Back to Journals » Journal of Inflammation Research » Volume 19
IL-17RA Promotes Cigarette Smoke-Induced Alveolar Epithelial Cell Pyroptosis in COPD via Dual Activation of the NLRP3/Caspase1/GSDMD and NF-κB/GSDME Pathways
Authors An X, Gu Y, Zhou J, Liu Y, Yu S, Zhang L, Ouyang Y
Received 10 November 2025
Accepted for publication 23 April 2026
Published 18 June 2026 Volume 2026:19 578571
DOI https://doi.org/10.2147/JIR.S578571
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Cynthia Koziol-White
Xuan An, Yanhui Gu, Jian Zhou, Yuting Liu, Shengyi Yu, Lanying Zhang, Yao Ouyang
Department of Respiratory and Critical Care Medicine, Affiliated Hospital of Zunyi Medical University, Zunyi, Guizhou, 563003, People’s Republic of China
Correspondence: Yao Ouyang, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) leads to persistent and irreversible airflow limitation due to lung tissue inflammation. Pyroptosis is closely involved in the pathogenesis of COPD and participates in the release of inflammatory factors. IL-17RA is a key receptor for IL-17A; however, the impact of IL-17RA on pyroptosis requires further investigation.
Methods: This study elucidates the promoting effect of IL-17RA on the progression of COPD by constructing IL-17RA alveolar type II (AT2) epithelial cell-specific knockout animal models and knockdown models of IL-17RA in AT2 epithelial cells. The process of cellular pyroptosis was observed through transmission electron microscopy. Molecular biology techniques and functional experiments were conducted both in vivo and in vitro to validate the activation of canonical and non-canonical pyroptosis pathways by IL-17RA.
Results: Our research findings indicate that the specific knockout of IL-17RA significantly alleviates damage to alveolar epithelial cells in a mouse model exposed to cigarette smoke, thereby delaying the progression of COPD in these mice. In this model, the absence of IL-17RA inhibits pyroptosis in cells and reduces the expression of proteins associated with pyroptosis. In the AT2 cell lines line, the knockdown of IL-17RA significantly suppresses the CSE-induced NLRP3/Caspase1/GSDMD and NFκB/GSDME pathways, thereby mitigating the occurrence of pyroptosis. In the AT2 cell lines line, IL-17A binds to IL-17RA, triggering the NLRP3/Caspase1/GSDMD pathway without activating the NFκB/GSDME pathway.
Conclusion: Our research emphasizes that IL-17RA promotes pyroptosis by activating the NLRP3/Caspase1/GSDMD and NFκB/GSDME pathways, and is involved in the progression of COPD, providing a promising target for the prevention and intervention of COPD in clinical settings.
Keywords: IL-17A, IL-17RA, GSDMD, GSDME, pyroptosis, COPD
Introduction
COPD are prevalent conditions that pose a significant threat to human health and remain unresolved in clinical treatment. The hallmark of COPD is persistent irreversible airflow limitation. In recent years, despite notable advancements in the diagnosis and treatment of COPD, the high incidence and mortality rates associated with the disease have not shown substantial improvement. Actively investigating the pathological mechanisms underlying COPD is crucial for identifying effective therapeutic targets.
Chronic inflammation significantly contributes to the onset and progression of COPD, primarily affecting the lung parenchyma and surrounding airways, which results in persistent respiratory symptoms and irreversible airflow limitation.1 The generation of inflammation involves two mechanisms: epithelial intrinsic and immune cell-driven inflammatory responses. In chronic smoking-related COPD, cigarette smoke directly enters alveolar type II epithelial cells, driving the epithelial intrinsic inflammatory response, which in turn amplifies damage through the secretion of cytokines and chemokines. This process also leads to the depletion of AT2 cells, impairing the repair function of alveolar cells and promoting the progression of COPD. Notably, AT2 serve as crucial functional and regenerative units, activating immune cell inflammation through the expression of pro-inflammatory factors. Notably, AT2 are pivotal in expressing pro-inflammatory factors.2 The persistence of chronic inflammation is associated with an increase in the population of helper T17 cells (Th17),3 which correlates with the onset of COPD.4 Enhanced Th17-type immune responses have been observed in both COPD patients and animal models, leading to elevated levels of the Th17-related inflammatory factor IL-17A in peripheral blood, bronchoalveolar lavage fluid, and lung tissue.5,6 IL-17A serves as a crucial inflammatory mediator for Th17 cells, exerting its effects by binding to IL-17RA, thereby activating the IL-17A signaling pathway and promoting the expression of downstream proteins such as NF-κB. The absence of IL-17RA can inhibit the biological functions of IL-17A.7 Previous studies have indicated a close relationship between IL-17A and COPD;8 However, the precise mechanism by which the IL-17A/IL-17RA axis regulates the progression of COPD through intrinsic pathways in AT2 cells remains unclear.
The process of pyroptosis is characterized by the release of significant amounts of pro-inflammatory factors. A key pathological feature of COPD is persistent airway inflammation, in which pyroptosis plays a crucial role in both its onset and progression.9 Previous studies have indicated that Th17-mediated autoimmunity can trigger pyroptosis,10 with the IL-17A signaling pathway being a known activator of this process.11 However, the specific role of IL-17A in promoting the progression of COPD and the associated chronic inflammation through pyroptosis has yet to be investigated.
Pyroptosis is a programmed necrosis process characterized by the continuous expansion of the cell membrane until it ruptures due to the action of gasdermin proteins. The ultimate effector of pyroptosis is a member of the gasdermin family,12,13 which includes GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and GSDMF. However, the role of gasdermin family members in the progression of COPD and their specific mechanisms remain unclear. Literature reports indicate that cigarette smoke extract activates the canonical pyroptosis signaling pathway involving NLRP3/Caspase1/GSDMD,14 and also induces GSDME lysis through Caspase-3.15 Notably, GSDMD and GSDME exhibit significant alterations in COPD.15,16 Additionally, it has been reported that IL-17A regulates the protein expressions of NLRP3 and GSDMD.17 However, the potential role of the interleukin-17A signaling pathway in AT2 cells in promoting the progression of COPD and airway inflammation through the regulation of the NLRP3/Caspase1 /GSDMD canonical pathway and the GSDME non-canonical pathway remains to be elucidated.
In this study, we demonstrate that cigarette smoke can promote the release of IL-17A, which binds to IL-17RA on AT2 cells and activates the classical pyroptosis pathway involving NLRP3/Caspase1/GSDMD. Meanwhile, cigarette smoke activates the non-classical pyroptosis pathway in AT2 cells through IL-17RA, leading to the synergistic promotion of pyroptosis in AT2.
Methods
Cell Culture
Human ATII were obtained from Shanghai Kisdeno Biotechnology Co., Ltd. The ATII cell line was cultured in modified Igor medium (DMEM, Gibco), supplemented with 10% fetal bovine serum (by volume) and 1% penicillin-streptomycin (Beijing Sobo Biotechnology Co., Ltd). All cell cultures were maintained in an incubator at 37°C with a 5% carbon dioxide concentration.
Drug
Cells in the logarithmic growth phase were inoculated into 6-well plates. When the cell confluence reached 50% to 70%, the complete culture medium was replaced, and the required drugs were added. MCC950 (Item No. HY-12815), VX-765 (Item No. HY-13205), IL-17A (Item No. HY-P70527) were all procured from MCE Corporation in New Jersey, USA.
Preparation of Cigarette Smoke Extract (CSE)
A cigarette (tar: 10 mg per cigarette, nicotine: 0.9 mg per cigarette, Huangguoshu (Long March) Cigarettes) was used to collect smoke through a direct cold trap, which was then passed into 10 mL of DMEM. After adjusting the pH of the CSE solution to 7.2–7.4, it was filtered through a 0.22 μm filter membrane to remove bacteria and larger particles. The optical density (OD) was measured at 0.5 (absorbance: 405 nm), and the resulting solution was defined as 100% CSE. This concentration can be adjusted as necessary for experimental requirements, and CSE should be used within 30 minutes of preparation. Each experiment was conducted with at least three independent repetitions, and each sample underwent mechanistic repeats to ensure the reproducibility of the results.14
Hoechst 33342/PI Fluorescence Staining
Gently wash the cells twice with pre-cooled PBS to eliminate any residual culture medium. Subsequently, add 4% paraformaldehyde (PFA) and fix the cells at room temperature for 15 minutes. After fixation, wash the cells with PBS three times, each wash lasting 5 minutes. Prepare PBS staining solutions containing Hoechst 33342 (1 μg/mL) and propidium iodide (PI) (5 μg/mL), cover the cells, and incubate in the dark for 15–20 minutes. Aspirate and discard the staining solution, then gently wash the cells twice with PBS to prevent over-rinsing, which may lead to cell shedding. Immediately observe the cells using a fluorescence microscope; Hoechst (Ex/Em ≈ 350/461 nm) stains all cell nuclei blue, while PI (Ex/Em ≈ 535/617 nm) selectively stains the nuclei of dead cells red.
Cell Counting Kit 8
Select cells in the logarithmic growth phase and digest them with trypsin. Adjust the cell density to 5000 cells per well and inoculate into 96-well plates with 100 μL per well, ensuring three duplicate wells. Gently shake the plates to achieve uniform cell distribution. To prevent evaporation, add 100 μL of PBS to the outermost circle of the culture plate. Incubate the 96-well plates in a 37 °C, 5% CO2 incubator for 12, 24, 36, and 48 hours, respectively. After reaching each time point, add 10 μL of CCK-8 reagent to each well and continue incubation for an additional 2 hours. Finally, measure the absorbance (OD value) at 450 nm.
