Back to Journals » Journal of Inflammation Research » Volume 15
Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis
Authors Chang X ![]() , Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y
, Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y
Received 8 April 2022
Accepted for publication 4 July 2022
Published 11 July 2022 Volume 2022:15 Pages 3859—3871
DOI https://doi.org/10.2147/JIR.S368501
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Zili You
Xingyu Chang,1,* Yuchen Kang,2,* Yuxuan Yang,1,* Yajie Chen,1 Yanyu Shen,1 Chenjun Jiang,1 Yi Shen3
1The First Clinical Medical College, Lanzhou University, Lanzhou, People’s Republic of China; 2The Second Clinical Medical College, Lanzhou University, Lanzhou, People’s Republic of China; 3Department of Orthopaedics, The Second Xiangya Hospital of Central South University, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yi Shen, Department of Orthopaedics, The Second Xiangya Hospital of Central South University, Changsha, People’s Republic of China, Tel +86-13517490910, Email [email protected]
Abstract: Osteoarthritis (OA) is one of the most common chronic joint diseases and is gradually becoming the main cause of disability and joint pain in the elderly worldwide. Pyroptosis is a regulated programmed cell death triggered by inflammasomes. It leads to cell swelling, lysis, and bioactive molecule secretion. Studies found that the damaged chondrocytes in OA joints had morphological characteristics of pyroptosis, and the cytokines associated with pyroptosis in synovial fluid increased, indicating that pyroptosis may have certain impacts on the pathological progression of OA. This review briefly summarizes the molecular mechanisms of pyroptosis and the epidemiology and pathogenesis of OA. Furthermore, we discussed the role of pyroptosis in articular cartilage and synovium during OA and reviewed the progress of pyroptosis-related molecules in the targeted therapy of OA joints, hoping to provide feasible directions for the diversified treatment of OA.
Keywords: pyroptosis, osteoarthritis, articular cartilage, synovium, NLRP3 inflammasome, GSDMD
Introduction
Pyroptosis was first referred to as caspase-1-dependent cell death in mouse macrophages and human monocytes infected with Salmonella Shigella.1–3 In 2001, this new type of cell death was named ‘pyroptosis’, which was taken from the Greek roots “pyro” and ‘ptosis’, meaning “fever” and “fall”, respectively.4 This name aptly indicates the proinflammatory attribute of pyroptosis and therefore clearly distinguishes it from apoptosis. After encapsulating numerous articles over the past decades, the Nomenclature Committee on Cell Death formally defined pyroptosis as a type of regulated cell death (RCD) that is heavily dependent on members of the gasdermin protein family; those proteins form pores in the plasma membrane, and this process is usually (but not always) caused by inflammatory caspases.5 In recent years, an increasing number of studies have found that pyroptosis occurs in vascular endothelial cells and glial cells. These studies broke down the stereotype that pyroptosis only appears in the mononuclear phagocyte lineage and provided a strong theoretical basis for studying its role in nonimmune diseases, such as cardiovascular diseases and neurodegenerative diseases.6,7
Osteoarthritis (OA) is the most common degenerative joint disease, affecting approximately 10% of men and 18% of women globally.8 Although OA is remarkably prevalent, elderly individuals are the most susceptible.9 OA occurs not only in small joints, such as hand joints but also in giant joints, such as the knee and hip joints. Its pathological changes usually include cartilage degradation, synovial inflammation, subchondral bone sclerosis, and osteophyte formation.10 At present, most patients with OA are treated with palliative therapy. In the early and middle stages of OA, anti-inflammatory and analgesic therapy combined with exercise rehabilitation is adopted. In the late stage, only joint replacement surgery is available. Therefore, it is urgent to understand OA pathogenesis and formulate corresponding countermeasures.
In this review, we summarized the molecular mechanisms of pyroptosis, provided a brief review of osteoarthritis, and explained the role of chondrocyte and synoviocyte pyroptosis in OA-related lesions. Moreover, we reviewed the progress of pyroptosis-related molecules in OA therapy. Since there is no effective targeted treatment for OA at present, exploring the role of pyroptosis and relevant molecules in OA is advantageous for clinical treatment.
Molecular Mechanisms of Pyroptosis
Pyroptosis is a new type of programmed cell death triggered by inflammasomes and executed by gasdermin family members.11 Inflammasomes are cytoplasmic protein multimers typically composed of a sensor (pattern recognition receptors; PRRs), an adaptor (apoptosis‐associated speck‐like protein; ASC), and an effector (pro-caspase). Sensors, which are specific pattern recognition receptors (PRRs), mainly include nucleotide-binding oligomerization domains (NOD) and leucine-rich repeat (LRR) receptors (NLRs), AIM2-like receptors (ALR), and pyrin. Apoptosis-associated speck-like protein (ASC) contains a pyrin domain (PYD) and a caspase activation and recruitment domain (CARD), acting as a binder for sensors and effectors. Caspases engaged in pyroptosis can also be called proinflammatory caspases. In the classical pyroptosis theory, these caspases include human caspase-1/4/5 and mouse caspase-11.12 Gasdermins are a recently discovered protein family with membrane pore-forming competence, in which gasdermin D (GSDMD) is thought to be the main executor of pyroptosis.13 This family contains six homologous proteins in humans, gasdermin A-E (GSDMDA-GSDMDE) and DFNB59. Mice do not express GSDMB but express three GSDMA homologs (GSDMA1-3) and four GSDMC homologs (GSDMC1-4).14 Most members are composed of a N-terminal pore-forming domain (PFD) and a C-terminal autoinhibitory domain (RD).15 Generally, full-length gasdermins are silent due to the inhibitory competence of RD. When gasdermins are cleaved between RD and PFD from the linker domain by specific caspases or granzymes, the N-terminal domain recovers its cytotoxicity and mediates pyroptosis, as well as the secretion of inflammatory mediators and cytokines.16 Numerous gasdermins have been reported to be deeply related to human diseases, but their specific mechanisms need to be further studied.17
Presently, pyroptosis pathways are roughly separated into the canonical inflammasome (caspases-1 dependent) pathway, the noncanonical inflammasome (caspases-4/5/11 dependent) pathway, and other caspase-dependent/independent pathways.
