Back to Journals » International Journal of General Medicine » Volume 18
Identification of Potential Therapeutic Targets for Sepsis Using Mendelian Randomization and Integrated eQTL/pQTL Analysis
Authors Yue H, Tian J, Yin W, Zhao H ![]()
Received 29 May 2025
Accepted for publication 2 October 2025
Published 9 October 2025 Volume 2025:18 Pages 6137—6152
DOI https://doi.org/10.2147/IJGM.S535716
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Haigui Yue,1 Jinping Tian,2,3 Wei Yin,4 Houyu Zhao2
1Department of Clinical Pharmacy, Taizhou Orthopedics Hospital, Taizhou, Zhejiang, People's Republic of China; 2Department of Oncology, The First Affiliated Hospital of Xinxiang Medical University, Weihui, People’s Republic of China; 3Department of Radiology, The Affiliated Hospital of Hubei Provincial Government, Wuhan, Hubei, People’s Republic of China; 4Department of Radiology, Xianning Central Hospital, The First Affiliated Hospital of Hubei University of Science and Technology, Xianning, Hubei, People’s Republic of China
Correspondence: Wei Yin, Email [email protected] Houyu Zhao, Email [email protected]
Background: Sepsis significantly contributes to global morbidity, yet effective treatments remain limited. Mendelian randomization (MR), integrated with genetic data, offers promise for uncovering novel therapeutic targets.
Methods: We utilized eQTL (eQTLGen) and pQTL (DECODE) data as exposures, and GWAS summaries for sepsis (UK Biobank, FinnGen) as outcomes. GEO datasets (GSE57065, GSE95233) underwent batch correction via PCA clustering using the “sva” R package. Differentially expressed genes (DEGs, |log2FC|> 1, adjusted P< 0.05) intersected with druggable genes were identified. MR analyses were performed using TwoSampleMR and MR-PRESSO, followed by drug-target predictions using DGIdb. Key genes (BCL6, PTX3, IL7R, BTN3A2, LGALS1) were validated experimentally through qRT-PCR and Western blot in a mouse sepsis model induced by cecal ligation and puncture (CLP).
Results: Intersection analyses yielded 398 therapeutic candidates. MR revealed 6 genes and 21 proteins significantly associated with sepsis risk, including protective (eg, HDC, IFI27) and harmful factors (eg, CTSO, BTN3A2). Furthermore, 13 druggable genes correlated with sepsis-related factors, such as BTN3A2 with diabetes, and IL7R, BCL6, PTX3, among others, linked to vitamin D deficiency and cancer. DGIdb identified 34 potential drugs targeting these hub genes, with KEGG and GO analyses highlighting immune regulation and FoxO signaling pathways. qRT-PCR and Western blot confirmed consistent downregulation (BCL6, PTX3, IL7R) and upregulation (BTN3A2, LGALS1) at both mRNA and protein levels in septic mice compared to controls, supporting MR-based predictions.
Conclusion: We identified and experimentally validated 6 sepsis-associated genes and 21 proteins, providing crucial insights into potential therapeutic targets and enhancing understanding of the molecular pathogenesis of sepsis.
Keywords: sepsis, drug target, Mendelian randomization, bioinformatics, CLP model
Graphical Abstract:

Introduction
Sepsis results from a disrupted host response to infection and immune imbalance, leading to severe organ dysfunction.1 With around 11 million annual deaths, representing 20% of global mortality, sepsis also imposes a significant financial burden, costing $27 billion in the United States in 2019 alone.2 This imposes a significant burden on healthcare systems worldwide. To date, the management of sepsis has primarily relied on a combination of antimicrobial therapy, infection source control, and organ support measures. However, despite advances in critical care, sepsis remains a leading cause of morbidity and mortality, highlighting the urgent need for more effective therapeutic strategies. Current approaches often fail to address the underlying dysregulation of the host immune response, which plays a central role in the progression of sepsis.3 Clinically effective and specific therapeutic agents are still lacking. Meanwhile, the rise of extensively drug-resistant pathogens is rapidly depleting the available arsenal of antimicrobial treatments for infected patients.4 The search for effective therapeutic agents or prophylactic targets for sepsis is urgent.
Developing effective drugs relies on accurately identifying therapeutic candidates and confirming their impact on disease progression. However, traditional drug discovery is both challenging and costly. Integrating genomics into this process can accelerate the identification of novel therapeutic targets.5 By combining genome-wide association study (GWAS) data with molecular quantitative trait locus (molQTL) data, such as expression quantitative trait locus (eQTL) or protein quantitative trait locus (pQTL), researchers can infer causal relationships between risk variants and target genes.6 Mendelian randomization (MR) offers a powerful alternative by mimicking randomized trials using genetic data, bypassing the need for extensive drug testing.7 This approach has gained widespread recognition and has successfully identified therapeutic targets for diseases such as multiple sclerosis, asthma, and small cell lung cancer.8 Leveraging MR to validate differentially expressed genes from the GEO database provides an efficient strategy for pinpointing candidate genes for further experimental study. In this context, the current study integrates differential gene expression data across tissues with MR methods to uncover novel therapeutic targets for sepsis.
Methods
Study Design
This study employed a case-control epidemiological study design. This investigation adhered closely to the Strengthening the Reporting of Observational Studies in Epidemiology–Mendelian Randomization (STROBE-MR) recommendations.9 Exposure datasets included eQTL summary statistics from the eQTLGen Consortium (https://eqtlgen.org/), comprising data on 16,989 genes measured across blood samples from 31,684 healthy individuals of European descent. Additionally, plasma-based protein quantitative trait loci (pQTL) data were derived from DECODE (https://www.decode.com/summarydata/), a collaborative initiative involving the UK Biobank (UKB) and several biopharmaceutical organizations. DECODE performed comprehensive plasma proteomic profiling on 54,219 participants from the UK Biobank, identifying a total of 14,287 robust genetic associations involving 2923 distinct plasma proteins. eQTLs were obtained from SMR-formatted cis-eQTLs provided by the eQTLGen consortium (https://www.eqtlgen.org), which included peripheral blood profiles of 31,684 individuals.3 To create genetic tools that can replace 398 druggable genes, we specifically selected cis-eQTLs located within ± 100 kb of the genomic position of each gene, resulting in a total of 1592 eQTLs corresponding to 265 druggable genes; (eQTL tool variables included in the analysis are provided in the attachment eQTL_drug_exposure.txt). Proteomic quantitative trait loci (pQTL) data are from the deCODE (https://www.decode.com/summarydata/) database. The data used in this study come from the plasma protein pQTL data of the deCODE database version 2021,4 which describes a GWAS study of 35,559 Europeans that measured plasma protein levels using 4907 aptamers; (the pQTL instrumental variables included in the analysis are provided in the appendix pQTL_drug_exposure.txt; 10.5281/zenodo.17283362). Conversely, outcome variables consisted of GWAS summary-level data related to sepsis and its associated risk factors, extracted from the FinnGen research database and UK Biobank consortium. All datasets included in our analysis were publicly available with existing ethical approvals from original sources, further ethical review for this study was approved by Xianning Central Hospital.