Western Blot
Tissues or cells were lysed using a RIPA lysis buffer containing 1% protease inhibitor mixture and 1% phosphatase inhibitor (P1260, Solarbio). The tissue/cell lysate mixture was centrifuged at 12,000 rpm at 4 °C for 15 minutes, and the supernatant was collected. Protein concentration was determined using the BCA protein detection kit (PC0020, Solarbio, Beijing). Subsequently, proteins were transferred to polyvinylidene fluoride (PVDF) membranes via SDS-PAGE gel electrophoresis (20 μg per sample) (Millipore, Billerica, Massachusetts, USA) (Yu et al, 2023). The PVDF membrane was blocked with 5% skimmed milk (AR0104, BOSTER Biotechnology, Shanghai) for 2 hours and then incubated overnight with the primary antibodies: anti-GSDMD (1:1000; PU224937, Abmart, China), anti-GSDME (1:1000; P79886, Abmart, China), anti-Caspase-1 (1:1000; P79884R2, Abmart, China), anti-Caspase-3 (1:1000; T40044, Abmart, China), anti-NLRP3 (1:1000; P60622R3, Abmart, China), anti-IL-18 (1:1000; TD6252, Abmart, China), anti-NF-κB p65 (1:1000; HY-P80765, MCE, China), anti-TNF-α (1:1000; A11534, ABclonal, China), anti-IL-18 (1:1000; A23076, ABclonal, China), anti-IL-17RA (1:1000; A10052, ABclonal, China), anti-IL-17A (1:1000; A12454, ABclonal, China), anti-IL-1β (1:1000; A22257, ABclonal, China), anti-IL-6 (1:1000; A0286, ABclonal, China), anti-GAPDH (1:100,000; A19056, ABclonal, China), and anti-tubulin (1:5000; A12289, ABclonal, China). The next day, the secondary antibody bound to the PVDF membrane was incubated with horseradish peroxidase (HRP) at 4 °C for 40 to 50 minutes. The ECL chemiluminescence reagent (MA0186, MeilunBio, China) was used to visualize the protein bands on the PVDF membrane. Exposure and development were performed using the ChemiDocTM imaging system (Bio-Rad, Hercules, California, USA), and quantitative analysis of the proteins was conducted using Image Lab software.
RT-qPCR
Add Trizol reagent and chloroform to the tissue or cells in sequence. Subsequently, centrifuge the mixture to remove the upper aqueous phase. Following this, add an equal volume of isopropyl alcohol and centrifuge again. Discard the supernatant and wash the RNA twice with 75% ethanol to facilitate precipitation. The RNA precipitate is then dissolved in DEPC buffer solution for concentration determination. The reaction conditions are set as follows: 37°C for 5 seconds and 85°C for 4 seconds. After a reaction time of 15 minutes, the reverse transcription products can be stored at −80°C. Perform real-time fluorescence quantitative PCR using the primers listed in Table 1.
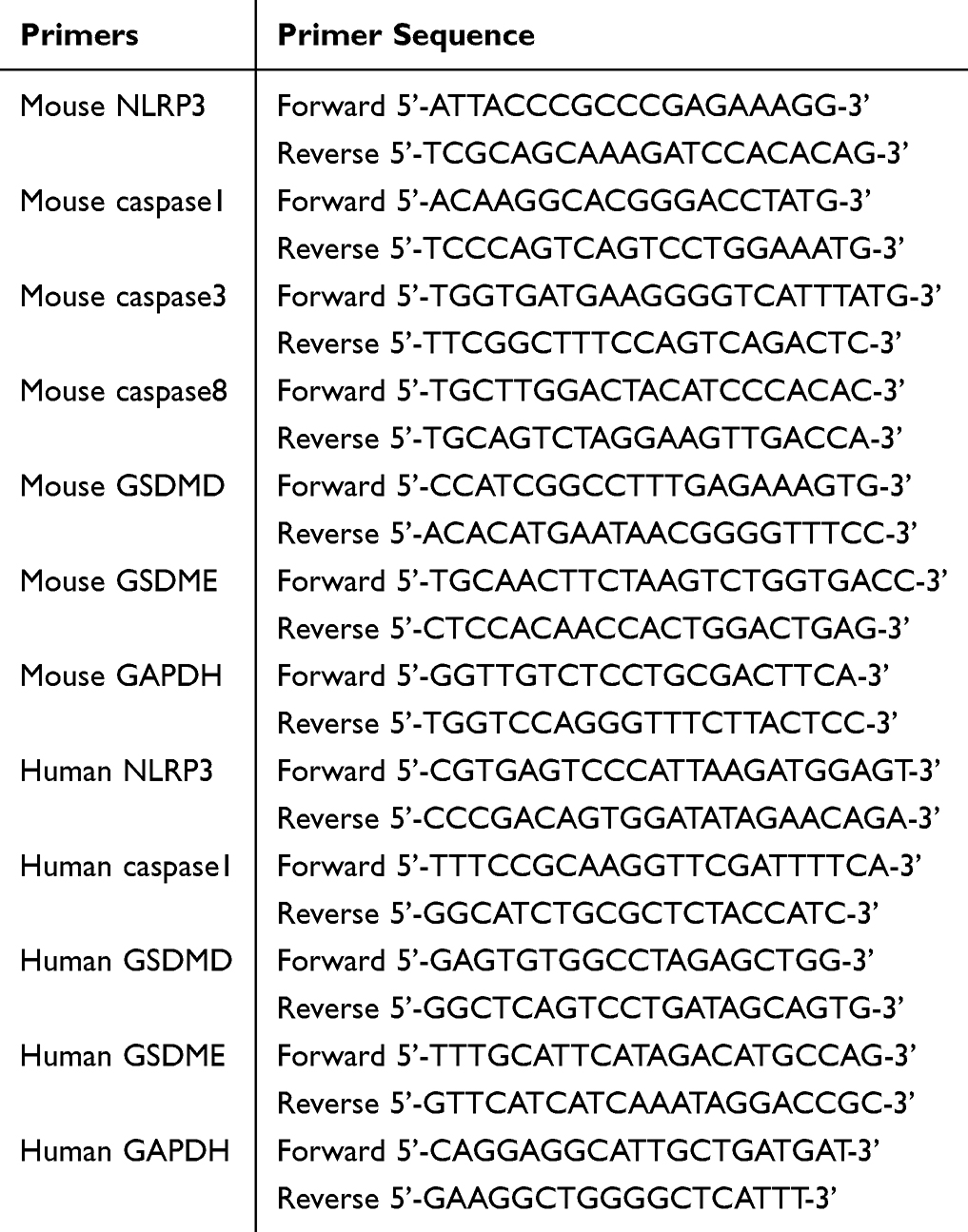 |
Table 1 PCR Experiment mRNA Primers |
Mouse Model of Chronic Obstructive Pulmonary Disease (COPD)
Male C57BL/6, IL-17RA(flox/flox) SFPTC-cre(+) and IL-17RA(flox/flox) SFPTC-cre(-) mice (6–8 weeks old, weight 20 ± 2 g) were maintained under specific pathogen-free conditions with a 12-hour light/dark cycle at 22 ± 1°C and 50 ± 10% humidity. Animals were provided ad libitum access to autoclaved food and water. After one week of acclimatization, C57BL/6, IL-17RA(flox/flox) SFPTC-cre(+) and IL-17RA(flox/flox) SFPTC-cre(-) mice were randomly divided into two experimental groups respectively (n = 6/group): Control group (Received ambient air exposure) and COPD group (Subjected to chronic cigarette smoke exposure). The total number of mice was 36. The COPD model was established via a standardized CS exposure protocol adapted from.18 Mice in the COPD group were placed in a 30-L plexiglass exposure chamber. Throughout the experiment, Changzheng Huangguoshu brand cigarettes, each containing 10 mg of tar, were used as the smoke source to expose the mouse to cigarette smoke. Each exposure lasted for 1 hour, involving 12 cigarettes per session, with a half-hour interval between exposures. This procedure was conducted twice daily for 5 consecutive days each week over a duration of 6 months. Control mice underwent identical handling in a separate chamber without CS exposure. After 24 weeks, mice were anesthetized by intraperitoneal injection of pentobarbital sodium (150 mg/kg). Finally, lungs were harvested, and divided for histopathology. All animals were tested and analyzed. All the mice were purchased from Cyagen Biotechnology Co., LTD. All animal studies comply with the ARRIVE reporting guidelines.
Ethics Approval and Consent to Participate
The animal study was approved by the Laboratory Animal Welfare and Ethics Committee (Zunyi Medical University). The study was conducted in accordance with the local legislation and institutional requirements.
Genetic Identification
The tails of one-week-old mouse were collected for identification purposes, specifically using lung tissue from mouse subjected to gene knockout. The genotype of the mouse was determined through PCR. The primer sequences necessary for identification were supplied by Cyagen (Suzhou) Biotechnology Co., Ltd. and synthesized by Hejin Biotechnology Co., Ltd. The detailed contents of these sequences are shown in Table 2: To begin the procedure, add 20 µL of Lysis Buffer M and 0.5 µL of Liquid Proteinase K to the mouse tissue. Shake and mix thoroughly, then allow the mixture to stand at room temperature for 20 minutes. Following this, inactivate the reaction by incubating at 98 °C for 30 minutes. Next, prepare the PCR amplification system according to the previously described method, and establish the PCR reaction conditions as per the identification protocol provided by Cyagen. For gel electrophoresis, prepare an agarose gel using 0.8 g of agarose powder in 40 mL of 1×TAE, and conduct the electrophoresis at 150 V for 30 minutes. The analysis was performed using Image Lab software, and photographs were taken to document and annotate the pertinent information of the samples.
