Back to Journals » Journal of Inflammation Research » Volume 14
Induction of IL-25 Expression in Human Nasal Polyp Epithelium by Influenza Virus Infection is Abated by Interferon-Alpha Pretreatment
Authors Hong H ![]() , Tan KS, Yan Y, Chen F, Ong HH, Oo Y, Liu J, Ong YK, Thong M, Sugrue R, Chow VT
, Tan KS, Yan Y, Chen F, Ong HH, Oo Y, Liu J, Ong YK, Thong M, Sugrue R, Chow VT ![]() , Wang DY
, Wang DY ![]()
Received 29 January 2021
Accepted for publication 8 June 2021
Published 28 June 2021 Volume 2021:14 Pages 2769—2780
DOI https://doi.org/10.2147/JIR.S304320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Haiyu Hong,1– 3,* Kai Sen Tan,3– 6,* Yan Yan,3,7,8,* Fenghong Chen,2,* Hsiao Hui Ong,3,5 Yukei Oo,4 Jing Liu,3,5 Yew Kwang Ong,3,9 Mark Thong,3,9 Richard Sugrue,10 Vincent T Chow,4,5 De Yun Wang3,5
1Allergy Center, Department of Otolaryngology, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, People’s Republic of China; 2Otorhinolaryngology Hospital, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, People’s Republic of China; 3Department of Otolaryngology, Yong Loo Lin School of Medicine, National University of Singapore, National University Health System, Singapore; 4Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore, National University Health System, Singapore; 5NUHS Infectious Diseases Translational Research Program, Yong Loo Lin School of Medicine, National University of Singapore, Singapore; 6Biosafety level 3 Core Facility, Yong Loo Lin School of Medicine, National University Health System, National University of Singapore, Singapore; 7Guangdong Provincial Key Laboratory of Biomedical and Guangdong Engineering Research Center of Molecular Imaging, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, People’s Republic of China; 8Central Laboratory, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, People’s Republic of China; 9Department of Otolaryngology Head & Neck Surgery, National University Hospital, National University Health System, Singapore; 10School of Biological Sciences, Nanyang Technological University, Singapore
*These authors contributed equally to this work
Correspondence: Vincent T Chow
Department of Microbiology and Immunology, National University of Singapore, 5 Science Drive 2, Singapore, 117545, Singapore
Email [email protected]
De Yun Wang
Department of Otolaryngology, National University of Singapore, National University Health System, 1E Kent Ridge Road, Singapore, 119228, Singapore
Email [email protected]
Background: Epithelial cytokines including IL-25, IL-33 and thymic stromal lymphopoietin (TLSP) are recently established as drivers of type 2 chronic inflammatory diseases such as chronic rhinosinusitis with nasal polyps (CRSwNP). Here, we further confirmed the increased expression of IL-25 in CRSwNP and investigated potential contributors of IL-25 in CRSwNP epithelium.
Methods: Sixty CRSwNP, 25 CRSsNP and 15 healthy control tissues were examined for IL-25 expression and for the accompanying type 2 inflammatory cytokines. We then tested different respiratory virus infections on human nasal epithelial cells (hNECs) for their ability to trigger IL-25 expression. In addition, we subjected hNECs generated from CRSwNP tissues to pretreatment with recombinant interferon-alpha (IFN-α) prior to viral infection to evaluate IFN effects on IL-25 induction.
Results: We confirmed that significantly enhanced levels of IL-25 were observed in CRSwNP tissues, and that IL-25 expression correlated with type 2 inflammatory cytokine expression. In vitro, we observed significantly elevated IL-25 in hNECs infected with influenza A virus as early as 24 hours post-infection (hpi), regardless of tissue origin, and IL-25 correlated positively with viral load. While other respiratory viruses exhibited increasing trends of IL-25, these were not significant at the time-points tested. IFN-α treatment of CRSwNP epithelium was found to exert bimodal effects, ie IFN-α treatment alone induced moderate IL-25 expression, whereas IFN-α pretreatment of hNECs before influenza infection significantly diminished IL-25 induction by active influenza virus infection.
Conclusion: We have authenticated the observation of elevated IL-25 in CRSwNP, which is correlated with type 2 inflammatory cytokines. Notably, we identified influenza virus infection as a potential contributor of IL-25 in both control and CRSwNP epithelium during active infection. This IL-25 induction can be abated by IFN-α pretreatment which ameliorated active influenza infection.
Trial Registration: Chictr.org.cn ChiCTR-BON-16010179, Registered 18 December 2016, http://www.chictr.org.cn/showproj.aspx?proj=17331. The authors agree on the sharing of deidentified participant data where it pertains to request directly related to the data in this article when contacted (Haiyu Hong; [email protected]).
Keywords: chronic rhinosinusitis with nasal polyps, interferon-alpha, interleukin 25, respiratory viruses, influenza virus, type 2 inflammation
Introduction
Chronic rhinosinusitis (CRS) is one of the most common diseases in the upper respiratory tract, with an estimated prevalence of about 10% globally.1–3 CRS is classified as CRS with or without nasal polyps (CRSwNP or CRSsNP), based on clinical examination of visible NP in the nasal meatus.3 CRSwNP is an inflammatory condition of the paranasal sinuses that lasts at least 12 weeks in duration, and the risk of CRSwNP exacerbation is increased by external pathogen infections, including bacterial, viral, and fungal infections.3 Although emerging evidence implicates numerous cytokines and chemokines in the interactions of host and environmental factors during occurrence and exacerbation of CRSwNP, factors that contribute to and perpetuate type 2 inflammation in existing CRSwNP are not fully elucidated.4
The nasal epithelium plays crucial roles in the host defenses by forming a physical barrier that mechanically clears foreign materials via its mucociliary functions.5 In addition, human nasal epithelial cells (hNECs) are also an important source of cytokines and chemokines, including the recently discovered trio of epithelial-derived cytokines (thymic stromal lymphopoietin or TSLP, IL-25, and IL-33) that are highly associated with type 2 inflammation. Among them, IL-25 (also known as IL-17E) is a member of the IL-17 cytokine family, and plays a variety of roles in different inflammatory disorders such as asthma, atopic dermatitis, pulmonary fibrosis, and CRSwNP.6,7 It was shown that intraperitoneal or intranasal administration of IL-25 protein results in the accumulation of eosinophils or production of type 2 inflammatory cytokines in the bronchoalveolar lavage fluid and the lung tissues.8 Conversely, blocking IL-25 decreases type 2 inflammatory cytokine production in a murine asthma model.9 These studies suggest that that IL-25 may be an important biomarker in the diagnosis and management of CRSwNP. However, the potential triggers in CRSwNP epithelium that can cause the expression of IL-25 are poorly understood.