The Canonical Inflammasome Pathway
The canonical inflammasome pathway begins with assembly, namely, the recognition of specific pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs). NLRs are further divided into NLRP1/3/6/9 and NLRC4 according to the N-terminal composition. The activation of NLRP1 is mediated by pathogenic enzymes. For instance, mouse NLRP1b was activated by lethal factors secreted by Bacillus anthracis.18 NLRP6/9 mainly respond to viral or bacterial infections and are activated in the innate immune response.19,20 Distinct from the former, NLRP3 is the most comprehensively studied intracellular sensor, detecting a wide range of endogenous danger signals and microenvironmental stimuli. Of note, the complete activation process of NLRP3 contains two steps, priming and activation. In addition, NIMA-related kinase (NEK7) is essential for NLRP3 activation. The initial priming step is NLRP3 transcription, involving Toll-like receptors (TLRs), IL-1 receptors and tumor necrosis factor receptors. The transmitted signal then passes through the NF-κB pathway to modulate NLRP3, procaspase-1β, and procaspase-18. The second step is to receive stimulation similar to other PRRs. NLRP3 reacts to bacterial, viral, and fungal infections, as well as endogenous DAMPs mediating sterile inflammation and environmental stimuli, which is a unique feature distinguishing it from most PRRs that only target one or several PAMPs or DAMPs.21 NLRC4 is generally activated only after the NAIP protein binds to an irritant, such as Salmonella typhimurium.22 Different kinds of NAIP give NLRC4 recognition specificity to diverse pathogens as well. Non-NLR PRRs, including AIM2 and pyrin, mainly play a role in sensing viral infection and toxic invasion. AIM2 binds to cytoplasmic electronegative double-stranded DNA (dsDNA) through the positively charged C-terminal HIN-200 domain.23 Pyrin binds to intracellular and extracellular toxins.24 Figure 1A depicts the stimuli they received, and Figure 1C describes the domain composition of PRRs. Subsequently, activated PRRs interact through the CARD-CARD domain to recruit ASC, which then binds to pro-caspase-1 via homotypic pairing. At this point, the assembly of inflammasomes is completed. Pro-caspase-1 cleaves and is activated to form the caspase-1 polymer (p10/p20 tetramer) after accepting the transduced dangerous signals. Activated caspase-1 recognizes pro-IL-1β and pro-IL-18 without bioactivity to mature.12 On the other hand, it cleaves the intermediate linker domain of GSDMD to release the GSDMD-NT fragment into the membrane. GSDMD-NT then binds with phospholipids to form GSDMD pores, mediating pyroptosis and the secretion of bioactive factors, including IL-1β, IL-18, and high mobility group protein B1 (HMGB1).13
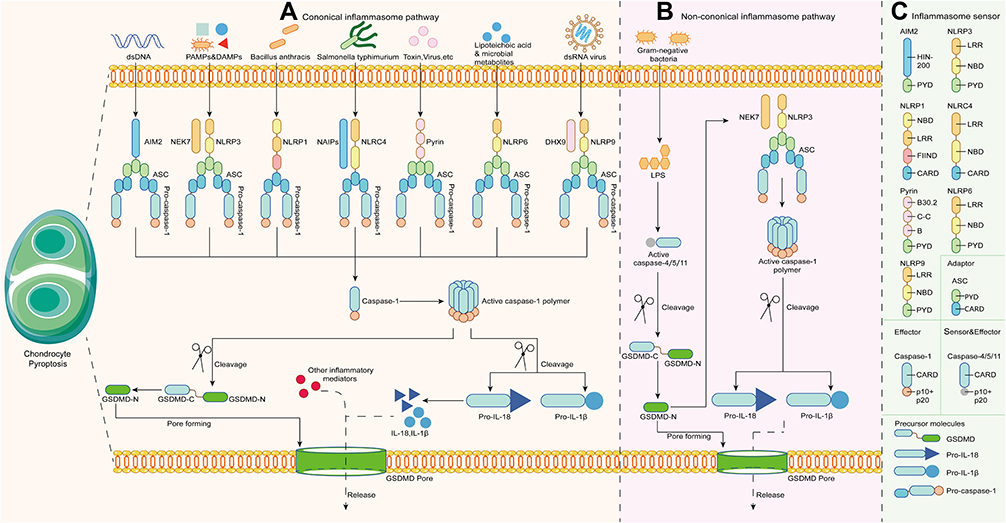 |
Figure 1 Molecular mechanisms of pyroptosis. (A) The canonical inflammasome pathway. (B) The noncanonical inflammasome pathway. (C) The components of inflammasome sensors, adaptors, effectors, and inflammatory precursor molecules in pyroptosis. |
The Noncanonical Inflammasome Pathway
In the noncanonical inflammasome pathway, the CARD domains in human caspase-4/5 and mouse caspase-11 precisely respond to the stimulation of lipopolysaccharide (LPS) from Gram-negative bacteria in the cytoplasm and oligomerize to become active (Figure 1B).25–27 Inflammasomes are not involved in this process. The activated caspase-4/5/11 polymer cleaves GSDMD at the FLTD site to mediate the formation of GSDMD pores and pyroptosis, which is intrinsically no different from the canonical inflammasome pathway.28 However, in the canonical inflammasome pathway, caspase-4/5/11 does not increase IL-1β and IL-18 levels, but rather acts on the NLRP3/caspase-1 axis.29 In addition, the ATP-dependent ion channel P2X7 regulates pyroptosis via the noncanonical pathway.30 Moreover, neutrophil pyroptosis induced by this pathway results in the formation of neutrophil extracellular traps (NETs), indicating that pyroptosis is associated with NETosis to jointly deal with pathogen infection.31
Other Newly Discovered Pyroptosis Pathways
In addition to the canonical inflammasome pathway and noncanonical inflammasome pathway, cell biologists have also found the caspase-3/8-dependent pathway and granzyme-mediated GSDMD-independent pathway. Caspase-3/8 were considered to be apoptosis-related caspases in the past and showed no activity in pyroptosis. However, Wang discovered that chemicals induce caspase-3 to cleave the linker of GSDME, producing a GSDME-NT fragment with the same pore-forming activity to mediate pyroptosis.32 Similarly, caspase-8, which modulates pyroptosis by cleaving GSDMC, makes the transformation between apoptosis and pyroptosis possible.33 Moreover, granzyme A and granzyme B directly and indirectly induce GSDMB and GSDME cleavage, respectively.34,35 These findings suggest that there is heterogeneity in pyroptosis. Different molecular mechanisms of pyroptosis could be an outcome of different stimuli, which play a specific role in the pathological process of infection, tumorigenesis, and inflammation.
Osteoarthritis
Epidemiology
Many systematic reviews and epidemiological surveys have revealed that the prevalence of OA depends significantly on age range, sex distribution, geographical location, involved joints, and means of definition among the investigated population. In Europe and North America, it was very rare for young and middle-aged people to suffer from OA, but there was also a low-rise trend with age.36 In elderly individuals, especially with 65 years of age as the baseline, the prevalence of symptomatic knee osteoarthritis (KOA) and hip osteoarthritis (HOA) increased almost linearly.36,37 In addition, the incidence of KOA or hand osteoarthritis in women was generally higher than that in men in the same age group.38,39 Moreover, it is also worth noting that the rate of radiographic osteoarthritis was always several percentage points above that of symptomatic osteoarthritis. This is related to the definition because OA diagnosis by imaging occurs before patients exhibit obvious clinical symptoms, such as joint pain, swelling, and stiffness. In addition to the well-known physiological problems, OA also induces psychological diseases in patients and places heavy economic burdens on families and society. Compared with normal individuals, OA patients are more likely to be isolated socially, resulting in cognitive impairment, depression, and even suicidal tendency, which is deeply correlated with the disease.40–42 Since there is currently no curative therapy, long-term treatment has brought excess financial expenses to the families of patients. The medical costs per patient in the United States were considerable, with average annual indirect expenses ranging from 238 dollars to 29,935 dollars, but patient quality of life has not improved.43
Risk Factors and Pathogenesis
In the past, OA was often thought to be the consequence of aging, but researchers are now gradually discovering that the onset of OA is the outcome of multiple factors in vivo and in vitro. The risk factors for OA are roughly divided into individual-level factors that alter a person’s susceptibility and joint-level factors that change biomechanical stability. The individual-level risk factors mainly include age, sex, genetics, and dietary habits. OA is primarily generated by a series of factors mediated by aging, including chondrosenescence and hindered proliferation,44 decreased proprioceptive sense,45 and weakened muscle strength.46 Genetics also contribute to OA, but the influence of genetic factors on different joints varies. In addition, poor living habits, such as alcoholism47 and in nutrition,48 are suspected to promote the risk of OA. However, the results of some clinical trials have been disappointing.49 Therefore, whether poor living habits play roles in the onset and development of OA remains to be discussed. The biomechanical factors of joints are principally joint shape,50 abnormal joint loading and injuries.51 In addition, metabolic disorders in joint tissue, including the deposition of oxidized low-density lipoprotein (Ox-LDL),52 cholesterol,53 and hydroxyapatite crystals,54 can also be attributed to biomechanical factors. The microlevel changes will manifest as macrolevel effects, such as joint degeneration. Whether there is a significant difference between the onset and development of OA is still controversial; therefore, other unclassified risk factors, such as obesity, that lead to OA or promote the progression of OA may exist as independent factors.