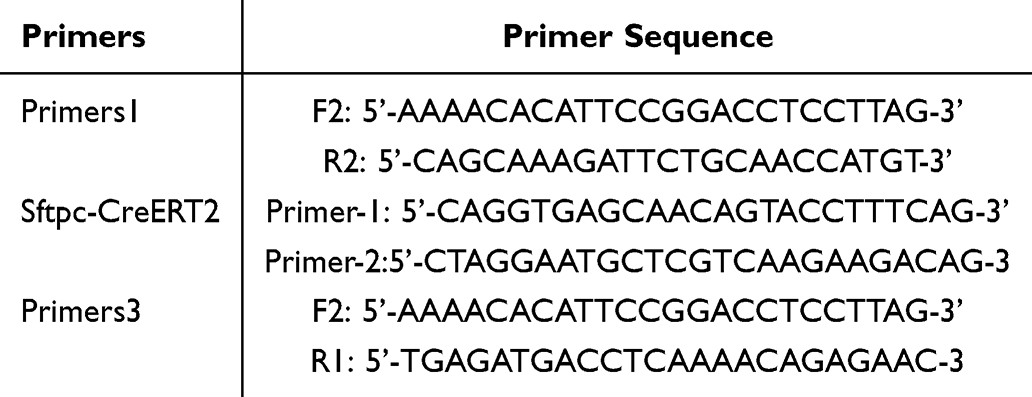 |
Table 2 Primer Sequences for Genotyping of Flox and Cre Mice |
Statistical Analysis
SPSS 29.0 statistical software was used to analyze all experimental data. The experimental data were expressed as mean ± standard deviation (mean ± SD). Data were analyzed by t-test for two-way comparisons; one-way ANOVA was used for multiple comparisons, and LSD test was used if the variance was uniform, and Dunnett’s T3 test was used if the variance was not uniform. p<0.05 indicated that the difference was statistically significant.
Result
IL-17RA−/− Mouse Improve the Cigarette Smoke Exposure Model of COPD
It has been reported that the cigarette smoke exposure model is the most suitable method for studying COPD.18 To investigate the role of IL-17RA in COPD in mouse, we specifically knocked out IL-17RA in mouse alveolar type II epithelial cells and established a model using the cigarette smoke exposure method (Figure 1A). Gene identification results from the tails of the mouse confirmed the successful knockout of IL-17RA (Figure 1B). Furthermore, gene identification results showed that IL-17RA was not knocked out in the bronchi and kidneys, indicating a successful specific knockout of IL-17RA in the lung tissue of the mouse (Figure 1C). In the cigarette smoke exposure model, lung tissue from mice in the WT group exhibited a uniform pink coloration, smooth surface, absence of collapse, and preserved elasticity. In contrast, lung tissue from the CS group appeared grayish-white with markedly reduced elasticity. Notably, mice in the IL-17RA−/−+CS group displayed a partial restoration of lung surface color toward pink, accompanied by a recovery of elastic properties. In the SFTPCcre cohort, mice exposed to cigarette smoke SFTPCcre+CS showed light pink lung tissue; however, elastic function remained diminished.(Figure 1D). Histological analysis by HE staining revealed that the alveolar architecture remained intact in the WT group. In contrast, the WT+CS group exhibited alveolar cavity enlargement, disruption of the alveolar septa, and significant inflammatory cell infiltration. Notably, in the IL-17RA−/−+CS group, the alveolar cavities appeared contracted, the alveolar septa showed signs of structural reconstruction, and inflammatory responses were markedly reduced. Similar pathological changes, including alveolar dilation and inflammatory cell infiltration, were observed in both the SFTPCcre and SFTPCcre+CS groups. (Figure 1E). Masson’s staining results showed that in the WT group, collagen was only distributed in the blood vessels and airway walls, in the WT CS group, interstitial collagen proliferated diffusely (blue), in the IL-17RA-/-+CS group, collagen deposition was reduced. Similarly, in the SFTPCcre group, collagen deposition around the airways was significant in the SFTPCcre CS group (Figure 1F). CT test results indicated that, compared to the WT group, the anterior-posterior diameters of mouse in the CS group significantly increased, while IL-17RA−/− mouse showed marked improvement (Figure 1G and I). Pulmonary function tests indicated that, compared to the WT group, the forced expiratory volume in the first second/forced vital capacity (FEV1/FVC), peak expiratory flow (PEF), maximal voluntary ventilation (MV), and dynamic lung compliance (CDyn) significantly decreased in the CS group, whereas IL-17RA−/− mouse exhibited significant recovery. Pulmonary function test was significantly decreased in the SFTPCcre+CS group relative to the SFTPCcre group (Figure 1J).
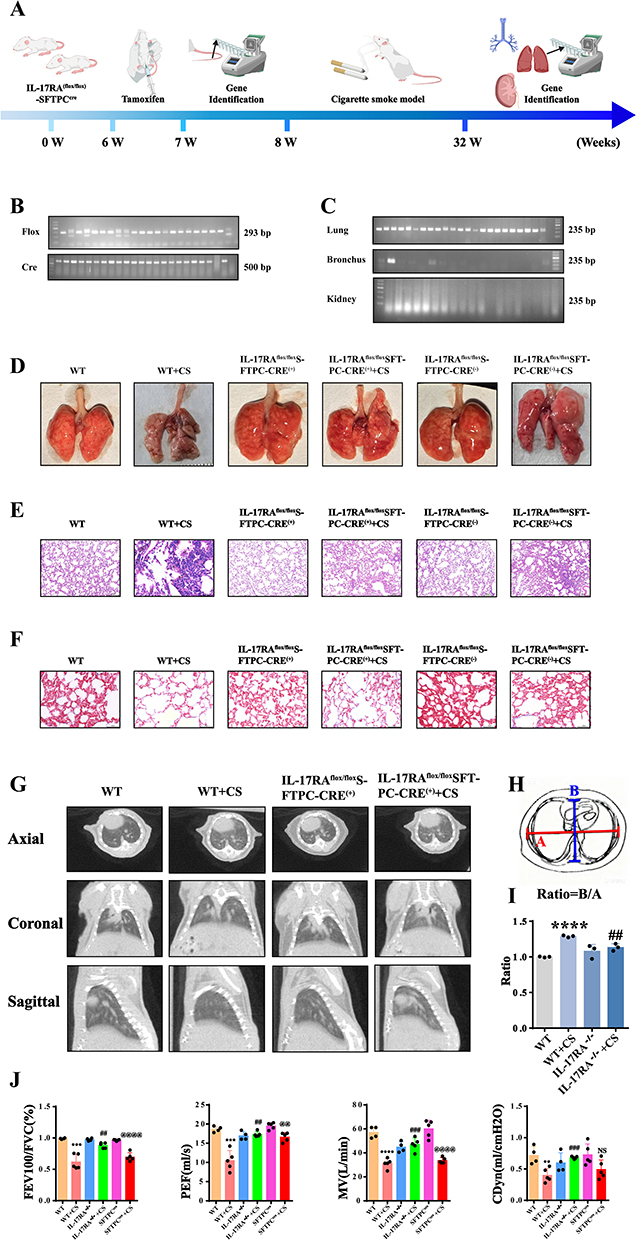 |
Figure 1 IL-17RA−/− -SFTPCCRE mouse improve the cigarette smoke exposure model of COPD. (A) Schematic diagram of IL-17RA−/− -SFTPCcre mouse COPD model establishment and detection. (B) 7-week-old IL-17RA−/− -SFTPCcre mouse FLOX and CRE gene detection diagram. (C) Genetic testing of the FLOX gene in the lungs, bronchi and kidneys of 6-month-old IL-17RA−/− -SFTPCcre mouse. (D) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS. SFTPCcre. SFTPCcre+CS mouse lung tissue. (n=5) (E) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS. SFTPCcre. SFTPCcre+CS mouse HE staining images.(n=5) (F) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS. SFTPCcre. SFTPCcre+CS mouse Masson staining images. (n=5) (G) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS mouse CT. (n=5) (H and I) Ratio of left and right diameters to front and rear diameters. (J) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS. SFTPCcre. SFTPCcre+CS mouse Pulmonary Function Results (FEV100/FVC. PEF. MV. CDyn) (n=5). (VS WT **P<0.01, ***P<0.001, ****P<0.0001, VS IL-17RA−/−+CS ## P<0.01, ### P<0.001, VS SFTPCcre @@ P<0.01, @@@@ P<0.0001). |
IL-17RA Promotes Cellular Pyroptosis, Facilitating the Progression of COPD in Mouse
To explore whether IL-17RA affects cell pyroptosis and thereby promotes the progression of COPD in a cigarette smoke-exposed mouse model, we further evaluated the indicators related to cell pyroptosis. Immunohistochemical results showed that the expression of N-GSDMD was significantly elevated in the cytoplasmic periphery of the cells in the CS group, while the expression of GSDME protein also increased. In IL-17RA−/− mouse, the expression of N-GSDMD and GSDME was significantly reduced (Figure 2A–D). Transmission electron microscopy observations revealed that some type II alveolar epithelial cells in the CS group had undergone pyroptosis. The nuclei appeared irregular in shape, with a uniform distribution of chromatin, sparse heterochromatin, intact and continuous nuclear membranes, and some nuclei exhibited widened perinuclear spaces. The rough endoplasmic reticulum was dilated, and the luminal spaces widened to form vesicle-like structures; a small amount of autophagy and lipid droplets were also observed in the cytoplasm. The cell membranes showed significant perforation, with some areas completely lost, leading to cytoplasmic contents spilling into the alveolar lumen. In IL-17RA−/− mouse, the nuclear membranes of some type II alveolar epithelial cells remained intact and continuous, with some nuclei showing widened perinuclear spaces. The cell membranes were also intact, and numerous short, thick microvilli were observed on the cell surface. A few small perforations were noted on the membranes of some cells, indicating a significant improvement in pyroptosis (Figure 2E). Further analysis using ELISA showed that pyroptosis-related inflammatory factors in serum and bronchoalveolar lavage fluid were elevated in the CS group compared to the WT group, with increased levels of IL-1β, IL-6, and IL-18, while IL-17RA−/− mouse exhibited significantly reduced levels in both serum and bronchoalveolar lavage fluid (Figure 2F and G).
