Back to Journals » Lung Cancer: Targets and Therapy » Volume 13
A Long Overdue Targeted Treatment for KRAS Mutations in NSCLC: Spotlight on Adagrasib
Authors Brazel D, Arter Z ![]() , Nagasaka M
, Nagasaka M ![]()
Received 25 July 2022
Accepted for publication 21 October 2022
Published 10 November 2022 Volume 2022:13 Pages 75—80
DOI https://doi.org/10.2147/LCTT.S383662
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Danielle Brazel,1 Zhaohui Arter,1 Misako Nagasaka1– 3
1Department of Medicine, University of California Irvine School of Medicine, Orange, CA, USA; 2Chao Family Comprehensive Cancer Center, Orange, CA, USA; 3St. Marianna University School of Medicine, Kawasaki, Japan
Correspondence: Misako Nagasaka, Department of Medicine, University of California Irvine School of Medicine, Chao Family Comprehensive Cancer Center, 101 The City Drive South, Orange, CA, 92868, USA, Email [email protected]
Abstract: KRASG12C is one of the most common oncogenes in non-small cell lung cancer (NSCLC) and is associated with a poor prognosis. Historically, KRAS mutations have been difficult to target due to lack of binding sites and exceptionally high affinity for guanosine triphosphate/guanosine diphosphate (GTP/GDP). Recently, KRASG12C selective inhibitors have shown promising results in Phase I/II studies. Here we discuss the mechanism of action, pharmacokinetic and pharmacodynamic properties, efficacy, and tolerability of adagrasib (MRTX849).
Keywords: MRTX849, KRAS, non-small cell lung cancer, targeted therapy
Introduction
Lung cancer is one of the most common cancers, accounting for 235,760 new cases and 131,880 deaths in the United States in 2021.1 Kirsten rat sarcoma viral oncogene (KRAS) mutation is the most common gain-of-function oncogene in NSCLC and typically presents as a single-driver mutation.2,3 KRAS serves as a binary switch for signal transduction of most receptor tyrosine kinases making KRAS a key mediator of RAS/MAPK signaling pathways and ultimately cell growth and proliferation.4,5
KRAS mutations occur more commonly in Caucasians and African Americans than in Asian patients5–7 and are more common in younger patients.8,9 Smoking is associated with both KRAS mutations and higher mutational burden compared to those who have never smoked.10,11 Although the prognostic impact remains controversial, a meta-analysis of 53 studies found that KRAS mutations correlate with poor prognosis (HR 1.40; p = 0.01).12 KRASG12C is the most common, accounting for 40–50% of KRAS mutations13 and is most commonly seen in lung (14%) and colon (4%) adenocarcinoma.14–16
KRASG12C is associated with shorter OS compared to other KRAS mutations (HR 1.39; 95% confidence interval [CI]1.03–1.89) and compared to no KRAS mutation (HR 1.32; 95% CI 1.03–1.69).17 Recent data show that central nervous system (CNS) metastases are present in 42% of KRASG12C-mutated NSCLC patients at the time of initial diagnosis18 and 33–40% during follow-up.19 From a retrospective observational study, patients with KRAS mutant NSCLC with baseline CNS metastases had a median overall survival of 9 months when brain metastases were treated with surgery and/or radiation and only 5 months when untreated.20
Until recently, KRAS mutations represented an unmet need in treatment of solid tumors despite being one of the most frequent oncogenic drivers. Historically, KRAS mutations have been difficult to target, attributed to the overall smooth surface structure with lack of binding sites, small dimension, and exceptionally high affinity for GTP/GDP.21,22 The potential of targeting KRASG12C was first recognized by Ostrem et al in 2013 who showed that binding an allosteric pocket could lock GDP-bound KRAS in its inactive state.23 Studies showed that mutations in codon 12 or 13 of RAS proteins impair GTP hydrolysis leaving RAS in the GTP-bound active state.24 This led to the development of sotorasib, one of the first in class KRASG12C off-state inhibitors and ultimately to FDA approval of sotorasib for patients with G12C mutant advanced metastatic lung cancer post one prior line of therapy.25 Here, we discuss adagrasib (MRTX849), another selective and potent KRASG12C inhibitor with similar mechanism of action to sotorasib.