Compared with normal joints, OA lesions basically cover the whole joint and feature articular cartilage degradation, synovial inflammation, subchondral bone remodeling, and increased sensory and vascular input (Figure 2A). Although the pathogenesis has not been fully revealed, researchers now generally believe that OA is caused by the imbalance between repair and destruction of joint tissue rather than the so-called wear and tear disease. Its complex pathogenesis is now widely acknowledged as the combined outcome of mechanical, metabolic, and inflammatory factors.55 Initially, the cartilage surface is altered due to the innate immune response or metabolic disorders, which are usually characterized by fibrous villi. Subsequently, the cartilage sheds due to gradual erosion. These changes enable chondrocytes to be hypertrophic and to present enhanced anabolic activity. Thus, proinflammatory mediators, such as IL-1β, IL-6, TNF-α, and matrix-degrading enzymes, are synthetized and infiltrated into synovial fluid to induce subsequent secondary reactions, including synovial inflammation. Meanwhile, damaged chondrocytes also secrete the prorepair cytokine TGF-β, improving the prognosis of synovial lesions.56 In subchondral bone, osteoblasts and osteoclasts interact with chondrocytes to enhance local bone formation and transformation, thus leading to subchondral bone pore formation and subchondral bone sclerosis. Meanwhile, this process is accompanied by more vascular and nerve invasion, resulting in expanded OA pain susceptibility.57 Figure 2B summarizes the pathogenic progression and the relationship between cytokines mediated by intercellular communications and OA lesions.
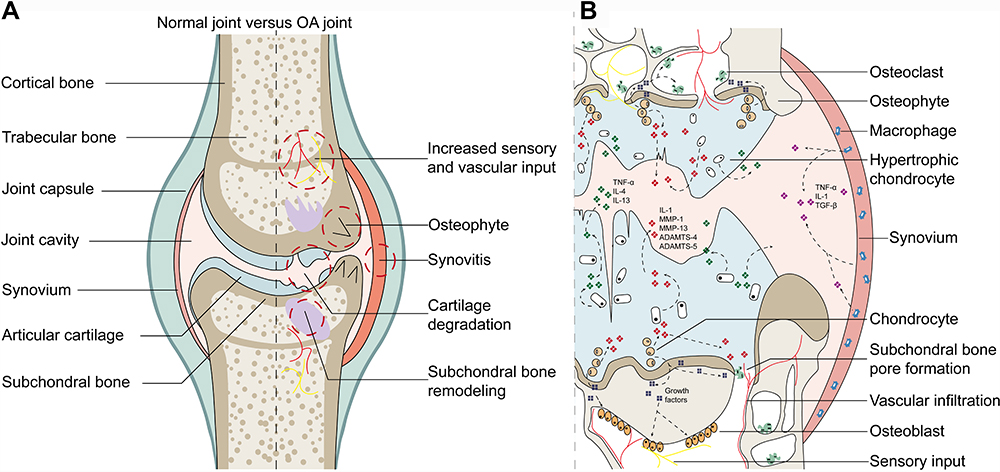 |
Figure 2 Macroscopic pathological changes and microscopic pathogenic relationships in OA joints. (A) Pathological changes in OA joints compared with normal joints. (B) Microscopic pathogenic interactions by cytokines in OA joints. |
Implications of Pyroptosis in Osteoarthritis
Chondrocyte Pyroptosis in the Articular Cartilage
Articular cartilage is composed of chondrocytes and the surrounding extracellular matrix (ECM). Under normal physiological conditions, joints rotate and collide almost frictionlessly, which is attributed to the special mechanical structure and chemical composition of the ECM. The solid components of cartilage ECM consist of type II collagen, proteoglycans, a small number of other types of collagens, and noncollagen proteins. They are cross-linked into a network to resist external mechanical stress.58 Chondrocytes are embedded in a narrow and thick matrix region; they usually show no proliferative activity but are essential for maintaining the low turnover stability of collagen metabolism in the absence of pathological impairment.59 Meanwhile, an increasing number of studies have suggested that cartilage degradation, the characteristic pathological change in OA, seems to be driven by ECM disruption caused by the imbalance of substance metabolism in chondrocytes.
Although whether pyroptosis is the cause or the result of OA cartilage degeneration is still unclear, many experiments have found that OA-related risk factors, including Ox-LDL and cholesterol promoted by obesity,52,53 hydroxyapatite crystals,54 and migrated LPS due to intestinal circulation disorder,60 are DAMPs or PAMPs that trigger pyroptosis simultaneously. These substances activate chondrocyte pyroptosis via the NLRP3 inflammasome pathway, stopping substance metabolism and releasing matrix metalloproteinases (MMPs), disintegrin and metalloproteinase with thrombospondin-like motifs (ADAMTS) enzymes, IL-1β, IL-18, and TNF-α.61,62 These molecules may be involved in chondrocyte pyroptosis in the progression of OA. MMP-1/3/13 and ADAMTS-4/5 have been proven to be the main factors for degrading cartilage ECM components,63,64 and their rise is partly attributed to IL-1β and TNF-α from pyroptotic chondrocytes.65,66 Therefore, the occurrence of chondrocyte pyroptosis disrupts the balance between anabolism and catabolism of the chondrocyte ECM, and their release makes catabolism dominant. In terms of the manifestations of different stages of cartilage degeneration in OA, the ECM regions lacking chondrocyte support are gradually separated from other parts resulting in the appearance of early surface fibrosis of OA cartilage.67 Additionally, secreted IL-1β, IL-18, and TNF-α diffuse into cartilage, synovial fluid, and even subchondral bone, causing secondary injuries to OA joints. IL-1β implies to be associated with cartilage destruction and the inflammatory cascade. IL-1β is involved in chondrocyte processes, including gene expression, cell proliferation and death. It influences the expression of MMP-1/11,65 and induces the production of reactive oxygen species (ROS),68 which eventually triggers chondrocyte pyroptosis and ECM decomposition in an adverse positive feedback loop. When the feedback process accelerates the aggravation of cartilage loss, pieces of cartilage debris fall off into synovial fluid, precipitating secondary injuries to the synovium. However, scholars believe that the changes in synoviocyte behaviors are the reasons for the destruction of OA cartilage.69 Although there is no conclusion at present, the specific etiology may be related to whether the patient’s joint impairments are internal or traumatic. Moreover, IL-1β released into synovial fluid boosts inflammatory-related compounds, cytokines, and chemokines, including inducible nitric oxide synthase (iNOS),70 cyclooxygenase-2 (COX-2),71 IL-6,72 IL-8,73 and chemokine CCL5,74 to mediate synovial inflammation. The bioeffect of TNF-α is similar to that of IL-1β, and they can be induced by each other.75 IL-18 has been fully studied previously. It increases inflammation-linked molecules, such as prostaglandin E2 (PGE2) and vascular endothelial growth factor (VEGF), promoting the inflammatory response and vascular invasion.76,77 Even though IL-18 can mediate production of MMPs by chondrocytes and chondrocyte apoptosis, those processes more likely to occur through IL-1-dependent and/or TNF-α-dependent pathways.78,79 In general, the direct role of pyroptosis in articular cartilage is to annihilate chondrocytes, disintegrate the original structurally stable cartilage ECM, and secrete proinflammatory factors to indirectly accelerate cartilage degeneration and prompt joint inflammation. Therefore, anti-IL-1β and anti-TNF-α may still be among the few effective treatments at present, whether from the perspective of inhibiting chondrocyte pyroptosis and ECM degradation or from the perspective of preventing the inflammatory response. Blocking intermediates of the pyroptosis pathway may be a promising research direction in the near future.