Data Retrieval and Preprocessing
We retrieved gene expression data from the publicly accessible Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) repository. Specifically, the datasets GSE57065 and GSE95233 (as listed in Table 1) were chosen. Raw expression files along with their respective annotation platforms were downloaded directly from GEO. Data normalization was executed with the “affy” package (version 1.79.2; available at http://bioconductor.org/packages/release/bioc/html/affy.html). The specific process was to first perform RMA background correction and quantile normalization using the quantiles method, and then use the medianpolish method to summarize the expression levels of the probe set to finally obtain the probe expression data. Subsequently, the GPL570 platform annotation file was used to perform Symbol annotation on the probe ID. Probe identifiers were mapped to corresponding gene symbols using the downloaded annotation files, and probes lacking gene-symbol annotations were eliminated. For genes represented by multiple probes, their mean expression values were calculated and recorded as final expression levels. Subsequently, all expression data were integrated using the ComBat function,10 as provided by the R package “sva”.11
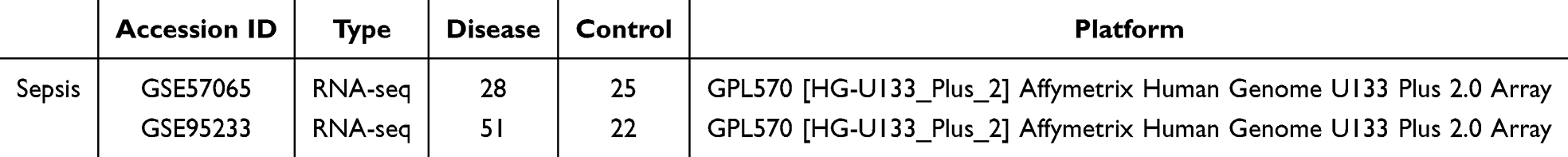 |
Table 1 Information for Selected Microarray Datasets for the Current Study |
Identification of Significantly Expressed Genes
Differential gene expression was assessed between sepsis and control samples using the “limma” R package (version 3.54.0).12 To enhance the reliability of results, p-values underwent multiple testing correction via the Benjamini–Hochberg method, controlling for false discovery rate (FDR). Genes with absolute log2 fold changes exceeding 1 and adjusted p-values less than 0.05 were identified as significantly differentially expressed genes (DEGs).
Determining Druggable Candidate Genes
Candidate genes potentially amenable to pharmacological targeting were identified using the Drug-Gene Interaction Database (DGIdb version 4.2.0; https://www.dgidb.org/) in conjunction with insights from a comprehensive review by Finan et al concerning gene “druggability”.13 DGIdb incorporates extensive drug-gene interaction data gathered from various sources, including scientific literature, online databases, and additional web-based platforms. Approximately 5883 potentially druggable genes were ultimately obtained for further analysis.
Exposure Datasets
Exposure variables included summarized eQTL datasets from the eQTLGen Consortium and pQTL data from the DECODE database.
Outcome Datasets
Sepsis-associated GWAS summary statistics were obtained from datasets released by the UK Biobank consortium,14 encompassing 26,052 cases alongside 487,214 control subjects. These GWAS data can also be retrieved from the IEU Open GWAS repository, under accession code ieu-b-4980, which covers a population of 486,484 participants, including 11,643 sepsis cases. Furthermore, we selected three clinically relevant sepsis risk factors for detailed analysis: vitamin D deficiency, cancer, and diabetes. Corresponding GWAS summary statistics for these traits were sourced from FinnGen, a nationwide Finnish GWAS consortium integrating genetic data from 13 biobanks and cohorts, which exhibit minimal overlap with the datasets used as exposures. Specifically, these data were represented by the following FinnGen identifiers: vitamin D deficiency (finn-b-E4_VIT_D_DEF), cancer (finn-b-C3_CANCER), and diabetes (finn-b-E4_DIABETES).
Selection and Validation of Instrumental Variables
Instrumental variables (IVs) were carefully chosen based on the foundational assumptions inherent in Mendelian randomization (MR): IVs must directly influence exposure, remain free from confounding, and affect the outcome solely via the exposure pathway. To strictly adhere to these principles, genetic variants initially underwent filtering according to a genome-wide significance criterion (P < 1e-5). Furthermore, linkage disequilibrium (LD) was assessed with a stringent threshold of r² < 0.001 and a clustering window of 10,000 kb. Finally, weak instrumental variants, identified by F-statistics below 10, were systematically excluded to enhance reliability.
Mendelian Randomization Analysis
We employed the TwoSampleMR package within R software (version 4.3.3) to execute the MR analyses. Five distinct MR approaches were implemented: inverse variance-weighted fixed effects (IVW-FE), inverse variance-weighted random effects (IVW-RE), weighted median, maximum likelihood, and both simple and weighted mode methods. This study uses the inverse variance weighted method (IVW) as the primary causal effect estimate. The IVW method is an ideal estimate. It is an effective analysis based on the basic premise that all genetic variants are valid instrumental variables and has a strong causal relationship detection capability. However, the IVW method specifically requires that genetic variants only affect the target outcome through the exposure in the study. Therefore, we used several other methods for reference to test the reliability and stability of the results. At the same time, we performed multi-effect tests when using the IVW method, so as to ensure the accuracy of the results as much as possible. To evaluate the stability and validity of our findings, sensitivity analyses were performed, carefully checking for signs of heterogeneity or horizontal pleiotropy. Pleiotropy refers to the influence of a single genetic variant on multiple traits. The intercept term of the MR-Egger method was observed using the mr_heterogeneity function to test for pleiotropy. p > 0.05 indicated no pleiotropy. Heterogeneity was assessed using the Cochran’s Q value obtained using the mr_pleiotropy_test function. Influential outliers were identified by creating scatter plots and leave-one-out plots, and potential outliers were detected and corrected using Mendelian randomization pleiotropy residual sum and outlier (MR-PRESSO).