Structural Characteristics and Mechanism of Action of Adagrasib
Adagrasib is a highly selective and potent oral small-molecule inhibitor of KRASG12C. Adagrasib irreversibly binds cysteine 12 in the KRASG12C switch II pocket to lock the molecule in its inactive GDP-bound form.26 This inactivation inhibits the rat sarcoma (RAS)/mitogen-activated protein kinase (MAPK) pathway for cellular proliferation. Through the use of a 2-fluoroacrylamide warhead, blood stability was achieved with half-life >50 hours. A unique co-crystal structure was generated through tetrahydropyridopyrimidine analogue design.
Pharmacodynamic Properties
Having high oral bioavailability, long half-life, extensive tissue distribution, and central nervous system penetration make adagrasib an optimal KRASG12C inhibitor.24 In preclinical models, adagrasib demonstrated potent inhibition of KRAS-dependent signal transduction with an IC50 (cellular half-maximal inhibitory concentration) of approximately 5 nM and cancer cell viability, with a >1000-fold selectivity for KRASG12C compared with wild-type KRAS.9,24
Pharmacokinetics
Adagrasib (MRTX849) demonstrates linear pharmacokinetics.27,28 A single dose of 30 mg/kg to H358 xenograft-bearing mice resulted in modified fraction of KRASG12C 74% at 6 hours and 47% at 72 hours.24 This extended effect is consistent with irreversible inhibition of KRASG12C by adagrasib (MRTX849). The half-life is 25 hours after a single dose and 63 hours once steady state is reached.28 Importantly, adagrasib demonstrates CNS penetration.29,30
Preclinical Studies
Hallin et al utilized LCMS-based KRASG12C protein modification assay to show greater efficacy of adagrasib (MRTX849) when preloaded with GDP rather than GTP, supporting the notion that adagrasib (MRTX849) binds and stabilizes the inactive GDP-bound form.24
In a panel of KRASG12C -mutant cell lines and patient-derived xenograft models, adagrasib demonstrated a wide variability of IC 50 values ranging from 10 to 973 nmol/L within in vitro cell viability studies.24 Adagrasib (MRTX849) showed >1000 fold selectivity in inhibition of KRASG12C compared to other proteins. At 100 mg/kg/day dosing, adagrasib (MRTX849) achieved tumor regression in 17 of 26 (65%) in vitro KRASG12C models. Conversely, this dose had no anti-tumor activity in non-KRASG12C models, demonstrating the KRASG12C-dependent mechanism of action.
Clinical Trials
The KRYSTAL-1 (NCT03785249) open-label phase I/Ib expansion cohort trial enrolled patients with advanced/metastatic solid tumors harboring KRASG12C mutations previously treated with chemotherapy and anti-programmed cell death protein (PD)-1 agents. Patients with active brain metastases were excluded from the study although patients were eligible if CNS metastases were adequately treated and deemed neurologically stable. In the Phase I expansion, the recommended Phase 2 dose was 600 mg twice a day,16 which is 2-folds above the concentration associated with efficacy in resistant models and 5-folds above that in sensitive models.28
Recently reported, the results of the updated analysis of the phase I/II KRYSTAL-1 study had 116 patients with KRASg12c-mutated NSCLC receiving adagrasib 600 mg twice a day with 112 patients having measurable disease at baseline.29 At a median follow-up of 12.9 months, objective response rate (ORR) was 42.9%. The median duration of response was 8.5 months (95% CI 6.2–12.8), and median progression-free survival (PFS) was 6.5 months (95% CI 4.7–8.4). At median follow-up of 15.6 months, overall survival (OS) was 12.6 months (95% CI 9.2–51.8). Importantly, adagrasib demonstrated CNS activity in 19 patients who were enrolled with treated, stable CNS metastases to have an objective response rate (ORR) of 33% and a disease control rate of 85%.29,30 Concordance between systemic and intracranial disease control was 88%.30 In patients with brain metastases, median duration of intracranial response was 11.2 months (95% CI 2.99- not evaluable) and median PFS was 5.4 months (95% CI 3.3–11.6).29
Safety, Tolerability, and Adverse Events
Treatment-related adverse events were common (97.4% of participants), and grade 3 or higher adverse events occurred in 44.8% of the patients.29 The drug discontinuation rate was 6.9% owing to treatment-related adverse events. The most common adverse events include diarrhea (70.7%), nausea (69.8%), fatigue (59.5%), and vomiting (56.9%).
