Back to Journals » ImmunoTargets and Therapy » Volume 9
Immunotherapeutic and Targeted Approaches in Multiple Myeloma
Authors Nadeem O, Tai YT ![]() , Anderson KC
, Anderson KC
Received 18 July 2020
Accepted for publication 22 September 2020
Published 14 October 2020 Volume 2020:9 Pages 201—215
DOI https://doi.org/10.2147/ITT.S240886
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Omar Nadeem, Yu-Tzu Tai, Kenneth C Anderson
Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
Correspondence: Omar Nadeem
Dana-Farber Cancer Institute, Harvard Medical School, 450 Brookline Ave, Boston, MA 02215, USA
Tel +1 617-632-3000
Email [email protected]
Abstract: The multiple myeloma (MM) therapeutic landscape has evolved significantly with the approval of numerous novel agents, including next generation proteasome inhibitors (PIs), immunomodulatory agents (IMIDs), and monoclonal antibodies (MoABs) targeting CD38 and SLAMF7. While these discoveries have led to an unprecedented improval in patient outcomes, the disease still remains incurable. Immunotherapeutic approaches have shown substantial promise in recent studies of chimeric antigen receptor T-cell (CAR T-cell) therapy, bispecific antibodies, and antibody drug conjugates targeting B-cell maturation antigen (BCMA). This review will highlight these novel and targeted therapies in MM, with particular focus on PIs, IMIDs, MoAb and BCMA-directed immunotherapy.
Keywords: immunotherapy multiple myeloma, chimeric antigen receptor T-cell therapy; CAR T-cell therapy, bispecific antibodies, immunomodulatory agents; IMID, monoclonal antibodies; MoAB
Introduction
Multiple myeloma (MM) is a plasma cell neoplasm with an incidence of approximately 30,000 new cases in the United States per year.1 Over the past several decades, there has been an unprecedented improvement in survival of MM patients due to discovery of numerous novel agents.2 These novel therapies have broad mechanism of actions including proteasome inhibition, immune modulation, and targeting of CD38 and SLAMF7 with monoclonal antibodies (MoAB). The use of alkylating agents, once a cornerstone of myeloma therapy, remains prevalent in the current era, particularly in the form of high dose chemotherapy and autologous stem cell transplantation (ASCT). More recently, there has been tremendous progress in immune-based therapy for myeloma, including chimeric antigen receptor T-cell (CAR T-cell) therapy, bispecific antibodies, and antibody drug conjugates targeting B-cell maturation antigen (BCMA). These agents are rapidly making their way through clinical development, and are expected to add to the growing arsenal of available MM therapies. This review will highlight these novel and targeted therapies in MM, with particular focus on proteasome inhibitors (PI), immunomodulatory agents (IMIDs), MoAb and BCMA-directed immunotherapy.
Background
MM accounts for approximately 1% of all cancers and represents 10% of all hematologic malignancies.1 There is an increased incidence in African Americans and first degree relatives,3,4 and approximately 10% of MM cases are familial. The monoclonal protein is most commonly IgG, followed by IgA and light chain only. IgD, IgM, and non-secretory myeloma are less common and comprise the rest of the cases.
The common clinical manifestations include bone pain due to osteolytic bone lesions, renal insufficiency due to cast nephropathy, anemia due to marrow infiltration and hypercalcemia (CRAB). Presence of concurrent immunoglobulin deposition or AL amyloidosis, although rare, usually leads to further deterioration of organ function, including cardiomyopathy and nephrotic range proteinuria.
Serum and urine protein electrophoresis (SPEP/UPEP) with immunofixation identifies the monoclonal protein in the vast majority of cases. Serum free light chain analysis is increasingly utilized for diagnosis and assessment of light chain myeloma. The international staging system (ISS) stratifies patients based on values of beta 2 microglobulin and albumin. The revised ISS (R-ISS) incorporates elevations in serum LDH and high-risk cytogenetics including deletion 17p, t(4;14), and t(14;16) into the ISS stage, and is now the gold standard staging system for myeloma5. A bone marrow biopsy and aspiration remains crucial for diagnosis, quantification of plasma cell burden, and cytogenetic analysis.
Imaging in the form of a skeletal survey was historically used for detection of osteolytic bone disease. In the current era, whole body MRI and FDG PET/CT offer far greater sensitivity for detection of bone lesions, and have now become standard of care as part of the diagnostic evaluation.6
MM treatment is classified into various phases including induction, consolidation, and maintenance therapy. The intent of induction therapy is to achieve initial disease control with the aim of reversing or stabilizing the end organ damage that has already occurred. Induction therapy is usually given for 4 to 6 cycles with each cycle lasting approximately 3 to 4 weeks. This is followed by consolidation therapy with the goal of further deepening the response, commonly with high dose melphalan chemotherapy followed by ASCT in transplant-eligible patients. This has historically led to significant improvement in progression-free survival (PFS) and overall survival (OS).7
Consolidation therapy is generally followed by maintenance therapy, the goal of which is to delay time to relapse and to subsequent treatment. Invariably, patients will ultimately experience relapse of their disease, and the decision to use specific therapies at that point depends on several factors including prior treatment history, toxicity, comorbid conditions, end organ function, healthcare costs, and patient preferences for parental or oral therapy. Patients will usually undergo several lines of therapy in sequence, with various combinations of approved agents with each subsequent relapse.
Immune Dysfunction in MM
The dysregulation of the immune system is a characteristic finding in plasma cell disorders. This is highlighted by an increased risk of infections in patients with newly diagnosed MM (NDMM),8 and lack of robust immune response to vaccinations in patients with MM.9 The disruption in the immune microenvironment in MM leads to evasion of immune recognition of MM cells and promotes tumor cell growth.10 The “control” of the innate immune system over an abnormal population of plasma cells is lost over time, as patients progress from the precursor stage of monoclonal gammopathy of undetermined significance (MGUS) to overt myeloma.11 Compromised immune component of the bone marrow niche including dysregulated regulatory T cells, myeloid derived suppressor cells, Th17 cells, tumor-associated macrophages, mesenchymal stromal cells, and osteoclasts confer immunosuppression and promote tumor immune escape; and therefore represents an ideal target for novel therapeutics.12,13 Importantly, extensive research has shown the role of plasmacytoid dendritic cells (DCs) in promoting MM cell survival and drug resistance,14 providing the framework for targeting plasmacytoid DCs/MM cells interaction in novel therapies.15,16 Similarly, increased expression of immune checkpoints, ie PD-1/PD-L1, in T cells and MM cells, promotes tumor immune evasion and has been also associated to progression from precursor stages.13,17 Therapeutic targeting by means of immune checkpoint inhibitor in MM will be discussed later in this review. The understanding of the complex interactions between the immune microenvironment and plasma cells has been the key driver of treatment advances over the past decade in MM therapeutics.
Proteasome Inhibition in MM
PIs have become a mainstay of myeloma therapy and are routinely used in combination therapy in NDMM and relapsed/refractory (RRMM) MM (Table 1). The mechanism of action of PIs involves the ubiquitin-protease system (UPS), which plays a critical role in maintenance of cell cycle progression, DNA repair, and degradation of intracellular misfolded proteins. The 26S proteasome complex has several alpha and beta subunits, including the catalytic rings B1, B2 and B5 subunits, targets of the various PIs.18,19 Substrate proteins are identified by polyubiquitination by the single ubiquitin-activating enzyme 1 (E1) and multiple ubiquitin-conjugating enzymes (E2) and ubiquitin-protein ligases (E3).19 In malignant cells, particularly MM, this process is dysregulated leading to excessive degradation of tumor suppressor p53, an inhibitor of nuclear factor -kB (NF-kB).20 This leads to a rapid proliferation of MM cells due to continual activation of NF-kB transcription pathway.