Synoviocyte Pyroptosis in Synovium
The synovium is composed of the synovial membrane and synovial fluid. The synovial membrane is divided into two layers. The outer layer is composed of various connective tissues rich in type I collagen, microvessels, lymph, and nerve fibers. The inner layer is composed of synoviocytes, which mainly consist of synovial macrophages and fibroblast-like synoviocytes (FLS) that have high biological activity. They are suffused in the joint cavity, regulating the entry and exit of small molecules. Meanwhile, they are also the primary secretion source of synovial components, such as lubricant and hyaluronic acid, playing an important role in maintaining the low friction feature of the synovium and nourishing chondrocytes.80
Studies have shown that synovial changes in OA joints can be divided into three categories, increased debris, inflammation, and fibrosis, but it is uncertain whether these characteristics appear at the same time or in stages.81 Nevertheless, synovial macrophages and FLS are closely related to those characteristics. As mentioned above, deciduous cartilage debris, secreted cytokines, and proteases secreted from pyroptotic chondrocytes are delivered into the synovial fluid. They act as DAMPs to interact with membrane TLRs of synovial macrophages and FLS to mediate subsequent pyroptosis, causing pathological changes in the synovium.82,83 Some researchers have shown that the communication between OA chondrocytes and synoviocytes is also realized via exosome-like vesicles.84 Pyroptotic synovial macrophages and FLS produce high mobility group protein B1 (HMGB1) and transforming growth factor-β (TGF-β), in spite of MMPs, ADAMTS, IL-1β, IL-18, and TNF-α, which are known to aggravate cartilage injuries and synovitis, as mentioned above.85,86 HMGB1 reacts on FLS and promotes the assembly of proinflammatory factors resulting in synovitis;87 HMGB1 also binds to LPS and is transported to synovial macrophages through RNA resulting in pyroptosis.88 TGF-β increases the expression levels of the fibrogenic markers procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2), collagen type I α1 chain (CLO1A1), and tissue inhibitor of metalloproteinases 1 (TIMP1) in OA FLS, which finally causes excessive deposition of ECM, namely, fibrosis.85 Moreover, hypoxia-inducible transcription factor-1α (HIF-1α)89 and adenosine triphosphate (ATP)90 induced by injured synovium stimulate FLS pyroptosis, thereby deteriorating synovitis and fibrosis. In conclusion, according to the current aftereffect, synoviocyte pyroptosis and secreted tissue-specific cytokines are associated with OA synovitis and fibrosis. Meanwhile, the interaction of pyroptosis and fibrosis with chondrocytes through cytokines causes cartilage loss and synovial changes to form an adverse positive feedback loop, similar to cartilage destruction. However, more experiments are needed to explore the distinct relationship between the two because pyroptosis is often linked to catabolism, while fibrosis is dominated by anabolism in the late stage of OA synovium. The transformation of metabolic intensity means that more types of cells or cytokines are engaged in the process.
Inhibiting the Pyroptosis-Associated Components for the Therapeutic Strategies of Osteoarthritis
NLRP3 Inflammasome
Inflammasomes are recognized as initiators and central processors of pyroptosis because they identify a variety of DAMPs and PAMPs, endowing proinflammatory caspases with biological activity. Therefore, if inflammasomes are blocked from being activated and assembled, cells may be protected from pyroptosis and the subsequent disorder. Currently, researchers are focusing on the NLRP3 inflammasome, which responds to diverse triggers causing cell disturbance, and the signaling pathways controlling it (Table 1).
 |
Table 1 Summary of NLRP3-Associated Inhibitors in Osteoarthritis |
As mentioned earlier, the NF-κB pathway is a necessary precondition for NLRP3 inflammasome priming. Thus, preventing the actuation or conduction of the NF-κB pathway is the preferred method for the directional inhibition of pyroptosis. Loganin,91 ursolic acid,92 and chondroitin sulfate,93 were established to regulate the NF-κB/NLRP3 inflammasome axis, which slowed cartilage degradation and improved synovitis and OA pain in rats. In 2021, Baek synthesized a novel multiprotein kinase inhibitor KMU-1170, a derivative of indoline-2-ketone. Then, they further found that it prevented almost all the phosphorylation processes in the NF-κB pathway to inhibit the mRNA transcription of the NLRP3 inflammasome.94 Compared with the former, KMU-1170ʹs mechanism has been thoroughly investigated. However, considering its inhibitory effects on various kinases in other signaling pathways, the experimental results obtained only in specific single-cell systems were not universal and need to be verified in other single-cell or multi-cell systems. Moreover, Franco-Trepat’s team discovered that the antidepressant amitriptyline could bind to TLR4, which fundamentally blocked the NF-κB signaling pathway, thus stopping NLRP3 activation in chondrocytes, synoviocytes, and osteoblasts in OA and eventually obstructing the secondary immune responses.95 Furthermore, when receiving priming signals, the NLRP3 inflammasome still needs second-stage signals to activate and assemble. ROS have been proven to be crucial in the activation and assembly of the NLRP3 inflammasome.96 Therefore, ameliorating the state of intracellular oxidative stress becomes another feasible strategy. NF-E2-related factor 2 (Nrf2), a transcription factor modulated by oxidative stress, enhances the expression of antioxidant stress proteins, including HO-1, after sensing abnormal ROS levels. They then eliminate excessive ROS, improving the intracellular imbalance condition and inhibiting NLRP3 inflammasome activation indirectly, thus delaying the progression of OA.97 The Nrf2/HO-1/ROS axis is clearly not the only pathway involved in pyroptosis. Both the antiallergic drug loratadine98 and licochalcone A (Lico A), a compound extracted from Glycyrrhiza species,99 are regulatory molecules of Nrf2 that alleviate the NLRP3 inflammasome stimulated by ROS. The former alleviates the NF-κB pathway via the Nrf2/HO-1 axis; the latter allows Nrf2 to enter the nucleus and directly alleviates the expression of the NLRP3 gene. There are many pharmacological agents, such as the NLRP3 inhibitor CY-09,66 chrysin,100 of and baicalein (via intra-articular injection),62 and physical therapy procedures, such as reducing the mechanical load of the knee joint61 and electroacupuncture,101 that have been proven to reduce NLRP3 inflammasome expression, inhibit chondrocyte pyroptosis, catabolism, and inflammatory responses, and ameliorate cartilage impairments. However, those studies did not explore the molecular mechanisms in animal models, and relevant research requires further consideration and improvement in the future.