Respiratory viruses are well-established triggers of exacerbation of chronic airway inflammatory diseases.10 In addition, as the primary target cells for respiratory viruses,10,11 the hNECs also play roles in mediating antiviral innate and adaptive immunity that orchestrate both the type 1 and type 2 inflammatory responses at the local mucosa site.12 Interestingly, while viral infection typically triggers type 1 inflammatory responses geared towards viral clearance, they have also been shown to trigger the expression of type 2 inflammatory cytokines.10,13,14 Therefore, it is interesting to interrogate their roles in inducing IL-25 in CRSwNP epithelium which may contribute to the increased accumulation of local IL-25 at the diseased site.
Recently, there is increasing evidence to support the association between respiratory viruses and CRSwNP, especially in Asian cohorts.15–17 Hence, our study aims to reaffirm the importance of IL-25 in CRSwNP pathogenesis; as well as to implicate respiratory viruses as potential triggers of IL-25 expression in local CRSwNP epithelium. We hypothesize that IL-25 expression is a part of the immune response repertoire in the nasal epithelium during virus infection due to its role in epithelial injury and barrier damage. This in turn contributes to the accumulation of IL-25 and its associated type 2 inflammatory cytokines in the local CRSwNP environment. At the same time, given that anti-infective agents are proposed as a means of managing chronic airway inflammatory diseases,16,18 we therefore investigated the antiviral effect of IFN-α and whether it can modulate IL-25 expression in hNECs infected with respiratory viruses.
Materials and Methods
This study was approved by the Ethical Committees of the First and Fifth Affiliated Hospitals of Sun Yat-sen University (clinical trial registration no. ChiCTR-BON-16010179 and NCT02110654), and National University of Singapore (National Healthcare Group Domain-Specific Board of Singapore, DSRB Reference no. D/11/228, and Institutional Review Board of the National University of Singapore, IRB code 13–509) in accordance with the Declaration of Helsinki. Written informed consent was obtained from each subject.
Patients and Tissue Samples
60 CRSwNP patients and 25 CRSsNP patients were recruited from the First and Fifth Affiliated Hospitals of Sun Yat-sen University (Guangzhou and Zhuhai, China). Diagnosis of CRSwNP and CRSsNP was carried on the basis of on disease history, nasal endoscopy, and computed tomography scan of the paranasal cavities, in accordance with the current European position paper on rhinosinusitis and nasal polyps.2,3 The atopic status was evaluated by skin prick test (SPT) and serum IgE levels (Phadia, Uppsala, Sweden) specific to common inhaled allergens (eg pollens, house dust mites, pets, molds, and cockroaches). For healthy controls, 15 subjects with traumatic optic neuropathy were enrolled, and uncinate process tissues were sampled during endoscopic optic nerve decompression. Peripheral blood samples were collected before surgery.
Immunohistochemical Staining
Sino-nasal tissues were embedded in paraffin and cut into 5-μm thick sections. Hematoxylin-eosin (HE) staining and immunochemistry using peroxidase-labeled streptavidin-biotin technique were performed for histological analyses, following published protocol.7
Real-Time Quantitative PCR
The mRNA expression profiles of target genes were determined using real-time quantitative PCR (RT-qPCR) as previously described.19 Briefly, total RNA was extracted from the tissue or cell samples using mirVana miRNA isolation kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions. Reverse transcription was performed to synthesize cDNA from 2 µg of total RNA using oligo(dT) 18 primer and M-MLV reverse transcriptase (Takara, Shiga, Japan). Expression of mRNA was determined by qPCR using the ABI PRISM 7500 Detection System (Applied Biosystems, Foster City, CA, USA) and SYBR Premix TaqTM (Takara). The mean threshold cycle (Ct) values were normalized to β-actin and PGK1 housekeeping genes for clinical specimens and hNEC samples, respectively. The relative mRNA levels of target genes were analyzed using the formula 2−ΔΔCt. Experiments were performed in triplicate for each datapoint. The primer sequences used in the study for IL-25, IL-17RB, IL-4, IL-5, IL-13, IFN-γ, IL-17A, intercellular adhesion molecule 1 (ICAM-1), interferon regulatory factor 7 (IRF7), retinoic acid-inducible gene I (RIG-I), Toll-like receptors (TLR) TLR3 and TLR7, β-actin, and PGK1 are listed in Table S1.
Luminex-Based Multiplex Assay
Cytokines and chemokines from polyp and uncinated process tissues were measured using Luminex-based multiplex assay. Briefly, 30 mg of each sample was homogenized in 0.5 mL lysis buffer supplemented with protease inhibitor (Millipore, Burlington, MA, USA) for 1 minute on ice and centrifuged for 20 minutes at 4000 rpm at 4°C. Supernatants were collected, and total protein concentrations in the supernatants were determined by standard BCA assay normalized to a concentration of 50 μg total protein per sample. All samples were analyzed for IL-25 (IL-17E), IL-4, IL-5, IL-13, IFN-γ, and IL-17A using Luminex-based Multiplex kits (Millipore), following the manufacturer’s protocol. IL-25 levels from supernatants (apical and basal) of influenza virus-infected hNECs were detected using the human IL-17E/IL-25 XL Magnetic Luminex performance assay (R&D Systems, Minneapolis, MN, USA) according to manufacturer’s protocol.
Virus Infection and Antiviral Pretreatment with Recombinant IFN-α Protein in hNECs Model
Infections with influenza virus, rhinovirus and respiratory syncytial virus (RSV) were performed as previously described.11,20,21 Briefly, in vitro differentiated hNECs cultured in Transwells were infected at multiplicity of infection (MOI) of 0·1 (influenza A/Aichi/2/1968/H3N2 strain), 2·5 (rhinovirus RV16 strain), and 3·0 (RSV A2 strain), and incubated at 33°C (rhinovirus), and 35°C (influenza and RSV) for 1 hour, respectively. The inocula were then removed, and infected hNECs were incubated for 24 or 48 hours. Pretreatment with recombinant human IFN-α 2A protein expressed in HEK293 cells (Sigma-Aldrich, St Louis, MO, USA) was performed 48 hours prior to viral infection. The cells were first subjected to pretreatment with IFN-α2A at four different concentrations (5, 25, 50, 100 ng/mL) for 48 hours to determine the optimal/effective concentration, before selecting 5 and 25 ng/mL for the main pretreatment experiments.
PrestoBlueTM Cell Viability Assay
PrestoBlueTM cell viability assay was conducted to check if any toxicity effect was induced by IFN-α2A treatment. Cell viability assay using PrestoBlueTM reagent (Life Technologies) was performed following the manufacturer’s protocol, as previously described.22 A “no cell” control (1× working solution without incubation with cells) was used as the baseline fluorescence value. The fluorescence reading from the mock treatment group was used as a negative control for normalization.
Virus Plaque Assay
Viral plaque assay was performed as previously described.20 Plaque-forming unit (PFU) values were calculated as follows: Number of plaques × Dilution factor = Number of PFU per 100 µL. The final data were presented as PFU per mL.