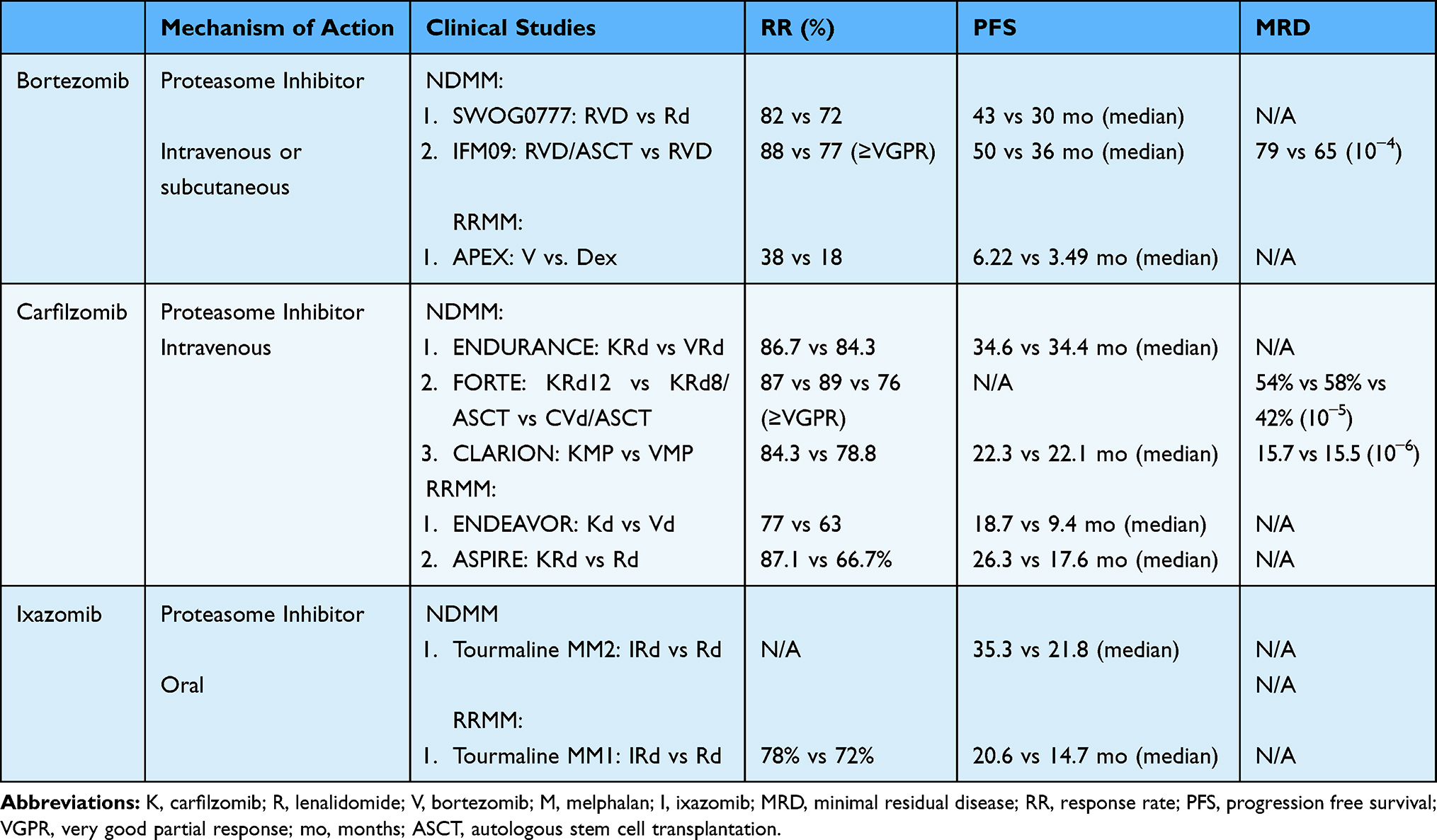 |
Table 1 Proteasome Inhibitors |
Bortezomib, carfilzomib and ixazomib are PIs that are currently approved for MM therapy in the United States. All three inhibit the B5 subunit, where bortezomib and ixazomib are peptide boronic acid based reversible inhibitors21 and carfilzomib is an an IV peptide epoxyketone irreversible inhibitor. Bortezomib is now most commonly administered via subcutaneous route due to a lower incidence of peripheral neuropathy compared to intravenous administration.22,23 Carfilzomib is administered intravenously and ixazomib is administered orally.
The exceptional sensitivity of MM plasma cells to PI is related to their heightened dependency on the protein quality control system due the high turnover of abnormal immunoglobulins.12,24 PIs overwhelm proteasome load, increase the accumulation of misfolded and unfolded proteins, thus leading to endoplasmic reticulum stress and eventually cell death.12 As proteins involved in several biological processes are substrate of the proteasome, its inhibition has multiple downstream effects, including decrease of cell proliferation and apoptosis induction, cell-cycle arrest, deficit in DNA repair mechanisms, and alteration of cellular metabolism. Proteasome inhibition affects both cellular and non cellular components of the host microenvironment via decreasing the expression of several adhesion molecules (such as ICAM1 and VCAM1), disrupting the interaction of MM cells with bone marrow accessory cells, inhibiting the secretion of cytokines (such as IL-6, VEGF, IGF-1) and angiogenesis, and modifying bone turnover and osteoclast activity.18 Most of these effects rely, at least in part, on the direct inhibition of the NF-kB pathway, one of the major pro-survival pathways in MM.
The role of PI in modulating the immune compartment is still largely undefined. Although preclinical studies have shown that PIs are non-selective inhibitors of the immunoproteasome,25 suggesting an immunosuppressive activity of this class of drugs, previous reports already showed the ability of bortezomib to enhance DC-mediated induction of immunity to MM in vitro by increasing the exposure of cell surface heat shock protein 90 on dying tumor cells.26 Moreover, our recent studies using of MM provide evidence, both in a syngeneic in vivo model and in MM patients, that bortezomib treatment triggers “immunogenic” cell death, which is able to prime an effective anti-MM immune response and disease control (Gulla et al, ASH 2019, abs 134).
Immune Modulation in MM
IMIDs are used in combination therapy in NDMM and RRMM. The approved agents include thalidomide, lenalidomide, and pomalidomide. Thalidomide was the first agent to demonstrate activity in RRMM in the 1990s.27 Thalidomide was initially used to treat pregnancy-associated nausea, which then led to development of several birth defects in infants including phocomelia and additional malformations.28 This led to its withdrawal from the market until it demonstrated activity in myeloma. Subsequent studies of thalidomide and dexamethasone confirmed its benefit leading to FDA approval.29 However, the dose-limiting toxicity of peripheral neuropathy with thalidomide limited its prolonged use. Lenalidomide and pomalidomide were subsequently studied with better tolerance and efficacy.
IMIDs have anti-myeloma properties through a variety of mechanisms. Thalidomide initially demonstrated anti-angiogenic properties, and lenalidomide and pomalidomide were noted to induce cell death via inhibition of NFkB, activation of caspases, and induction of apoptosis.30–32 The IMIDs also lead to decrease in cell surface adhesion molecules, thereby disrupting the stromal and myeloma cell interactions and decreasing IL-6 production.33 IMIDs also enhance and stimulate T cells via the B7-CD28 pathway, triggering tyrosine phosphorylation of CD28 on T cells leading to activation of NFkB.34 IMIDs also enhance tumor antigen uptake by DCs, trigger cytolytic CD8+ T and NK cells, and decrease regulatory T cells (Treg).
Further studies ultimately revealed cereblon (CRBN) as a major target for IMIDs in MM. It was first demonstrated that inactivation of CRBN led to myeloma cell death and resistance to IMIDs.35 CRBN is within the CRL4 complex which has E3 ubiquitin ligase activity. Ikaros (IKZF1) and aiolos (IKZF3) were subsequently identified as key transcription factors and CRBN interacting proteins. These proteins are critical in longevity of plasma cells, and IMID binding to CRBN leads to enhanced affinity for IKZF1 and IKZF3, with subsequent ubiquitination and proteasomal degradation of these transcription factors.36 Most recently, it has been shown that IMIDs can enhance T and NK cell cytotoxic activity in (1) Zap70-mediated CRBN independent, as well as (2) CRBN-mediated ZAP-70 independent mechanisms, providing the framework for developing novel therapeutics to activate ZAP-70 and thereby enhance T and NK anti-MM cytotoxicity.37
Although lenalidomide and pomalidomide share several mechanisms of action and CRBN as a major binding target, clinical efficacy of pomalidomide in lenalidomide-refractory patients suggests that distinct biological effects may underlie its activity in vivo. For instance, preclinical studies have demonstrated differential requirement of CRBN level in the context of IMiD treatment.38,39 The expression level of CRBN protein in tumor cells from MM patients treated with pomalidomide and dexamethasone revealed a weaker association with patient response and survival as compared to single agent pomalidomide, suggesting the involvement of tumor microenvironment in mediating the antitumor effect with the addition of dexamethasone.38,39 Pomalidomide induces rapid changes in the composition of the tumor microenvironment and immune cells with activation of both innate and adaptive immunity, strengthening the hypothesis of the immune-mediated effect of pomalidomide in a lenalidomide-resistant setting. Pomalidomide treatment leads also to an increase of immune checkpoint Tim-3 and BTLA4 in vivo, thus suggesting a potential synergistic effect in combination with immune checkpoint blockade.39
Thalidomide
Thalidomide was studied in RRMM and demonstrated significant activity and responses across a wide range of therapeutic doses.27 This led to FDA approval through a special restricted distribution program (REMS), due to its association with birth defects that originally prompted its withdrawal from the market.40 Thalidomide has since been a part of many pivotal myeloma studies both in NDMM,41 maintenance post-ASCT,42 and RRMM.