Gsdmd
GSDMD performs pyroptosis along with integrating a large number of endogenous and exogenous signals from the inflammasome. Once GSDMD pores cannot be formed normally, most of the previously activated inflammasomes and caspases along with their biomolecular signals are blocked. This serves to stop pyroptosis, by stopping GSDMD from triggering pyroptosis on a massive scale and weakening the subsequent inflammatory cascades indirectly. Therefore, compared with specific types of inflammasome inhibitors or directional anti-IL-1β/anti-IL-18 pharmaceutical preparations, targeted GSDMD blocking has superior selectivity. Thus, theoretically, GSDMD blocking may be the most attractive intervention objective from this time forth.
At this time, the strategies of targeted inhibition of GSDMD can be separated into two aspects: alleviating GSDMD from being cleaved by proinflammatory caspases and preventing GSDMD-NT oligomerization and/or membrane insertion.102 Yang synthesized the GSDMD-derived inhibitor N-acetyl-phenylalanine-leucine-threonine-aspartate-chloromethyl ketone (Ac-FLTD-CMK) by locating the cleavage sites where GSDMD binds to caspase-1/4/5/11. They found that it replaced the full-length GSDMD and bound to the above caspases, preventing cleavage.28 In addition, experimenters detected that disulfiram and necrosulfonamide could not oligomerize GSDMD-NT by covalently modifying human GSDMD cyc191 and mouse GSDMD cyc192, showing efficient inhibition of pore formation in vivo and in vitro.103,104 The results also suggested that other FDA-approved Cys-modified drugs, such as dimethyl fumarate, had no activity on GSDMD, indicating that the existence of the Cys reactive site itself was not the root cause of the blockage of pore formation.104 Therefore, testing the specific mechanisms of some anti-inflammatory drugs at the pyroptotic molecular level may gain new insights and perspectives. The chemical LDC7559 was found to have comparable efficacy to other drugs targeting GSDMD, which may be due to its binding to GSDMD-NT.105 Furthermore, cell biologists have explored whether the cell can remove GSDMD pores in the cell membrane, by forming vesicles that encapsulate the GSDMD pores and release them into the extracellular environment to achieve repair.106 GSDMD deficiency has been demonstrated to inhibit neonatal onset system inflammatory disease, familial Mediterranean fever, and sepsis in animal models.107–109 In addition, another experiment explored the role of GSDMD in posttraumatic osteoarthritis. After knocking out GSDMD, cartilage alteration, synovitis, and subchondral bone sclerosis were alleviated, but the experiment also revealed that GSDMD deficiency had no inhibitory impact on serum transfer-induced rheumatoid arthritis.110 Similarly, GSDMD deletion had no influence on gouty arthritis induced by uric acid crystals.109 Even twin studies revealed that GSDMD removal played an opposite role in dextran sulfate-induced colitis in mice.111,112 This may be associated with caspase-8, a type of apoptosis-related caspase, which was found to be involved in pyroptosis. In conclusion, GSDMD is another promising breakthrough for alleviating/treating pyroptosis-related diseases. However, due to the lack of thorough research on the molecular mechanisms of pyroptosis in other specific cell types, except immune cells, and considering that targeted inhibitors of GSDMD will not affect inflammasome-relevant events in the early stage of pyroptosis, more experiments are needed to confirm the exact molecular mechanisms and consequences of targeted inhibitors of GSDMD in different diseases.
Conclusion and Perspective
Pyroptosis is a new form of proinflammatory programmed cell death that was recently found to be closely related to cartilage and synovial lesions in OA joints. The DAMP level in the synovial fluid of the OA joint rises sharply, triggering chondrocyte and synoviocyte pyroptosis mediated by the canonical inflammasome pathway. Then, cartilage matrix-degrading enzymes and proinflammatory mediators are released into the synovial fluid and act on cartilage and synovium, accelerating cartilage degeneration and synovial inflammation in turn. Moreover, pyroptosis may be related to OA pain. The dense neural networks around vessels may expand to the synovium in pathological conditions, and excessive inflammatory mediators increase injury input.113 Concurrently, targeted inhibition of pyroptosis-associated molecules has made great progress in ameliorating OA joints. The effects of those molecules against the NLRP3 inflammasome and GSDMD on OA cartilage and synovial tissues were especially prominent.
However, our understanding of the role of pyroptosis in OA is still not complete. Considering that the pathological progress in OA also includes subchondral bone sclerosis and osteophyte formation, these features may be the consequence of communication between pyroptotic chondrocytes and osteoclasts or osteoblasts under imbalanced metabolic conditions. However, only a few researchers have conducted studies in that arena, and no current experiments have confirmed that pyroptosis is directly related to subchondral bone sclerosis and osteophyte formation.97 Moreover, research on pyroptosis in OA remains at the relevant molecular level. In addition, IL-1β and IL-18 secreted from pyroptotic cells may not be effective blocking targets, although they are associated with rapidly rising proinflammatory cytokine levels in the synovium and are the key mediators of cell communication in different joint tissues. Nasi found that the absence of IL-1β and the NLRP3 inflammasome did not improve cartilage erosion in a meniscectomy model of osteoarthritis.114 The enthusiasm regarding the use of anti-IL-1 to treat OA is declining because many controlled randomized clinical trials have had disappointing results.115 Therefore, cytokine therapy may not be the final solution for OA treatment, or researchers may have aimed at the wrong cytokines. Finally, the inhibition of pyroptosis-related molecules, such as the NLRP3 inflammasome and GSDMD, could be a new direction in the clinic. Blocking NLRP3 transcription, activation, or GSDMD-NT expression relieved cartilage loss and synovitis in animal models, but more preclinical studies are needed to explore side effects. In addition, the NLRP1 inflammasome is also involved in OA progression but is not dominant.116 Other inflammasomes, such as NLRC4, NLRP9, and pyrin, have not been studied to explore their role in OA. In addition, caspase-1 inhibition is also a potential therapeutic strategy, but only one current experiment explored OA cartilage amelioration in a caspase-1-inhibited state. Therefore, caspase-1 direct inhibitors may also be a promising research direction in the future, under circumstances that can be distinguished from the former two inhibitors.117
Copyright Corrections
All the figures are original.
Funding
This work was financially supported by the National Natural Science Foundation of China (81873669, 81800209); Gansu science and technology planning project (21JR7RA445); National Innovation and Entrepreneurship training program (202210730175); Lanzhou University Innovation and Entrepreneurship training program (20220060133, 20220060024).
Disclosure
The authors declare no conflict of interests.