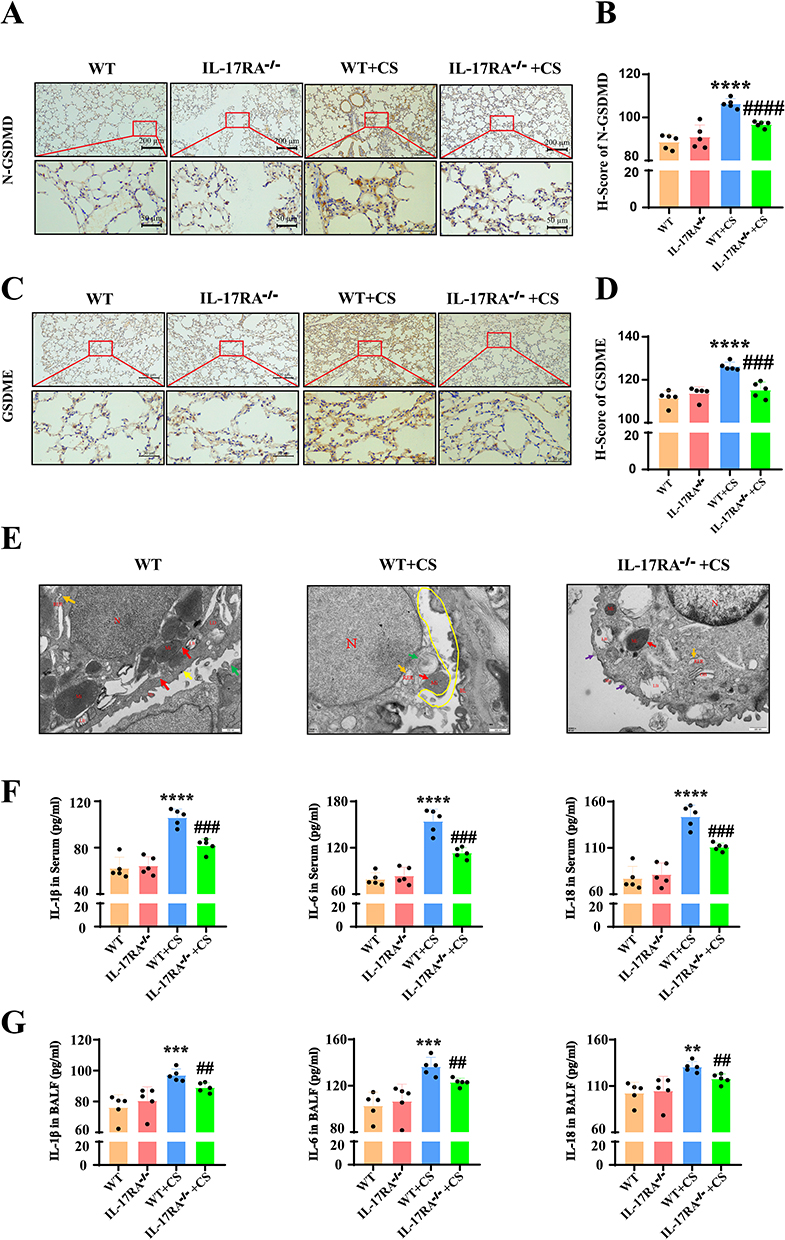 |
Figure 2 IL-17RA promotes cellular pyroptosis, facilitating the progression of COPD in mouse. (A and B) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS mouse Immunohistochemical staining (A) and positive statistics (B) of N-GSDMD. (n=5) (C and D) WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS mouse Immunohistochemical staining (C) and positive statistics (D) of GSDME. (n=5) (E) WT. WT+CS. IL-17RA−/−+CS mouse Transmission electron microscope image.(n=3) (F) The content of inflammatory factors such as IL-1β, IL-6 and IL-18 in WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS mouse serum. (n=5) (G) The content of inflammatory factors such as IL-1β, IL-6 and IL-18 in WT. WT+CS. IL-17RA−/−. IL-17RA−/−+CS mouse alveolar lavage fluid. (n=5) (VS WT **P<0.01, ***P<0.001, ****P<0.0001, VS WT+CS ##P<0.01, ###P<0.001, ####P<0.0001). |
IL-17RA Activates Proteins Related to the Canonical/Non-Canonical Pathways of Pyroptosis
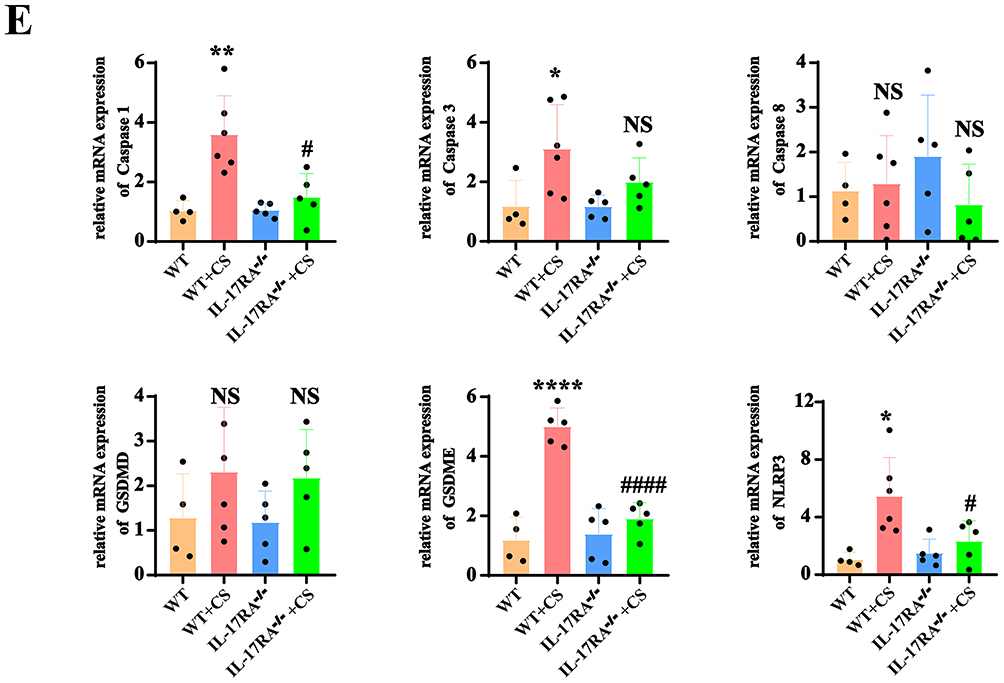
It has been reported that the canonical pyroptosis pathway is triggered by the NLRP3/Caspase1/GSDMD pathway, while the non-canonical pyroptosis pathway is activated by the NFκB/GSDME pathway. Therefore, we examined the expression of proteins related to both canonical and non-canonical pyroptosis pathways. WB experimental results indicated that compared to the WT group, the expressions of NLRP3, Caspase 1, caspase 1 p33, Caspase 3, GSDMD, N-GSDMD, GSDME, IL-6, IL-18, IL-17A, and TNF-α proteins were elevated in the CS group mouse model (Figure 3A and B), while their expressions were reduced in the lung tissues of IL-17RA−/− mouse except caspase3 and GSDMD. These results indicate that the IL-17RA protein regulates the cleavage of the GSDMD protein. (Figure 3C and D). Compared to the WT group mice, there was no significant change in protein expression in the IL-17RA−/− group mice. Similarly, there was no significant change in protein expression when comparing IL-17RA−/−+Air to IL-17RA−/−+CS (Supplementary Material 1). PCR results showed that compared to the WT group, the mRNA levels of NLRP3, caspase 1, and GSDME were increased in the CS group mouse model, while they were decreased in IL-17RA−/− mouse. However, no significant changes were observed in the mRNA levels of caspase 3, caspase 8, and GSDMD (Figure 3E). These results suggest that IL-17RA activates proteins associated with the canonical/non-canonical pyroptosis pathways.
Figure 3 IL-17RA activates proteins related to the canonical/non-canonical pathways of pyroptosis. (A and B) Expression (A) and Statistical chart (B) of NLRP3, Caspase 1, Caspase 1 p33, Caspase 3, GSDMD, N-GSDMD, GSDME, IL-6, IL-18, IL-17A, TNFα protein in WT and WT + CS mouse. (n=5) (C and D) Expression (C) and Statistical chart (D) of NLRP3, Caspase 1, Caspase 1 p20, Caspase 1 p33. GSDMD, N-GSDMD, GSDME, IL-6, IL-1β, IL-18, TNFα, IL-17A protein in WT+CS and IL-17RA−/− + CS mouse. (n=5) (E) mRNA expression of Caspase 1. Caspase 3. Caspase 8. GSDMD. GSDME. NLRP3. (n=5) (VS WT *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, VS WT+CS #P<0.05, ####P<0.0001). Figure 3 Continued. Figure 3 Continued.