Most recently, the Phase III CASSIOPEIA trial evaluated the combination of daratumumab, bortezomib, thalidomide, and dexamethasone (D-VTD) versus bortezomib, thalidomide, and dexamethasone (VTD) in NDMM. This trial demonstrated improvement in stringent complete responses in favor of D-VTD at day 100 post-ASCT (29% vs. 20%, OR 1.60, 95% CI 1.21–2.12, p=0.0010). The rates of minimal residual disease (MRD) negativity at 10–5, assessed by multiparametric flow cytometry, were significantly higher in favor of D-VTD (64% vs. 44%, p<0.0001). This trial led to the approval of D-VTD combination in NDMM.
The major toxicity associated with thalidomide is development of peripheral neuropathy, particularly with prolonged use.43,44 Hence, its use has been diminished by newer generation IMIDs such as lenalidomide and pomalidomide.
Lenalidomide
Lenalidomide is used in various settings, including in NDMM, as maintenance therapy post-SCT, and in RRMM. The landmark FIRST trial45 evaluated the role of lenalidomide and dexamethasone (Rd) versus melphalan, prednisone, and thalidomide (MPT), which was the established standard of care therapy at that time in transplant-ineligible myeloma. Two different schedules of Rd were studied, which included treatment duration of 18 months and treatment until disease progression. This trial demonstrated improvement in PFS in favor of both lenalidomide arms versus MPT (25.5 months-continuous Rd, 20.7 months 18 cycles of Rd, 21.2 months with MPT [HR 0.72 Rd continuous vs. MPT]). This established Rd as the standard first line therapy for transplant-ineligible myeloma.
This trial established the Rd backbone as a highly effective and well-tolerated treatment approach, and further studies were conducted with an addition of a third drug to this platform. The SWOG S0777 trial,46 described above, led to further improvement in PFS and OS of RVD platform versus Rd. Further dose modification of RVD was studied in transplant-ineligible myeloma (RVD-lite) and demonstrated similar outcomes and less toxicity with a weekly bortezomib administration schedule and lower dose lenalidomide.22 This triplet therapy remains the standard of care induction therapy for NDMM to date, and excellent long outcomes with RVD regimen have been recently reported in a large single center real-world analysis, with a median PFS of 65 months (95% CI, 58.7 to 71.3 months).47
Lenalidomide maintenance therapy has been shown to be very effective post-transplantation due to modulation of the immune system, along with its anti-angiogenic and anti-proliferative effects.48 Several positive phase III trials of lenalidomide maintenance demonstrated improvement in PFS, leading to its approval for use post-ASCT.49–51 A meta-analysis of lenalidomide post-ASCT revealed significant improvement in both PFS (52.8 months for the lenalidomide group and 23.5 months for the placebo or observation group (hazard ratio, 0.48; 95% CI, 0.41 to 0.55) and OS (Median OS not been reached for the lenalidomide maintenance group, whereas it was 86.0 months for the placebo or observation group [hazard ratio, 0.75; 95% CI, 0.63 to 0.90; P = 0.001]).52 Of note, there is a higher incidence of secondary malignancies associated with lenalidomide use post-ASCT. However, the improvement in myeloma-related outcomes are substantially greater, and lenalidomide maintenance remains standard of care in the current era.
In the RRMM setting, lenalidomide is currently approved and commonly used as part of combination therapy with daratumumab,53 elotuzumab,54 ixazomib55 and carfilzomib.56 Due to the increased used of lenalidomide in the newly diagnosed and maintenance settings, many patients are lenalidomide-refractory at the time of relapse. Immune modulation in lenalidomide-refractory patients plays a key role, as one study demonstrated re-sensitivity to lenalidomide using a low dose cyclophosphamide and prednisone backbone in previously refractory patients.57
Common adverse effects of lenalidomide include hematologic toxicity, risk of venous thromboembolism, gastrointestinal side effects, and rash.
Pomalidomide
Pomalidomide is a more potent IMID that is approved for treatment of RRMM. Pomalidomide is currently approved to be used in combination with dexamethasone, elotuzumab, and daratumumab.58–60
The recently reported phase III OPTIMISMM trial61 evaluated the combination of pomalidomide, bortezomib, and dexamethasone (PVd) versus bortezomib and dexamethasone in lenalidomide-treated RRMM with 1–3 prior lines of therapy. This study demonstrated significant improvement in PFS in favor PVd compared to Vd (median 11.20 months [95% CI 9.66–13.73] vs. 7.10 months [5.88–8.48]; hazard ratio 0.61, 95% CI 0.49–0.77; p<0.0001). The overall response rate was significantly improved in both lenalidomide-refractory and non-refractory patients, making PVd an attractive option for patients who are experiencing a disease relapse on lenalidomide maintenance therapy.
Pomalidomide is metabolized in the liver, making it the preferred IMID in patients with renal insufficiency. Adverse effects of pomalidomide include hematologic toxicity, gastrointestinal side effects, and rash.
CELMoD Agents
Iberdomide is a novel cereblon E3 ligase immunomodulatory agent currently under study. This agent binds to CRBN with much higher potency with far greater degradation of Aiolos and Ikaros.62 Clinical studies of iberdomide have yielded promising results in RRMM63 with an overall response rate in combination with dexamethasone of 32.3% in this highly refractory population.64
Another CELMoD agent CC-92480 is currently under study in combination with dexamethasone in patients with RRMM (Richardson et al, ASCO 2020, abs 8500). This is also a potent cereblon E3 ligase modulator with rapid degradation of Ikaros and Aiolos and has potent antiproliferative and tumoricidal activity. CC-92480 has demonstrated an overall response rate of 21.1% in a highly refractory population (median number of cycles = 6). The recommended Phase 2 dose of 1mg days 1 to 21 of 28 day cycle has been identified for future study. In patients treated with the 1mg dose, a response rate of 48% was observed. Treatment associated adverse events mainly included myelosuppression.
Monoclonal Antibodies in MM
MoABs have transformed the landscape of MM treatment since the approval of daratumumab and elotuzumab in 2015 targeting of CD38 and SLAMF7, respectively. Isatuximab, a novel CD38 MoAB, was also recently approved for use in RRMM (Table 2).