Prediction of Candidate Therapeutic Drugs
We conducted an analysis of potential drug-gene interactions using the Drug Signatures Database (DSigDB, accessible via http://dsigdb.tanlab.org/DSigDBv1.0/).15 DSigDB, an extensive resource comprising 22,527 gene sets, 17,389 unique compounds, and data on approximately 19,531 genes, facilitated the identification of candidate drugs. Evaluating these protein-drug interactions provides valuable insights into the therapeutic potential of the identified gene targets.
Functional Enrichment Analysis (GO/KEGG)
To elucidate the biological significance and molecular pathways associated with our target genes, we performed pathway enrichment analysis using the KEGG database via the ClusterProfiler package within R software.
Protein-Protein Interaction (PPI) Network Analysis
To further characterize and clarify the biological interactions among selected proteins, we constructed a protein-protein interaction (PPI) network using the GeneMANIA platform (available at https://genemania.org/). By mapping the complex interactions of the 13 genes identified, this analysis offered insights into their potential cooperative roles and functional interrelations within cellular mechanisms.
Animal Model of Sepsis
All experimental procedures strictly complied with guidelines established by the Chinese Animal Welfare Agency. Additional ethical approval was also granted by the Animal Research Ethics Committee of Xianning Central Hospital (The First Affiliated Hospital of Hubei University of Science and Technology) under Approval No. 2024-A15.
We utilized the well-established cecal ligation and puncture (CLP) technique to induce polymicrobial sepsis. Briefly, male C57BL/6 mice aged eight weeks were randomly assigned into two groups: sham-operated (n = 6) and CLP-induced sepsis (n = 6). Anesthesia was administered using intraperitoneal injection of pentobarbital sodium (50 mg/kg). In CLP mice, the cecum was tied securely just below the ileocecal junction using 5–0 silk sutures, punctured twice with a sterile 22-gauge needle, and gently pressed to release a small quantity of feces. Sham mice underwent abdominal incision only, without cecal ligation or puncture. Following the procedure, warmed sterile saline (37°C, 50 μL/g body weight) was administered subcutaneously for fluid resuscitation. After 24 hours, liver, lung tissues, and peripheral blood mononuclear cells (PBMCs) were harvested. Tissue samples were snap-frozen immediately in liquid nitrogen and preserved at −80°C until further use. PBMCs were isolated from EDTA-collected blood by centrifugation at 1300 × g for 10 minutes at 4°C using density gradient separation techniques.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from liver, lung, and PBMC samples utilizing the FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme, Nanjing, China). Reverse transcription was then performed on 1 μg total RNA per sample using the ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan). Subsequently, quantitative PCR was conducted using the SYBR Green Master Mix (Vazyme) on a LightCycler 96 Real-Time PCR system (Hoffmann-La Roche Ltd., Shanghai, China). The relative quantification of gene expression was determined via the 2−ΔΔCt method, normalized against GAPDH expression. Target genes included BCL6, PTX3, IL7R, BTN3A2, and LGALS1. All assays were executed in triplicate.
Western Blot Analysis
Western blot experiments were conducted to measure the expression of BCL6, PTX3, IL7R, BTN3A2, and LGALS1 proteins in liver, lung, and PBMC samples. Tissue samples and cells were lysed using RIPA buffer containing protease inhibitors, and protein concentrations were measured via a BCA assay kit (Beyotime, China). Equal protein amounts (approximately 20–40 μg per sample) were separated using SDS-PAGE gels and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). Membranes underwent blocking with 5% skimmed milk in TBST for one hour at ambient temperature and were subsequently incubated overnight (4°C) with primary antibodies specific to BCL6 (ab33901), PTX3 (ab90806), IL7R (ab314106), BTN3A2 (Cat No. 15148-1-AP, Proteintech), LGALS1 (ab150427), and the internal control GAPDH (60004-1-Ig, Proteintech). Following washing steps, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Proteintech) at room temperature for one hour. Bands were visualized using an enhanced chemiluminescence (ECL) detection system (Tanon, China), and quantified via ImageJ software, with protein levels normalized to GAPDH.
Statistical Analysis
All statistical calculations and graphical outputs were performed using GraphPad Prism version 8.0 (GraphPad Software, USA), R software (version 4.2.0), and Python (version 3.11.2). Data from continuous variables were expressed as mean ± standard deviation (SD) according to normality test results. Comparisons between two experimental groups were conducted via Student’s t-tests. Comparisons involving more than two groups were analyzed by one-way ANOVA, with Tukey’s post-hoc correction for multiple comparisons. Statistical significance was defined as a p-value of less than 0.05.
Results
Elimination of Batch Effects and Selection of Genetic Instrument Variants
First, we used PCA clustering plots to assess the batch effects in the GEO datasets (GSE57065 and GSE95233). The results indicated the presence of batch effects in both datasets (Figure 1A). Therefore, the combat method in the sva package was used to de-batch the data. The combat function is a function based on the Bayesian framework to adjust the batch effect of data. After normalization and batch effect removal, the PCA clustering plots demonstrated that the batch effects in GSE57065 and GSE95233 had been eliminated (Figure 1B). Subsequently, we identified 398 druggable genes by intersecting the druggable genes with the differentially expressed genes (DEGs) from GSE57065 and GSE95233 (Figure 1C and D).