Cytospin and Viral Staining
Cytospin and viral staining were performed as previously described.20 Briefly, at each time-point, single-cell suspensions (1–2 × 105 cells) were dissociated from Transwells using 0.5× Trypsin/EDTA solution (Gibco, Carlsbad, CA, USA) at 37°C. Dissociated cells were fixed in 4% paraformaldehyde at room temperature for 10 minutes, followed by 2 rounds of washing with 1× DPBS, and centrifuged at 3000 rpm for 5 minutes. Cytospin preparations (2 × 104 cells per slide) were prepared at 500 rpm for 5 minutes with mild acceleration with Shandon Cytospin 3 Cytocentrifuge (Thermo Fisher Scientific, Waltham, MA, USA). Mouse anti-influenza A nucleoprotein antibody (Abcam, Cambridge, MA, USA) was used at 1:200 dilution for immunofluorescence (IF) staining to visualize viral replication in host cells, while rabbit anti-IL-25 antibody (Abcam) was used at 1:250 dilution to analyze IL-25 protein expression. Alexa Fluor 488 (anti-rabbit) and Alexa Fluor 594 (anti-mouse)-labeled IgG (H1L) secondary antibodies (Life Technologies) were both used at 1:500 dilutions for IF staining. ProLong AntiFade mounting medium with DAPI (Life Technologies) was used for mounting of the stained slides.
Statistical Analyses
Clinical data were expressed as median and interquartile range (IQRs) and analyzed using the Kruskal–Wallis H-test and nonparametric Mann–Whitney U-test, unless otherwise stated. For in vitro infection assays, the data were expressed as the means and standard errors of the mean (SEM). One-way analysis of variance (ANOVA) and paired or unpaired Student’s t-test were used for the statistical analyses. For in vitro pretreatment assays, two-way ANOVA was employed to assess the differences between treatment and infection groups. A p-value of less than 0.05 was considered statistically significant.
Results
Increased IL-25 in CRSwNP Promotes Type 2 Inflammation in NP Tissues
Sixty CRSwNP patients, 25 CRSsNP patients, and 15 healthy control subjects were enrolled in this study. The demographic data of all subjects are shown in Table S2. Our results revealed that both protein and mRNA levels of IL-25 were significantly elevated in CRSwNP patients when compared to CRSsNP patients and healthy controls (p<0·05, Figure 1A–C). In addition, LUMINEX cytokine analysis showed that the protein levels of type 2 inflammatory cytokines (IL-4, IL-5, and IL-13) were only significantly increased in CRSwNP patients, affirming that IL-25 was more strongly associated with type 2 inflammation and was significantly more prevalent in CRSwNP (p<0·05, Figure 1D). The findings corroborated our previous study which confirmed the role of polyp IL-25 in inducing type 2 inflammatory cytokines.23 Moreover, we also further showed that IL-25 protein expression was positively correlated with expression of type 2 inflammatory cytokines, CT scores and sensitization to allergens, but not endoscopic scores (p<0·05, Figure 1E–H), indicating a strong correlation between IL-25, CRSwNP and type 2 inflammation, particularly allergy-driven ones. In addition, we further demonstrated that the mRNA and protein levels of IL-25 were well correlated with each other, indicating that the mRNA levels are truly indicative of the IL-25 protein expression (Figure 1I).
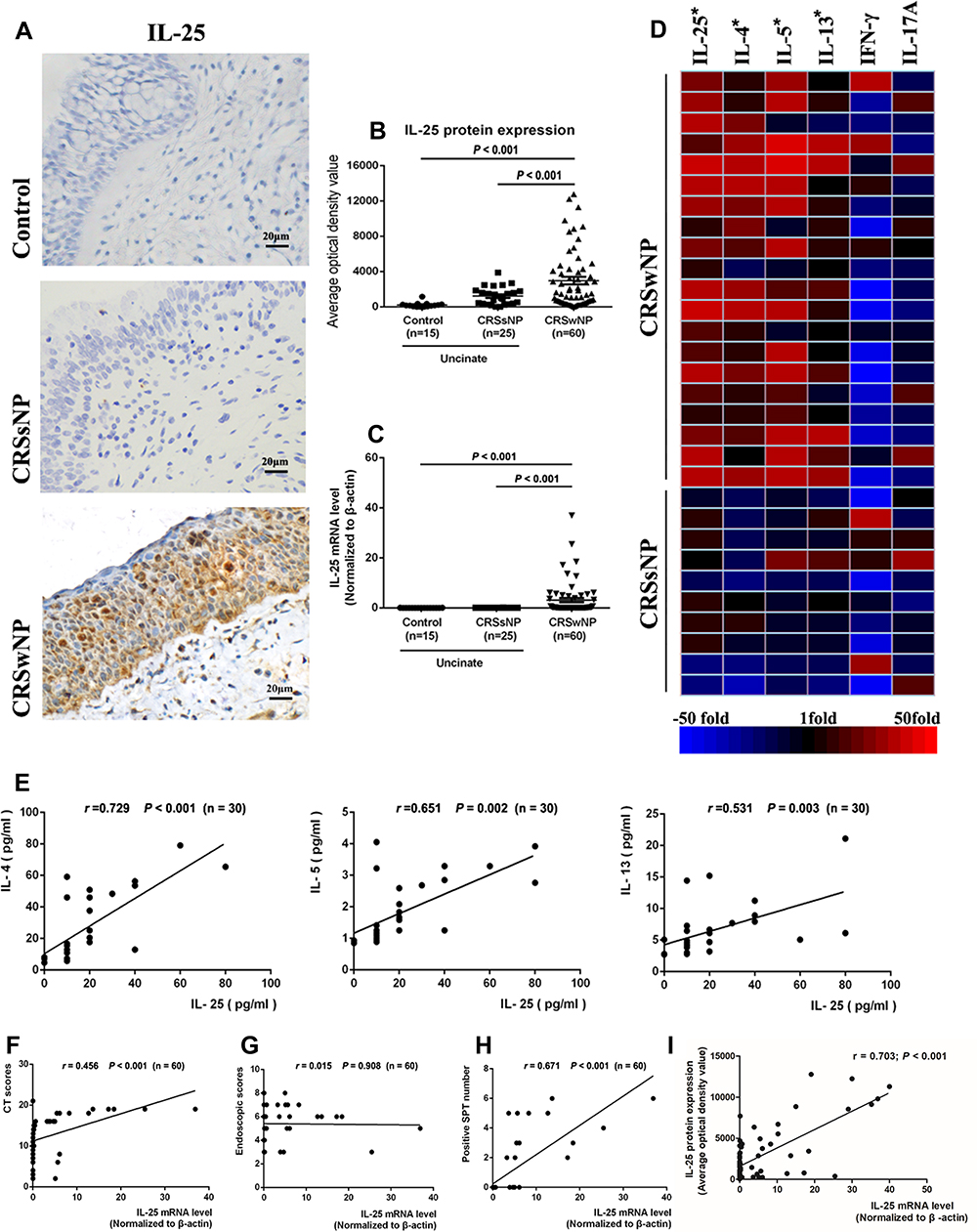 |