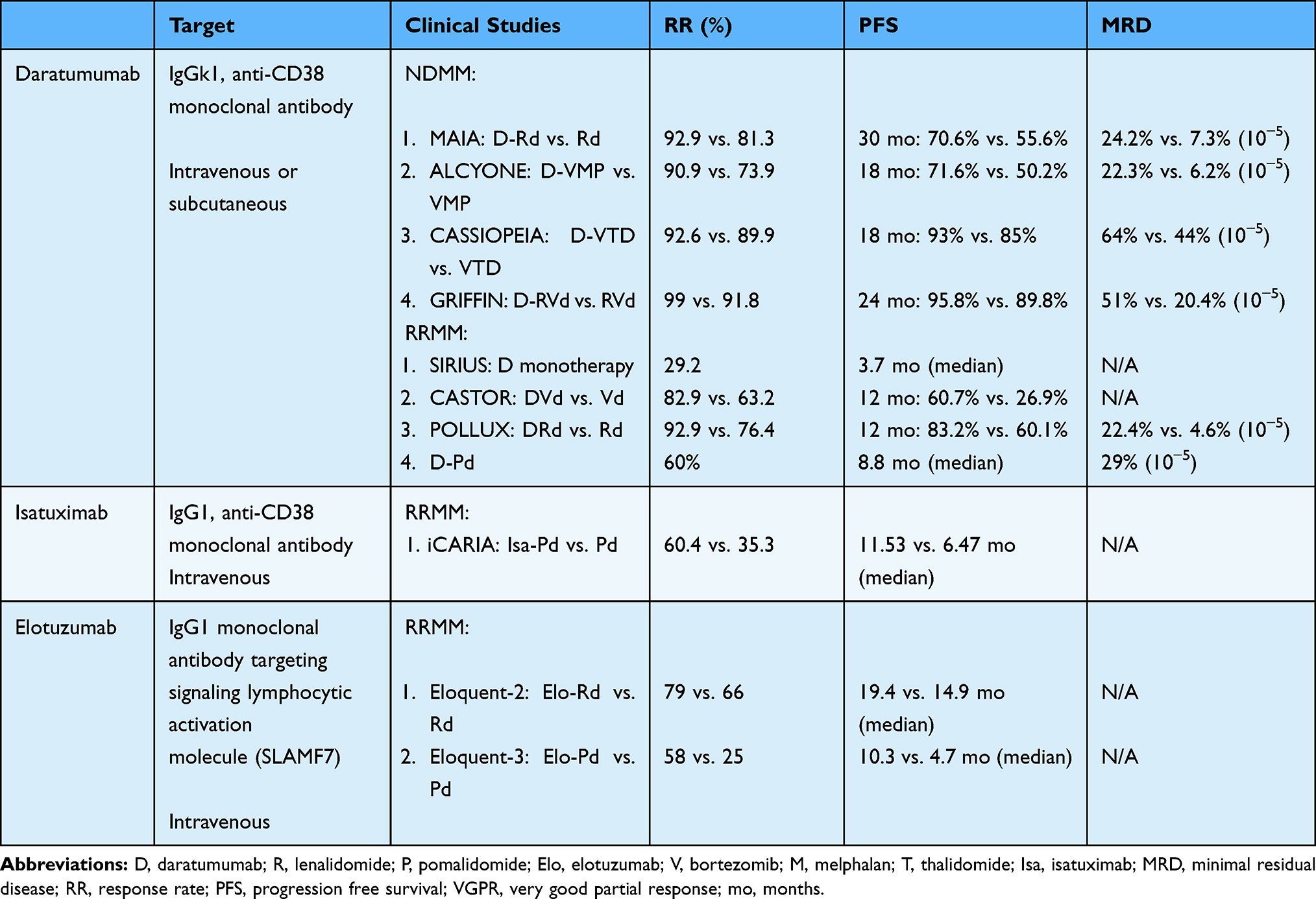 |
Table 2 Monoclonal Antibodies |
MoABs are able to deliver highly-directed activation of the immune system towards the clonal cells, particularly if the target antigen is either solely or highly expressed on the malignant cells. The mechanisms by which MoABs lead to cell death include activation of the immune system through antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC). Furthermore, MoABs can also lead to direct cell death by binding the target receptor inducing apoptosis.65
CD38 is type II single-chain transmembrane glycoprotein highly expressed in plasma cells and various other normal cells including other hematologic cells (NK cells, platelets, B cells), neuronal cells, pancreas islet cells, skeletal and cardiac muscle cells, and bronchial cells. The function of CD38 includes catabolism of extracellular nucleotides via ectoenzymatic activity, and transduction of intracellular growth signals66 The expression of CD38 on myeloma cells is extremely high, and preclinical studies demonstrated various Fc dependent processes leading to myeloma cell death including ADCC, ADCP and CDC.67,68 To date, off-target effects have not been noted to a great degree in patients treated with CD38-directed MoAB.
Daratumumab
Daratumumab is an IgG1k anti-CD38 monoclonal antibody currently approved for treatment of NDMM and RRMM. Mechanisms of action of daratumumab include: 1) CDC: via binding of both targeted antigen CD38 and C1q, with subsequent initiation of complement cascade and formation of a membrane attack complex (MAC) leading to MM cell lysis; 2) ADCC: the Fc fragment of daratumumab can bind an FcR-bearing effector cells, such as NK, thus inducing MM cytotoxicity; 3) ADCP: daratumumab induces macrophage phagocytosis by allowing the binding of its Fc fragment with an FcR-bearing macrophage; and 4) tumor cell apoptosis upon FcγR cross-linking.69 Along with these effects mediated by the Fc fragment, daratumumab also has immunomodulatory effects that increase host-anti MM response as it induces killing of CD38-positive immune suppressor cells, including Tregs, Bregs and MDSCs.70 These effects partly explain the corresponding increase in the number and clonality of T cells, and their activation and killing capacity as shown by higher levels of granzyme B observed after daratumumab treatment.70
The pivotal SIRIUS trial evaluated the efficacy of daratumumab monotherapy, yielding an overall response rate of 29.2% in a highly refractory population.71 Subsequent POLLUX53 (daratumumab, lenalidomide, and dexamethasone vs. lenalidomide and dexamethasone) and CASTOR72 (daratumumab, bortezomib, and dexamethasone vs. bortezomib and dexamethasone) trials solidified the role of daratumumab combinations in RRMM, with marked improvement in PFS, response rates, and MRD negativity rates with the triplet combinations. Daratumumab is also approved in combination with pomalidomide and dexamethasone in RRMM due to encouraging results in a Phase 1b study with a median PFS of 8.8 (95% CI, 4.6–15.4) months in a highly refractory population.58 The confirmatory phase III APOLLO study utilizing subcutaneous daratumumab in combination with pomalidomide and dexamethasone has demonstrated improvement in PFS compared to pomalidomide and dexamethasone in RRMM (NCT03180736).
In NDMM, the MAIA trial73 evaluated the combination of daratumumab, lenalidomide and dexamethasone versus lenalidomide and dexamethasone in transplant-ineligible myeloma. This trial led to substantial improvement in PFS in favor of daratumumab, lenalidomide and dexamethasone (estimated PFS at 30 months: 70.6% vs. 55.6% [hazard ratio for disease progression or death, 0.56; 95% CI, 0.43 to 0.73; P<0.001]) leading to its FDA approval for this indication. This is an increasingly used combination in this population, with a contrasting safety profile compared to the RVd combination, including a markedly lower incidence of peripheral neuropathy. This is particularly attractive for the older population that may have pre-existing peripheral neuropathy or in whom development of peripheral neuropathy can lead to significant deterioration of quality of life.
Daratumumab has also been studied in a four-drug combination strategy in various trials leading to impressive results. The phase III ALCYONE trial74 evaluated the combination of daratumumab, bortezomib, melphalan, and prednisone versus bortezomib, melphalan, and prednisone in a transplant-ineligible population, leading to improvement in PFS with the four-drug combination.
In transplant-eligible MM, daratumumab, bortezomib, thalidomide and dexamethasone (Dara-VTD) was compared to bortezomib, thalidomide and dexamethasone (VTD) in the phase IIII CASSIOPEIA trial, demonstrating improvement in stringent complete response rate in favor of Dara-VTD, as summarized above.75 Daratumumab, bortezomib, lenalidomide, and dexamethasone (Dara-RVD) is compared to RVD in the Phase II GRIFFIN study76 in the context of transplantation. This trial has demonstrated improvement in stringent complete response with Dara-RVD combination (42.4% vs. 32%, OR 1.57; 95% CI, 0.87–2.82, p = 0.068), with very high levels of MRD-negativity rates (51% vs. 24%, p<0.001). These encouraging results may favor utilizing a daratumumab-based four drug platforms as standard of care induction therapy for transplant-eligible NDMM in the near future.
Daratumumab is given as an intravenous infusion, and was recently approved for subcutaneous use based on the phase III COLUMBA study demonstrating comparable efficacy.77 The subcutaneous formulation allows for far greater ease of use, particularly due to the reduced risk of infusion-related reactions associated with intravenous formulation with the first and second doses. The phase II PLEIDUS trial (NCT03412565) studied varies subcutaneous daratumumab combinations including lenalidomide and dexamethasone (DRd), bortezomib, melphalan and prednisone (D-VMP), and bortezomib, lenalidomide and dexamethasone (D-RVd). These combinations demonstrated comparable efficacy and safety to the intravenous formulation, leading to FDA approval.
Isatuximab
Isatuximab is an IgG1 anti-CD38 monoclonal antibody that is currently approved for treatment of RRMM with pomalidomide and dexamethasone. Isatuximab binds to a different epitope than daratumumab and spares NK cells, unlike daratumumab which depletes NK cells.78 It further inhibits regulatory T and B cells highly expressing CD38 to block inhibition on effector T cell function, which in turn may ameliorate immunosuppressive tumor microenvironment79,80 The phase III ICARIA-MM trial evaluated the combination of isatuximab, pomalidomide, and dexamethasone vs. pomalidomide and dexamethasone in patients with RRMM with 2 prior lines of therapy.81 This study demonstrated improvement of PFS in favor of the triplet combination (11.53 mo vs. 6.47 mo, HR 0.596, p = 0.001). The approval of isatuximab adds another anti-CD38 option for management of patients with RRMM. Isatuximab is also currently under investigation in other combination trials in both NDMM and RRMM.