 |
Figure 1 Elimination of batch effects and selection of genetic instrument variants. (A and B) Principal component analysis of two sepsis datasets; (C) The volcano plot of DEGs with the two datasets; (D) Venn plots. Two circles respectively represent the differential expression analysis results and druggable genes. |
MR Analysis Between Drug Available Genes and Sepsis
We performed a two-sample MR analysis on sepsis patients, including 26,052 cases and 487,214 controls from the UK Biobank consortium. The meta-analysis was considered both the IVW fixed effect model and the IVW random effect model. Since the assumptions of the two effect models are slightly different, in order to ensure the accuracy of the results, we selected the results that were significant in both effect models. At Bonferroni significance (p<5e-8, IVW), 6 genes (Figure 2) and 21 proteins (Figure 3) were significantly related to the risk of sepsis. The results showed that a reduced risk of sepsis was related with HDC, IFI27, SIGLEC9, IL18R1, BCL6, CAMK1D, CCNB1, CD247, CDKN2C, DPP4, ELANE, GMNN, GZMK, IL7R, LCN2, LY9, MAP2K6, NUP210, PDGFC, PFKFB3, PLN3, PSMA2, PTX3, and SIRPG, while CTSO, BTN3A2, and LGALS1 was related with increased sepsis risk. In the sensitivity analysis, there was no heterogeneity and horizontal pleiotropy. Steiger filtering was also verified.
 |
Figure 2 MR analysis between drug available genes and sepsis. |
 |
Figure 3 MR analysis between drug available proteinsand sepsis. |
Correlation Between Druggable Genes and Risk Factors of Sepsis
The GWAS summary data of three risk factors for sepsis were downloaded, including vitamin D deficiency, cancer and diabetes. We then explored the associations between 27 druggable genes and sepsis risk factors. Using 27 druggable genes as exposure variables and three risk factors as outcome variables, MR analysis was performed. As results, BTN3A2 was associated with diabetes; BCL6, CAMK1D, CCNB1, CD247, GMNN, PTX3, NUP210, PDGFC, PFKFB3, IR7R and PLIN3 were associated with Vitamin D Deficiency; IR7R and LY9 were associated with Malignant tumor (Table 2).
 |
Table 2 Correlation Between Druggable Genes and Risk Factors of Sepsis |
Potential Drugs for Sepsis and Enrichment Analysis
Using the DGIdb database, we analyzed potential drugs for sepsis and obtained 34 predicted drugs corresponding to 12 hub genes. The diagram revealed the relationships between drugs and targets (Figure 4A). Table 3 presents the potential druggability results of the targets and the interactions between the screened genes and existing drugs. There was a possibility that most potential drugs could interact with the hub gene, either in unknown ways or by inhibiting it. Simultaneously, the STRING database was employed to conduct Protein-Protein Interaction (PPI) network analysis on 11 genes, with the network confidence threshold set at 0.15. This facilitated the construction and visualization of the PPI network, from which outlier genes were subsequently removed. The outcome of this process is illustrated in Figure 