Cigarette Smoke Extract Activates IL-17RA to Trigger Pyroptosis
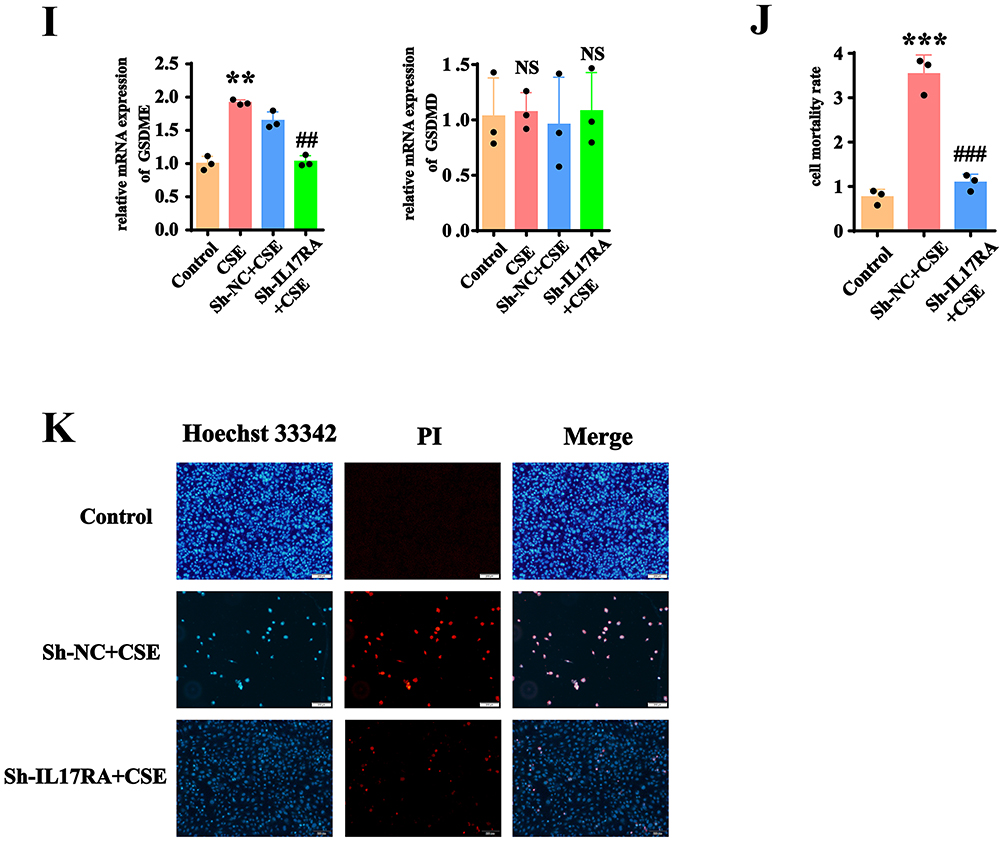
To validate the mechanism of IL-17RA activation in cell pyroptosis, we utilized immortalized human alveolar type II epithelial cells. The use of cigarette smoke extract (CSE) demonstrated through CCK8 results that at a concentration of 5% CSE, AT2 cell lines underwent cell death (Figure 4A). CCK8 experiments indicated that after exposure to 8% CSE for 12h, 24h, 36h, and 48h, the viability of AT2 cell lines significantly decreased (Figure 4B). Therefore, we decided to use an 8% CSE concentration and conduct the subsequent experiments for 24 hours. WB results showed that at an 8% CSE concentration, the expressions of NLRP3 and N-GSDMD increased, while the total protein level of GSDMD did not exhibit significant changes (Figure 4C and D). To verify whether cigarette smoke extract activates IL-17RA to trigger cell pyroptosis, we knocked down IL-17RA using lentivirus, followed by validation through WB, RT-qPCR, and fluorescence selection (Figure 4E–G). Upon the addition of 8% CSE, WB results indicated that the levels of pyroptosis-related proteins N-GSDMD, GSDME, IL-1β, IL-17A, IL-18, and TNFα significantly increased; however, after knocking down IL-17RA, the protein expressions decreased. Notably, the total protein level of GSDMD did not show significant changes (Figure 4H). Concurrently, RT-qPCR results demonstrated that the expression of GSDME increased after the addition of CSE, and decreased following IL-17RA knockdown, while GSDMD consistently showed no significant changes (Figure 4I). These results indicate that CSE activates IL-17RA to regulate the cleavage process of GSDMD protein and the transcription process of GSDME protein. The results of LDH and PI/Hoechst 33342 experiments indicated that after the addition of CSE, pyroptosis occurred in the cells, and after knockdown of IL-17RA, pyroptosis decreased (Figure 4J and K). The results indicated that in the AT2 cell lines line, CSE activates IL-17RA to trigger pyroptosis.
Figure 4 Cigarette smoke extract activates IL-17RA to trigger pyroptosis. (A) Cell morphology of AT2 cell lines under different CSE concentrations. (B) Cell viability of AT2 cell lines under different CSE concentrations. (C and D) Expression (C) and Statistics (D) of NLRP3, GSDMD, and N-GSDMD proteins under different CSE concentrations. (VS 0% *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001) (E–G) Proteins (E), mRNAs (VS Control NS>0.05 VS Sh-NC #P<0.05) (F) and fluorescence (VS Sh-NC *P<0.05, **P<0.01) (G) images in the AT2 cell lines line with knocked-down IL-17RA gene. (H) Expression and statistics of GSDMD. N-GSDMD. GSDME. IL-1β. IL-17A. IL-18. TNFα protein in Control, CSE, sh-NC+CSE, sh-IL-17RA Cells. (VS Control *P<0.05, **P<0.01, ***P<0.001, VS Sh-NC+CS #P<0.05) (I) mRNA Expression of GSDMD. N-GSDMD. GSDME. IL-1β. IL-17A. IL-18. TNFα in Control, CSE, sh-NC+CSE, sh-IL-17RA mouse. (J) The LDH kit is used to detect the cell mortality rate. (K) Hoechst 33342/PI fluorescence expression diagram. Figure 4 Continued. Figure 4 Continued.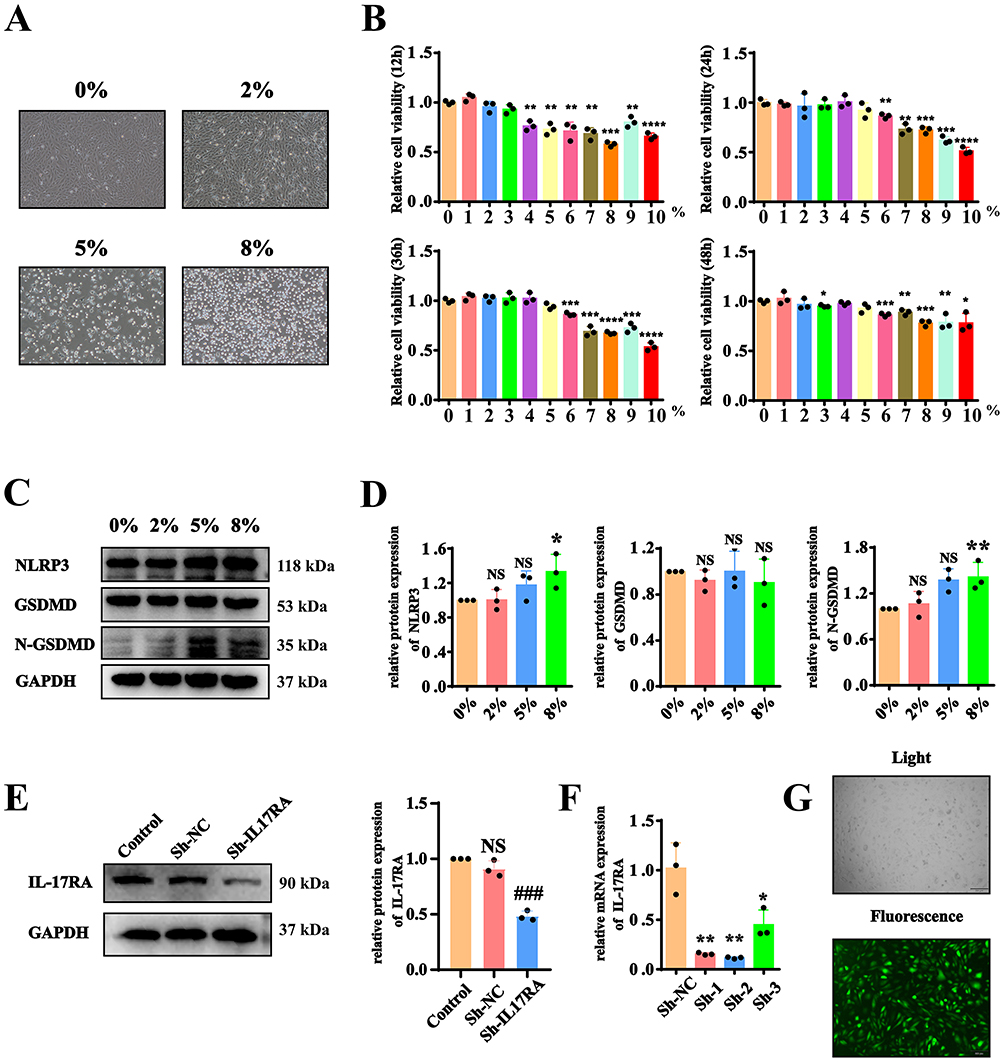
CSE Activates IL-17RA, Triggering Cell Pyroptosis Through the NLRP3/Caspase1/GSDMD and NFκB/GSDME Pathways
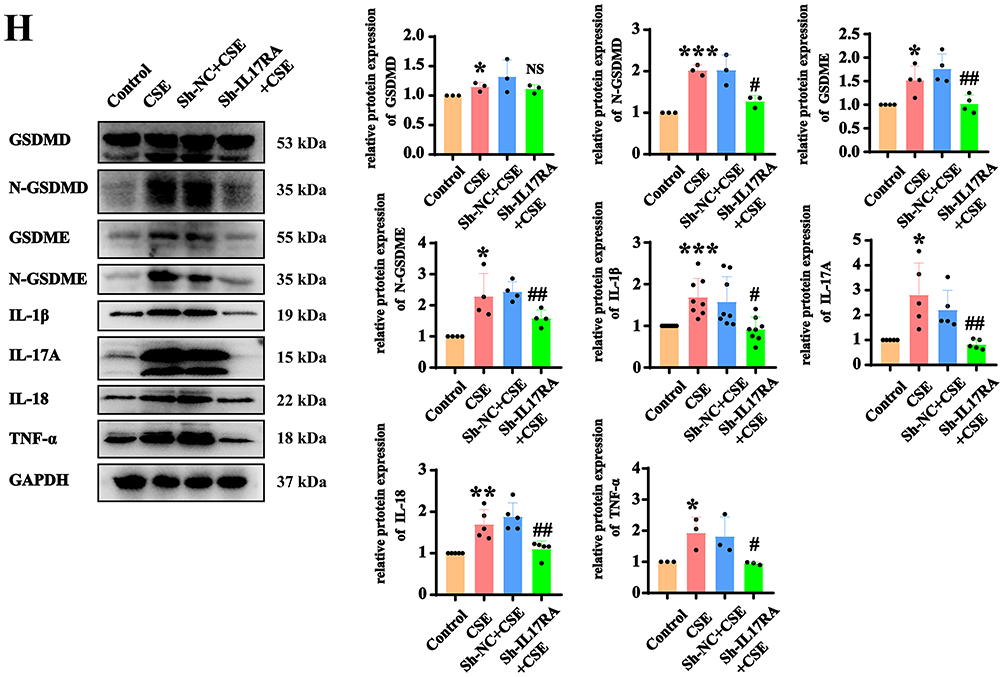