Figure 1 Increased IL-25 and type 2 inflammatory profiles in CRSwNP tissues (A and B) The expression of IL-25 in CRSwNP, CRSsNP and healthy control tissues (400× magnification) and its quantification via optical density of IL-25 positive staining. (C) IL-25 mRNA levels in CRSwNP, CRSsNP and control tissues. (D) Heatmap of selected cytokines as determined by Luminex-based multiplex assay, showing up- or down-regulation in CRSwNP and CRSsNP samples, compared to controls. (E) Correlation of levels of IL-25 and type 2 inflammatory cytokines IL-4, IL-5, and IL-13 in CRSwNP patients. (F–H) Correlation of IL-25 mRNA expression with CT score, endoscopic score, and number of sensitizations to common environmental allergens (indicated by SPT) in CRSwNP patients. (I) Correlation of IL-25 protein and mRNA levels from CRSwNP tissues. Data are expressed as median values (IQRs). Comparison of median values was performed using Kruskal–Wallis test and Mann-Whitney U-test. p < 0.05 is considered statistically significant. |
Influenza Virus Infection of hNECs in vitro Induce IL-25 Expression
It has been established that respiratory virus infection of an inflamed airway can exacerbate symptoms of chronic airway inflammatory diseases.24 Therefore, we investigated common respiratory viral infections (influenza, rhinovirus, RSV) of in vitro differentiated hNECs derived from control or CRSwNP tissues to investigate their ability to induce IL-25 (Table S3). Interestingly, among the respiratory viruses tested at the time-points in this study, H3N2 influenza virus stimulated significantly increased expression of IL-25 in hNECs derived from control and CRSwNP samples alike, when compared to rhinovirus (RV16) and RSV (A2) which exhibited weaker, non-significant increase of IL-25 (Figure 2A and B; Figure S1). As shown in Figure 3A, we observed significant fold changes of IL-25 expression at 24 hours and 48 hours post-infection (hpi) with influenza (p<0.05), which also positively correlated with IL-17RB (r=0.578, p<0.05, Figure 3B). Additionally, we also observed the same magnitude of significant increase in secreted IL-25 protein following influenza infection of hNECs in both apical and basal supernatants (Figure 3C). This corroborated with the tissue data, and confirmed strong IL-25 correlation at mRNA and protein levels. Therefore, for the subsequent investigation, we used IL-25 mRNA levels as a surrogate for IL-25 protein expression levels.
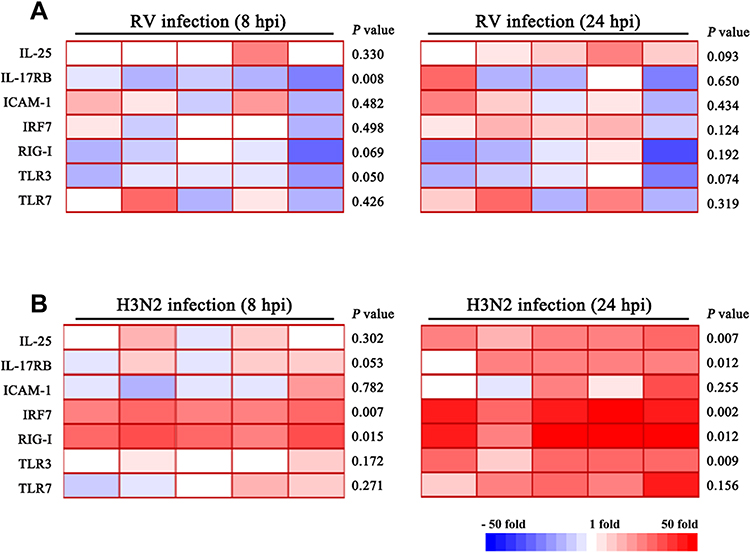 |
Figure 2 Heatmap of expression profiles of IL-25 and related innate immune factors in response to influenza and rhinovirus infection at 8 and 24 hpi. Cytokine mRNA level changes in (A) rhinovirus infection and (B) influenza infection. Greater significant changes in cytokine levels were observed in influenza compared to rhinovirus infection at early stages of infection of hNECs. Changes in expression of cytokines at various time-points of virus infections were determined by RT-qPCR (n=5). Red color denotes up-regulation, while blue color denotes down-regulation of the corresponding cytokine mRNA. Comparison of means was performed by Student’s t-test between infected and uninfected hNECs. p < 0.05 is considered statistically significant. |
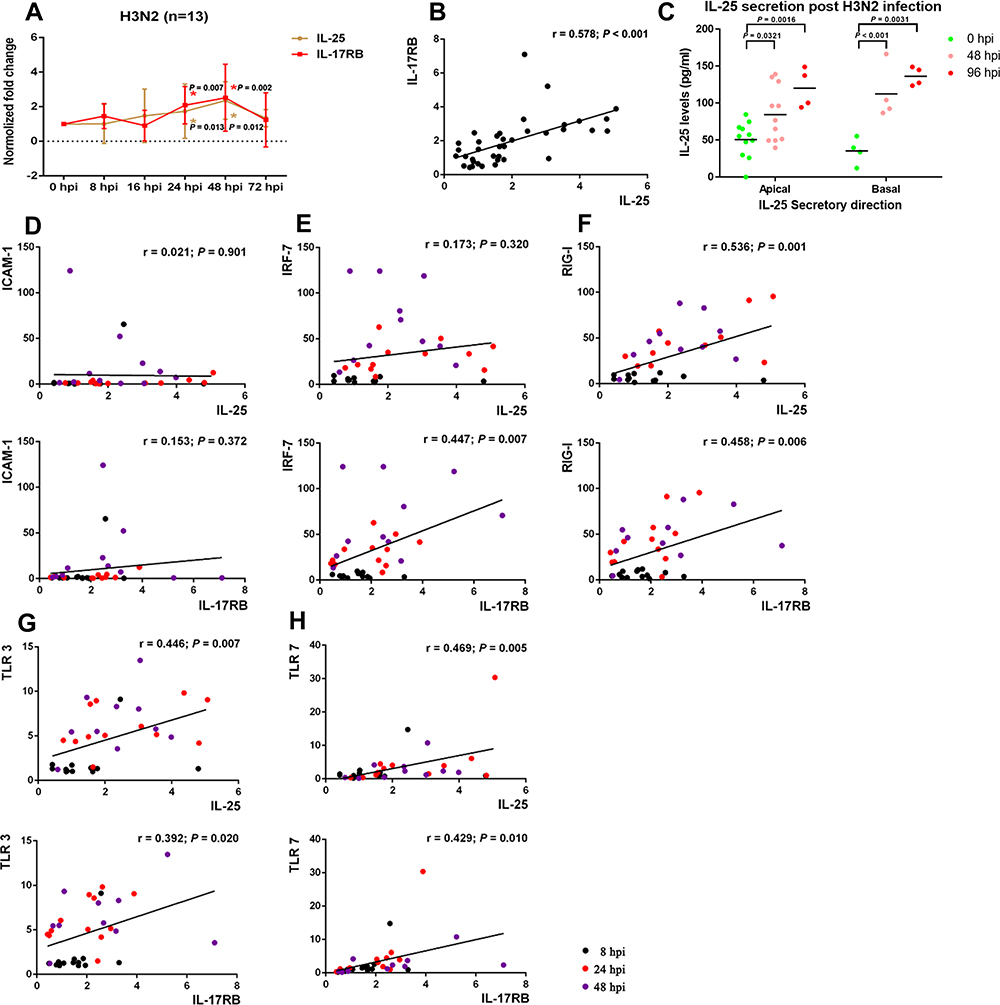 |
Figure 3 Significantly increased expression of IL-25 and IL-17RB upon influenza infection of hNECs. (A) The mRNA levels of IL-25 and IL-17RB at different time-points post-infection (n=13). (B) Correlation of IL-25 and IL-17RB mRNA expression following H3N2 influenza infection of hNECs. Comparison of means was performed by Student’s t-test between infected and uninfected hNECs. (C) Apical and basal secretions of IL-25 protein in supernatants of influenza virus-infected hNECs as detected by Luminex assay at different time-points post-infection. Comparison of means was performed by one-way ANOVA with Dunnett’s multiple comparison post-hoc test. (D–H) Correlation of mRNA levels of IL-25 and IL-17RB with the five most reactive markers as determined by RT-qPCR. p < 0.05 is considered statistically significant. |
