Back to Journals » Drug Design, Development and Therapy » Volume 20
Key Considerations for Targeting KRAS in Pancreatic Cancer: Potential Impact on the Treatment Paradigm
Authors Eslinger C, Sonbol MB ![]() , George B, Babiker H, Borad MJ, Bekaii-Saab TS
, George B, Babiker H, Borad MJ, Bekaii-Saab TS
Received 20 January 2026
Accepted for publication 24 March 2026
Published 27 March 2026 Volume 2026:20 559325
DOI https://doi.org/10.2147/DDDT.S559325
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Anastasios Lymperopoulos
Cody Eslinger,1 Mohamad Bassam Sonbol,1 Ben George,2 Hani Babiker,3 Mitesh J Borad,1 Tanios S Bekaii-Saab1
1Department of Hematology-Oncology, Mayo Clinic, Phoenix, AZ, USA; 2Department of Oncology, Mayo Clinic, Rochester, MN, USA; 3Department of Hematology-Oncology, Mayo Clinic, Jacksonville, FL, USA
Correspondence: Tanios S Bekaii-Saab, Department of Hematology-Oncology, Mayo Clinic, 5881 E Mayo Blvd, Phoenix, AZ, 85054, USA, Email [email protected]
Abstract: Pancreatic ductal adenocarcinoma (PDAC) is among the most lethal solid malignancies, characterized by aggressive biology and a paucity of effective treatments. Activating mutations in KRAS occur in more than 90% of cases and are fundamental to tumor initiation, progression, therapeutic resistance, and immune exclusion, establishing KRAS as the dominant oncogenic driver in PDAC. Long considered undruggable, KRAS has recently become a viable therapeutic target with the development of allele-specific inhibitors as well as pan-RAS(ON) agents capable of broadly suppressing mutant RAS signaling. Preclinical models and early-phase clinical trials demonstrate meaningful antitumor activity, with emerging evidence of tumor microenvironment remodeling and delayed resistance. Combination strategies integrating KRAS-directed therapies with chemotherapy, vertical pathway inhibition, immunotherapy, and emerging approaches such as KRAS degradation and RNA-targeted approaches are being explored to improve the depth and durability of response. Together, these advances signal a paradigm shift toward molecularly guided treatment strategies in PDAC and offer a promising path forward in a disease with substantial unmet clinical need.
Plain Language Summary: Pancreatic ductal adenocarcinoma is one of the most aggressive and deadly cancers, with limited treatment options and poor long-term survival. Most patients are diagnosed at an advanced stage, and even with modern chemotherapy, outcomes remain disappointing. There is an urgent need for new and more effective therapies. More than 90% of pancreatic cancers contain mutations in a gene called KRAS, which drives cancer growth and resistance to treatment. For many years, this was thought to be impossible to target with drugs. Recent scientific advances have changed this view, leading to the development of new treatments that directly inhibit KRAS or block its signaling pathways. Early clinical trials of targeted therapies have shown encouraging results, including tumor shrinkage and disease control in some patients. Newer drugs can target specific mutations or multiple variants at once. However, responses are often temporary, as cancers can develop resistance through alternative growth pathways. To improve the durability of responses, researchers are testing combination strategies that pair KRAS-targeted therapies with chemotherapy, other targeted drugs, or immunotherapy. In addition, innovative approaches such as KRAS protein degradation, RNA-based therapies, gene editing, and targeted vaccines are being explored. Together, these advances represent a shift toward precision medicine in pancreatic cancer and offer new hope in a disease with few effective treatment options.
Keywords: KRAS, pancreatic cancer, chemotherapy, targeted therapy, next generation sequencing
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most aggressive solid tumors, with limited therapeutic options despite advances in surgical techniques and systemic therapy. Globally, PDAC ranks as the seventh leading cause of cancer-related death, and incidence continues to rise, particularly in developed countries.1 Even in patients who undergo resection following neoadjuvant chemotherapy, long-term survival remains dismal, with 5-year survival rates below 15%.2 In the metastatic setting, first-line combination regimens such as FOLFIRINOX and gemcitabine plus nab-paclitaxel have modestly improved median overall survival (OS) to approximately 8–11 months, yet nearly all patients ultimately experience disease progression.3–5 The biology of PDAC is characterized by an immunosuppressive microenvironment, rapid development of chemoresistance, and early systemic dissemination, highlighting the urgent need for novel therapeutic strategies.6
Among the molecular drivers of PDAC, activating mutations in the Kirsten rat sarcoma (KRAS) gene represent the most common and defining genetic alteration, occurring in more than 90% of cases (Figure 1).7–9 Historically considered nonactionable, KRAS has long been recognized as a central oncogenic driver through its regulation of downstream signaling pathways such as MAPK and PI3K/AKT.10 Recent progress in direct KRAS inhibition has resulted in the development of both selective and multi-select RAS inhibitors, including agents targeting the G12C variant, with regulatory approvals spanning lung cancer, colorectal cancer, and PDAC.11,12 Although KRAS G12C is relatively rare in PDAC (1–3%), other variants such as G12D and G12V predominate, and novel small-molecule inhibitors against these alleles are entering clinical trials (Figure 2).8,9,13 Therapeutic strategies include allele-specific inhibitors as well as broader RAS pathway blockade, alongside parallel approaches targeting downstream signaling networks, addressing resistance mechanisms, and integrating targeted therapies with chemotherapy or immunotherapy.14 In this review, we aim to synthesize current advances in pancreatic cancer biology and targeted therapeutic development, highlighting emerging strategies, clinical progress, and key challenges that will shape future treatment paradigms. This expanding body of research establishes oncogenic KRAS signaling as a central focus in pancreatic cancer and a promising guide for future therapies.
 |
Figure 1 Mutations associated with pancreatic cancer. |
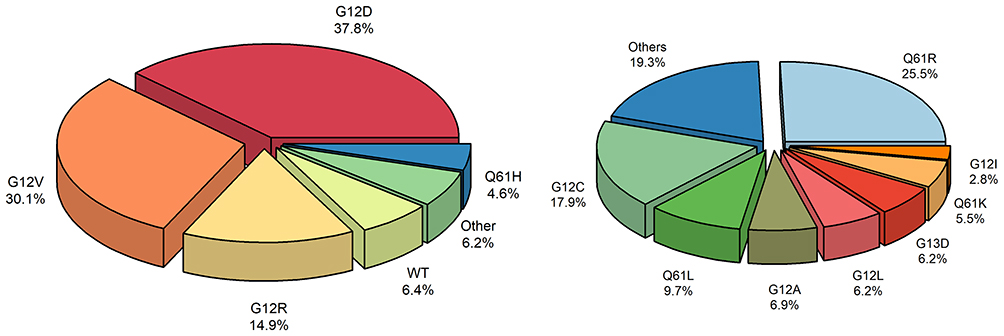 |
Figure 2 Distribution of common KRAS variants in pancreatic cancer (left) and composition of less frequent “Other” KRAS mutations (right). |
KRAS Biology and Molecular Landscape in PDAC
The genomic landscape of PDAC is characterized by a small set of core driver mutations, among which KRAS remains the most dominant and defining. Unlike alterations in TP53, SMAD4, and CDKN2A, which typically accumulate later in the course of disease, KRAS mutations represent an initiating event in pancreatic carcinogenesis and are detectable in the earliest pre-malignant lesions, such as pancreatic intraepithelial neoplasia (PanIN).15,16 This temporal primacy underscores why KRAS has been regarded as the central oncogenic driver of PDAC biology.
