor F, Bashir S, Bokhari SN. Serum surfactant protein D during acute exacerbations of chronic obstructive pulmonary disease. Dis Markers. 2009;27(6):287–294. doi:10.1155/2009/759304
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Mechanistic Regulation of Wnt Pathway-Related Progression of Chronic Obstructive Pulmonary Disease Airway Lesions
Liu M, Huo Y, Cheng Y
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:871-880
Published Date: 15 May 2023
Abhd2, a Candidate Gene Regulating Airway Remodeling in COPD via TGF-β
Lv MY, Jin LL, Sang XQ, Shi WC, Qiang LX, Lin QY, Jin SD
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:33-50
Published Date: 5 January 2024
LAMC2 Drives Airway Remodeling in COPD via EMT Regulation Through the AKT Pathway
Wang Z, Shi J, Zhang Y, Luo Y, Rao Y, Qu J, Gai X, Sun Y
International Journal of Chronic Obstructive Pulmonary Disease 2026, 21:580964
Published Date: 27 March 2026