To validate that IL-17RA activates cell pyroptosis through the NLRP3/Caspase1/GSDMD canonical pathway and the NFκB/GSDME non-canonical pathway, WB results indicated that after the addition of CSE, the protein expressions of NLRP3, Caspase-1, Caspase-1 P20 and nuclear NF-κB p65 increased significantly, while the protein expression decreased markedly following the knockdown of IL-17RA (Figure 5A and B). It is reported that the active caspase-1 protein cleaves the GSDMD protein. To further investigate whether CSE activates caspase-1 and thereby induces cleavage of GSDMD. The Caspase-1 activity assay demonstrated that the activity of Caspase-1 was significantly enhanced after the addition of CSE, whereas the activity was notably reduced following the knockout of IL-17RA (Figure 5C). RT-qPCR results showed that the mRNA levels of NLRP3 and Caspase-1 underwent significant increased after the addition of CSE, and the mRNA expression decreased significantly after the knockdown of IL-17RA (Figure 5D). Furthermore, the addition of the NLRP3-specific inhibitor MCC950 and the Caspase-1-specific inhibitor VX765, along with results from PI/Hoechst 33342 and LDH assays, indicated that in the CSE group, cell pyroptosis increased, while the knockdown of IL-17RA led to cell recovery (Figure 5E–H). Therefore, we confirm that CSE activates IL-17RA, thereby triggering the NLRP3/Caspase1/GSDMD canonical pathway and the GSDME non-canonical pathway.
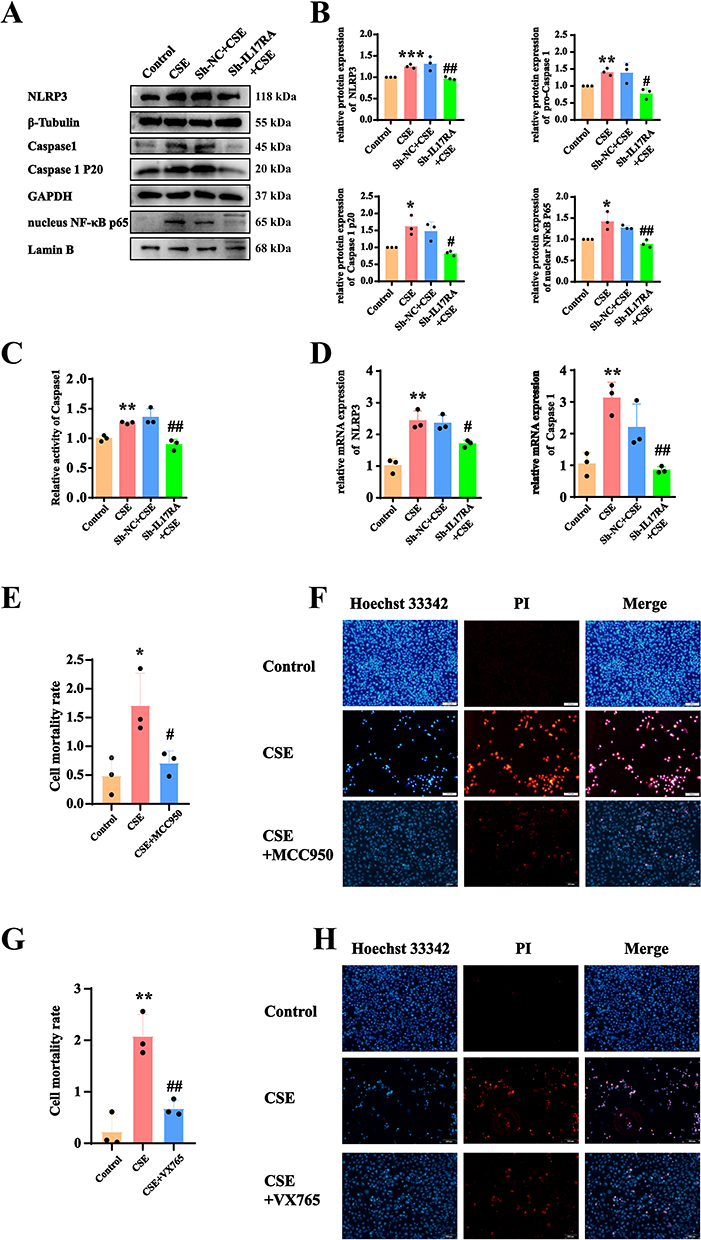 |
Figure 5 CSE activates IL-17RA, triggering cell pyroptosis through the NLRP3/Caspase1/GSDMD and NFκB/GSDME pathways. (A and B) Expression (A) and statistics (B) of NLRP3, Caspase 1, Caspase 1 P20, nuclear NFκB p65 protein in Control, CSE, sh-NC+CSE, sh-IL-17RA AT2 cell lines. (C) To detect the effects of CSE and IL-17RA knockdown on the activity of caspase1 protein. (D) mRNA Expression of NLRP3, Caspase 1 in Control, CSE, sh-NC+CSE, sh-IL-17RA AT2 cell lines. (VS Control *P<0.05, **P<0.01, ***P<0.001, VS Sh-NC+CS #P<0.05, ##P<0.01) (E and F) The LDH kit (E) and Hoechst/PI fluorescence experiments (F) were used to detect the effects of CSE and MCC950 (a NLRP3-specific inhibitor) on pyroptosis of AT2 cell lines. (VS Control *P<0.05, **P<0.01 VS CSE #P<0.05, ##P<0.01) (G and H) The LDH kit (G) and Hoechst/PI fluorescence experiments (H) were used to detect the effects of CSE and VX765 (a Caspase 1-specific inhibitor) on pyroptosis of AT2 cell lines. |
IL-17A Binds to IL-17RA to Activate the NLRP3/Caspase1/GSDMD Pyroptosis Pathway Rather Than the GSDME Pathway
IL-17A is the most important ligand for IL-17RA. Therefore, we further examined whether cigarette smoke activates canonical and non-canonical pyroptosis through the binding of IL-17A to IL-17RA. ELISA results indicated that the expression of IL-17A in the serum and bronchoalveolar lavage fluid of CS group mouse was elevated compared to the WT group, while IL-17A levels were significantly reduced in the serum and bronchoalveolar lavage fluid of IL-17RA−/− mouse (Figure 6A and B). Consequently, we stimulated AT2 cell lines with recombinant IL-17A, and WB experiments demonstrated a concentration-dependent response of NLRP3, Caspase1, Caspase 1 P20, and N-GSDMD to IL-17A, whereas GSDMD and GSDME showed no significant changes (Figure 6C and D). To further validate whether IL-17A activates the canonical pyroptosis pathway, we added the NLRP3-specific inhibitor MCC950, which revealed that following IL-17A addition, the protein expressions of Caspase 1, Caspase 1 P20, N-GSDMD, IL-1β, and IL-18 were elevated (Figure 6E). Additionally, the application of the Caspase1-specific inhibitor VX765 showed that after IL-17A addition, N-GSDMD, IL-1β, and IL-18 protein expressions were increased (Figure 6F). Therefore, we conclude that IL-17A promotes pyroptosis through the activation of the NLRP3/Caspase1/GSDMD pathway via IL-17RA, rather than through the NFκB/GSDME pathway (Figure 7).