The distribution of KRAS variants in PDAC reflects a distinct mutational pattern compared to other solid tumors. G12D and G12V account for nearly 70% of KRAS-mutant PDAC, while G12R represents 13–20%, a variant uniquely enriched in pancreatic cancer.17 These variants differ in downstream signaling, metabolic dependencies, and tumor microenvironment remodeling, which may influence both tumor biology and therapeutic sensitivity. For example, G12R exhibits impaired PI3K-p110α signaling, reduced MAPK pathway activation, and distinct metabolic features compared to G12D or G12V.18
Aberrant KRAS signaling drives tumor initiation, progression, and therapeutic vulnerabilities by locking KRAS in a constitutively active, GTP-bound state, independent of upstream receptor tyrosine kinases (Figure 3).19,20 This leads to persistent activation of downstream effectors, most prominently the RAF-MEK-ERK mitogen-activated protein kinase (MAPK) cascade and the PI3K-AKT-mTOR pathway, which promote uncontrolled proliferation, survival, and metabolic adaptation of tumor cells. Furthermore, mutant KRAS signaling remodels the tumor microenvironment, promoting fibrotic desmoplasia and immune exclusion, processes which collectively sustain PDAC growth and underlie its therapeutic recalcitrance.21 This stromal remodeling drives cancer-associated fibroblast expansion and dense extracellular matrix deposition, creating a fibrotic barrier that limits drug penetration.
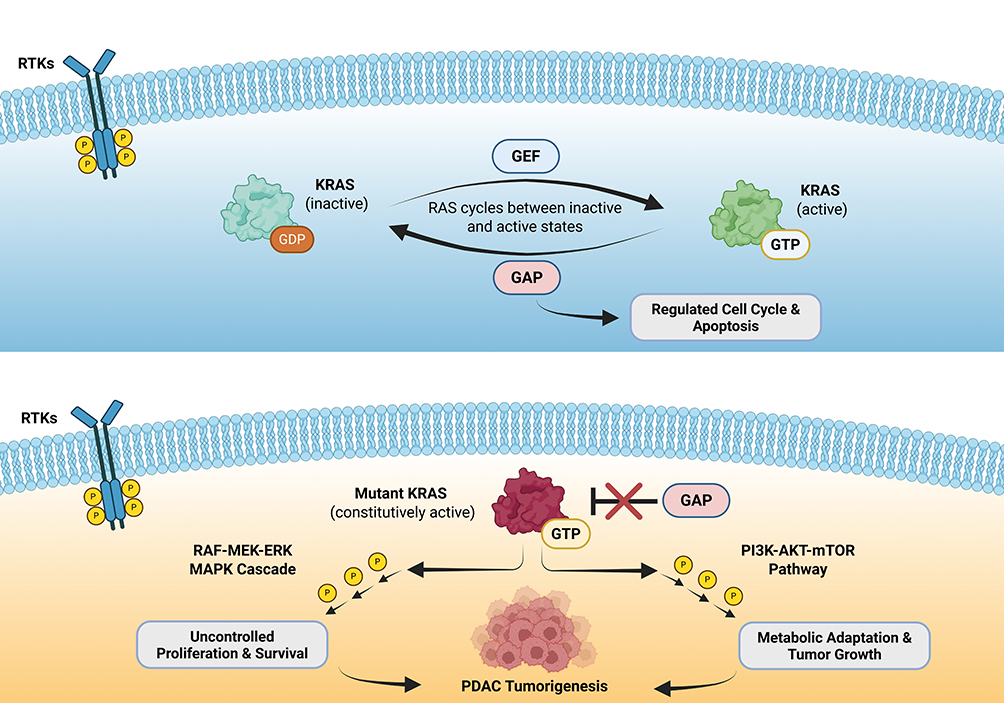 |
Figure 3 KRAS signaling under physiologic conditions (top) compared with oncogenic KRAS-driven signaling in pancreatic tumorigenesis (bottom). Red cross (bottom) indicates mutant KRAS prevents normal GAP-mediated GTP hydrolysis. Abbreviations: GEF, guanine nucleotide exchange factors; GAP, GTPase activating proteins. |
Therapeutic targeting of KRAS is challenged by both structural features and diverse resistance mechanisms. On-target resistance often arise from secondary KRAS mutations (codons 12, 13, or 61, or switch-II pocket), which impair inhibitor binding, while off-target resistance can occur via activation of bypass pathways through amplification or mutation of receptor tyrosine kinases (eg, MET), downstream effectors (eg, BRAF, MAP2K1), or parallel RAS isoforms (NRAS, HRAS).22,23 In a cohort of patients treated with adagrasib (KRAS G12C inhibitor), nearly half developed acquired resistance, with many harboring multiple coincident alterations ranging from secondary KRAS mutations to bypass signaling through MET amplification, oncogenic fusions (ALK, RET, BRAF, RAF1, and FGFR3), or tumor suppressor loss (NF1 and PTEN).22 Co-occurring mutations in genes such as TP53, CDKN2A, and SMAD4 further disrupt cell cycle control, apoptosis, and DNA repair, synergizing with KRAS to enhance malignant transformation.24 Clinically, patients harboring multiple concurrent alterations in these key driver genes experience worse prognosis and may exhibit variable response to KRAS-directed therapies.25
KRAS as a Prognostic Marker
KRAS mutations are a hallmark of PDAC and carry important prognostic implications. Population-based analyses and institutional cohorts report median OS of approximately 10 months for patients with KRAS-mutant PDAC, compared with approximately 20 months in the rare KRAS–wild-type subset.26 Notably, a small proportion of KRAS–wild-type tumors harbor actionable alterations such as NRG1 gene fusions, for which the bispecific antibody zenocutuzumab has received FDA approval, highlighting a clinically relevant exception within this molecular subgroup.27 More granular data indicate that specific KRAS variants may confer differential prognostic risk. For example, KRAS G12D, the most prevalent mutation in PDAC, has been associated with worse clinical outcomes, including shorter progression-free survival (PFS) and OS, compared to other variants in multiple retrospective studies.28 Conversely, G12R tumors display distinct molecular features that may contribute to relatively better prognosis in select cohorts.18 These include reduced MAPK pathway activation, altered PI3K signaling, and unique metabolic dependencies, along with a less fibrotic tumor microenvironment and lower PD-L1 expression.29 Preclinical models demonstrate limited tumorigenesis and attenuated KRAS transcriptional output compared with G12D. Clinically, patients with G12R-mutant PDAC consistently show longer OS than those with G12D across metastatic and resectable settings, highlighting allele-specific differences with potential therapeutic implications.