Elotuzumab
Elotuzumab is a humanized IgG1 monoclonal antibody targeting the glycoprotein signaling lymphocytic activation molecule (SLAMF7), which is a cell surface glycoprotein receptor and a member of the signaling lymphocyte activating molecule family. This receptor is highly expressed on plasma cells and NK cells, and to a lesser degree, on CD8+ cells and DCs.82 Elotuzumab inhibits SLAMF7 directed MM cell adhesion to bone marrow stromal cells, and also leads to activation of ADCC and NK-cell mediated cytotoxicity.83 Elotuzumab has also been shown to suppress function of soluble SLAMF7, which has been shown to promote growth of MM cells due to homophilic interaction with surface SLAMF7 and leading to activation of the SHP-2 and ERK signaling pathways.84 Elotuzumab is currently approved in combination with lenalidomide and dexamethasone in patients with RRMM with 1–3 prior lines of therapy or with pomalidomide and dexamethasone in patients with 2 prior lines of therapy.
Elotuzumab, lenalidomide, and dexamethasone (Elo-LD) was compared to lenalidomide and dexamethasone (Ld) in patients with RRMM in the phase III ELOQUENT-2 trial.54 This trial demonstrated that the median PFS was higher with the triplet combination (19.4 months vs. 14.9 months). In the subsequent ELOQUENT-3 trial, elotuzumab, pomalidomide, and dexamethasone was compared to pomalidomide and dexamethasone in patients with 2 prior lines of therapy. This trial also led to improvement in patients treated with the triplet combination (median PFS 10.3 months vs. 4.7 months). The overall response rate was also higher at 53% in the elotuzumab group as compared with 26% in the control group (odds ratio, 3.25; 95% CI, 1.49 to 7.11).59
Checkpoint Inhibition in MM
The discovery and blockade of PD-1/PD-L1 axis has been one of the biggest breakthroughs in cancer therapeutics in the past decade, with several labeled indications in hematologic malignancies85 and solid tumors.86,87 PD-L1 expression on malignant PCs increases from precursor conditions such as MGUS and smoldering myeloma to NDMM with the highest levels observed in RRMM, suggesting a role of the PD-1/PD-L1 axis in promoting tumor immune evasion.88 Mechanistically, interaction of PCs with the MM microenvironment and secretion of proinflammatory cytokines (such as IL-6) in the bone marrow milieu contribute to the upregulation of PD-1 on MM cell surface and inhibition of T cell immune response.88 Its tumorigenic potential is also independent on the T cell evasion mechanism, as PD-L-1 overexpression can also induce reverse signaling to MM cells by activating the PI3K/AKT pathway and MM survival.88
Initial preclinical studies led to substantial activity with blockage of the PD-L1/PD-1 axis in MM.89 Further phase II studies of PD-1 inhibitors in MM revealed impressive overall response rates of greater than 50% in combination with IMIDs in a highly refractory RRMM population.90 However, immune-related toxicities and deaths occurred on further combination studies with checkpoint inhibitors and IMIDs, leading to a FDA clinical hold on all MM trials. The future of checkpoint inhibitor use in MM is uncertain.
Anti-BCMA Therapy in MM
BCMA is highly and universally expressed in plasma cells with minimal expression in other tissues, making it an ideal therapeutic target in MM.91 BCMA is a member of the tumor necrosis factor family, with ligands including B-cell activating factor (BAFF) and proliferation-inducing ligand (APRIL).92–94 BCMA-mediated upregulation of NFkB pathway and other tumor survival pathways leads to myeloma cell proliferation and suppression of the immune system.95,96 Increased soluble serum BCMA levels have been associated with shortened survival and progression from precursor states to overt myeloma.97
BCMA targeted therapies in myeloma include CAR T-cell therapy, bispecific antibodies, and antibody drug conjugates (Table 3). All of these treatment modalities have their advantages and disadvantages including frequency and mode of administration, need for hospitalization, toxicities, and efficacy.
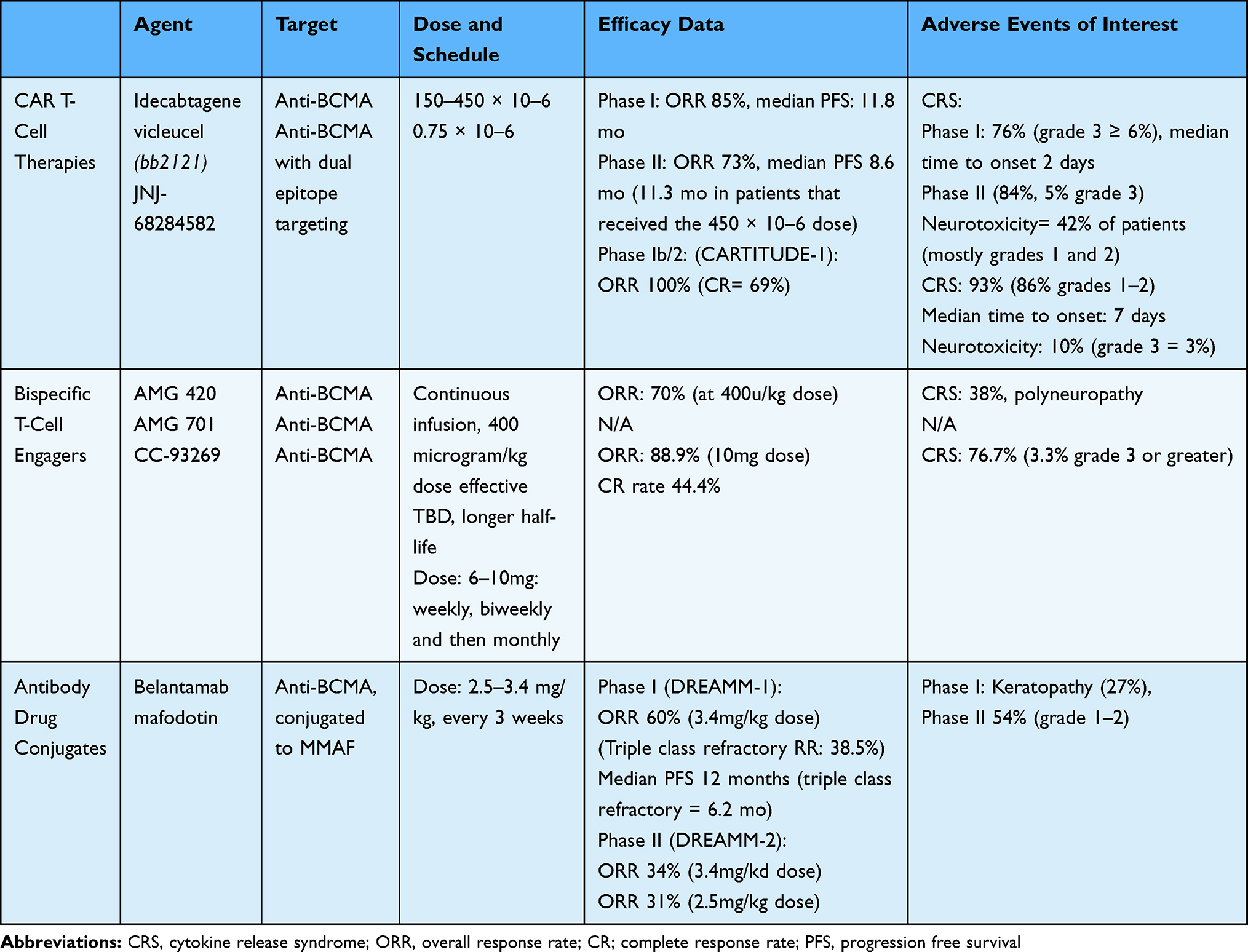 |
Table 3 Anti-BCMA Therapies |
CAR T-Cell Therapies
CAR T-cell therapy is currently approved for treatment of relapsed B-cell lymphoma and acute lymphoblastic leukemia.98,99 CAR T-cells are produced after autologous collection of the patient’s own white blood cells followed by stimulation of T cells and gene transfer of DNA encoding the CAR, commonly via a lentiviral vector. CAR T-cell construct utilizes an engineered CAR against the target antigen on the tumor cells (CD19 in lymphoma/leukemia and BCMA in MM). In case of MM, the anti-BCMA scFV are linked to CD3-zeta intracellular signaling domain. Costimulatory domains such as CD28 and 4–1BB are utilized for enhanced proliferation.100
Idecabtagene Vicleucel (bb2121)
Idecabtagene vicleucel (ide-cel; bb2121) is an anti-BCMA CAR T-cell product produced by transduction with lentiviral vector containing anti-BCMA scFV, a 4–1BB costimulatory domain, and CD3-zeta signaling domain. Initial preclinical studies of bb2121 in mouse models demonstrated significant activity, prompting further study.101 A Phase I trial in patients with heavily pretreated RRMM included patients with a median of 7 lines of therapy.102 The dose of bb2121 chosen for expansion ranged from 150 × 10–6 to 450 × 10–6. The overall response rate in this cohort was 85%, with a complete response rate of 45%. This translated to a median PFS of 11.8 months in the entire cohort (95% confidence interval, 6.2 to 17.8). MRD negativity was demonstrated in patients that achieved a partial response or better (at a level of 10–4). This led to a median PFS of 17.7 months in MRD-negative patients. Treatment-related adverse events were as expected, including hematologic toxicity and cytokine release syndrome (CRS), the latter observed in 76% of patients (grade 3 or greater in 6%). Median time to onset of CRS was 2 days (range 1 to 25 days). Neurotoxicity was seen in 42% of patients, and mostly low grade.