4B, culminating in the identification of 11 genes. KEGG analysis revealed that these genes were significantly enriched in pathways such as the “FoxO signaling pathway”, “PDK-Akt signaling pathway”, “Human immunodeficiency virus 1 infection”, “Primary immunodeficiency”, and “Fructose and mannose metabolism” (Figure 4C). Additionally, GO analysis demonstrated that these genes were significantly enriched in terms including “adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains”, “negative regulation of adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains”, “negative regulation of lymphocyte-mediated immunity”, “positive regulation of fibroblast proliferation”, and “cell cycle DNA replication”, among others (Figure 4D).
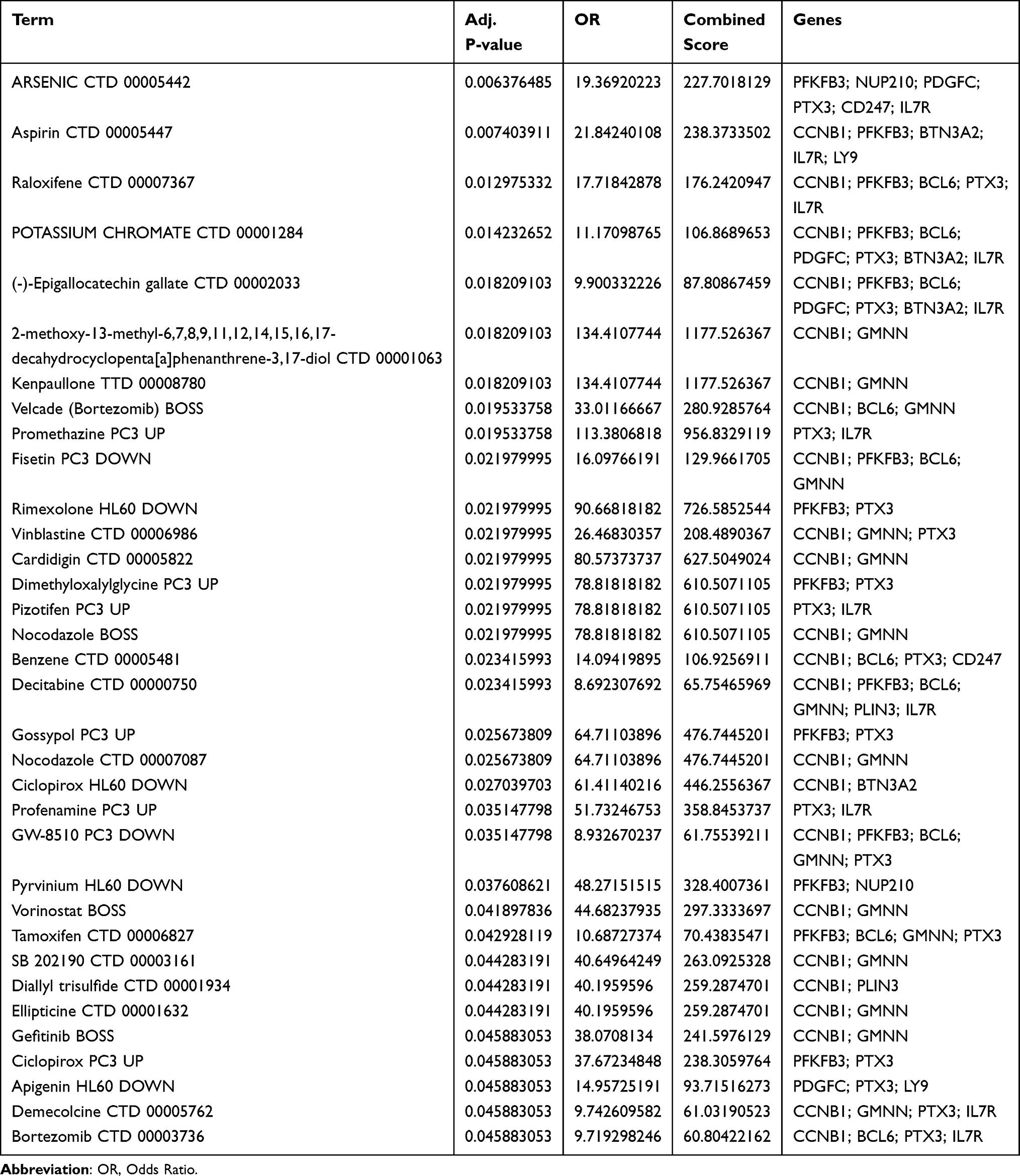 |
Table 3 Interactions Between Target Genes and Existing Drugs |
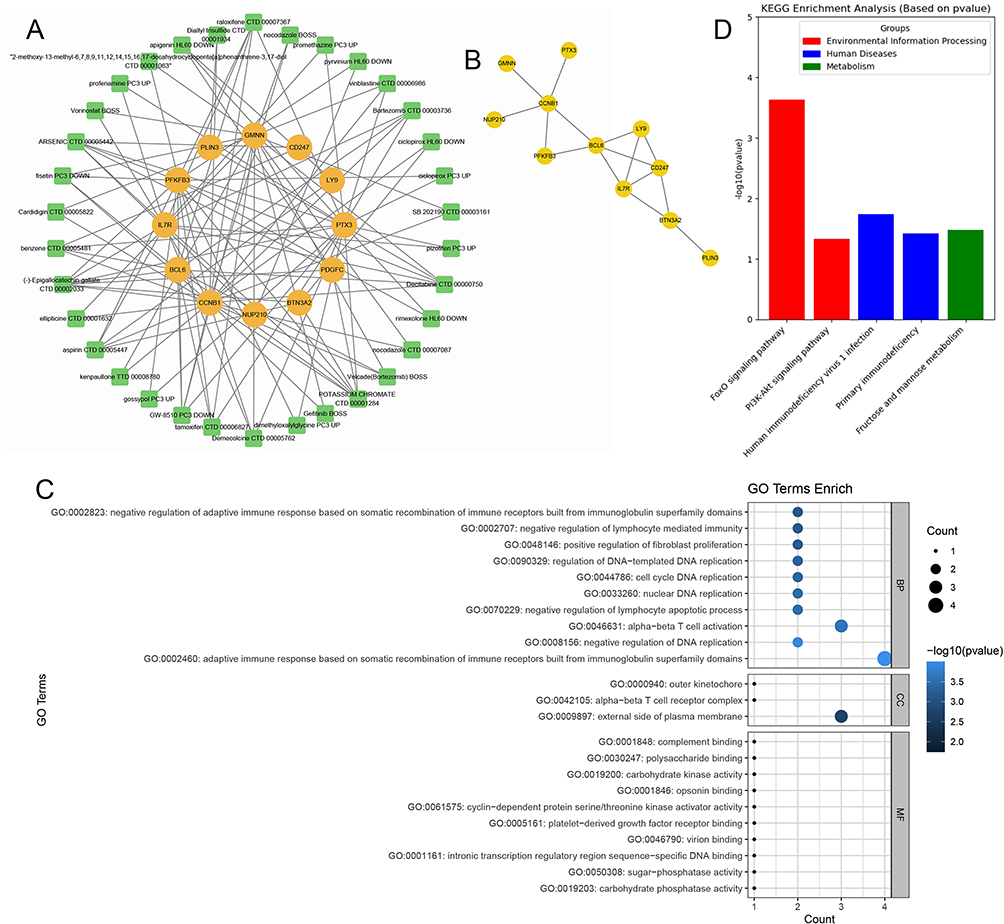 |
Figure 4 Potential drugs for sepsis and enrichment analysis. (A) Candidate drug prediction by analyzing DGIdb database; (B) PPI networks; (C and D) GO and KEGG analysis. |
Expression of BCL6, PTX3, IL7R, BTN3A2, and LGALS1 Levels in Sepsis Model
To validate the expression profiles derived from our MR analysis, qRT-PCR and Western blot were conducted using liver, lung, and PBMC samples from both sham and CLP groups. At the mRNA level, compared to the sham-operated controls, the CLP group exhibited significantly decreased expression of BCL6, PTX3, and IL7R (p < 0.05), whereas BTN3A2 and LGALS1 expression were significantly increased (p < 0.05, Figure 5A–C). These findings were consistently validated at the protein level by Western blotting, further confirming the robustness of our results. Specifically, Western blot analysis showed marked downregulation of BCL6, PTX3, and IL7R proteins and significant upregulation of BTN3A2 and LGALS1 proteins in the liver, lung, and PBMC samples from CLP mice, compared with the sham group (p < 0.05, Figure 5D–F).
 |
Figure 5 mRNA and protein expression of BCL6, PTX3, IL7R, BTN3A2, and LGALS1 in liver, lung, and PBMC samples from sham and CLP groups. (A–C) Relative mRNA expression levels of BCL6, PTX3, IL7R, BTN3A2, and LGALS1 determined by qRT-PCR in liver (A), lung (B), and PBMC (C). (D–F) Relative protein expression levels of BCL6, PTX3, IL7R, BTN3A2, and LGALS1 assessed by Western blotting in liver (D), lung (E), and PBMC (F). Data are presented as mean ± SD. *P < 0.05 compared with the sham group. |
Discussion
To our knowledge, this study is the first to employ a two-sample Mendelian randomization (MR) approach to investigate the causal relationships between druggable genes and sepsis risk, while further examining their associations with sepsis-related risk factors such as vitamin D deficiency, cancer, and diabetes. The findings revealed 27 druggable genes significantly associated with sepsis, 13 of which were also linked to established risk factors for the condition. Additionally, a murine sepsis model was established, through which differential expression of these potential candidate genes was validated.