References
1. Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci USA. 1996;93(18):9833–9838. doi:10.1073/pnas.93.18.9833
2. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–169. doi:10.1038/358167a0
3. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA. 1999;96(5):2396–2401. doi:10.1073/pnas.96.5.2396
4. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114. doi:10.1016/s0966-842x(00)01936-3
5. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
6. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11(6). doi:10.15252/emmm.201810248
7. Ji N, Qi Z, Wang Y, et al. Pyroptosis: a new regulating mechanism in cardiovascular disease. J Inflamm Res. 2021;14:2647–2666. doi:10.2147/jir.S308177
8. Woolf AD, Pfleger B. Burden of major musculoskeletal conditions. Bull World Health Organ. 2003;81(9):646–656.
9. Vina ER, Kwoh CK. Epidemiology of osteoarthritis: literature update. Curr Opin Rheumatol. 2018;30(2):160–167. doi:10.1097/bor.0000000000000479
10. Abramoff B, Caldera FE. Osteoarthritis: pathology, diagnosis, and treatment options. Med Clin North Am. 2020;104(2):293–311. doi:10.1016/j.mcna.2019.10.007
11. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
12. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50(6):1352–1364. doi:10.1016/j.immuni.2019.05.020
13. He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
14. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–157. doi:10.1038/s41577-019-0228-2
15. Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. doi:10.1038/nature18590
16. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi:10.1038/nature18629
17. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017;38(4):261–271. doi:10.1016/j.it.2017.01.003
18. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. 2019;364(6435). doi:10.1126/science.aau1330
19. Ghimire L, Paudel S, Jin L, Jeyaseelan S. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020;13(3):388–398. doi:10.1038/s41385-020-0256-z
20. Mullins B, Chen J. NLRP9 in innate immunity and inflammation. Immunology. 2021;162(3):262–267. doi:10.1111/imm.13290
21. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
22. Sundaram B, Kanneganti TD. Advances in understanding activation and function of the NLRC4 inflammasome. Int J Mol Sci. 2021;22(3):1048. doi:10.3390/ijms22031048
23. Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. doi:10.1038/nature07725
24. Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The pyrin inflammasome in health and disease. Front Immunol. 2019;10:1745. doi:10.3389/fimmu.2019.01745
25. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi:10.1038/nature10558
26. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi:10.1038/nature13683
27. Viganò E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun. 2015;6:8761. doi:10.1038/ncomms9761
28. Yang J, Liu Z, Wang C, et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc Natl Acad Sci USA. 2018;115(26):6792–6797. doi:10.1073/pnas.1800562115
29. Aglietti RA, Estevez A, Gupta A, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113(28):7858–7863. doi:10.1073/pnas.1607769113
30. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43(5):923–932. doi:10.1016/j.immuni.2015.10.009
31. Burgener SS, Schroder K. Neutrophil extracellular traps in host defense. Cold Spring Harb Perspect Biol. 2020;12(7):a037028. doi:10.1101/cshperspect.a037028
32. Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi:10.1038/nature22393
33. Newton K, Wickliffe KE, Maltzman A, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575(7784):679–682. doi:10.1038/s41586-019-1752-8
34. Zhou Z, He H, Wang K, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368(6494). doi:10.1126/science.aaz7548
35. Liu Y, Fang Y, Chen X, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5(43). doi:10.1126/sciimmunol.aax7969
36. Safiri S, Kolahi AA, Smith E, et al. Global, regional and national burden of osteoarthritis 1990–2017: a systematic analysis of the global burden of disease study 2017. Ann Rheum Dis. 2020;79(6):819–828. doi:10.1136/annrheumdis-2019-216515
37. Pereira D, Peleteiro B, Araújo J, Branco J, Santos RA, Ramos E. The effect of osteoarthritis definition on prevalence and incidence estimates: a systematic review. Osteoarthritis Cartilage. 2011;19(11):1270–1285. doi:10.1016/j.joca.2011.08.009
38. Vos T, Allen C, Arora M. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388(10053):1545–1602. doi:10.1016/s0140-6736(16)31678-6
39. Prieto-Alhambra D, Judge A, Javaid MK, Cooper C, Diez-Perez A, Arden NK. Incidence and risk factors for clinically diagnosed knee, Hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann Rheum Dis. 2014;73(9):1659–1664. doi:10.1136/annrheumdis-2013-203355
40. Kye SY, Park K. Suicidal ideation and suicidal attempts among adults with chronic diseases: a cross-sectional study. Compr Psychiatry. 2017;73:160–167. doi:10.1016/j.comppsych.2016.12.001
41. Veronese N, Stubbs B, Solmi M, et al. Association between lower limb osteoarthritis and incidence of depressive symptoms: data from the osteoarthritis initiative. Age Ageing. 2017;46(3):470–476. doi:10.1093/ageing/afw216
42. Siviero P, Veronese N, Smith T, et al. Association between osteoarthritis and social isolation: data from the EPOSA study. J Am Geriatr Soc. 2020;68(1):87–95. doi:10.1111/jgs.16159
43. Xie F, Kovic B, Jin X, He X, Wang M, Silvestre C. Economic and humanistic burden of osteoarthritis: a systematic review of large sample studies. Pharmacoeconomics. 2016;34(11):1087–1100. doi:10.1007/s40273-016-0424-x
44. Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. 2021;17(1):47–57. doi:10.1038/s41584-020-00533-7
45. van der Esch M, Knoop J, Hunter DJ, et al. The association between reduced knee joint proprioception and medial meniscal abnormalities using MRI in knee osteoarthritis: results from the Amsterdam osteoarthritis cohort. Osteoarthritis Cartilage. 2013;21(5):676–681. doi:10.1016/j.joca.2013.02.002
46. Reid KF, Price LL, Harvey WF, et al. Muscle power is an independent determinant of pain and quality of life in knee osteoarthritis. Arthritis Rheumatol. 2015;67(12):3166–3173. doi:10.1002/art.39336
47. Muthuri SG, Zhang W, Maciewicz RA, Muir K, Doherty M. Beer and wine consumption and risk of knee or hip osteoarthritis: a case control study. Arthritis Res Ther. 2015;17(1):23. doi:10.1186/s13075-015-0534-4
48. Beaudart C, Lengelé L, Leclercq V, et al. Symptomatic efficacy of pharmacological treatments for knee osteoarthritis: a systematic review and a network meta-analysis with a 6-month time horizon. Drugs. 2020;80(18):1947–1959. doi:10.1007/s40265-020-01423-8
49. MacFarlane LA, Cook NR, Kim E, et al. The effects of vitamin D and marine omega-3 fatty acid supplementation on chronic knee pain in older US adults: results from a randomized trial. Arthritis Rheumatol. 2020;72(11):1836–1844. doi:10.1002/art.41416