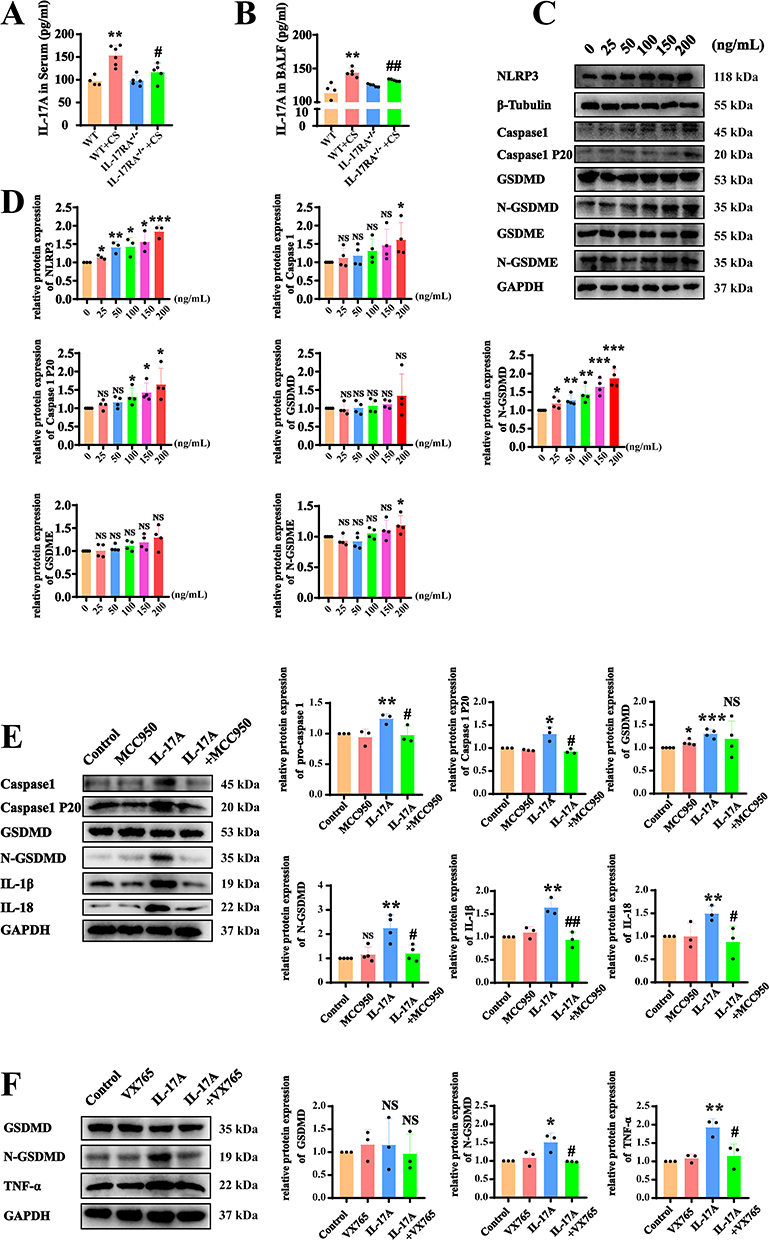 |
Figure 6 IL-17A binds to IL-17RA to activate the NLRP3/Caspase1/GSDMD pyroptosis pathway rather than the GSDME pathway. (A and B) ELISA experiment was used to detect the expression of IL-17A in serum (A) and bronchoalveolar lavage fluid (B) (VS WT **P<0.01, VS WT+CS #P<0.05, ##P<0.01) (C and D). The protein expression (C) and Statistical chart (D) of NLRP3, Caspase 1, Caspase 1 P20, GSDMD, N-GSDMD, GSDME, N-GSDME in AT2 cell lines in response to IL-17A recombinant protein at different concentrations. (VS 0% *P<0.05, **P<0.01, ***P<0.001) (E) Detect the protein expression of Caspase 1, Caspase 1 P20, GSDMD, N-GSDMD, IL-1β, IL-18 in AT2 cell lines in response to the recombinant protein of IL-17A and MCC950. (F) Detect the protein expression of GSDMD, N-GSDMD and TNFα in AT2 cell lines in response to the recombinant protein of IL-17A and VX765. (VS Control *P<0.05, **P<0.01, ***P<0.001, VS IL-17A #P<0.05, ##P<0.01). |
 |
Figure 7 Schematic diagram of IL-17RA drives cigarette smoke-induced alveolar epithelial cell pyroptosis in COPD via dual activation of the NLRP3/Caspase1/GSDMD and NF-κB/GSDME pathways. |
Discussion
COPD is a respiratory condition that severely impacts the quality of life of patients and is the third leading cause of death worldwide. Its pathophysiological characteristics include persistent and irreversible airflow limitation and inflammatory cell infiltration.1,19 We demonstrated that IL-17RA gene knockout mice delay the progression of COPD, and that knocking out IL-17RA in the AT2 cell line can alleviate cellular pyroptosis. Furthermore, we found that IL-17A and IL-17RA can activate the pyroptosis process in AT2 cells, which is associated with the progression of COPD. Mechanistic studies indicate that IL-17RA promotes pyroptosis in AT2 cells by activating the NLRP3/Caspase1 /GSDMD pathway and the NFκB/GSDME pathway, while IL-17A promotes pyroptosis in AT2 cells solely through the activation of the NLRP3/Caspase1/GSDMD pathway.
Airway inflammation is one of the important causes promoting the progression of COPD.20 Previous literature has indicated the significance of Th17 cytokines, with IL-17 neutralizing antibodies alleviating structural changes in COPD.21 IL-17A is significantly overexpressed in the lung tissues of clinical COPD patients.22 IL-17RA is the main shared receptor subunit necessary for the activity of IL-17 family cytokines,23 and IL-17RA is essential for COPD induced by cigarette smoke.24 AT2 epithelial cells are the core cells that promote emphysema and the release of inflammatory factors. Our results further demonstrate that the specific knockout of IL-17RA in alveolar type II epithelial cell mouse models significantly alleviates the progression of COPD, and the knockdown of IL-17RA in the AT2 cell lines line reduces cell death. This study provides an experimental basis for precisely targeting the IL-17RA protein in alveolar type II epithelial cells, thereby contributing to the treatment of COPD.
Pyroptosis is typically induced by the gasdermin family and is accompanied by the release of inflammatory cytokines such as IL-1β and IL-18.25 Consistent with previous studies, we verified that cigarette smoke activates the NLRP3/Caspase1/GSDMD canonical pyroptosis pathway,14 while also activating the GSDME non-canonical pyroptosis pathway.26 However, the upstream proteins that activate pyroptosis have not been fully elucidated. Our study further demonstrates both in vivo and in vitro that the knockout of IL-17RA inhibits the NLRP3/Caspase1/GSDMD canonical pyroptosis pathway, while also suppressing the NFκB/GSDME non-canonical pyroptosis pathway. Consequently, the levels of pyroptosis-related inflammatory cytokines (IL-1β, IL-6) are reduced in the lung tissues, serum, and bronchoalveolar lavage fluid of mice. This indicates that IL-17RA is a key protein in the pathway of pyroptosis activated by cigarette smoke, providing new insights into the critical proteins involved in the pyroptosis process induced by cigarette smoke.
IL-17RA is one of the important receptors activated by IL-17A,27 which promotes airway remodeling and accelerates the pathological process of COPD.28 Consistent with previous studies, IL-17A levels are elevated in lung tissue, serum, and bronchoalveolar lavage fluid in COPD mice.29 Our study further demonstrates that the expression of IL-17A protein is significantly reduced after the knockout of the IL-17RA gene. In other diseases, IL-17A activates downstream pyroptosis processes through IL-17RA;30 however, this has not been reported in COPD. Using recombinant IL-17A protein, we found that it induces pyroptosis in AT2 cell lines. Mechanistically, we examined the activation of the canonical pyroptosis pathway NLRP3/Caspase1/GSDMD by recombinant IL-17A protein, and interestingly, it does not activate the non-canonical pyroptosis pathway NFκB/GSDME. According to the literature, IL-17A can activate downstream pathways through IL-17RD as an alternative co-receptor,31 while IL-17F can also activate downstream pathways via IL-17RA.32 Thus, the relationship between IL-17A and IL-17RA is not unique. We speculate that the activation of the NFκB/GSDME non-canonical pathway by IL-17RA is not mediated by IL-17A but may involve other inflammatory factors. Our study provides new insights into the distinct roles of IL-17A and IL-17RA in COPD.
Several limitations of this study should be acknowledged. First, while our smoke exposure model effectively recapitulated aspects of acute epithelial injury, the potential for sex-specific differences in the inflammatory or pyroptotic responses was not evaluated, as all experiments were conducted using male animals and cells. Given that sex hormones can modulate both immune responses and susceptibility to smoke-induced injury, future studies incorporating both sexes are warranted to determine the generalizability of our findings. Second, the precise mechanisms by which IL-17RA activates NLRP3 and NFκB have not been fully elucidated. Third, whether IL-17RA activates classical and non-classical pyroptosis pathways requires further validation in COPD patients and relevant mouse models; accordingly, we maintain a conservative stance regarding the translational and therapeutic implications of our findings. Fourth, although we observed differences in downstream pyroptosis activated by IL-17RA and IL-17A, it remains to be confirmed whether IL-17A activates the canonical pyroptosis pathway in cells through its binding to IL-17RA. Finally, the specific factors involved in the activation of atypical pyroptosis pathways by IL-17RA warrant further investigation.
Data Sharing Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
The animal study was approved by the Laboratory Animal Welfare and Ethics Committee (Zunyi Medical University), with the approval number (zyfy-an-2024-0654). The study was conducted in accordance with the local legislation and institutional requirements. All animal studies comply with the ARRIVE reporting guidelines.