Previous studies utilizing MR integrated with eQTL and pQTL data have identified 6 genes linked to 28-day sepsis mortality, including C5, IGFLR1, COL6A2, FN1, PPP3R1 and IL18R1.16 However, in our study, The genes HDC, IFI27, SIGLEC9, IL18R1, BCL6, and CAMK1D were associated with a reduced risk of sepsis. HDC encodes histidine decarboxylase, which is involved in histamine production, a key mediator of immune responses. Histamine has been shown to modulate sepsis outcomes by regulating cytokine release and immune cell activity.17 IFI27, an interferon-stimulated gene, plays a role in antiviral responses and has been linked to immune dysregulation.18 SIGLEC9 is a member of the sialic acid-binding immunoglobulin-like lectin family, which modulates immune cell signaling and has been implicated in immunosuppression.19 The IL-18 receptor 1 (IL-18R1) serves as the principal signaling receptor for IL-18, facilitating IL-18-induced pro-inflammatory reactions.20 Studies have shown that IL-18 plays a crucial role in various chronic inflammatory diseases, including CD and UC.21 IL-18R1 polymorphisms have been associated with CD susceptibility, particularly in specific populations.22 Furthermore, IL-18 and its receptor play a critical role in regulating intestinal immune responses by influencing T cell differentiation and function.23
Conversely, CTSO, BTN3A2, and LGALS1 were associated with an increased risk of sepsis. CTSO, BTN3A2, and LGALS1 are genes involved in immune regulation and cellular processes, potentially playing significant roles in the pathogenesis of sepsis. CTSO, a lysosomal cysteine protease, participates in protein degradation and antigen processing, which could influence the hyperactivated immune response seen in sepsis. Dysregulation of CTSO may contribute to tissue damage through excessive protease activity, degrading extracellular matrix components and cellular proteins. BTN3A2, a member of the butyrophilin family, modulates T cell responses and may regulate γδ T cells, which are critical in innate immunity. In sepsis, BTN3A2 could help balance pro-inflammatory and anti-inflammatory responses, potentially mitigating immune dysregulation.24,25 LGALS1, a galectin family member, binds to β-galactoside sugars and plays a role in immune modulation, cell adhesion, and apoptosis. Its anti-inflammatory properties, such as promoting T cell apoptosis and suppressing pro-inflammatory cytokines, could help control the cytokine storm characteristic of sepsis. Additionally, LGALS1’s role in maintaining endothelial function might reduce vascular leakage and organ failure, common complications in sepsis.26–28
The analysis of potential drugs for sepsis using the DGIdb database identified 34 drugs targeting 12 hub genes, providing valuable insights into potential therapeutic strategies for sepsis. Among these, arsenic, benzene, and potassium chromate are well-known toxic and carcinogenic agents. In addition, nocodazole, demecolcine, and SB 202190 exhibit potent cytotoxicity and are restricted to laboratory use only. Acetylsalicylic acid (aspirin), known for its anti-inflammatory and antiplatelet effects, has demonstrated potential in sepsis treatment through direct antibacterial action. Studies indicate that early aspirin administration may reduce both in-hospital and 90-day mortality in septic patients.29 Vorinostat, a hydroxamate-based Zn²⁺-dependent pan-HDAC inhibitor, is among the broad-spectrum and selective HDACis that have been extensively studied for their role in inducing ACD.30 Bortezomib (BTZ), a proteasome inhibitor approved for first-line multiple myeloma therapy, has been demonstrated to alleviate symptoms of various autoimmune diseases (ADs) in animal models and human patients. It has shown efficacy in systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, neuromyelitis optica spectrum disorder, chronic inflammatory demyelinating polyneuropathy, and autoimmune hematologic diseases that are refractory to conventional treatments.31 Bortezomib pretreatment enhanced viability and attenuated inflammatory cytokine release in LPS-stimulated RAW 264.7 cells. In a CLP mouse model, administration of 0.01 mg/kg bortezomib markedly improved survival and reduced lung inflammation.32 Tamoxifen, a non-steroidal selective estrogen receptor modulator (SERM), has revolutionized breast cancer treatment and prevention since its FDA approval in 1977.33 Epigallocatechin gallate (EGCG) EGCG exhibits potent antioxidant and anti-inflammatory activities. Microbiological analysis revealed that oral EGCG administration restored LPS-induced gut microbiota dysbiosis in mice, reversed the Firmicutes/Bacteroidetes ratio, and significantly decreased Enterobacteriales abundance.34 Fisetin, a dietary flavonoid derived from berries and Fabaceae plants, demonstrates neuroprotective and antioxidant properties. In LPS-stimulated BMDMs, fisetin (3–10 μM) dose-dependently suppressed the expression of IL-6, TNF-α, IL-1β, and iNOS. It also inhibited the phosphorylation of p38 MAPK, MK2, and TAK1 by attenuating TAK1–TAB interaction.35 DMOG, a prolyl hydroxylase inhibitor, upregulated HIF-1α, leading to reduced plasma inflammatory mediators and oxidative stress markers, alleviated intestinal mucosal damage, and decreased intestinal permeability.36 GW-8510, Roscovitine, and Kenpaullone are inhibitors of cyclin-dependent kinases (CDKs), while Pyrvinium inhibits the Wnt/β-catenin pathway and modulates mitochondrial function. Pretreatment with this antagonist attenuated sepsis-induced cardiac dysfunction and structural damage by inhibiting mPTP opening and reducing calcium overload in heart tissue.37
The KEGG and GO enrichment analyses highlighted the involvement of these genes in critical pathways such as the FoxO signaling pathway, PI3K-Akt signaling pathway, and primary immunodeficiency. The FoxO signaling pathway regulates immune cell function and oxidative stress responses, which are dysregulated in sepsis.38,39 The PI3K-Akt signaling pathway is crucial for cell survival and metabolism, and modulation of this pathway has been demonstrated to improve outcomes in septic models.40,41 Primary immunodeficiency pathways underscore the importance of restoring immune function in sepsis patients. In the present study, qRT-PCR and Western blot analyses confirmed that key genes associated with these pathways, including BCL6, PTX3, IL7R, BTN3A2, and LGALS1, exhibited significant differential expression in liver, lung, and PBMC samples from septic mice. Specifically, consistent with the prediction from our bioinformatics and MR analyses, the expression levels of BCL6, PTX3, and IL7R were significantly downregulated, whereas BTN3A2 and LGALS1 were significantly upregulated, suggesting a potential mechanistic involvement of these genes in sepsis-induced immune dysregulation. Notably, the downregulation of BCL6 and IL7R, critical immune regulators associated with T-cell and B-cell functions, aligns with impaired adaptive immune responses commonly observed in sepsis. Meanwhile, the upregulation of BTN3A2 and LGALS1 further highlights the intricate balance of immune activation and suppression during sepsis progression. These findings provide experimental validation of the functional importance of identified signaling pathways and genes, offering valuable insights into their roles in sepsis pathogenesis and potential as therapeutic targets.