A follow up phase II study of bb2121was recently presented (Munshi et al, ASCO 2020, abs 8503, NCT03361748). This study enrolled 128 heavily pretreated patients with ≥ 3 prior regimens (median 6) and administered bb2121 doses ranging from 150 × 10–6 (4), 300 × 10–6 (70) and 450 × 10–6 (54). The overall response rate was 73%, with a median PFS of 8.6 months. In patients who received the 450 × 10–6 dose, median PFS was 11.3 months. Toxicities were manageable, with 84% of patients experiencing CRS (5% grade 3, 1 patient with grade 4 and 1 patient with grade 5). Neurotoxicity was observed in 18% of patients (3% grade 3).
JNJ-68284582
JNJ-68284582 is a dual epitope targeting CAR T-cell construct with two BCMA-targeting single domain antibodies. CD3 and 4–1BB are costimulatory domains, and the dual epitope targeting is meant to increase avidity to target antigen. Original trials from China (LCAR-B38M, Legend) demonstrated promising efficacy, with an overall response rate of 88% (68% complete response). MRD negativity was observed in 63% of the patients and median PFS was 15 months.103
Phase 1b/2 study (CARTITUDE-1) of JNJ-68284582 demonstrated an overall response rate of 100% in a heavily pretreated RRMM population.104 Complete response rate was 69%, and a high rate of MRD negativity was observed. The recommended phase 2 dose of 0.75x10-6 CAR T-cells was chosen, and is currently under investigation in subsequent studies. The toxicity profile of JNJ-68284582 is similar to other CAR T-cell products, with CRS rate of 93% (86% grades 1–2). The median time to CRS is slightly longer compared to bb2121 (median onset 7 days). Neurotoxicity was observed in 10% of patients, with only 3% grade 3 or greater neurotoxicity.
Descartes-08
Descartes-08 is RNA-generated anti-BCMA CD8 CAR T cell, as CD8 T cells are the main executive T effector cells to lyse tumor target cells.105 It has been demonstrated that Descartes-08 CAR T cells effectively depleted MM cell lines and patient MM cells, regardless of drug resistant status of MM cells and the presence of the immunosuppressive bone marrow microenvironment. Specifically, CAR-specific suppression of myeloma maintained throughout the duration of treatment in a mouse model of disseminated human MM, and repeated dosing was feasible to better control MM cell in vivo. The magnitude of cytolytic and cytokine responses correlated with the duration of anti-BCMA CAR expression. This may be more cost-effective, time-saving, and decrease the risk of severe CRS compared to virus-generated counterparts. An ongoing clinical trial of Descartes-08 in patients with RRMM (NCT03448978) has shown efficacy in a plasma cell leukemia patient.
Bispecific T-Cell Engagers
Bispecific antibodies are another method to engage the patient’s own T cells towards recognizing antigens on tumor cells. BCMA-directed bispecific antibodies bind BCMA on the MM cells and CD3 on T cells, and there are several current products in development.
AMG 420
AMG 420 is an anti-BCMA BiTE106 that has demonstrated significant activity in RRMM.107 However, further development of this has been halted, as its extremely short half-life requires continuous infusion through a pump and is not readily feasible for use in the clinic. However, the study did demonstrate a high response rate of 70% at the 400 microgram/kg dose (50% MRD negativity rate). Durability of response was also observed in some patients beyond 1 year. CRS rate was 38%, mostly grades 1 or 2, and peripheral polyneuropathy was seen in patients.
AMG 701
AMG 701 is an anti-BCMA Bite with longer half life which is under clinical development (NCT03287908). In preclinical studies, it has been shown that lenalidomide or pomalidomide combined with AMG 701 allows for enhanced MM cytotoxicity at lower doses, suggesting a more favorable therapeutic index (Cho et al, Blood Adv 2020).
CC-93269
CC-93269 is another anti-BCMA bispecific antibody in development. It is a humanized 2+1 IgG1-based T cell engager with a much easier schedule of administration that utilizes weekly, biweekly, followed by monthly dosing.108 Preliminary data of this construct has demonstrated an overall response rate of 88.9% in a phase I study at a dose of 10mg given intravenously. Complete response rates were also very high at 44.4%. CRS was seen in 76.7% of patients (3.3% being grade 3 or greater). There was 1 death reported on study related to CC-93269, which was attributed to CRS.
Antibody Drug Conjugates
Antibody drug conjugates (ADC) are yet another method to deliver targeted therapy to the tumor cells. These MoABs are conjugated to toxins such as tubulin polymerization inhibitor monomethyl auristatin F (MMAF) via a linker. The ADCs are then internalized, and the toxins are released, leading to DNA damage and cell death.109
Belantamab Mafodotin
Belantamab mafodotin is an anti-BCMA humanized monoclonal antibody drug conjugate (ADC) conjugated to MMAF, which is internalized leading to several mechanisms of myeloma cell death including apoptosis, ADCC, ADCP and immunogenic cell death.109 Belantamab mafodotin was recently FDA approved for RRMM patients who have had 4 prior therapies.
In the phase I DREAMM-1 study, belantamab mafodotin demonstrated a 60% overall response rate, with a dose of 3.4mg/kg intravenous administered every 3 weeks. The median PFS was 12 months and the median duration of response was 14.3 months,110 which was highly encouraging in a refractory RRMM population. The response rate in patients that were triple-class refractory to anti-CD38 antibody, PI and IMIDs was 38.5% with a median PFS of 6.2 months.
In the subsequent phase II DREAMM-2 study of 196 patients, two different dosing schedules were studied (2.5mg/kg or 3.4 mg/kg every 3 weeks). The ORR was 34% for patients treated at the 3.4mg/kg dose and 31% in patients treated with 2.5mg/kg dose.111 These results are again encouraging, and the 2.5mg/kg dose is being developed in multiple combination studies. (NCT04162210, NCT04091126).
The major toxicity concern with belantamab mafodotin is corneal toxicity due to changes in the corneal epithelium. Keratopathy of grade 3 or greater was observed in 27% of patients in DREAMM-2 study, with grades 1–2 keratopathy seen in up to 54% of patients. Vision returned to baseline or near baseline in 77–85% of patients. Methods for preventing and treating this toxicity are currently under study.
Conclusion and Future Directions
MM therapy continues to improve, with marked advancements in the previous two decades due to the bench to bedside translation of PIs, IMIDs, and MoAB.
The discovery of BCMA as a target has led to further development of immunotherapy in MM, and we are now entering a new era of myeloma therapeutics. These immunotherapies include CAR T-cell therapy, bispecific antibodies, and the recently approved antibody-drug conjugate, belantamab mafodotin. There are unique advantages and disadvantages to all of these approaches, and it is unclear at the present time if one is substantially superior to the other. The CAR T-cell therapies are particularly attractive due to a single infusion, leading to deep responses in a highly refractory population. The disadvantages include manufacturing time and resources, need for hospitalization, toxicities including CRS, neurotoxicity, cytopenias, and lack of durability beyond 1 year. The bispecific antibodies are attractive due to their “off-the-shelf” capabilities and continuous administration schedule. The toxicities overlap with CAR T-cell products, and it is unclear whether continuous administration will lead to more durable responses. The antibody-drug conjugates also are attractive due to their ease of use, but the corneal toxicity is a real-world and practical concern that needs further understanding and optimal management techniques. Additional immunotherapies currently under study include allogeneic CAR T-cells (NCT04171843), NK-cell therapies (NCT04309084), and dual targeting CAR T-cells targeting both plasma cells and cancer associated fibroblasts in the tumor microenvironment, with the intent to reduce resistance and improve durability of response (Sakemura et al, ASH 2019, abs 835).