Within the cohort of 19 patients with brain metastases, treatment-related adverse events occurred in 96% of patients with grade 3 or higher events in 36% of patients.30 The most common adverse events were nausea (80%) and diarrhea (80%). Adverse events led to drug discontinuation in 4% of patients.
Other Agents
AMG-510 was the first selective KRASG12C inhibitor to receive accelerated approval for clinical practice. The phase I portion of the clinical trial produced 32% confirmed response with 56% of patients with stable disease.31 More than 90% of patients who received the highest dose of 960mg daily achieved disease control. In the Phase II portion, 960 mg daily dosing was utilized and resulted in 37.1% of patients with confirmed response and 80.6% of patients with disease control. Both phase I and phase II had similar PFS, in 6.3 months and 6.8 months, respectively. In both phases, grade 4 adverse events occurred in only one patient. The most common adverse events were elevations in LFTs, which downtrended upon dose reduction and steroid treatment (NCT03600883). Given the favorable safety profile, further studies are ongoing in different settings.
A newer generation KRASG12C inhibitor JNJ-74699157 (ARS-3248) is currently being studied in the phase I setting (NCT04006301) as well as KRASG12C inhibitors LY3499446 (NCT04165031), GDC-6036 (NCT04449874), and D-1553 (NCT04585035). Furthermore, the ras “on” inhibitors are under development. These inhibitors require a chaperone protein called cyclophilin. The drugs make a binary complex with cyclophilin that in turn attaches to the KRAS on-state. Revolution Medicine has introduced the first in class RAS-ON inhibitors and these include RMC6291, a G12C on-inhibitor, RMC9805, a G12D on-inhibitor, RMC8839, a G13C on-inhibitor, and RMC6236, a tricomplex RAS-“Multi”-ON inhibitor which is expected to work beyond G12C including G12D, G12V and G12R.32
Future Directions
While the accelerated approval of sotorasib and the clinical activity demonstrated by adagrasib were major breakthroughs in the field of KRAS, unfortunately, the development of resistance to adagrasib and other KRAS G12C inhibitors is inevitable and can occur “on target” as well as both upstream sites (EGFR, HER2, FGFR) and downstream sites (MAPK/MEK pathway). Resistance mechanisms may also occur by means of bypass mechanisms such as MET amplifications and oncogenic fusions such as ALK and RET.33 Some notable “on-target” resistance mutations include r68, H85, and Y96. In particular, the Y96D mutation has been described as a mechanism of resistance to sotorasib, disrupting the hydrogen bond between Y96 and the carboxyl group of sotorasib, thereby affecting the switch pocket.34 Another study exposed 142 Ba/F3 cells, transduced with KRASG12C via retroviruses, to sotorasib or adagrasib, and looked for secondary resistance mutations.35 Twelve secondary mutations were identified in 124 clones. Y96D and Y96S mutations were found to be resistant to both KRAS G12C inhibitors. The addition of BI-3406, a SOS1 inhibitor, plus trametinib was able to provide activity against these resistances.