50. Saberi Hosnijeh F, Zuiderwijk ME, Versteeg M, et al. Cam deformity and acetabular dysplasia as risk factors for hip osteoarthritis. Arthritis Rheumatol. 2017;69(1):86–93. doi:10.1002/art.39929
51. Wink AE, Gross KD, Brown CA, et al. Varus thrust during walking and the risk of incident and worsening medial tibiofemoral MRI lesions: the multicenter osteoarthritis study. Osteoarthritis Cartilage. 2017;25(6):839–845. doi:10.1016/j.joca.2017.01.005
52. Yin Y, Li X, Sha X, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. 2015;35(4):804–816. doi:10.1161/atvbaha.115.305282
53. Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 2019;60(4):721–727. doi:10.1194/jlr.S091280
54. Pazár B, Ea HK, Narayan S, et al. Basic calcium phosphate crystals induce monocyte/macrophage IL-1β secretion through the NLRP3 inflammasome in vitro. J Immunol. 2011;186(4):2495–2502. doi:10.4049/jimmunol.1001284
55. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–1759. doi:10.1016/s0140-6736(19)30417-9
56. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(7):412–420. doi:10.1038/nrrheum.2016.65
57. Hsia AW, Emami AJ, Tarke FD, et al. Osteophytes and fracture calluses share developmental milestones and are diminished by unloading. J Orthop Res. 2018;36(2):699–710. doi:10.1002/jor.23779
58. Eyre D. Collagen of articular cartilage. Arthritis Res. 2002;4(1):30–35. doi:10.1186/ar380
59. Guilak F, Nims RJ, Dicks A, Wu CL, Meulenbelt I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 2018;71–72:40–50. doi:10.1016/j.matbio.2018.05.008
60. Huang Z, Kraus VB. Does lipopolysaccharide-mediated inflammation have a role in OA? Nat Rev Rheumatol. 2016;12(2):123–129. doi:10.1038/nrrheum.2015.158
61. He Z, Nie P, Lu J, et al. Less mechanical loading attenuates osteoarthritis by reducing cartilage degeneration, subchondral bone remodelling, secondary inflammation, and activation of NLRP3 inflammasome. Bone Joint Res. 2020;9(10):731–741. doi:10.1302/2046-3758.910.Bjr-2019-0368.R2
62. Bai H, Yuan R, Zhang Z, et al. Intra-articular Injection of baicalein inhibits cartilage catabolism and NLRP3 inflammasome signaling in a posttraumatic OA model. Oxid Med Cell Longev. 2021;2021:6116890. doi:10.1155/2021/6116890
63. Wang M, Sampson ER, Jin H, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15(1):R5. doi:10.1186/ar4133
64. Fosang AJ, Beier F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pract Res Clin Rheumatol. 2011;25(6):751–766. doi:10.1016/j.berh.2011.11.010
65. Zhang L, Ma S, Su H, Cheng J. Isoliquiritigenin inhibits IL-1β-induced production of matrix metalloproteinase in articular chondrocytes. Mol Ther Methods Clin Dev. 2018;9:153–159. doi:10.1016/j.omtm.2018.02.006
66. Zhang Y, Lin Z, Chen D, He Y. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem Biophys Res Commun. 2021;553:119–125. doi:10.1016/j.bbrc.2021.03.055
67. Burr DB. Anatomy and physiology of the mineralized tissues: role in the pathogenesis of osteoarthrosis. Osteoarthritis Cartilage. 2004;12(Suppl A):S20–30. doi:10.1016/j.joca.2003.09.016
68. Sun FF, Hu PF, Xiong Y, Bao JP, Qian J, Wu LD. Tricetin protects rat chondrocytes against IL-1β-induced inflammation and apoptosis. Oxid Med Cell Longev. 2019;2019:4695381. doi:10.1155/2019/4695381
69. Thomson A, Hilkens CMU. Synovial macrophages in osteoarthritis: the key to understanding pathogenesis? Front Immunol. 2021;12:678757. doi:10.3389/fimmu.2021.678757
70. Ma Z, Wang Y, Piao T, Liu J. Echinocystic acid inhibits IL-1β-induced COX-2 and iNOS expression in human osteoarthritis chondrocytes. Inflammation. 2016;39(2):543–549. doi:10.1007/s10753-015-0278-y
71. Eitner A, Müller S, König C, et al. Inhibition of inducible nitric oxide synthase prevents IL-1β-induced mitochondrial dysfunction in human chondrocytes. Int J Mol Sci. 2021;22(5):2477. doi:10.3390/ijms22052477
72. Guerne PA, Carson DA, Lotz M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J Immunol. 1990;144(2):499–505.
73. Aigner T, McKenna L, Zien A, Fan Z, Gebhard PM, Zimmer R. Gene expression profiling of serum- and interleukin-1 beta-stimulated primary human adult articular chondrocytes–a molecular analysis based on chondrocytes isolated from one donor. Cytokine. 2005;31(3):227–240. doi:10.1016/j.cyto.2005.04.009
74. Pulsatelli L, Dolzani P, Piacentini A, et al. Chemokine production by human chondrocytes. J Rheumatol. 1999;26(9):1992–2001.
75. Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. doi:10.1155/2014/561459
76. Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol. 1999;162(2):1096–1100.
77. Futani H, Okayama A, Matsui K, et al. Relation between interleukin-18 and PGE2 in synovial fluid of osteoarthritis: a potential therapeutic target of cartilage degradation. J Immunother. 2002;25(Suppl 1):S61–4. doi:10.1097/00002371-200203001-00009
78. Bao J, Chen Z, Xu L, Wu L, Xiong Y. Rapamycin protects chondrocytes against IL-18-induced apoptosis and ameliorates rat osteoarthritis. Aging. 2020;12(6):5152–5167. doi:10.18632/aging.102937
79. Dai SM, Shan ZZ, Nishioka K, Yudoh K. Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis: direct effect on chondrocytes may not be pivotal. Ann Rheum Dis. 2005;64(5):735–742. doi:10.1136/ard.2004.026088
80. Martel-Pelletier J, Barr AJ, Cicuttini FM, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072. doi:10.1038/nrdp.2016.72
81. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19(1):18. doi:10.1186/s13075-017-1229-9
82. Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51(2):249–257. doi:10.1016/j.bone.2012.02.012
83. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11(1):35–44. doi:10.1038/nrrheum.2014.162
84. Ni Z, Kuang L, Chen H, et al. The exosome-like vesicles from osteoarthritic chondrocyte enhanced mature IL-1β production of macrophages and aggravated synovitis in osteoarthritis. Cell Death Dis. 2019;10(7):522. doi:10.1038/s41419-019-1739-2
85. Zhang L, Xing R, Huang Z, et al. Inhibition of synovial macrophage pyroptosis alleviates synovitis and fibrosis in knee osteoarthritis. Mediators Inflamm. 2019;2019:2165918. doi:10.1155/2019/2165918
86. Xiao Y, Ding L, Yin S, et al. Relationship between the pyroptosis of fibroblast‑like synoviocytes and HMGB1 secretion in knee osteoarthritis. Mol Med Rep. 2021;23(2). doi:10.3892/mmr.2020.11736
87. García-Arnandis I, Guillén MI, Gomar F, Pelletier JP, Martel-Pelletier J, Alcaraz MJ. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1β in osteoarthritic synoviocytes. Arthritis Res Ther. 2010;12(4):R165. doi:10.1186/ar3124
88. Kim HM, Kim YM. HMGB1: LPS delivery vehicle for caspase-11-mediated Pyroptosis. Immunity. 2018;49(4):582–584. doi:10.1016/j.immuni.2018.09.021