To confirm that the induction of IL-25 was associated with virus infection and its host response, we also evaluated the correlation of IL-25 and IL-17RB transcripts and the five most sensitive host response markers16,20,25 at time-points of 8, 24, and 48 hpi after influenza infection. While ICAM1 and IRF7 were negatively correlated with IL-25 and IL-17RB (Figure 3D and E), the expression of viral sensors RIG-I, TLR3 and TLR7 was positively correlated with IL-25 and IL-17RB (p<0.05, Figure 3F–H), signifying that the expression profiles of IL-25 and IL-17RB were associated with the induction of antiviral response in hNECs, particularly with influenza which induced strong antiviral responses as early as 24 hpi (Figure 2).
Induced Expression of IL-25 Correlates with Influenza Virus Replication in hNECs Derived from CRSwNP Tissues
In order to evaluate the relationship between IL-25 expression with replication dynamics of H3N2 influenza virus in NP epithelium, recombinant protein of IFN-α, an FDA-approved antiviral drug for hepatitis C (INTRON®A), was used to inhibit virus replication in infected hNECs derived from CRSwNP donors.16,26 The effective concentration of human recombinant IFN-α protein and pretreatment duration were screened during pilot experiments, and low doses of 5 and 25 ng/mL were employed for the inhibition of virus replication throughout the antiviral study, as shown by fold changes in virus titers (Figure S2). The viability of the hNECs following low-dose IFN-α treatment was reduced which was expected with IFN treatment, but this decreased viability was not significantly different than that of the infected cells (Figure S3). The heatmap of mRNA levels of pathogen sensors is shown in Figure 4A – we observed significant reduction of IL-25 expression at 24 and 48 hpi (Figure 4B), but not IL-17RB (Figure 4C) in CRSwNP hNECs pretreated with IFN-α. The results revealed that IL-25 was consistently elevated following influenza infection, but not IL-17RB which showed a different trend as in previous results (Figure 3A). The discrepancies suggested that IL-17RB expression may be more susceptible to batch variation, and its expression may change more dynamically and may not be entirely dependent on influenza infection. IFN-α treatment alone increased IL-25 expression, which was relatively lower than influenza-induced IL-25 expression. Interestingly, the increased IL-25 by influenza infection was abated by the pretreatment of CRSwNP hNECs with IFN-α. This suggests bimodal effects of IFN-α, where its treatment alone moderately enhanced IL-25, whereas IFN-α pretreatment of hNECs instead suppressed the IL-25 expression induced by influenza. The bimodal effects may also explain the different levels of inhibition between 5 and 25 ng/mL of IFN-α at different time-points, in which the higher concentration treatment initially led to increased IL-25 (24 hpi), followed by stronger modulation of influenza-induced IL-25 at the later time-point (48 hpi). Congruent with this, we showed that the reduction of IL-25 was accompanied by efficient inhibition of influenza virus replication by a low dose of IFN-α (Figure 4D), suggesting a virus-dependent induction of IL-25 in differentiated CRSwNP epithelium. The induced expression of IL-25 by influenza infection in hNECs was further illustrated in Figure S4 (24 hpi) – indicated by the co-localization of IL-25 and influenza viral-nucleoprotein on cytospin slides.