In order to overcome these resistances, utilization of aforementioned upstream and downstream pathways as well as other mechanisms (ie, checkpoint inhibitors) to develop combination strategies to enhance the activity of existing KRAS inhibitors is being evaluated. Indeed, a combination study of RMC4630 (SHP2 inhibitor) and sotorasib (NCT05054725) and similar studies are ongoing. SHP2 has a role in recycling KRAS between inactive and active form.36 As G12C inhibitors target the inactive KRAS (or GDP-KRAS), therefore, SHP2 inhibition is anticipated to keep more GDP bound for drugs like sotorasib or adagrasib to work. Another potential target is Son of Sevenless (SOS), which is one of the partners of SHP2 and is a protein that is part of the RAS complex and plays a role in converting KRAS GDP to GTP. Attempts have been made to target SOS as well as pre-clinical and clinical studies in combination with G12C and SOS inhibitors are ongoing although SOS is a tricky target and has toxicity associated with its inhibition. In fact, KRYSTAL 14, a Phase 1 study (NCT04975256) combining adagrasib with BI1701963 (SOS1 inhibitor) and a few other studies utilizing BI1701963 (NCT04111458, NCT04835714, NCT04627142) have been abandoned or terminated due to concerns of toxicities. One must carefully watch the outcome of G12C inhibitor combinations with SOS inhibitors. Going further downstream in the KRAS pathway, a new approach of “ras clamp” strategies that simultaneously block both RAF and MEK (VS-6766/VerastemR) is also emerging. This strategy shows impressive blockade of MEK phosphorylation and studies with the clamp inhibitors are ongoing (NCT05074810, NCT05375994).
Multiple combination strategies are being developed to increase the efficacy of KRAS inhibition. In the meantime, equally important would be to identify signatures and co-mutations that could predict outcomes to KRASG12C inhibitors, so that therapy could be tailored, either intensified or de-intensified for individual patients. For example, tumors with KRAS and p53 mutations carry inflammatory signatures and have been postulated to benefit from immune checkpoint inhibitor combination therapies, while tumors with mutations in KRAS and STK11 are thought to be immune inert. These observations merge the KRAS and immunotherapy fields together and multiple