In conclusion, these available immunotherapies are likely to change the landscape of MM therapy as we enter a new decade. Future studies will incorporate these approaches into earlier lines of therapy, and will combine them with other targeted myeloma agents. Correlative science studies of patient samples will define both mechanisms of action and resistance, allowing for informed combination therapies to achieve sustained MRD negativity on the one hand, while restoring host anti-MM immunity on the other.
Disclosure
Dr Omar Nadeem reports personal fees from Takeda, Celgene, Janssen, Sanofi, outside the submitted work. Dr Kenneth C Anderson reports personal fees from Bristol Myers Squibb, served at the advisory board for Millennium-Takeda, Sanofi-Aventis, Janssen, served at the scientific advisory board for Gillead and Precision Biosciences, during the conduct of the study; he is also the scientific founder for Oncopep and C4 Therapeutics, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122–1128. doi:10.1038/leu.2013.313
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33.
4. Lynch HT, Sanger WG, Pirruccello S, Quinn-Laquer B, Weisenburger DD. Familial multiple myeloma: a family study and review of the literature. J Natl Cancer Inst. 2001;93(19):1479–1483. doi:10.1093/jnci/93.19.1479
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol. 2015;33(26):2863–2869. doi:10.1200/JCO.2015.61.2267
6. Lecouvet FE, Boyadzhiev D, Collette L, et al. MRI versus (18)F-FDG-PET/CT for detecting bone marrow involvement in multiple myeloma: diagnostic performance and clinical relevance. Eur Radiol. 2020;30(4):1927–1937.
7. Koreth J, Cutler CS, Djulbegovic B, et al. High-dose therapy with single autologous transplantation versus chemotherapy for newly diagnosed multiple myeloma: a systematic review and meta-analysis of randomized controlled trials. Biol Blood Marrow Transplant. 2007;13(2):183–196. doi:10.1016/j.bbmt.2006.09.010
8. Sorrig R, Klausen TW, Salomo M, Vangsted A, Gimsing P. Risk factors for infections in newly diagnosed multiple myeloma patients: a Danish retrospective nationwide cohort study. Eur J Haematol. 2019;102(2):182–190.
9. Robertson JD, Nagesh K, Jowitt SN, et al. Immunogenicity of vaccination against influenza, Streptococcus pneumoniae and Haemophilus influenzae type B in patients with multiple myeloma. Br J Cancer. 2000;82(7):1261–1265. doi:10.1054/bjoc.1999.1088
10. Pratt G, Goodyear O, Moss P. Immunodeficiency and immunotherapy in multiple myeloma. Br J Haematol. 2007;138(5):563–579. doi:10.1111/j.1365-2141.2007.06705.x
11. Romano A, Conticello C, Cavalli M, et al. Immunological dysregulation in multiple myeloma microenvironment. Biomed Res Int. 2014;2014:198539. doi:10.1155/2014/198539
12. Gulla A, Anderson KC. Multiple myeloma: the (r)evolution of current therapy and a glance into future. Haematologica. 2020. doi:10.3324/haematol.2020.247015
13. Holthof LC, Mutis T. Challenges for immunotherapy in multiple myeloma: bone marrow microenvironment-mediated immune suppression and immune resistance. Cancers (Basel). 2020;12:4. doi:10.3390/cancers12040988
14. Chauhan D, Singh AV, Brahmandam M, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell. 2009;16(4):309–323. doi:10.1016/j.ccr.2009.08.019
15. Ray A, Song Y, Chauhan D, Anderson KC. Blockade of ubiquitin receptor Rpn13 in plasmacytoid dendritic cells triggers anti-myeloma immunity. Blood Cancer J. 2019;9(8):64. doi:10.1038/s41408-019-0224-6
16. Ray A, Das DS, Song Y, et al. A novel agent SL-401 induces anti-myeloma activity by targeting plasmacytoid dendritic cells, osteoclastogenesis and cancer stem-like cells. Leukemia. 2017;31(12):2652–2660. doi:10.1038/leu.2017.135
17. Costello C. The future of checkpoint inhibition in multiple myeloma? Lancet Haematol. 2019;6(9):e439–e440. doi:10.1016/S2352-3026(19)30149-8
18. Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017;36(4):561–584.
19. Okazuka K, Ishida T. Proteasome inhibitors for multiple myeloma. Jpn J Clin Oncol. 2018;48(9):785–793.
20. Chauhan D, Hideshima T, Anderson KC. Targeting proteasomes as therapy in multiple myeloma. Adv Exp Med Biol. 2008;615:251–260.
21. Williamson MJ, Blank JL, Bruzzese FJ, et al. Comparison of biochemical and biological effects of ML858 (salinosporamide A) and bortezomib. Mol Cancer Ther. 2006;5(12):3052–3061. doi:10.1158/1535-7163.MCT-06-0185
22. O’Donnell EK, Laubach JP, Yee AJ, et al. A phase 2 study of modified lenalidomide, bortezomib and dexamethasone in transplant-ineligible multiple myeloma. Br J Haematol. 2018;182(2):222–230. doi:10.1111/bjh.15261
23. Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, Phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–440. doi:10.1016/S1470-2045(11)70081-X
24. Bianchi G, Oliva L, Cascio P, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113(13):3040–3049. doi:10.1182/blood-2008-08-172734
25. Ettari R, Zappala M, Grasso S, Musolino C, Innao V, Allegra A. Immunoproteasome-selective and non-selective inhibitors: a promising approach for the treatment of multiple myeloma. Pharmacol Ther. 2018;182:176–192. doi:10.1016/j.pharmthera.2017.09.001
26. Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109(11):4839–4845. doi:10.1182/blood-2006-10-054221
27. Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565–1571. doi:10.1056/NEJM199911183412102
28. Lenz W, Knapp K. Thalidomide embryopathy. Arch Environ Health. 1962;5(2):100–105. doi:10.1080/00039896.1962.10663250
29. Palumbo A, Bertola A, Falco P, et al. Efficacy of low-dose thalidomide and dexamethasone as first salvage regimen in multiple myeloma. Hematol J. 2004;5(4):318–324. doi:10.1038/sj.thj.6200403
30. Hideshima T, Chauhan D, Shima Y, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96(9):2943–2950. doi:10.1182/blood.V96.9.2943
31. Mitsiades N, Mitsiades CS, Poulaki V, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99(12):4525–4530. doi:10.1182/blood.V99.12.4525
32. Holstein SA, McCarthy PL. Immunomodulatory drugs in multiple myeloma: mechanisms of action and clinical experience. Drugs. 2017;77(5):505–520.
33. Gupta D, Treon SP, Shima Y, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15(12):1950–1961. doi:10.1038/sj.leu.2402295
34. LeBlanc R, Hideshima T, Catley LP, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood. 2004;103(5):1787–1790. doi:10.1182/blood-2003-02-0361
35. Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–4779. doi:10.1182/blood-2011-05-356063
36. Zhu YX, Braggio E, Shi CX, et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood. 2014;124(4):536–545. doi:10.1182/blood-2014-02-557819
37. Hideshima T, Ogiya D, Liu J, et al. Immunomodulatory drugs activate NK cells via both Zap-70 and cereblon-dependent pathways. Leukemia. 2020. doi:10.1038/s41375-020-0809-x
38. Ocio EM, Fernandez-Lazaro D, San-Segundo L, et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia. 2015;29(3):705–714. doi:10.1038/leu.2014.238
39. Sehgal K, Das R, Zhang L, et al. Clinical and pharmacodynamic analysis of pomalidomide dosing strategies in myeloma: impact of immune activation and cereblon targets. Blood. 2015;125(26):4042–4051. doi:10.1182/blood-2014-11-611426
40. Vargesson N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today. 2015;105(2):140–156.
41. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127(21):2569–2574. doi:10.1182/blood-2016-01-693580
42. Attal M, Harousseau JL, Leyvraz S, et al. Maintenance therapy with thalidomide improves survival in patients with multiple myeloma. Blood. 2006;108(10):3289–3294. doi:10.1182/blood-2006-05-022962
43. Glasmacher A, Hahn C, Hoffmann F, et al. A systematic review of phase-II trials of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2006;132(5):584–593. doi:10.1111/j.1365-2141.2005.05914.x
44. Palumbo A, Facon T, Sonneveld P, et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood. 2008;111(8):3968–3977. doi:10.1182/blood-2007-10-117457
45. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371(10):906–917. doi:10.1056/NEJMoa1402551
46. Durie BGM, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet. 2017;389(10068):519–527. doi:10.1016/S0140-6736(16)31594-X
47. Joseph NS, Kaufman JL, Dhodapkar MV, et al. Long-term follow-up results of lenalidomide, bortezomib, and dexamethasone induction therapy and risk-adapted maintenance approach in newly diagnosed multiple myeloma. J Clin Oncol. 2020;38(17):1928–1937.
48. Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22–32. doi:10.1038/leu.2009.236
49. Palumbo A, Cavallo F, Gay F, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med. 2014;371(10):895–905. doi:10.1056/NEJMoa1402888
50. McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770–1781. doi:10.1056/NEJMoa1114083
51. Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782–1791. doi:10.1056/NEJMoa1114138
52. McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol. 2017;35(29):3279–3289. doi:10.1200/JCO.2017.72.6679
53. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319–1331. doi:10.1056/NEJMoa1607751
54. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621–631. doi:10.1056/NEJMoa1505654
55. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621–1634. doi:10.1056/NEJMoa1516282
56. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142–152. doi:10.1056/NEJMoa1411321
57. Nijhof IS, Franssen LE, Levin MD, et al. Phase 1/2 study of lenalidomide combined with low-dose cyclophosphamide and prednisone in lenalidomide-refractory multiple myeloma. Blood. 2016;128(19):2297–2306. doi:10.1182/blood-2016-07-729236
58. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974–981. doi:10.1182/blood-2017-05-785246
59. Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med. 2018;379(19):1811–1822. doi:10.1056/NEJMoa1805762
60. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123(12):1826–1832.
61. Richardson PG, Oriol A, Beksac M, et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): a randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(6):781–794. doi:10.1016/S1470-2045(19)30152-4
62. Amatangelo M, Bjorklund CC, Kang J, Polonskaia A, Viswanatha S, Iberdomide TA. (CC-220) has synergistic anti-tumor and immunostimulatory activity against multiple myeloma in combination with both bortezomib and dexamethasone, or in combination with daratumumab in vitro. Blood. 2018;132(Supplement 1):1935. doi:10.1182/blood-2018-99-113383
63. A Study to Determine Dose, Safety, Tolerability and Efficacy of CC-220 Monotherapy, and in Combination With Other Treatments in Subjects With Multiple Myeloma. https://ClinicalTrials.gov/show/NCT02773030.
64. Lonial S, NWCJvd D, Popat R, et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. 2019;37(15_suppl):8006.
65. Tai YT, Anderson KC. Antibody-based therapies in multiple myeloma. Bone Marrow Res. 2011;2011:924058. doi:10.1155/2011/924058
66. D’Agostino M, Boccadoro M, Smith EL. Novel immunotherapies for multiple myeloma. Curr Hematol Malig Rep. 2017;12(4):344–357.
67. van der Veer MS, de Weers M, van Kessel B, et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica. 2011;96(2):284–290. doi:10.3324/haematol.2010.030759
68. de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–1848. doi:10.4049/jimmunol.1003032
69. Sanchez L, Wang Y, Siegel DS, Wang ML. Daratumumab: a first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J Hematol Oncol. 2016;9(1):51. doi:10.1186/s13045-016-0283-0
70. van de Donk N, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. 2018;9:2134. doi:10.3389/fimmu.2018.02134
71. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387(10027):1551–1560. doi:10.1016/S0140-6736(15)01120-4
72. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754–766. doi:10.1056/NEJMoa1606038
73. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104–2115.
74. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518–528. doi:10.1056/NEJMoa1714678
75. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394(10192):29–38. doi:10.1016/S0140-6736(19)31240-1
76. Voorhees P, Kaufman JL, Laubach JP, et al. e. Depth of Response to Daratumumab (DARA), Lenalidomide, Bortezomib, and Dexamethasone (RVd) Improves over Time in Patients with Transplant-Eligible Newly Diagnosed Multiple Myeloma (NDMM): griffin Study Update. In. ASH Annual Meeting - Abstract 6912019.
77. Mateos MV, Nahi H, Legiec W, et al. Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Lancet Haematol. 2020;7(5):e370–e380.
78. Deckert J, Wetzel MC, Bartle LM, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20(17):4574–4583. doi:10.1158/1078-0432.CCR-14-0695
79. Feng X, Zhang L, Acharya C, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23(15):4290–4300. doi:10.1158/1078-0432.CCR-16-3192
80. Zhang L, Tai YT, Ho M, et al. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017;7(3):e547. doi:10.1038/bcj.2017.24
81. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet. 2019;394(10214):2096–2107. doi:10.1016/S0140-6736(19)32556-5
82. Ishida T. Therapeutic antibodies for multiple myeloma. Jpn J Clin Oncol. 2018;48(11):957–963. doi:10.1093/jjco/hyy133
83. Collins SM, Bakan CE, Swartzel GD, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62(12):1841–1849. doi:10.1007/s00262-013-1493-8
84. Kikuchi J, Hori M, Iha H, et al. Soluble SLAMF7 promotes the growth of myeloma cells via homophilic interaction with surface SLAMF7. Leukemia. 2020;34(1):180–195. doi:10.1038/s41375-019-0525-6
85. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi:10.1056/NEJMoa1411087
86. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi:10.1056/NEJMoa1910231
87. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(13):1270–1271. doi:10.1056/NEJMoa1504030
88. Tamura H, Ishibashi M, Sunakawa-Kii M. Inokuchi K. PD-L1-PD-1 pathway in the pathophysiology of multiple myeloma. Cancers (Basel). 2020;12:4. doi:10.3390/cancers12040924
89. Gorgun G, Samur MK, Cowens KB, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. 2015;21(20):4607–4618. doi:10.1158/1078-0432.CCR-15-0200
90. Badros A, Hyjek E, Ma N, et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130(10):1189–1197.
91. Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy. 2015;7(11):1187–1199. doi:10.2217/imt.15.77
92. Cho SF, Anderson KC, Tai YT. Targeting B Cell Maturation Antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. 2018;9:1821. doi:10.3389/fimmu.2018.01821
93. Tai YT, Anderson KC. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin Biol Ther. 2019;19(11):1143–1156. doi:10.1080/14712598.2019.1641196
94. Cho SF, Lin L, Xing L, et al. BCMA-targeting therapy: driving a new era of immunotherapy in multiple myeloma. Cancers (Basel). 2020;12:6. doi:10.3390/cancers12061473
95. Tai YT, Acharya C, An G, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127(25):3225–3236. doi:10.1182/blood-2016-01-691162
96. Tai YT, Lin L, Xing L, et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: therapeutic implications. Leukemia. 2019;33(2):426–438. doi:10.1038/s41375-018-0242-6
97. Sanchez E, Li M, Kitto A, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol. 2012;158(6):727–738. doi:10.1111/j.1365-2141.2012.09241.x
98. Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919
99. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
100. Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017;130(24):2594–2602. doi:10.1182/blood-2017-06-793869
101. Friedman KM, Garrett TE, Evans JW, et al. Effective Targeting of Multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther. 2018;29(5):585–601. doi:10.1089/hum.2018.001
102. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi:10.1056/NEJMoa1817226
103. Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141. doi:10.1186/s13045-018-0681-6
104. Madduri D, Usmani SZ, Jagannath S, et al. A Phase 1b/2 study of JNJ-4528, a CAR-T Cell Therapy Directed Against B-Cell Maturation Antigen (BCMA), in Patients with Relapsed and/or Refractory Multiple Myeloma (R/R MM) Abstract 577. ASH Annual Meeting 2019 2019.
105. Lin L, Cho SF, Xing L, et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia. 2020. doi:10.1038/s41375-020-0951-5
106. Hipp S, Tai YT, Blanset D, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017;31(8):1743–1751. doi:10.1038/leu.2016.388
107. Topp MS, Duell J, Zugmaier G, et al. Treatment with AMG 420, an Anti-B-Cell Maturation Antigen (BCMA) Bispecific T-Cell Engager (BiTE®) Antibody Construct, Induces Minimal Residual Disease (MRD) Negative Complete Responses in Relapsed and/or Refractory (R/R) Multiple Myeloma (MM) Patients: results of a First-in-Human (FIH) Phase I Dose Escalation Study. Blood. 2018;132(Supplement 1):1010.
108. Costa LJ, Wong SW, Bermudez A, et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): interim Results of a Phase 1 Multicenter Trial ASH Annual Meeting - Abstract 1432019.
109. Tai YT, Mayes PA, Acharya C, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128–3138. doi:10.1182/blood-2013-10-535088
110. Trudel S, Lendvai N, Popat R, et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019;9(4):37. doi:10.1038/s41408-019-0196-6
111. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.