89. Zhang L, Zhang L, Huang Z, et al. Increased HIF-1α in knee osteoarthritis aggravate synovial fibrosis via fibroblast-like synoviocyte pyroptosis. Oxid Med Cell Longev. 2019;2019:6326517. doi:10.1155/2019/6326517
90. Shi J, Zhao W, Ying H, et al. Estradiol inhibits NLRP3 inflammasome in fibroblast-like synoviocytes activated by lipopolysaccharide and adenosine triphosphate. Int J Rheum Dis. 2018;21(11):2002–2010. doi:10.1111/1756-185x.13198
91. Hu J, Zhou J, Wu J, et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-κB activity and pyroptosis in chondrocytes. J Ethnopharmacol. 2020;247:112261. doi:10.1016/j.jep.2019.112261
92. Wang C, Gao Y, Zhang Z, et al. Ursolic acid protects chondrocytes, exhibits anti-inflammatory properties via regulation of the NF-κB/NLRP3 inflammasome pathway and ameliorates osteoarthritis. Biomed Pharmacother. 2020;130:110568. doi:10.1016/j.biopha.2020.110568
93. Stabler TV, Huang Z, Montell E, Vergés J, Kraus VB. Chondroitin sulphate inhibits NF-κB activity induced by interaction of pathogenic and damage associated molecules. Osteoarthritis Cartilage. 2017;25(1):166–174. doi:10.1016/j.joca.2016.08.012
94. Baek HS, Hong VS, Kim SH, Lee J, Kim S. KMU-1170, a novel multi-protein kinase inhibitor, suppresses inflammatory signal transduction in THP-1 cells and human osteoarthritic fibroblast-like synoviocytes by suppressing activation of NF-κB and NLRP3 inflammasome signaling pathway. Int J Mol Sci. 2021;22(3):1194. doi:10.3390/ijms22031194
95. Franco-Trepat E, Alonso-Pérez A, Guillán-Fresco M, et al. Amitriptyline blocks innate immune responses mediated by toll-like receptor 4 and IL-1 receptor: preclinical and clinical evidence in osteoarthritis and gout. Br J Pharmacol. 2022;179(2):270–286. doi:10.1111/bph.15707
96. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi:10.1038/nm.3893
97. Chen Z, Zhong H, Wei J, et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res Ther. 2019;21(1):300. doi:10.1186/s13075-019-2085-6
98. Gao F, Zhang S. Loratadine alleviates advanced glycation end product-induced activation of NLRP3 inflammasome in human chondrocytes. Drug Des Devel Ther. 2020;14:2899–2908. doi:10.2147/dddt.S243512
99. Yan Z, Qi W, Zhan J, et al. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J Cell Mol Med. 2020;24(22):13046–13057. doi:10.1111/jcmm.15905
100. Liao T, Ding L, Wu P, et al. Chrysin attenuates the NLRP3 inflammasome cascade to reduce synovitis and pain in KOA rats. Drug Des Devel Ther. 2020;14:3015–3027. doi:10.2147/dddt.S261216
101. Wang Z, Chen M, Wang B, et al. Electroacupuncture alleviates osteoarthritis by suppressing NLRP3 inflammasome activation in guinea pigs. Evid Based Complement Alternat Med. 2020;2020:5476064. doi:10.1155/2020/5476064
102. Xia S, Hollingsworth L, Wu H. Mechanism and regulation of gasdermin-mediated cell death. Cold Spring Harb Perspect Biol. 2020;12(3):a036400. doi:10.1101/cshperspect.a036400
103. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aat2738
104. Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745. doi:10.1038/s41590-020-0669-6
105. Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aar6689
106. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–960. doi:10.1126/science.aar7607
107. Xiao J, Wang C, Yao JC, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. 2018;16(11):e3000047. doi:10.1371/journal.pbio.3000047
108. Kanneganti A, Malireddi RKS, Saavedra PHV, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J Exp Med. 2018;215(6):1519–1529. doi:10.1084/jem.20172060
109. Rashidi M, Simpson DS, Hempel A, et al. The pyroptotic cell death effector gasdermin D Is activated by gout-associated uric acid crystals but is dispensable for cell death and il-1β release. J Immunol. 2019;203(3):736–748. doi:10.4049/jimmunol.1900228
110. Yang T, Sun K, Wang C, et al. Gasdermin D deficiency attenuates arthritis induced by traumatic injury but not autoantibody-assembled immune complexes. Arthritis Res Ther. 2021;23(1):286. doi:10.1186/s13075-021-02668-8
111. Ma C, Yang D, Wang B, et al. Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci Adv. 2020;6(21):eaaz6717. doi:10.1126/sciadv.aaz6717
112. Bulek K, Zhao J, Liao Y, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. J Clin Invest. 2020;130(8):4218–4234. doi:10.1172/jci138103
113. Mapp PI, Walsh DA. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat Rev Rheumatol. 2012;8(7):390–398. doi:10.1038/nrrheum.2012.80
114. Nasi S, Ea HK, So A, Busso N. Revisiting the Role Of Interleukin-1 Pathway In Osteoarthritis: Interleukin-1α and −1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front Pharmacol. 2017;8:282. doi:10.3389/fphar.2017.00282
115. Vincent TL. IL-1 in osteoarthritis: time for a critical review of the literature. F1000Res. 2019;8:934. doi:10.12688/f1000research.18831.1
116. Zhao LR, Xing RL, Wang PM, et al. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP‑induced pyroptosis in knee osteoarthritis. Mol Med Rep. 2018;17(4):5463–5469. doi:10.3892/mmr.2018.8520
117. Ramesova A, Vesela B, Svandova E, Lesot H, Matalova E. Caspase-1 inhibition impacts the formation of chondrogenic nodules, and the expression of markers related to osteogenic differentiation and lipid metabolism. Int J Mol Sci. 2021;22(17):9576. doi:10.3390/ijms22179576
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Characterization of Cathepsin B in Mediating Silica Nanoparticle-Induced Macrophage Pyroptosis via an NLRP3-Dependent Manner
Ma L, Han Z, Yin H, Tian J, Zhang J, Li N, Ding C, Zhang L
Journal of Inflammation Research 2022, 15:4537-4545
Published Date: 8 August 2022
α-Chaconine Facilitates Chondrocyte Pyroptosis and Nerve Ingrowth to Aggravate Osteoarthritis Progression by Activating NF-κB Signaling

Zhang Z, Fu F, Bian Y, Zhang H, Yao S, Zhou C, Ge Y, Luo H, Chen Y, Ji W, Tian K, Yue M, Du W, Jin H, Tong P, Wu C, Ruan H
Journal of Inflammation Research 2022, 15:5873-5888
Published Date: 17 October 2022
Inhibition of Dectin-1 Alleviates Neuroinflammatory Injury by Attenuating NLRP3 Inflammasome-Mediated Pyroptosis After Intracerebral Hemorrhage in Mice: Preliminary Study Results
Ding Z, Zhong Z, Wang J, Zhang R, Shao J, Li Y, Wu G, Tu H, Yuan W, Sun H, Wang Q
Journal of Inflammation Research 2022, 15:5917-5933
Published Date: 19 October 2022
A Bibliometric and Knowledge Map Analysis of Osteoarthritis Signaling Pathways from 2012 to 2022
Li B, Zheng J
Journal of Pain Research 2022, 15:3833-3846
Published Date: 6 December 2022
The Mechanism of Pyroptosis and Its Application Prospect in Diabetic Wound Healing
Al Mamun A, Shao C, Geng P, Wang S, Xiao J
Journal of Inflammation Research 2024, 17:1481-1501
Published Date: 6 March 2024