The prognostic impact of KRAS mutations is further influenced by co-occurring alterations in tumor suppressor genes and other driver pathways. Loss-of-function mutations in TP53, CDKN2A, and SMAD4 frequently accompany KRAS alterations and are associated with more aggressive tumor biology, higher metastatic burden, and increased chemoresistance.24 Patients with 0–2 driver gene alterations (KRAS, TP53, CDKN2A, SMAD4) have the most favorable outcomes, with a median OS of approximately 26.7 months in resected patients, whereas those with alterations in all four key genes experience the poorest prognosis, with median survival of 17.8 months and five-year survival of 8.2%.30 This high-risk molecular subset underscores the heterogeneity of PDAC and supports the rationale for therapies tailored to specific variants and co-mutations, motivating ongoing clinical evaluation of KRAS-targeted therapies.
KRAS-Directed Therapy in Development and Clinical Trials
Overview
KRAS-directed therapies in PDAC encompass allele-specific inhibitors, pan-RAS(ON) blockade, and combination strategies. Allele-specific and pan-RAS agents have shown preclinical and early clinical activity, while combinations with chemotherapy, vertical pathway inhibitors, or immunotherapy aim to enhance efficacy, delay resistance, and remodel the tumor microenvironment. Together, these approaches provide a translational framework for targeting KRAS-driven PDAC. Summaries of key KRAS inhibitors of clinical and translational relevance are provided below (Tables 1–3).
 |
Table 1 Allele-Specific Treatment and Pan-RAS Inhibitors in Pancreatic Cancer |
 |
Table 2 Combination Treatment Approaches (RAS Inhibition + Chemotherapy / Immunotherapy / Targeted Agents) in Pancreatic Cancer |
 |
Table 3 Novel Treatment Approaches in Pancreatic Cancer |
Allele-Specific Inhibitors
KRAS G12C
Although rare in PDAC, the KRAS G12C variant has served as an important clinical proof-of-concept for direct KRAS targeting. In the Phase 1/2 CodeBreaK 100 study, which primarily enrolled heavily pretreated patients, sotorasib demonstrated a median PFS of 4.0 months and median OS of 6.9 months in KRAS G12C–mutant PDAC, with an objective response rate (ORR) of approximately 21%.31 Treatment was well tolerated, with the majority of adverse events being grade 1–2, most commonly diarrhea, nausea, fatigue, and transaminase elevations. Grade ≥3 treatment-related adverse events occurred in a minority of patients and rarely led to discontinuation. Adagrasib, another KRAS G12C inhibitor, has shown comparable or modestly improved activity in PDAC in the KRYSTAL-1 trial, including responses in heavily pretreated patients, with a similar toxicity profile.32
Other next-generation G12C inhibitors, including divarasib, olomorasib, and glecirasib, have also entered clinical development. Divarasib (GDC-6036) is a highly selective, irreversible KRAS G12C inhibitor designed to improve potency and target engagement, and has demonstrated encouraging response rates and manageable toxicity profiles in early-phase trials, particularly in non–small cell lung cancer and colorectal cancer.33 Similar response rates were observed with olomorasib (LY3537982) (ORR 46%, DCR 92%) in the G12C-mutated PDAC cohort.34 Glecirasib has likewise shown antitumor activity in phase 1/2 studies, with evidence of target inhibition and disease control across multiple tumor types.35 However, experience in pancreatic cancer remains limited, with small patient numbers and predominantly refractory populations. Ongoing studies are increasingly focused on combination strategies, including pairing G12C inhibitors with EGFR, SHP2, or downstream pathway inhibitors, to enhance depth and durability of response in this challenging disease context (Table 1).
KRAS G12D
The KRAS G12D mutation is the most prevalent KRAS alteration in pancreatic cancer, making it a central focus of ongoing therapeutic development. Zoldonrasib (RMC-9805), an oral covalent inhibitor designed to engage the active GTP-bound form of KRAS G12D, has also advanced into clinical testing (NCT06040541). Preliminary results presented at ASCO 2025 from the G12D–mutant PDAC cohort (40 evaluable patients) demonstrated encouraging antitumor activity, with an ORR of approximately 30% and disease control achieved in 80% of patients.36 Pharmacodynamic analyses showed on-target activity, including reductions in KRAS G12D circulating tumor DNA (ctDNA) in responding patients. Treatment was well tolerated, with predominantly grade 1–2 adverse events, most commonly gastrointestinal symptoms; grade ≥3 treatment-related toxicities were uncommon, and no unexpected safety signals were observed, supporting continued clinical development.
In parallel, several additional KRAS G12D–directed agents have entered early clinical development. HRS-4642 is a high-affinity, selective, non-covalent G12D inhibitor currently in Phase I/II trials in combination with EGFR inhibitor nimotuzumab in the refractory setting (NCT06773130) as well as the first line setting with chemotherapy (NCT06770452). Early dose-escalation cohorts have demonstrated preliminary antitumor activity, and preclinical studies suggest synergistic effects when combined with proteasome inhibition with carfilzomib.37 TSN1611 targets both GTP-bound (active) and GDP-bound (inactive) G12D and has demonstrated good tolerability with preliminary tumor shrinkage in refractory G12D–mutant tumors, including PDAC, non-small cell lung cancer, and colorectal cancer, in early phase I/II studies (NCT06385925).38
Another candidate, QTX3034, is being investigated as a G12D-preferential multi-KRAS inhibitor in early-phase studies (NCT06021052). Initial reports confirm on-target activity with emerging signs of clinical benefit, though longer follow-up is needed to define its role in pancreatic cancer.39 Together, these efforts represent the first generation of KRAS G12D inhibitors to enter the clinic and mark a pivotal shift toward allele-specific therapy in pancreatic cancer (Table 1). Beyond small-molecule inhibition, immune-based strategies targeting KRAS G12D are also under investigation. Proof-of-concept for KRAS-targeted cellular therapy was demonstrated by Leidner et al, who reported regression of metastatic PDAC following adoptive transfer of autologous T cells engineered to recognize KRAS G12D presented by HLA-C*08:02.40 This landmark case highlighted the feasibility of targeting intracellular oncogenic drivers through mutation-specific T-cell responses and provided a foundation for ongoing T-cell receptor (TCR)-based therapeutic development.
KRAS G12V
Targeting the G12V variant, which accounts for approximately one-third of KRAS mutations in PDAC, represents a critical unmet need. KRAS G12V-selective small-molecule inhibitors, including JAB-23000 and QTX3544, have shown potent preclinical activity by selectively inhibiting G12V-driven signaling and tumor growth in vitro and in vivo.41,42 While small-molecule inhibitors tailored to G12V are in early-stage development, novel approaches are also being explored. A G12V-specific TCR therapy is now in Phase I testing (NCT06767046). This strategy engineers autologous T cells to recognize the KRAS G12V peptide presented by HLA-A*11:01, thereby directing a highly specific cytotoxic immune response against tumor cells harboring the mutation.43 These early efforts illustrate both small-molecule and immune-based therapeutic avenues for G12V-specific targeting, paving the way for future clinical expansions in PDAC (Table 1).