studies are being done to evaluate the combination of G12C inhibitors and immunotherapy (NCT04613596, NCT04185883, NCT04449874, NCT04699188, NCT04956640). Furthermore, analysis is needed to determine additional biomarkers, ideally from circulating tumor DNA, that could correlate with treatment efficacy and resistance to identify appropriate target populations. Table 1 summarizes select ongoing studies utilizing adagrasib.
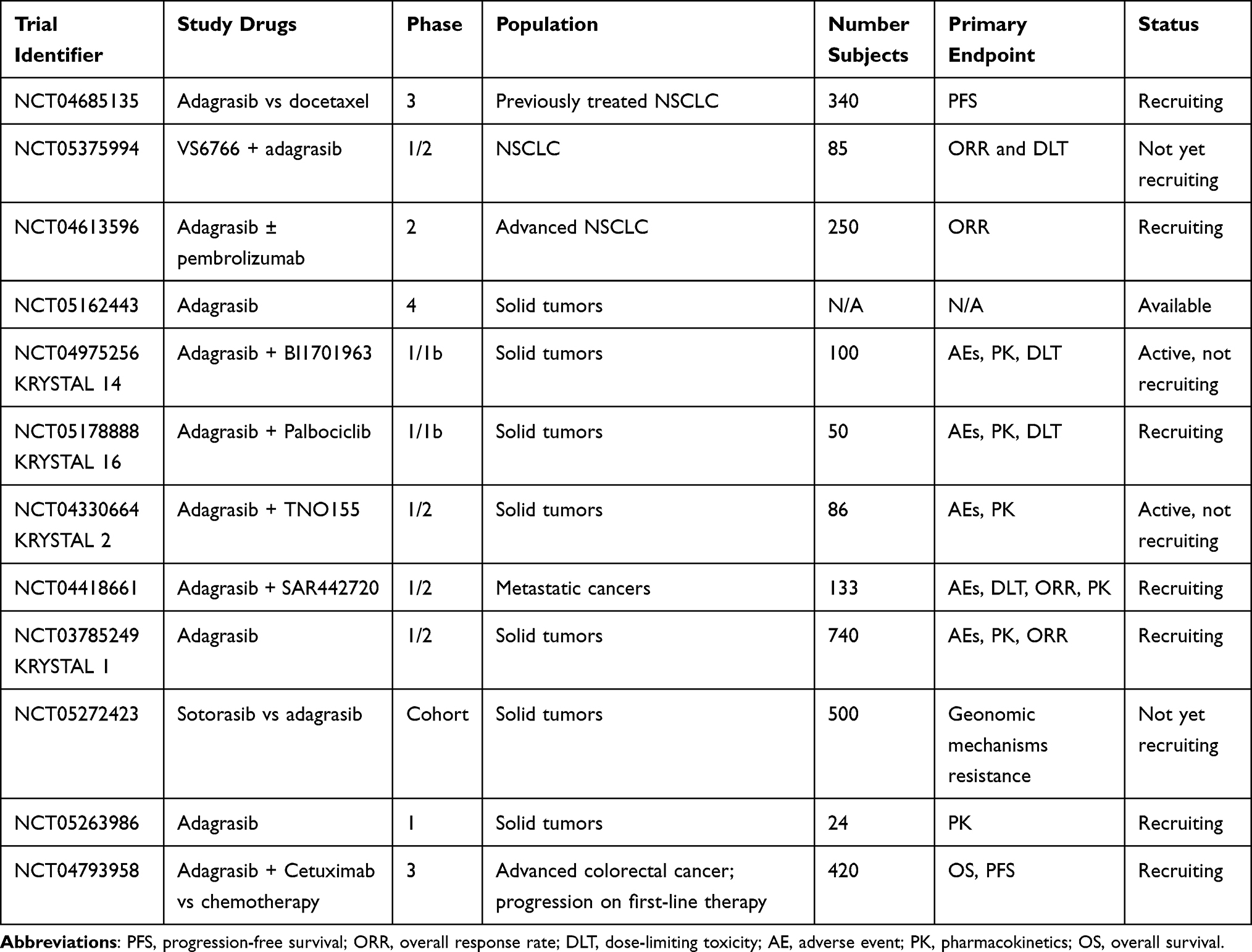 |
Table 1 Ongoing Trials of MRTX849/Adagrasib |
Conclusion
As shown by Janne et al, adagrasib (MRTX849) was shown to have significant clinical activity in heavily pretreated patients and even demonstrated objective responses in patients without response to prior treatments.29 Based on these promising results from the phase I/II KYRSTAL-1 trial, expansion cohorts for KRASG12C–mutated NSCLC and CRC are ongoing. While having two new potent drugs of sotorasib and adagrasib in the field of KRAS was a recent major breakthrough, more needs to be done to overcome the inevitable resistance of KRAS inhibition and to come up with more efficacious and better tolerated strategies with focus on individualized genomic approach.
Abbreviations
CNS, central nervous system; CI, confidence interval; GDP, guanosine diphosphate; GTP, guanosine triphosphate; IC50, cellular half-maximal inhibitory concentration; KRAS, Kirsten rat sarcoma virus; MAPK, mitogen-activated protein kinase; NR, not reached; NSCLC, non-small cell lung cancer; ORR, objective response rate; OS, overall survival; PD-1, programmed cell death 1; PFS, progression-free survival; RAS, rat sarcoma.
Ethics Approval and Consent to Participate
This report did not meet criteria for IRB approval.
Data Sharing Statement
Not applicable to this study.
Funding
No funding was secured for this report.
Disclosure
Dr Misako Nagasaka reports personal fees from AstraZeneca, Caris Life Sciences, Daiichi Sankyo, Takeda, Novartis, EMD Serono, Blueprint Medicines, Janssen, Pfizer, Lilly, Genentech, and Tempus; non-financial support from An Heart, outside the submitted work. The authors declare that they have no other competing interests in this work.
References
1. American Cancer Society. Cancer facts and figures 2021; 2021. Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2021/cancer-facts-and-figures-2021.pdf.