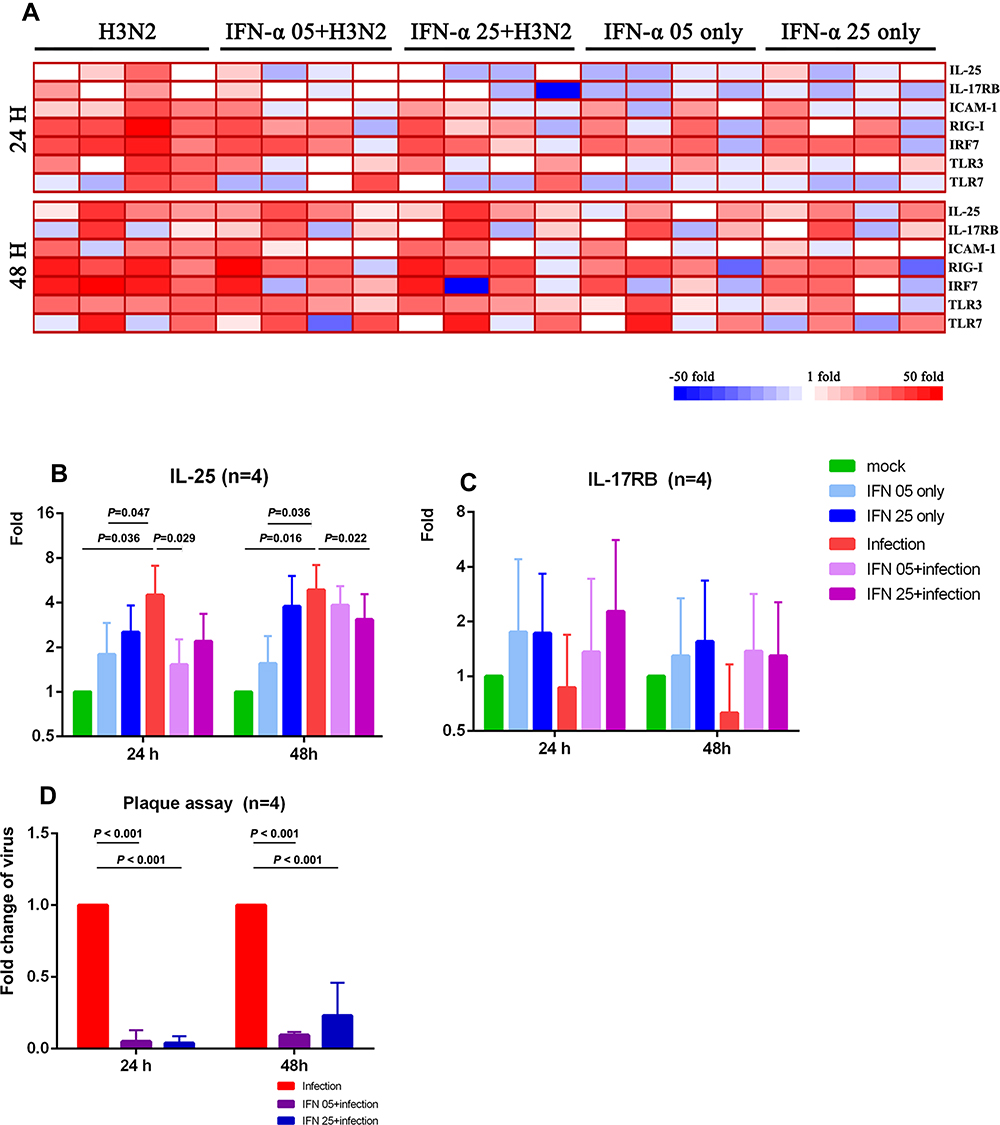 |
Figure 4 Expression of IL-25, IL-17RB, and host factors following inhibition of viral replication by pretreatment with IFN-α (A) Changes of mRNA expression of IL-25, IL-17RB and innate antiviral factors at 24 and 48 hpi with IFN-α pretreatment, together with controls. Fold change of (B) IL-25 and (C) IL-17RB mRNAs shown individually following influenza infection, and pretreatment with IFN-α, together with controls (n=4). Comparison of means was performed by two-way ANOVA. (D) Fold changes in viral replication titers (by viral plaque assay) following pretreatment with IFN-α that accompanied the reduction in IL-25 expression. Comparison of means was performed by Student’s t-test between infected and uninfected hNECs. p < 0.05 is considered statistically significant. |
Discussion
The epithelial-derived cytokine IL-25, acting through the receptor IL-17RB, has been implicated in promoting type 2 inflammatory responses in airway inflammation in concert with IL-33 and TSLP.12,27,28 We have recently found that IL-25, part of the epithelial “alarmins”, was highly produced locally in CRSwNP tissues,23 which is further verified in this study. However, the potential sources of such local IL-25 accumulation in CRSwNP patients remain relatively uncharacterized, especially given that IL-25 levels can differ between patients.29 Respiratory viral infections are increasingly associated with chronic allergic and inflammatory diseases since their presence often exacerbates the type 2 inflammation in inflamed airways – despite their conventional activation of type 1 inflammation for viral clearance.16,18,24,30,31 Therefore, to investigate the ability of viral infections to induce expression of IL-25 that is strongly associated with type 2 inflammation, we infected in vitro differentiated hNECs from control and CRSwNP donors, and quantitatively characterized the induction of IL-25. Interestingly, we demonstrated that among the viruses tested, influenza virus contributed the most to IL-25 expression in control and CRSwNP epithelium alike. The increased IL-25 correlated with the antiviral response genes induced by viral infections, with influenza infection inducing the strongest antiviral responses, as well as the highest IL-25 expression.
Respiratory virus infections are known to aggravate chronic allergic diseases via epithelial cytokines IL-25, IL-33 and TSLP, but most studies focus on the induction of type 2 inflammation in asthmatic patients, with emphasis on the lower airway.32,33 To date, there are limited reports that document detailed analysis of the epithelial cytokines during respiratory virus infections with regards to upper airway diseases such as CRSwNP. In our study, we demonstrated that respiratory virus infections, especially influenza, can contribute towards IL-25 accumulation in upper airway epithelium, which may help predispose or perpetuate type 2 inflammation in CRSwNP pathogenesis. This finding is interesting considering that influenza infection is a less common exacerbator of type 2 inflammation, due to the relatively strong type 1 inflammation it induces to counteract type 2 responses.20,21 Nevertheless, influenza virus may induce severe exacerbation under specific circumstances. Therefore, we selected influenza virus for further studies due to its ability to induce the highest IL-25 expression in the model of differentiated hNECs derived from CRSwNP tissues.
CRSwNP is a complex disease but the consensus is that it is generally classified as a type 2 inflammatory disease with eosinophilic mucosal inflammation, with type 2 inflammatory cytokines such as IL-5 and IL-13 being treatment targets.34–36 As the first line of mechanical and immunological defense against external stimuli, the airway epithelium is an important local source of pro-inflammatory cytokines, including the type 2 inflammation promoting IL-25, IL-33, and TSLP.37,38 Therefore, the modulation of these cytokines as a strategy for management of chronic allergic airway diseases such as asthma and CRSwNP has been intensively researched.7,18,32,33,39 In recent years, it was shown that proper management of viral infections may lead to better outcomes during exacerbations of chronic allergic and inflammatory airway diseases.10,16,18 These studies highlighted that viral infections are a constant trigger of CRSwNP exacerbations. In view of the ever-present threat of emerging viruses, as exemplified by the recently emerging SARS-CoV-2 virus, it is hence desirable to identify interventions that can appropriately alleviate the key triggers of exacerbation in CRSwNP management.16
Studies of airway epithelium in asthmatic patients have pointed out deficiencies or dysregulations in the expression of antiviral molecules and type 2 inflammatory cytokines32,40–42 highlighting the importance of antiviral responses under type 2 inflammatory conditions in the airways. IL-25 has been accepted as the primary mediator of airway inflammation in asthmatic patients by promoting type 2 inflammation. Several studies explored the role of IL-25 in promoting type 2 inflammatory response in CRSwNP patients.7,43 Hence, we conducted this study which identified influenza virus as a potential factor that can contribute IL-25 expression and accumulation in CRSwNP epithelium. Interestingly, when influenza virus replication was inhibited by pretreatment with recombinant IFN-α protein, a reduction of IL-25 expression in the CRSwNP hNECs was observed. This indicates that IFN-α does not additively induce IL-25 with influenza infection; but instead decreases the viral load and modulates overall IL-25 expression in infected hNECs via reduction of active infection. This bimodal effect of IFN-α has hitherto not been reported, and thus offers insights into IFN’s role in modulating IL-25 in the presence or absence of active infection. This finding also suggests that the reduction of IL-25 using broad-spectrum antivirals may constitute a viable strategy in the management of CRSwNP exacerbations. This is further supported by the randomized controlled clinical trial by Gao et al26 who reported that the usage of IFN-α nasal spray may prevent infections caused by influenza A and B viruses, parainfluenza viruses, and adenovirus, without severe adverse effects. Thus, nasal spray containing IFNs may be useful for preventing viral infections that can contribute to the accumulation of IL-25 in CRSwNP.