Pan-RAS(ON) Blockade
While allele-specific KRAS inhibitors have demonstrated clinical activity, their efficacy is inherently limited to individual mutant alleles, and adaptive resistance frequently emerges through bypass signaling or co-occurring RAS isoforms.23 These challenges have motivated the development of pan-RAS strategies designed to inhibit multiple RAS mutants simultaneously, broadening applicability across PDAC patients and potentially overcoming mechanisms of resistance observed with single-allele inhibition.
Daraxonrasib (RMC-6236) is a multi-selective RAS(ON) inhibitor that targets active RAS independent of the specific mutant allele. Updated phase 1 data in previously treated metastatic KRAS G12X-mutated PDAC show encouraging clinical activity, with ORRs of 36% at 300 mg versus 27% in patients with broader RAS mutations, and 29% versus 25% across the 160–300 mg dose range.44 Disease control was seen in 91–95% across groups and doses. Median PFS was 8.5 months and median OS 14.5 months, exceeding historical benchmarks for second-line chemotherapy. Importantly, responses were observed in patients previously exposed to chemotherapy and in tumors harboring multiple KRAS variants, suggesting the ability of daraxonrasib to overcome allele-specific resistance mechanisms. On the strength of these findings, daraxonrasib has received FDA Breakthrough Therapy designation for previously treated metastatic PDAC harboring KRAS G12X mutations, and a global Phase 3 trial (RASolute 302) is underway to confirm efficacy and survival benefit (NCT06625320).
Multiple other pan-RAS candidates are advancing in parallel. ADT-007 and its orally bioavailable prodrug ADT-1004 is a pan-RAS inhibitor currently under clinical investigation in PDAC and other RAS-driven cancers, with superior efficacy over sotorasib and adagrasib in tumor resistant models.45,46 AMG-410 is a non-covalent, cycling-independent pan-KRAS inhibitor now in phase I (NCT07094113) that binds both GTP- and GDP-bound KRAS via the switch II allosteric pocket while selectively sparing HRAS and NRAS, a mechanistic distinction that may reduce off-tumor toxicity and improve combinability relative to broader pan-RAS blockade.47 ERAS-4001 is another emerging pan-KRAS inhibitor that targets both wild-type and mutant KRAS proteins and inhibits downstream effector signaling, including RAF-mediated pathway activation, with potent preclinical activity across KRAS-mutant tumor models.48 Finally, ADT-030, a dual pan-RAS/β-catenin inhibitor, has demonstrated potent preclinical efficacy in KRAS-driven PDAC and is positioned for clinical assessment.49 Collectively, these agents highlight the translational potential of pan-RAS blockade, offering a strategy to overcome resistance mechanisms associated with allele-specific inhibition and representing a rapidly expanding frontier in targeted therapy for PDAC (Table 1).
Combination Approaches
With Chemotherapy
Early-phase clinical studies support the combination of KRAS-targeted therapy with chemotherapy in PDAC. The siRNA-based therapy siG12D-LODER™, a biodegradable implant releasing KRAS G12D-targeting siRNA over several months, was evaluated with gemcitabine or modified FOLFIRINOX in patients with locally advanced disease. The treatment was generally well tolerated, and most evaluable patients achieved stable disease or partial responses, with encouraging survival outcomes. These results provide proof-of-concept that sustained KRAS inhibition can enhance the efficacy of cytotoxic therapy.50
Building on this foundation, ongoing trials are evaluating G12C inhibitors in combination with chemotherapy. A phase I/II study (NCT05251038) is assessing sotorasib (960 mg daily) with either liposomal irinotecan/5-FU/leucovorin or gemcitabine/nab-paclitaxel in patients with KRAS G12C-mutated PDAC, aiming to define safety, tolerability, and preliminary efficacy following encouraging single-agent activity in heavily pretreated patients.51 Additionally, the RAMP 205 trial is investigating the combination of avutometinib (a RAF/MEK clamp) and defactinib (a FAK inhibitor) with gemcitabine/nab-paclitaxel as first-line therapy in metastatic PDAC (NCT05669482). Preliminary results from 54 patients suggest the regimen is tolerable, with manageable adverse events and early signals of efficacy, supporting continued evaluation to determine the recommended Phase II dose.52 Together, these studies support continued exploration of KRAS-targeted strategies alongside standard chemotherapy to enhance antitumor activity, delay resistance, and improve outcomes in PDAC (Table 2).
With Targeted Agents
Vertical pathway inhibition aims to overcome adaptive resistance by simultaneously targeting upstream and downstream connections of KRAS signaling, including SHP2, SOS1, and the MEK/ERK cascade.53 This approach directly addresses the rapid feedback reactivation that limits the durability of KRAS-targeted monotherapy in PDAC. Preclinical studies have demonstrated that dual or multi-node blockade can suppress compensatory signaling, reduce proliferation, and induce apoptosis more effectively than single-agent KRAS inhibition alone.54 Combination strategies that improved outcomes in colorectal cancer (eg, G12C inhibitor plus EGFR blockade) are also being explored in PDAC, but long term outcomes are immature.55 These allele-specific and vertical inhibition strategies provide a rational framework for enhancing efficacy, delaying resistance, and expanding the therapeutic potential of KRAS-directed therapy (Table 2).
With Immunotherapy
Preclinical studies have demonstrated that KRAS inhibition, regardless of specific allele, can remodel the tumor microenvironment by increasing infiltration of cytotoxic CD8+ T cells, reducing suppressive myeloid populations, and enhancing antigen presentation, thereby promoting immune-mediated tumor cell killing.56 These findings provide a strong mechanistic rationale for combining KRAS-targeted therapies with immunomodulatory agents, including PD-(L)1 or CTLA-4 checkpoint inhibitors.
Combining KRAS inhibitors with other immunomodulatory agents, such as STING or CD40 agonists, has shown in preclinical PDAC models to enhance dendritic cell activation, promote T-cell priming, and drive synergistic tumor regression, providing a rationale for emerging combination trial designs.57,58 First-in-class allele-agnostic KRAS inhibitors such as BI-2493 have demonstrated robust preclinical activity in PDAC models when combined with immunotherapy, increasing intratumoral effector T cells and decreasing myeloid cell infiltration.59 This immune reprogramming sensitizes tumors to checkpoint blockade, although adaptive resistance mechanisms, such as YAP activation, may emerge. MEK inhibitors, which target KRAS downstream signaling, can further enhance these effects. In preclinical studies and early-phase trials, MEK inhibition combined with immunotherapy and radiotherapy or autophagy blockade has improved T-cell infiltration, increased antigen presentation, and augmented antitumor immunity.60
Furthermore, trametinib combined with pembrolizumab and stereotactic body radiotherapy demonstrated improved OS in patients with locally recurrent PDAC, supporting the integration of immune priming strategies with KRAS pathway blockade.61 Complementing these strategies, mutant KRAS-targeted vaccines paired with dual checkpoint blockade (eg, ipilimumab/nivolumab or balstilimab/botensilimab) are in phase I trials for resected and metastatic PDAC, offering a novel mechanism with potential for innovative drug development and delivery approaches (NCT04117087, NCT06411691) (Table 2).