2. Imielinski M, Berger A, Hammerman P, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi:10.1016/j.cell.2012.08.029
3. Lee B, Lee T, Lee SH, et al. Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6595 lung cancers. Oncotarget. 2016;7(17):23874–23884. doi:10.18632/oncotarget.8074
4. Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2(3):344–358. doi:10.1177/1947601911411084
5. Tímár J. The clinical relevance of KRAS gene mutation in non-small-cell lung cancer. Curr Opin Oncol. 2014;26(2):138–144. doi:10.1097/CCO.0000000000000051
6. Dearden S, Stevens J, Wu Y-L, et al. Mutation incidence and coincidence in non small-cell lung cancer, meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24(9):2371–2376. doi:10.1093/annonc/mdt205
7. Yang H, Liang S-Q, Schmid RA, et al. New Horizons in KRAS-mutant lung cancer, dawn after darkness. Front Oncol. 2019;9:953. doi:10.3389/fonc.2019.00953
8. Matikas A, Mistriotis D, Georgoulias V, et al. Targeting KRAS mutated non-small cell lung cancer, A history of failures and a future of hope for a diverse entity. Crit Rev Oncol Hematol. 2017;110:1–12. doi:10.1016/j.critrevonc.2016.12.005
9. Shepherd FA, Domerg C, Hainaut P, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and kras mutation subtype in early-stage resected non–small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31(17):2173–2181. doi:10.1200/JCO.2012.48.1390
10. Redig AJ, Chambers ES, Lydon CA, et al. Genomic complexity in KRAS mutant non-small cell lung cancer (NSCLC) from never/light-smokers v smokers. J Clin. 2016;34(15_suppl):9087. doi:10.1200/JCO.2016.34.15_suppl.9087
11. Ferrer I, Zugazagoitia J, Herbertz S, et al. KRAS-Mutant non-small cell lung cancer, From biology to therapy. Lung Cancer. 2018;124:53–64. doi:10.1016/j.lungcan.2018.07.013
12. Mascaux C, Iannino N, Martin B, et al. The role of RAS oncogene in survival of patients with lung cancer, a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92(1):131–139. doi:10.1038/sj.bjc.6602258
13. Jacobs F, Cani M, Malapelle U, et al. Targeting KRAS in NSCLC, old failures and new options for “Non-G12c”Patients. Cancers. 2021;13(24):6332. doi:10.3390/cancers13246332
14. Nassar AH, Adib E, Kwiatkowski DJ. Distribution of KRASG12C somatic mutations across race, sex, and cancer type. N Engl J Med. 2021;384(2):185–187. doi:10.1056/NEJMc2030638
15. Salem M, El-Refai S, Sha W, et al. O-3 Characterization of KRAS mutation variants and prevalence of KRAS-G12C in gastrointestinal malignancies. Ann Oncol. 2021;32:S218. doi:10.1016/j.annonc.2021.05.007
16. Ou SH, Jänne PA, Leal TA, et al. First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRASG12C solid tumors (KRYSTAL-1). J Clin. 2022;40(23):2530.
17. Finn SP, Addeo A, Dafni U, et al. Prognostic impact of KRAS G12C mutation in patients with NSCLC, results from the European thoracic oncology platform lungscape project. J Thorac Oncol. 2021;16(6):990–1002. doi:10.1016/j.jtho.2021.02.016
18. Wu MY, Zhang EW, Strickland MR, et al. Clinical and imaging features of non-small cell lung cancer with G12C KRAS mutation. Cancers. 2021;3(14):3572.