Author Contributions
Xuan An: Writing–original draft, Visualization, Formal analysis, Data curation. Yanhui Gu: Visualization, Formal analysis, Data curation. Jian Zhou: Methodology, Formal analysis, Writing – review & editing Yuting Liu: Methodology, Writing – review & editing. Shengyi Yu: Methodology, Writing – review & editing. Lanying Zhang: Methodology, Writing – review & editing Yao Ouyang: Writing–review & editing, Conceptualization, Supervision, Funding acquisition.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by the National Natural Science Foundation of China (8206010332), Zunyi Science and Technology Plan Project (ZUN SHI KE HE HZ ZI [2022] NO.255).
Disclosure
The authors declare no competing interests.
References
1. Xu J, Zeng Q, Li S, Su Q, Fan H. Inflammation mechanism and research progress of COPD. Front Immunol. 2024;15:1404615. doi:10.3389/fimmu.2024.1404615
2. Ruaro B, Salton F, Braga L, et al. The history and mystery of alveolar epithelial type II cells: focus on their physiologic and pathologic role in lung. Int J Mol Sci. 2021;22(5):2566. doi:10.3390/ijms22052566
3. Thomas R, Qiao S, Yang X. Th17/Treg imbalance: implications in lung inflammatory diseases. Int J Mol Sci. 2023;24(5):4865. doi:10.3390/ijms24054865
4. Ponce-Gallegos MA, Pérez-Rubio G, Ambrocio-Ortiz E, et al. Genetic variants in IL17A and serum levels of IL-17A are associated with COPD related to tobacco smoking and biomass burning. Sci Rep. 2020;10(1):784. doi:10.1038/s41598-020-57606-6
5. Ding T, Zhao S, Gu Y, et al. IL-17A regulates airway remodelling in COPD through the PI3K/AKT/mTOR pathway. Sci Rep. 2025;15(1):16546. doi:10.1038/s41598-025-00458-9
6. Doe C, Bafadhel M, Siddiqui S, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138(5):1140–19. doi:10.1378/chest.09-3058
7. Goepfert A, Barske C, Lehmann S, et al. IL-17-induced dimerization of IL-17RA drives the formation of the IL-17 signalosome to potentiate signaling. Cell Rep. 2022;41(3):111489. doi:10.1016/j.celrep.2022.111489
8. Ritzmann F, Lunding LP, Bals R, Wegmann M, Beisswenger C. IL-17 cytokines and chronic lung diseases. Cells. 2022;11(14):2132. doi:10.3390/cells11142132
9. Khawas S, Sharma N. Cell death crosstalk in respiratory diseases: unveiling the relationship between pyroptosis and ferroptosis in asthma and COPD. Mol Cell Biochem. 2025;480(3):1305–1326. doi:10.1007/s11010-024-05062-5
10. Takeuchi Y, Ohara D, Watanabe H, et al. Dispensable roles of Gsdmd and Ripk3 in sustaining IL-1β production and chronic inflammation in Th17-mediated autoimmune arthritis. Sci Rep. 2021;11(1):18679. doi:10.1038/s41598-021-98145-y
11. Li LL, Dai B, Sun YH, Zhang TT. The activation of IL-17 signaling pathway promotes pyroptosis in pneumonia-induced sepsis. Ann Transl Med. 2020;8(11):674. doi:10.21037/atm-19-1739
12. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. 2023;69:101781. doi:10.1016/j.smim.2023.101781
13. Feng Y, Li M, Yangzhong X, et al. Pyroptosis in inflammation-related respiratory disease. J Physiol Biochem. 2022;78(4):721–737. doi:10.1007/s13105-022-00909-1
14. Wang L, Meng J, Wang C, Wang Y, Yang C, Li Y. Hydrogen sulfide attenuates cigarette smoke‑induced pyroptosis through the TLR4/NF‑κB signaling pathway. Int J Mol Med. 2022;49(5). doi:10.3892/ijmm.2022.5112
15. Cristaldi M, Buscetta M, Cimino M, et al. Caspase-8 activation by cigarette smoke induces pro-inflammatory cell death of human macrophages exposed to lipopolysaccharide. Cell Death Dis. 2023;14(11):773. doi:10.1038/s41419-023-06318-6
16. Hou T, Tang Y, Wang L, Peng L, Ci X. Dihydromyricetin alleviates lipid peroxidation-induced Pyroptosis by inhibiting xCT ubiquitination and degradation in experimental COPD model. Phytomedicine. 2025;144:156929. doi:10.1016/j.phymed.2025.156929
17. Feng WQ, Zhang YC, Xu ZQ, et al. IL-17A-mediated mitochondrial dysfunction induces pyroptosis in colorectal cancer cells and promotes CD8 + T-cell tumour infiltration. J Transl Med. 2023;21(1):335. doi:10.1186/s12967-023-04187-3
18. Shu J, Li D, Ouyang H, et al. Comparison and evaluation of two different methods to establish the cigarette smoke exposure mouse model of COPD. Sci Rep. 2017;7(1):15454. doi:10.1038/s41598-017-15685-y
19. de Oca MM, Perez-Padilla R, Celli B, et al. The global burden of COPD: epidemiology and effect of prevention strategies. Lancet Respir Med. 2025;13(8):709–724. doi:10.1016/S2213-2600(24)00339-4
20. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50(4):1602434. doi:10.1183/13993003.02434-2016
21. Riani Moreira A, Uchoa da Silva C, de Paula Costa Mattos L, et al. IL-17-neutralizing antibody mitigates functional and structural changes in cigarette smoke-induced COPD model. Front Immunol. 2025;16:1641300. doi:10.3389/fimmu.2025.1641300
22. Yang T. Expression profile of IL-17 in lung tissues of patients with lung cancer and COPD and clinical significance. Cell Mol Biol. 2022;68(9):135–139. doi:10.14715/cmb/2022.68.9.21
23. Willis CR, Siegel L, Leith A, et al. IL-17RA signaling in airway inflammation and bronchial hyperreactivity in allergic asthma. Am J Respir Cell Mol Biol. 2015;53(6):810–821. doi:10.1165/rcmb.2015-0038OC
24. Chen K, Pociask DA, McAleer JP, et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One. 2011;6(5):e20333. doi:10.1371/journal.pone.0020333
25. Huang Y, Liang T, Liu J, Yu H, Li J, Han L. Dietary Zinc activates the Nrf2 signaling pathway to inhibit pyroptosis and attenuate the lung inflammatory response in COPD. Cytotechnology. 2025;77(2):62. doi:10.1007/s10616-025-00725-7
26. La Mensa A, Buscetta M, Woldhuis RR, et al. Caspase inhibition restores collagen Iα1 and fibronectin release in cigarette smoke extract-exposed human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2025;328(2):L239–l52. doi:10.1152/ajplung.00214.2024
27. Huang W, Zhou Y, Pan C, Zhang X, Zhao H, Shen L. Molecular modeling and rational design of disulfide-stapled self-inhibitory peptides to target IL-17A/IL-17RA interaction. J Mol Recognit. 2023;36(8):e3045. doi:10.1002/jmr.3045
28. Chen X, Chen L, Chen G, et al. Interleukin-17A promotes airway remodeling in chronic obstructive pulmonary disease by activating C-X-C motif chemokine ligand 12 secreted by lung fibroblasts. Chronic Obstr Pulm Dis. 2024;11(5):482–495. doi:10.15326/jcopdf.2024.0495
29. Huang L, Xu J, Zhou H, Li H, Cao W, Pu J. NR1D1 mitigates IL-17a-induced small airway remodeling in biomass smoke-induced COPD. Toxicol Lett. 2025;409:74–86. doi:10.1016/j.toxlet.2025.05.002
30. Lima C, Falcao MAP, Andrade-Barros AI, et al. Natterin an aerolysin-like fish toxin drives IL-1β-dependent neutrophilic inflammation mediated by caspase-1 and caspase-11 activated by the inflammasome sensor NLRP6. Int Immunopharmacol. 2021;91:107287. doi:10.1016/j.intimp.2020.107287
31. Su Y, Huang J, Zhao X, et al. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci Immunol. 2019;4(36). doi:10.1126/sciimmunol.aau9657
32. Ely LK, Fischer S, Garcia KC. Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol. 2009;10(12):1245–1251. doi:10.1038/ni.1813
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis
Chang X, Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y
Journal of Inflammation Research 2022, 15:3859-3871
Published Date: 11 July 2022
The Mechanism of Pyroptosis and Its Application Prospect in Diabetic Wound Healing
Al Mamun A, Shao C, Geng P, Wang S, Xiao J
Journal of Inflammation Research 2024, 17:1481-1501
Published Date: 6 March 2024
APAF1 Silencing Ameliorates Diabetic Retinopathy by Suppressing Inflammation, Oxidative Stress, and Caspase-3/GSDME-Dependent Pyroptosis
Ding Y, Chen L, Xu J, Feng Y, Liu Q
Diabetes, Metabolic Syndrome and Obesity 2024, 17:1635-1649
Published Date: 10 April 2024
SIRT2 Promotes NLRP3-Mediated Microglia Pyroptosis and Neuroinflammation via FOXO3a Pathway After Subarachnoid Hemorrhage
Sun JQ, Sheng B, Gao S, Liu XZ, Cui Y, Peng Z, Chen XX, Ding PF, Zhuang Z, Wu LY, Hang CH, Li W
Journal of Inflammation Research 2024, 17:11679-11698
Published Date: 27 December 2024
NLRP3 Inflammasome-Mediated Pyroptosis in Diabetic Nephropathy: Pathogenic Mechanisms and Therapeutic Targets
Chen Y, Chen R, Ji X, Zeng Z, Guan C
Journal of Inflammation Research 2025, 18:8399-8418
Published Date: 25 June 2025