Novel Approaches
Emerging KRAS-targeted strategies in PDAC include PROTAC degraders, RNA-based therapies, CRISPR approaches, and mutation-specific vaccines. These modalities aim to overcome therapeutic resistance and the dense, fibrotic stroma that limits drug delivery. Preclinical studies show promising antitumor activity, though clinical safety and efficacy remain limited. These approaches provide valuable insights into KRAS biology that may guide future treatment modalities.
PROTACs
Proteolysis-targeting chimeras (PROTACs) represent an emerging therapeutic strategy designed to eliminate oncogenic KRAS proteins rather than simply inhibiting their enzymatic function. These bifunctional molecules recruit an E3 ubiquitin ligase to a target protein, inducing ubiquitination and subsequent proteasomal degradation. This mechanism has the potential to address inhibitor-resistant alleles and expand therapeutic reach to proteins historically regarded as “undruggable”.62 Recent preclinical studies have generated KRAS G12D-selective degraders, including ZJK-807 and RP03707, which preferentially degrade mutant KRAS G12D, attenuate RAS–MAPK signaling, and reduce tumor growth in cell-based and xenograft PDAC models while largely sparing wild-type RAS function.63,64 Pan-KRAS degraders, such as MCB-36, have also been reported, demonstrating activity across multiple mutant alleles, including those associated with inhibitor resistance, and showing capacity to influence tumor signaling and the immune microenvironment.65 Early clinical translation is underway with first-in-human studies of KRAS-directed degraders. ASP3082, a KRAS G12D-targeting PROTAC, is currently being evaluated in a phase 1 dose-escalation trial for advanced solid tumors including PDAC, supported by preclinical evidence of selective degradation and antitumor activity (NCT05382559). Several challenges remain for clinical development, particularly enhancing cellular permeability and oral bioavailability, reducing off-target toxicities, and achieving tumor-selective delivery. Investigational strategies to address these barriers include nanoparticle-based formulations and conditionally activated prodrugs designed to localize PROTAC activity to the tumor microenvironment (Table 3).62
CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/ CRISPR-associated protein 9 (Cas9) systems have been extensively applied in PDAC research both as functional genomics tools and as therapeutic modalities.66 RNA-targeting CRISPR systems such as CasRx have demonstrated potent allele-specific silencing capabilities. In orthotopic KRAS G12D pancreatic tumors, AAV8-delivered CasRx guided by mutation-specific sgRNAs effectively suppressed mutant KRAS expression, abrogated downstream signaling, and significantly prolonged survival without detectable toxicity.67 These effects extended to patient-derived xenografts, where CasRx treatment inhibited tumor growth and enhanced sensitivity to gemcitabine (Table 3).
RNA Interference
RNA interference (RNAi) offers a complementary strategy for directly suppressing mutant KRAS in PDAC. As noted earlier, the siG12D-LODER phase I trial demonstrated the clinical feasibility of KRAS-targeted RNAi, with acceptable safety and evidence of disease stabilization in locally advanced PDAC.50 Preclinical platforms continue to advance the field, including cRGD-modified polymersomes delivering KRAS G12D siRNA that achieved approximately 90% gene knockdown and durable tumor regressions in xenograft models. Additional nanoparticle systems, including tLyp-1–targeted lipid carriers that increase intratumoral accumulation and iRGD-based delivery vectors that enhance stromal penetration, have each demonstrated improved delivery and antitumor activity in PDAC models.68,69 Gold nanoparticle–polyethylenimine (PEI) complexes have also shown efficient KRAS silencing with minimal toxicity in PDAC models.70 These advances highlight RNAi as a feasible strategy for KRAS-directed therapy, though challenges in stability, delivery, and stromal penetration remain significant barriers to clinical translation (Table 3).
KRAS Vaccines
Multiple vaccine platforms have been developed to elicit mutation-specific T cell responses against common KRAS variants, most frequently G12D, G12V, G12R, and G13D. Synthetic long peptide (SLP) vaccines incorporating pooled mutant KRAS epitopes have demonstrated favorable safety and consistent immunogenicity, inducing mutation-specific T cell responses in approximately 85% of patients with high risk features, including those with genetic predisposition or premalignant lesions.71 Lymph node–targeted amphiphile vaccines such as ELI-002 7P further enhance antigen delivery and immune priming, achieving near-universal T cell responses with evidence of minimal residual disease reduction in early-phase trials, which has led to ongoing randomized Phase 2 evaluation (AMPLIFY-7P) in the adjuvant setting.72 Earlier peptide-based approaches, including the TG01 RAS-neoantigen vaccine administered with adjuvant chemotherapy, have also demonstrated robust immune responses and encouraging survival outcomes compared with historical controls.73 Overall, KRAS vaccines have been well tolerated with predominantly low-grade adverse events and reproducible mutant KRAS-specific T cell responses; however, definitive clinical benefit has not yet been established, and ongoing randomized studies will determine whether vaccine-induced immunity translates into meaningful improvements in disease-free survival (DFS) and OS in PDAC.
Key Takeaways and Outlook
KRAS mutations are central drivers of pancreatic cancer, shaping tumor growth, therapeutic resistance, and the immunosuppressive microenvironment. While allele-specific inhibitors targeting G12C, G12D, and G12V, as well as pan-RAS(ON) agents, have shown promising activity in preclinical models and early-phase trials, clinical responses remain limited and variable. The aggressive biology of pancreatic cancer, its high mutational complexity, and the profound treatment resistance emphasize the urgent need for novel strategies beyond conventional chemotherapy.
Ongoing research is exploring rational combinations to overcome these challenges. Vertical pathway inhibition, immune-directed therapies, and KRAS-targeted vaccines represent distinct approaches to enhance activity, delay resistance, and remodel the tumor microenvironment. Early-phase studies integrating these strategies provide a translational framework for future clinical testing. Together, these efforts underscore the potential of KRAS-directed therapy to address a critical unmet need in a disease where current treatments yield poor long-term outcomes.