One limitation of this study is that the in vitro hNEC model was unable to show a direct link to enhanced type 2 inflammation due to the lack of immune cells in the system. While co-culture of the CRSwNP hNECs with immune cells is possible, the experimental system needs to be established with hNECs and immune cells from the same donor to prevent allogenic activation. Given that influenza virus stimulated the highest local epithelial IL-25 production, further experiments are warranted on IFN-α to account for its downstream effectors and interactions with cells such as group 2 innate lymphoid cells (ILC2s). It will therefore be interesting to include the co-culture of ILC2s to further explore the effects of influenza and other viral infections (rhinovirus, RSV) on the activation of type 2 inflammation. In addition, while we were able to correlate active viral infection and antiviral responses with IL-25 induction in CRSwNP epithelium, we are unable to further discern the mechanisms directly leading to the IL-25 induction during active influenza infection. Therefore, future studies should focus on studying the factors (eg TLR7, RIG-I) associated with IL-25 induction and evaluating their effects on IL-25 expression levels (eg by knockdown experiments of these factors).
Conclusions
In conclusion, our data, together with other studies, have demonstrated that respiratory viruses (especially influenza virus) can directly induce the expression of IL-25 in active virus-infected nasal epithelial cells, which may contribute to the accumulation of IL-25 in CRSwNP epithelium (based on the working hypothesis summarized in Figure S5). Using influenza infection, we have also provided several lines of evidence that active viral replication is positively correlated with the expression of IL-25, and that pretreatment with low doses of IFN-α abates influenza-dependent IL-25 production in CRSwNP epithelium. We also reported for the first time, potential bimodal functions of IFN-α which modulates IL-25 expression with active influenza infection, but by itself may moderately increase IL-25 expression (though not to the extent of active infection). Also noteworthy was the strong correlation between mRNA and protein levels of IL-25 in our study, indicating that IL-25 mRNA from epithelial tissue can serve as a marker for IL-25 secretion. Our findings may aid in designing novel strategies for the optimal prevention and management of chronic inflammatory upper airway diseases such as CRSwNP via the broad-spectrum amelioration of its viral triggers.
Acknowledgments
We thank Prof. Jianbo Shi and Prof. Huabin Li for their discussion and helpful comments on this study. We thank all ENT surgeons in the Department of Otolaryngology Head & Neck Surgery, National University Hospital, Singapore. We thank M.C. Phoon, and S.H. Lau for assistance in IFN-α pretreatment and viral titration techniques. We thank A.K. Andiappan and Singapore Immunology Network, A*STAR, Singapore for their assistance in Luminex assay, and Z.Z.R. Lew for assistance in the revision experiments.
Author Contributions
HH, DYW, VC, and YY conceived and designed the study; HH, YO, JL, KST, and FC carried out all in vivo and in vitro experiments; FC, YKO, MT, and HH collected and processed the clinical nasal mucosa samples. KST, HHO, YO, RS and VC performed the in vitro viral infection experiments. HH, YY, KST, and FC did the statistical analyses. All authors contributed to data interpretation. HH, KST, VC and DYW wrote the original and/or revised draft of the manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (No. 82071018), Natural Science Foundation of Guangdong Province, China (No. 2018A030313399), and the National Medical Research Council, Singapore (NMRC/CIRG/1362/2013; NMRC/CIRG/1458/2016; and MOH-OFYIRG19may-0007). Kai Sen Tan is a recipient of fellowship support from European Allergy and Clinical Immunology (EAACI) Research Fellowship 2019.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Shi JB, Fu QL, Zhang H, et al. Epidemiology of chronic rhinosinusitis: results from a cross-sectional survey in seven C hinese cities. Allergy. 2015;70(5):533–539. doi:10.1111/all.12577
2. Fokkens WJ, Lund VJ, Mullol J, et al. European position paper on rhinosinusitis and nasal polyps 2012. Rhinol Suppl. 2012;23:1–298.
3. Fokkens WJ, Lund VJ, Hopkins C, et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology. 2020;58(Suppl S29):1–464. doi:10.4193/Rhin20.600
4. Chin D, Harvey RJ. Nasal polyposis: an inflammatory condition requiring effective anti-inflammatory treatment. Curr Opin Otolaryngol Head Neck Surg. 2013;21(1):23–30. doi:10.1097/MOO.0b013e32835bc3f9
5. Steelant B, Seys SF, Boeckxstaens G, Akdis CA, Ceuppens JL, Hellings PW. Restoring airway epithelial barrier dysfunction: a new therapeutic challenge in allergic airway disease. Rhinology. 2016;54(3):195–205. doi:10.4193/Rhin15.376
6. Angkasekwinai P, Park H, Wang YH, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204(7):1509–1517. doi:10.1084/jem.20061675
7. Shin HW, Kim DK, Park MH, et al. IL-25 as a novel therapeutic target in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2015;135(6):1476–1485 e1477. doi:10.1016/j.jaci.2015.01.003
8. Fort MM, Cheung J, Yen D, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985–995. doi:10.1016/s1074-7613(01)00243-6
9. Ballantyne SJ, Barlow JL, Jolin HE, et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120(6):1324–1331. doi:10.1016/j.jaci.2007.07.051
10. Tan KS, Lim RL, Liu J, et al. Respiratory viral infections in exacerbation of chronic airway inflammatory diseases: novel mechanisms and insights from the upper airway epithelium. Front Cell Dev Biol. 2020;8:99. doi:10.3389/fcell.2020.00099
11. Jumat MR, Yan Y, Ravi LI, et al. Morphogenesis of respiratory syncytial virus in human primary nasal ciliated epithelial cells occurs at surface membrane microdomains that are distinct from cilia. Virology. 2015;484:395–411. doi:10.1016/j.virol.2015.05.014
12. Lloyd CM, Saglani S. Epithelial cytokines and pulmonary allergic inflammation. Curr Opin Immunol. 2015;34:52–58. doi:10.1016/j.coi.2015.02.001
13. Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol. 2010;125(6):1178–1187. doi:10.1016/j.jaci.2010.04.021
14. Grissell TV, Powell H, Shafren DR, et al. Interleukin-10 gene expression in acute virus-induced asthma. Am J Respir Crit Care Med. 2005;172(4):433–439. doi:10.1164/rccm.200412-1621OC
15. Cho GS, Moon BJ, Lee BJ, et al. High rates of detection of respiratory viruses in the nasal washes and mucosae of patients with chronic rhinosinusitis. J Clin Microbiol. 2013;51(3):979–984. doi:10.1128/jcm.02806-12
16. Tan KS, Yan Y, Ong HH, Chow VTK, Shi L, Wang DY. Impact of respiratory virus infections in exacerbation of acute and chronic rhinosinusitis. Curr Allergy Asthma Rep. 2017;17(4):24. doi:10.1007/s11882-017-0693-2
17. Peng Y, Zi XX, Tian TF, et al. Whole-transcriptome sequencing reveals heightened inflammation and defective host defence responses in chronic rhinosinusitis with nasal polyps. Eur Respir J. 2019;54(5). doi:10.1183/13993003.00732-2019
18. Edwards MR, Walton RP, Jackson DJ, et al. The potential of anti-infectives and immunomodulators as therapies for asthma and asthma exacerbations. Allergy. 2018;73(1):50–63. doi:10.1111/all.13257
19. Wen W, Liu W, Zhang L, et al. Increased neutrophilia in nasal polyps reduces the response to oral corticosteroid therapy. J Allergy Clin Immunol. 2012;129(6):1522–1528 e1525. doi:10.1016/j.jaci.2012.01.079
20. Yan Y, Tan KS, Li C, et al. Human nasal epithelial cells derived from multiple individuals exhibit differential responses to H3N2 influenza virus infection in vitro. J Allergy Clin Immunol. 2016;138(1):276–281. doi:10.1016/j.jaci.2015.11.016
21. Tan KS, Ong HH, Yan Y, et al. In vitro model of fully differentiated human nasal epithelial cells infected with rhinovirus reveals epithelium-initiated immune responses. J Infect Dis. 2018;217(6):906–915. doi:10.1093/infdis/jix640
22. Wang W, Yan Y, Li CW, et al. Live human nasal epithelial cells (hNECs) on chip for in vitro testing of gaseous formaldehyde toxicity via airway delivery. Lab Chip. 2014;14(4):677–680. doi:10.1039/c3lc51208h
23. Hong HY, Chen FH, Sun YQ, et al. Local IL-25 contributes to Th2-biased inflammatory profiles in nasal polyps. Allergy. 2018;73(2):459–469. doi:10.1111/all.13267
24. Murray CS, Poletti G, Kebadze T, et al. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61(5):376–382. doi:10.1136/thx.2005.042523
25. Deng Y, Yan Y, Tan KS, et al. MicroRNA-146a induction during influenza H3N2 virus infection targets and regulates TRAF6 levels in human nasal epithelial cells (hNECs). Exp Cell Res. 2017;352(2):184–192. doi:10.1016/j.yexcr.2017.01.011
26. Gao L, Yu S, Chen Q, et al. A randomized controlled trial of low-dose recombinant human interferons alpha-2b nasal spray to prevent acute viral respiratory infections in military recruits. Vaccine. 2010;28(28):4445–4451. doi:10.1016/j.vaccine.2010.03.062
27. Kouzaki H, Matsumoto K, Kato T, Tojima I, Shimizu S, Shimizu T. Epithelial cell-derived cytokines contribute to the pathophysiology of eosinophilic chronic rhinosinusitis. J Interferon Cytokine Res. 2016;36(3):169–179. doi:10.1089/jir.2015.0058
28. Hong H, Liao S, Chen F, Yang Q, Wang DY. Role of IL-25, IL-33, and TSLP in triggering united airway diseases toward type 2 inflammation. Allergy. 2020;75(11):2794–2804. doi:10.1111/all.14526
29. Hong H, Chen F, Sun Y, et al. Nasal IL-25 predicts the response to oral corticosteroids in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2018;141(5):1890–1892. doi:10.1016/j.jaci.2017.10.050
30. Hamilos DL. Host-microbial interactions in patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2014;133(3):640–653.e644. doi:10.1016/j.jaci.2013.06.049
31. Chan RW, Chan MC, Nicholls JM, Malik Peiris JS. Use of ex vivo and in vitro cultures of the human respiratory tract to study the tropism and host responses of highly pathogenic avian influenza A (H5N1) and other influenza viruses. Virus Res. 2013;178(1):133–145. doi:10.1016/j.virusres.2013.03.003
32. Beale J, Jayaraman A, Jackson DJ, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. 2014;6(256):256ra134. doi:10.1126/scitranslmed.3009124
33. Jackson DJ, Makrinioti H, Rana BM, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373–1382. doi:10.1164/rccm.201406-1039OC
34. Akdis CA, Bachert C, Cingi C, et al. Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2013;131(6):1479–1490. doi:10.1016/j.jaci.2013.02.036
35. Kortekaas Krohn I, Bobic S, Dooley J, et al. Programmed cell death-1 expression correlates with disease severity and IL-5 in chronic rhinosinusitis with nasal polyps. Allergy. 2017;72(6):985–993. doi:10.1111/all.13136
36. Bachert C, Mannent L, Naclerio RM, et al. Effect of subcutaneous dupilumab on nasal polyp burden in patients with chronic sinusitis and nasal polyposis: a randomized clinical trial. JAMA. 2016;315(5):469–479. doi:10.1001/jama.2015.19330
37. Gern JE. Virus/Allergen interaction in asthma exacerbation. Ann Am Thorac Soc. 2015;12 Suppl 2(Suppl 2):S137–143. doi:10.1513/AnnalsATS.201503-153AW
38. Bulek K, Swaidani S, Aronica M, Li X. Epithelium: the interplay between innate and Th2 immunity. Immunol Cell Biol. 2010;88(3):257–268. doi:10.1038/icb.2009.113
39. Valizadeh A, Khosravi A, Zadeh LJ, Parizad EG. Role of IL-25 in immunity. J Clin Diagn Res. 2015;9(4):OE01–04. doi:10.7860/JCDR/2015/12235.5814
40. Laza-Stanca V, Message SD, Edwards MR, et al. The role of IL-15 deficiency in the pathogenesis of virus-induced asthma exacerbations. PLoS Pathog. 2011;7(7):e1002114. doi:10.1371/journal.ppat.1002114
41. Contoli M, Message SD, Laza-Stanca V, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12(9):1023–1026. doi:10.1038/nm1462
42. Wark PA, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937–947. doi:10.1084/jem.20041901
43. Lam M, Hull L, Imrie A, et al. Interleukin-25 and interleukin-33 as mediators of eosinophilic inflammation in chronic rhinosinusitis. Am J Rhinol Allergy. 2015;29(3):175–181. doi:10.2500/ajra.2015.29.4176
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.