19. Cui W, Franchini F, Alexander M, et al. Real world outcomes in KRAS G12C mutation positive non-small cell lung cancer. Lung Cancer. 2020;146:310–317. doi:10.1016/j.lungcan.2020.06.030
20. Tomasini P, Serdjebi C, Khobta N, et al. EGFR and KRAS mutations predict the incidence and outcome of brain metastases in non-small cell lung cancer. Int J Mol Sci. 2016;17(12):2132. doi:10.3390/ijms17122132
21. Dang CV, Reddy EP, Shokat KM, et al. Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer. 2017;17(8):502–508. doi:10.1038/nrc.2017.36
22. Lindsay CR, Jamal-Hanjani M, Forster M, et al. KRAS, Reasons for optimism in lung cancer. Eur J Cancer. 2018;99:20–27. doi:10.1016/j.ejca.2018.05.001
23. Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi:10.1038/nature12796
24. Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) Inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10(1):54–71. doi:10.1158/2159-8290.CD-19-1167
25. Nakajima EC, Drezner N, Li X, et al. FDA approval summary: sotorasib for KRAS G12C -mutated metastatic NSCLC. Clin Cancer Res. 2022;28(8):1482–1486. doi:10.1158/1078-0432.CCR-21-3074
26. Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate MRTX849, a covalent KRAS G12C inhibitor for the treatment of Cancer. J Med Chem. 2020;63(13):6679–6693. doi:10.1021/acs.jmedchem.9b02052
27. Riely GJ, Ou S-HI, Rybkin I, et al. 99O_PR KRYSTAL-1, Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021;16(4):S751–S752. doi:10.1016/S1556-0864(21)01941-9
28. Janne PA, Papadopoulos K, Ou I, et al. A phase 1 clinical trial evaluating the pharmacokinetics (PK), safety, and clinical activity of MRTX849, a mutant-selective small molecule KRAS G12C inhibitor, in advanced solid tumors.
29. Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in non–small-cell lung cancer harboring a KRAS G12C mutation. N Engl J Med. 2022;387:120–131. doi:10.1056/NEJMoa2204619
30. Sabari JK, Velcheti V, Shimizu K, et al. Activity of Adagrasib (MRTX849) in brain metastases, preclinical models and clinical data from patients with KRASG12C-mutant non–small cell lung cancer. Clin Cancer Res. 2022;28(15):3318–3328.
31. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384(25):2371–2381. doi:10.1056/NEJMoa2103695
32. Nagasaka M, Azmi AS. Clinical progress of KRAS-targeted therapies, what next? Future Med Chem. 2022;14(15):1107–1110.
33. Awad MM, Liu S, Rybkin II, et al. Acquired resistance to KRAS G12C inhibition in cancer. N Engl J Med. 2021;384(25):2382–2393. doi:10.1056/NEJMoa2105281
34. Tanaka N, Lin JJ, Li C, et al. Clinical acquired resistance to KRAS(G12C) Inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021;11(8):1913–1922. doi:10.1158/2159-8290.CD-21-0365
35. Koga T, Suda K, Fujino T, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies insights from in vitro experiments. J Thorac Oncol. 2021;16(8):1321–1332. doi:10.1016/j.jtho.2021.04.015
36. Zhang J, Zhang F, Niu R. Functions of Shp2 in cancer. J Cell Mol Med. 2015;19(9):2075–2083. doi:10.1111/jcmm.12618
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Path to Personalized Treatment in KRAS-Mutant Non-Small Cell Lung Cancer: A Review of Targeted Therapies and Immunotherapy
Shu CL, Liu YL
Cancer Management and Research 2022, 14:3485-3492
Published Date: 16 December 2022
Comparative Cost Analysis for Direct Medical Costs of Protocol Administration of Non-Small Cell Lung Cancer Treatment Regimens in Curative Intent: A Micro-Costing Study in Jordan
Madae’en SS, Salem AA, Ararawi NS, Ramzi EJ, Aloueedat RF, Saabenh AM, Allouzi DA, Abuoudeh RH, Hnaif OE, Musa LM, Alshdaifat SH, Al-Tanashat AJ, Almasa’afeh HY, Abuallaban SM
ClinicoEconomics and Outcomes Research 2025, 17:455-471
Published Date: 12 July 2025
Key Considerations for Targeting KRAS in Pancreatic Cancer: Potential Impact on the Treatment Paradigm
Eslinger C, Sonbol MB, George B, Babiker H, Borad MJ, Bekaii-Saab TS
Drug Design, Development and Therapy 2026, 20:559325
Published Date: 27 March 2026
Targeted Therapy–Induced Interstitial Lung Disease in NSCLC: Mechanisms, Clinical Signatures, and a Precision Medicine Roadmap
Wang JF, Jiang LL, Wang MC, Li YL
Drug Design, Development and Therapy 2026, 20:600434
Published Date: 8 April 2026