Disclosure
M Sonbol reports consulting (self): Novartis; consulting (institution): Boehringer Ingelheim; research support (institution): Taiho, Eli Lilly. B. George reports consultant: Ipsen, Foundation Medicine, Taiho Oncology, Boston Scientific, Roche/Genentech, Astra Zeneca, Pfizer, Astellas, Amgen; research support (intitution): Roche/Genentech, Hoffman La-Roche, Taiho Oncology, Toray, Hutchison Medipharma, Mirati Therapeutics, CARsgen, Glyconex, Helix Biopharma, Pfizer, Tvardi Therapeutics, Faeth Therapeutics, BionTech, Transcenta, Legend Biotech, Elicio Therapeutics, Obsidian. H. Babiker reports consulting or advisory role: Endocyte, Celgene, Idera, Myovant Sciences, Novocure, Ipsen, Caris MPI, Incyte, Guardant Health; research funding (institution): Spirita Oncology, Novocure, AstraZeneca, JSI, Incyte, Qurient, HiFiBiO Therapeutics, Revolution Health Care, Elevation Oncology, Dragonfly Therapeutics, Zelbio, BMS, Mirati Therapeutics, Strategia. T. Bekaii-Saab reports research funding (institution): Agios, Arys, Arcus, Atreca, Boston Biomedical, Bayer, Eisai, Celgene, Lilly, Ipsen, Clovis, Seattle Genetics, Genentech, Novartis, Mirati, Merus, Abgenomics, Incyte, Pfizer, BMS; consulting (institution): Servier, Ipsen, Arcus, Pfizer, Seattle Genetics, Bayer, Genentech, Incyte, Eisai, Merus, Merck KgaA, and Merck; consulting (self): Stemline, AbbVie, Blueprint Medicines, Boehringer Ingelheim, Janssen, Daiichi Sankyo, Natera, TreosBio, Celularity, Caladrius Biosciences, Exact Science, Sobi, Beigene, Kanaph, Astra Zeneca, Deciphera, Zai Labs, Exelixis, MJH Life Sciences, Aptitude Health, Illumina, Foundation Medicine and Sanofi. Glaxo SmithKline, Xilio; IDMC/DSMB: The Valley Hospital, Fibrogen, Suzhou Kintor, Astra Zeneca, Exelixis, Merck/Eisai, PanCan, and 1Globe; Scientific Advisory Board: Imugene, Immuneering, Xilis, Replimune, Artiva and Sun Biopharma; Royalties: UpToDate; Inventions/Patents: WO/2018/183488: HUMAN PD1 PEPTIDE VACCINES AND USES THEREOF – Licensed to Imugene, WO/2019/055687: METHODS AND COMPOSITIONS FOR THE TREATMENT OF CANCER CACHEXIA – Licensed to Recursion. M Borad reports grants, personal fees from Revolution Med, grants from Incyte, grants from Arvinas, grants from Vividion, grants from Tango Therapeutics, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–15. doi:10.3322/caac.21834
2. Park W, Chawla A, O’Reilly EM. Pancreatic cancer: a review. JAMA. 2021;326(9):851–862. doi:10.1001/jama.2021.13027
3. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi:10.1056/NEJMoa1011923
4. Hoff DDV, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi:10.1056/NEJMoa1304369
5. Nichetti F, Rota S, Ambrosini P, et al. NALIRIFOX, FOLFIRINOX, and gemcitabine with nab-paclitaxel as first-line chemotherapy for metastatic pancreatic cancer: a systematic review and meta-analysis. JAMA Network Open. 2024;7(1):e2350756. doi:10.1001/jamanetworkopen.2023.50756
6. Argentiero A, Andriano A, Caradonna IC, de Martino G, Desantis V. Decoding the intricate landscape of pancreatic cancer: insights into tumor biology, microenvironment, and therapeutic interventions. Cancers. 2024;16(13). doi:10.3390/cancers16132438
7. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. doi:10.1038/nature16965
8. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
9. Varghese AM, Perry MA, Chou JF, et al. Clinicogenomic landscape of pancreatic adenocarcinoma identifies KRAS mutant dosage as prognostic of overall survival. Nat Med. 2025;31(2):466–477. doi:10.1038/s41591-024-03362-3
10. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. doi:10.1038/nrd4389
11. Hong DS, Fakih MG, Strickler JH, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207–1217. doi:10.1056/NEJMoa1917239
12. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. doi:10.1038/s41586-019-1694-1
13. Safyan RA, Chiorean EG. Updates on molecular targets and clinical trials with targeted therapies for pancreatic cancer. Surg Oncol. 2025;102268. doi:10.1016/j.suronc.2025.102268
14. Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol. 2018;15(11):709–720. doi:10.1038/s41571-018-0105-0
15. Hosoda W, Chianchiano P, Griffin JF, et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J Pathol. 2017;242(1):16–23. doi:10.1002/path.4884
16. Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–653. doi:10.1172/jci59227
17. Ardalan B, Ciner A, Baca Y, et al. Distinct molecular and clinical features of specific variants of KRAS Codon 12 in pancreatic adenocarcinoma. Clin Cancer Res. 2025;31(6):1082–1090. doi:10.1158/1078-0432.Ccr-24-3149
18. McIntyre CA, Grimont A, Park J, et al. Distinct clinical outcomes and biological features of specific KRAS mutants in human pancreatic cancer. Cancer Cell. 2024;42(9):1614–1629.e5. doi:10.1016/j.ccell.2024.08.002
19. Pantsar T. The current understanding of KRAS protein structure and dynamics. Comput Struct Biotechnol J. 2020;18:189–198. doi:10.1016/j.csbj.2019.12.004
20. Di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144(6):1220–1229. doi:10.1053/j.gastro.2013.01.071
21. Ma J, Fu S, Tan J, et al. Mechanistic FOUNDATIONS of KRAS-driven tumor ecosystems: integrating crosstalk among immune, metabolic, microbial, and stromal microenvironment. Adv Sci. 2025;12(30):e02714. doi:10.1002/advs.202502714
22. Awad MM, Liu S, Rybkin II, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med. 2021;384(25):2382–2393. doi:10.1056/NEJMoa2105281
23. Dilly J, Hoffman MT, Abbassi L, et al. Mechanisms of resistance to oncogenic KRAS inhibition in pancreatic cancer. Cancer Discov. 2024;14(11):2135–2161. doi:10.1158/2159-8290.Cd-24-0177
24. Yachida S, White CM, Naito Y, et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin Cancer Res. 2012;18(22):6339–6347. doi:10.1158/1078-0432.Ccr-12-1215
25. Herting CJ, Karpovsky I, Lesinski GB. The tumor microenvironment in pancreatic ductal adenocarcinoma: current perspectives and future directions. Cancer Metastasis Rev. 2021;40(3):675–689. doi:10.1007/s10555-021-09988-w
26. Lena J, Alamé M, Italiano A, et al. Extensive molecular profiling of KRAS wild-type as compared to KRAS mutated pancreatic ductal adenocarcinoma on 318 patients. Eur J Cancer. 2025;216:115197. doi:10.1016/j.ejca.2024.115197
27. Schram AM, Goto K, Kim D-W, et al. Efficacy of zenocutuzumab in NRG1 fusion–positive cancer. N Engl J Med. 2025;392(6):566–576. doi:10.1056/NEJMoa2405008
28. Norton C, Shaw MS, Rubnitz Z, et al. KRAS mutation status and treatment outcomes in patients with metastatic pancreatic adenocarcinoma. JAMA Network Open. 2025;8(1):e2453588. doi:10.1001/jamanetworkopen.2024.53588
29. Burge RA, Le Roux O, Popow O, et al. KRASG12R-mutant pancreatic cancer features limited ERK/MAPK transcriptional activity and a distinctive tumor microenvironment. Cancer Res. 2026. doi:10.1158/0008-5472.Can-25-2630
30. Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4(3):e173420. doi:10.1001/jamaoncol.2017.3420
31. Strickler JH, Satake H, George TJ, et al. Sotorasib in KRAS p.G12C–mutated advanced pancreatic cancer. N Engl J Med. 2023;388(1):33–43. doi:10.1056/NEJMoa2208470
32. Bekaii-Saab TS, Yaeger R, Spira AI, et al. Adagrasib in advanced solid tumors harboring a KRAS(G12C) mutation. J Clin Oncol. 2023;41(25):4097–4106. doi:10.1200/jco.23.00434
33. Sacher A, LoRusso P, Patel MR, et al. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N Engl J Med. 2023;389(8):710–721. doi:10.1056/NEJMoa2303810
34. Hollebecque A, Kuboki Y, Murciano-Goroff YR, et al. Efficacy and safety of LY3537982, a potent and highly selective KRAS G12C inhibitor in KRAS G12C-mutant GI cancers: results from a phase 1 study. J Clin Oncol. 2024;42(3_suppl):94. doi:10.1200/JCO.2024.42.3_suppl.94
35. Li J, Shen L, Gu Y, et al. Preliminary activity and safety results of KRAS G12C inhibitor glecirasib (JAB-21822) in patients with pancreatic cancer and other solid tumors. J Clin Oncol. 2024;42(3_suppl):604. doi:10.1200/JCO.2024.42.3_suppl.604
36. Spira AI, Papadopoulos KP, Kim DW, et al. Preliminary safety, antitumor activity, and circulating tumor DNA (ctDNA) changes with RMC-9805, an oral, RAS(ON) G12D-selective tri-complex inhibitor in patients with KRAS G12D pancreatic ductal adenocarcinoma (PDAC) from a phase 1 study in advanced solid tumors. J Clin Oncol. 2025;43(4_suppl):724. doi:10.1200/JCO.2025.43.4_suppl.724
37. Zhou C, Li C, Luo L, et al. Anti-tumor efficacy of HRS-4642 and its potential combination with proteasome inhibition in KRAS G12D-mutant cancer. Cancer Cell. 2024;42(7):1286–1300.e8. doi:10.1016/j.ccell.2024.06.001
38. Fu S, Shen L, Lu S, et al. The first-in-human phase 1/2 study of TSN1611, a highly selective KRAS G12D inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2025;43(16_suppl):3083. doi:10.1200/JCO.2025.43.16_suppl.3083
39. Zhang YW, Rominger D, Vo ED, et al. Abstract LB320: discovery and characterization of QTX3034, a potent, selective, and orally bioavailable allosteric KRAS inhibitor. Cancer Res. 2023;83(8_Supplement):LB320. doi:10.1158/1538-7445.Am2023-lb320
40. Leidner R, Sanjuan Silva N, Huang H, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med. 2022;386(22):2112–2119. doi:10.1056/NEJMoa2119662
41. Zhou X, Ji Y, Zhou J. Multiple strategies to develop small molecular KRAS directly bound inhibitors. Molecules. 2023;28(8). doi:10.3390/molecules28083615
42. Zhang YW, Vo ED, Gan P, et al. Abstract LB163: discovery and characterization of QTX3544, a potent, selective, and orally bioavailable allosteric G12V preferring multi KRAS inhibitor. Cancer Res. 2024;84(7_Supplement):LB163. doi:10.1158/1538-7445.Am2024-lb163
43. Xu X, Guo S, Gu H, et al. Identification and validation of a T cell receptor targeting KRAS G12V in HLA-A*11:01 pancreatic cancer patients. JCI Insight. 2025;10(2). doi:10.1172/jci.insight.181873
44. Garrido-Laguna I, Wolpin BM, Park W, et al. Safety, efficacy, and on-treatment circulating tumor DNA (ctDNA) changes from a phase 1 study of RMC-6236, a RAS(ON) multi-selective, tri-complex inhibitor, in patients with RAS mutant pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. 2025;43(4_suppl):722. doi:10.1200/JCO.2025.43.4_suppl.722
45. Filis P, Salgkamis D, Matikas A, Zerdes I. Breakthrough in RAS targeting with pan-RAS(ON) inhibitors RMC-7977 and RMC-6236. Drug Discov Today. 2025;30(1):104250. doi:10.1016/j.drudis.2024.104250
46. Bandi DSR, Nagaraju GP, Sarvesh S, et al. ADT-1004: a first-in-class, oral pan-RAS inhibitor with robust antitumor activity in preclinical models of pancreatic ductal adenocarcinoma. Mol Cancer. 2025;24(1):76. doi:10.1186/s12943-025-02288-9
47. Lanman BA, Wurz RP, Verma R, et al. Abstract ND01: AMG 410: an H/NRAS-sparing pan-KRAS inhibitor with dual GTP(on)/GDP(off)-state activity for the treatment of diverse KRAS-mutant tumors. Cancer Res. 2025;85(8_Supplement_2):ND01. doi:10.1158/1538-7445.Am2025-nd01
48. Bae JH, Lin B, Lew ED, et al. Abstract 4367: ERAS-4001 is a pan-KRAS inhibitor with robust anti-tumor activity in KRAS altered solid tumors. Cancer Res. 2025;85(8_Supplement_1):4367. doi:10.1158/1538-7445.Am2025-4367
49. Piazza GA, Bandi RD, Ganji PN, et al. Evaluation of a novel Pan-RAS/β-Catenin inhibitor, ADT-030, for treatment of pancreatic cancer. J Clin Oncol. 2024;42(16_suppl):e15084. doi:10.1200/JCO.2024.42.16_suppl.e15084
50. Golan T, Khvalevsky EZ, Hubert A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560–24570. doi:10.18632/oncotarget.4183
51. Mahalingam D, Burns MC, Kalyan A, et al. A phase Ib/II study of sotorasib combined with chemotherapy for second-line treatment of KRAS p. G12C–mutated advanced pancreatic cancer. J Clin Oncol. 2022;40(16_suppl):TPS4194. doi:10.1200/JCO.2022.40.16_suppl.TPS4194
52. Lim K-H, Safyan RA, Perez K, et al. Avutometinib/defactinib and gemcitabine/nab-paclitaxel combination in first-line metastatic pancreatic ductal adenocarcinoma: updated safety and efficacy of a phase 1b/2 study (RAMP 205). J Clin Oncol. 2025;43(16_suppl):e16403. doi:10.1200/JCO.2025.43.16_suppl.e16403
53. Tanaka N, Ebi H. Mechanisms of resistance to KRAS Inhibitors: cancer cells’ strategic use of normal cellular mechanisms to adapt. Cancer Sci. 2025;116(3):600–612. doi:10.1111/cas.16441
54. Ma Y, Schulz B, Trakooljul N, et al. Inhibition of KRAS, MEK and PI3K demonstrate synergistic anti-tumor effects in pancreatic ductal adenocarcinoma cell lines. Cancers. 2022;14(18). doi:10.3390/cancers14184467
55. Fakih MG, Salvatore L, Esaki T, et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N Engl J Med. 2023;389(23):2125–2139. doi:10.1056/NEJMoa2308795
56. Mahadevan KK, McAndrews KM, LeBleu VS, et al. KRAS(G12D) inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8(+) T cells. Cancer Cell. 2023;41(9):1606–1620.e8. doi:10.1016/j.ccell.2023.07.002
57. Broderick C, Mezzadra R, Sisso EM, et al. A RAS(ON) multi-selective inhibitor combination therapy triggers long-term tumor control through senescence-associated tumor-immune equilibrium in pancreatic ductal adenocarcinoma. Cancer Discov. 2025;15(8):1717–1739. doi:10.1158/2159-8290.Cd-24-1425
58. Jing W, McAllister D, Vonderhaar EP, et al. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immun Ther Cancer. 2019;7(1):115. doi:10.1186/s40425-019-0573-5
59. McAndrews KM, Paradiso F, Stalnecker CA, et al. An allele-agnostic mutant-KRAS inhibitor suppresses tumor maintenance signals and reprograms tumor immunity in pancreatic cancer. Sci Transl Med. 2025;17(814):eadt5511. doi:10.1126/scitranslmed.adt5511
60. Raufi A, Wong W, Lee SM, Manji GA. MEKiAUTO: a phase I/II open-label study of combination therapy with the MEK inhibitor cobimetinib, Immune-checkpoint blockade with atezolizumab, and the AUTOphagy inhibitor hydroxychloroquine in KRAS-mutated advanced malignancies. J Clin Oncol. 2021;39(3_suppl):TPS450. doi:10.1200/JCO.2021.39.3_suppl.TPS450
61. Zhu X, Cao Y, Liu W, et al. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: an open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2022;23(3):e105–e115. doi:10.1016/s1470-2045(22)00066-3
62. Anaya YA, Barragan M, Bracho RP, et al. Proteolysis-targeting chimeras in cancer therapy: targeted protein degradation for next-generation treatment. Cancer. 2025;131(21):e70132. doi:10.1002/cncr.70132
63. Ji X, Li H, Wu G, et al. Discovery and characterization of RP03707: a highly potent and selective KRAS(G12D) PROTAC. J Med Chem. 2025;68(10):10238–10254. doi:10.1021/acs.jmedchem.5c00428
64. Liu Z, Zheng H, Tian Y, et al. ZJK-807: a selective PROTAC degrader of KRAS(G12D) overcoming resistance in pancreatic cancer. J Med Chem. 2025;68(19):20103–20129. doi:10.1021/acs.jmedchem.5c01034
65. Feng J, Xiao X, Xia X, et al. A pan-KRAS inhibitor and its derived degrader elicit multifaceted anti-tumor efficacy in KRAS-driven cancers. Cancer Cell. 2025;43(10):1866–1884.e12. doi:10.1016/j.ccell.2025.07.006
66. Won EJ, Park H, Yoon TJ, Cho YS. Gene Therapy Using Nanocarriers for Pancreatic Ductal Adenocarcinoma: applications and Challenges in Cancer Therapeutics. Pharmaceutics. 2022;14(1). doi:10.3390/pharmaceutics14010137
67. Jiang W, Li H, Liu X, et al. Precise and efficient silencing of mutant Kras(G12D) by CRISPR-CasRx controls pancreatic cancer progression. Theranostics. 2020;10(25):11507–11519. doi:10.7150/thno.46642
68. Lo JH, Hao L, Muzumdar MD, et al. iRGD-guided tumor-penetrating nanocomplexes for therapeutic siRNA Delivery to pancreatic cancer. Mol Cancer Ther. 2018;17(11):2377–2388. doi:10.1158/1535-7163.Mct-17-1090
69. Anthiya S, Öztürk SC, Yanik H, et al. Targeted siRNA lipid nanoparticles for the treatment of KRAS-mutant tumors. J Control Release. 2023;357:67–83. doi:10.1016/j.jconrel.2023.03.016
70. Küçükekmekci B, Budak Yıldıran FA. Investigation of the efficacy of siRNA-mediated KRAS gene silencing in pancreatic cancer therapy. PeerJ. 2024;12:e18214. doi:10.7717/peerj.18214
71. Haldar SD, Judkins C, Ferguson A, et al. A phase I study of a mutant KRAS-targeted long peptide vaccine in patients at high risk of developing pancreatic cancer. J Clin Oncol. 2023;41(4_suppl):TPS758. doi:10.1200/JCO.2023.41.4_suppl.TPS758
72. Devoe CE, Pant S, Wainberg ZA, et al. AMPLIFY-7P, a first-in-human safety and efficacy trial of adjuvant mKRAS-specific lymph node targeted amphiphile ELI-002 7P vaccine in patients with minimal residual disease–positive pancreatic and colorectal cancer. J Clin Oncol. 2024;42(16_suppl):2636. doi:10.1200/JCO.2024.42.16_suppl.2636
73. Palmer DH, Valle JW, Ma YT, et al. TG01/GM-CSF and adjuvant gemcitabine in patients with resected RAS-mutant adenocarcinoma of the pancreas (CT TG01-01): a single-arm, phase 1/2 trial. Br J Cancer. 2020;122(7):971–977. doi:10.1038/s41416-020-0752-7
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Long Overdue Targeted Treatment for KRAS Mutations in NSCLC: Spotlight on Adagrasib
Brazel D, Arter Z, Nagasaka M
Lung Cancer: Targets and Therapy 2022, 13:75-80
Published Date: 10 November 2022
Breast Cancer: An Overview of Current Therapeutic Strategies, Challenge, and Perspectives
Wang J, Wu SG
Breast Cancer: Targets and Therapy 2023, 15:721-730
Published Date: 20 October 2023
Roles of SPOCK1 in the Formation Mechanisms and Treatment of Non-Small-Cell Lung Cancer and Brain Metastases from Lung Cancer
Zhang X, Zhang X, Yin H, Li Q, Fan B, Jiang B, Xie A, Guo D, Hao H, Zhang B
OncoTargets and Therapy 2025, 18:35-47
Published Date: 16 January 2025
Diagnosis, Prognosis, and Treatment of Triple-Negative Breast Cancer: A Review
Jie H, Ma W, Huang C
Breast Cancer: Targets and Therapy 2025, 17:265-274
Published Date: 17 March 2025
Comparative Cost Analysis for Direct Medical Costs of Protocol Administration of Non-Small Cell Lung Cancer Treatment Regimens in Curative Intent: A Micro-Costing Study in Jordan
Madae’en SS, Salem AA, Ararawi NS, Ramzi EJ, Aloueedat RF, Saabenh AM, Allouzi DA, Abuoudeh RH, Hnaif OE, Musa LM, Alshdaifat SH, Al-Tanashat AJ, Almasa’afeh HY, Abuallaban SM
ClinicoEconomics and Outcomes Research 2025, 17:455-471
Published Date: 12 July 2025