Subsequently, we identified the differentially expressed genes in septic mice. IL7R sustains T-cell survival; in septic shock, recombinant IL-7 restored lymphocyte counts and signaling in a randomized trial, supporting pathway augmentation as a rescue strategy. Accordingly, ex-vivo IL-7 treatment of septic-patient PBMCs (readouts: pSTAT5, proliferation, IFN-γ) will test sufficiency.42 PFKFB3 drives glycolytic reprogramming of myeloid and endothelial cells that fuels hyper-inflammation in sepsis; dampening this axis reduces inflammatory injury in preclinical work. We will probe PFKFB3 by CRISPR/siRNA or small-molecule inhibition in primary human monocytes/macrophages and endothelium (ECAR, cytokines, barrier assays), then benchmark efficacy/toxicity in endotoxemia or CLP models.43 PTX3—a humoral pattern-recognition molecule—rises early in sepsis and tracks severity/mortality, yet may also exert protective, inflammation-modulating effects in bacterial sepsis models; thus, gain/loss-of-function studies (eg, Ptx3-deficient mice or recombinant PTX3) plus prospective patient stratification by PTX3 trajectories are warranted.44
All targets will undergo cis-colocalization and MR sensitivity (Egger intercept, Cochran’s Q, MR-PRESSO) as analytical validation.
This study has several limitations. First, the GWAS and QTL data used were primarily derived from European populations, which limits the generalizability of the findings to other ethnic groups. Furthermore, although alterations in the expression of certain genes were validated through animal experiments, the specific regulatory mechanisms and their potential as therapeutic targets require further functional studies and preclinical research. Future research should delve deeper into the precise mechanisms by which these pathways and related genes modulate immune responses during sepsis, as well as their clinical translational potential.
Conclusion
In this study, we identified 27 genes significantly associated with sepsis risk through Mendelian randomization analysis, including 24 protective and 3 risk-enhancing genes. Furthermore, we linked 13 druggable genes to known sepsis risk factors and predicted potential therapeutic agents based on these targets. Through qRT-PCR and Western blot validations in mouse models, we experimentally confirmed significant differential expression of representative genes, including BCL6, PTX3, IL7R, BTN3A2, and LGALS1, highlighting their potential mechanistic roles in sepsis pathophysiology. These integrated bioinformatic and experimental findings offer deeper insights into the molecular mechanisms underlying sepsis and provide valuable evidence supporting the future development of targeted therapeutic strategies.
Declarations
All authors declare that the results/data/figures in this manuscript have not been published, nor are they under consideration for publication elsewhere.
Data Sharing Statement
The datasets analyzed during the current study are available in public databases such as GEO (https://www.ncbi.nlm.nih.gov/geo/), DECODE database (https://www.decode.com/) and FinnGen research database (https://www.finngen.fi/en).
Ethics Approval and Consent to Participate
Database data were approved by the Institutional Review Board of Xianning Central Hospital (Approval No. 2024-B01); animal experiments were approved by the Animal Research Ethics Committee of Xianning Central Hospital (Approval No. 2024-A15).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation Project of Hubei Province (2022CFB551).
Disclosure
Wei Yin and Houyu Zhao are co-correspondence authors for this study. All authors declare that they have no competing interests in this work.
References
1. Hawiger J, Veach RA, Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost. 2015;13(10):1743–1756. doi:10.1111/jth.13061
2. Farrah K, McIntyre L, Doig CJ, et al. Sepsis-associated mortality, resource use, and healthcare costs: a propensity-matched cohort study. Crit Care Med. 2021;49(2):215–227. doi:10.1097/CCM.0000000000004777
3. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304–377. doi:10.1007/s00134-017-4683-6
4. Monard C, Abraham P, Schneider A, et al. New targets for extracorporeal blood purification therapies in sepsis. Blood Purif. 2023;52(1):1–7. doi:10.1159/000524973
5. Namba S, Konuma T, Wu K-H, et al. A practical guideline of genomics-driven drug discovery in the era of global biobank meta-analysis. Cell Genom. 2022;2(10):100190. doi:10.1016/j.xgen.2022.100190
6. Kreitmaier P, Katsoula G, Zeggini E. Insights from multi-omics integration in complex disease primary tissues. Trends Genet. 2023;39(1):46–58. doi:10.1016/j.tig.2022.08.005
7. Davey SG. Capitalizing on mendelian randomization to assess the effects of treatments. J R Soc Med. 2007;100(9):432–435. doi:10.1177/014107680710000923
8. Lin J, Zhou J, Xu Y. Potential drug targets for multiple sclerosis identified through mendelian randomization analysis. Brain. 2023;146(8):3364–3372. doi:10.1093/brain/awad070
9. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): explanation and elaboration. BMJ. 2021;375:n2233. doi:10.1136/bmj.n2233
10. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi:10.1093/biostatistics/kxj037
11. Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. doi:10.1093/bioinformatics/bts034
12. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
13. Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383). doi:10.1126/scitranslmed.aag1166
14. Bycroft C, Freeman C, Petkova D, et al. The UK biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. doi:10.1038/s41586-018-0579-z
15. Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. doi:10.1093/bioinformatics/btv313
16. Tong H, Zhao Y, Cui Y, et al. Multi-omic studies on the pathogenesis of sepsis. J Transl Med. 2025;23(1):361. doi:10.1186/s12967-025-06366-w
17. Hattori M, Yamazaki M, Ohashi W, et al. Critical role of endogenous histamine in promoting end-organ tissue injury in sepsis. Intensive Care Med Exp. 2016;4(1):36. doi:10.1186/s40635-016-0109-y
18. Xu N, Guo H, Li X, et al. A five-genes based diagnostic signature for sepsis-induced ARDS. Pathol Oncol Res. 2021;27:580801. doi:10.3389/pore.2021.580801
19. Ming T, Dong M, Song X, et al. Integrated analysis of gene co-expression network and prediction model indicates immune-related roles of the identified biomarkers in sepsis and sepsis-induced acute respiratory distress syndrome. Front Immunol. 2022;13:897390. doi:10.3389/fimmu.2022.897390
20. Thomas JM, Huuskes BM, Sobey CG, et al. The IL-18/IL-18R1 signalling axis: diagnostic and therapeutic potential in hypertension and chronic kidney disease. Pharmacol Ther. 2022;239:108191. doi:10.1016/j.pharmthera.2022.108191
21. Harrison OJ, Srinivasan N, Pott J, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) treg cell function in the intestine. Mucosal Immunol. 2015;8(6):1226–1236. doi:10.1038/mi.2015.13
22. Kim SW, Kim ES, Moon CM, et al. Genetic polymorphisms of IL-23R and IL-17A and novel insights into their associations with inflammatory bowel disease. Gut. 2011;60(11):1527–1536. doi:10.1136/gut.2011.238477
23. Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev. 2018;281(1):138–153. doi:10.1111/imr.12616
24. Cai P, Lu Z, Wu J, et al. BTN3A2 serves as a prognostic marker and favors immune infiltration in triple-negative breast cancer. J Cell Biochem. 2020;121(3):2643–2654. doi:10.1002/jcb.29485
25. Lin Y, Zhou H, Li S. BTN3A2 expression is connected with favorable prognosis and high infiltrating immune in lung adenocarcinoma. Front Genet. 2022;13:848476. doi:10.3389/fgene.2022.848476
26. Chen Q, Han B, Meng X, et al. Immunogenomic analysis reveals LGALS1 contributes to the immune heterogeneity and immunosuppression in glioma. Int J Cancer. 2019;145(2):517–530. doi:10.1002/ijc.32102
27. Qin H, Peng M, Cheng J, et al. A novel LGALS1-depended and immune-associated fatty acid metabolism risk model in acute myeloid leukemia stem cells. Cell Death Dis. 2024;15(7):482. doi:10.1038/s41419-024-06865-6
28. Ruvolo PP, Ma H, Ruvolo VR, et al. LGALS1 acts as a pro-survival molecule in AML. Biochim Biophys Acta Mol Cell Res. 2020;1867(10):118785. doi:10.1016/j.bbamcr.2020.118785
29. Huang C, Tong Q, Zhang W, et al. Association of early aspirin use with 90-day mortality in patients with sepsis: an PSM analysis of the MIMIC-IV database. Front Pharmacol. 2024;15:1475414. doi:10.3389/fphar.2024.1475414
30. Patra S, Praharaj PP, Klionsky DJ, et al. Vorinostat in autophagic cell death: a critical insight into autophagy-mediated, -associated and -dependent cell death for cancer prevention. Drug Discov Today. 2022;27(1):269–279. doi:10.1016/j.drudis.2021.08.004
31. Liu J, Zhao R, Jiang X, et al. Progress on the application of bortezomib and bortezomib-based nanoformulations. Biomolecules. 2021;12(1):51. doi:10.3390/biom12010051
32. Han SH, Kim JS, Woo JH, et al. The effect of bortezomib on expression of inflammatory cytokines and survival in a murine sepsis model induced by cecal ligation and puncture. Yonsei Med J. 2015;56(1):112–123. doi:10.3349/ymj.2015.56.1.112
33. Scully EP, Aga E, Tsibris A, et al. Impact of tamoxifen on vorinostat-induced human immunodeficiency virus expression in women on antiretroviral therapy: AIDS clinical trials group A5366, the MOXIE trial. Clin Infect Dis. 2022;75(8):1389–1396. doi:10.1093/cid/ciac136
34. Ma Y, Liu G, Tang M, et al. Epigallocatechin gallate can protect mice from acute stress induced by LPS while stabilizing gut microbes and serum metabolites levels. Front Immunol. 2021;12:640305. doi:10.3389/fimmu.2021.640305
35. Zhang HF, Zhang H-B, Wu X-P, et al. Fisetin alleviates sepsis-induced multiple organ dysfunction in mice via inhibiting p38 MAPK/MK2 signaling. Acta Pharmacol Sin. 2020;41(10):1348–1356. doi:10.1038/s41401-020-0462-y
36. Lei X, Teng W, Fan Y, et al. The protective effects of HIF-1alpha activation on sepsis induced intestinal mucosal barrier injury in rats model of sepsis. PLoS One. 2022;17(5):e0268445. doi:10.1371/journal.pone.0268445
37. Sen P, Gupta K, Kumari A, et al. Wnt/beta-catenin antagonist pyrvinium exerts cardioprotective effects in polymicrobial sepsis model by attenuating calcium dyshomeostasis and mitochondrial dysfunction. Cardiovasc Toxicol. 2021;21(7):517–532. doi:10.1007/s12012-021-09643-4
38. Crossland H, Constantin‐Teodosiu D, Gardiner SM, et al. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol. 2008;586(22):5589–5600. doi:10.1113/jphysiol.2008.160150
39. Wang Y, Chen M-Q, Dai L-F, et al. Fangji fuling decoction alleviates sepsis by blocking MAPK14/FOXO3A signaling pathway. Chin J Integr Med. 2024;30(3):230–242. doi:10.1007/s11655-023-3601-8
40. Pan T, Sun S, Chen Y, et al. Immune effects of PI3K/Akt/HIF-1alpha-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit Care. 2022;26(1):29. doi:10.1186/s13054-022-03893-6
41. Tian J, Li Y, Mao X, et al. Effects of the PI3K/Akt/HO-1 pathway on autophagy in a sepsis-induced acute lung injury mouse model. Int Immunopharmacol. 2023;124(Pt B):111063. doi:10.1016/j.intimp.2023.111063
42. Francois B, Jeannet R, Daix T, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. 2018;3(5). doi:10.1172/jci.insight.98960
43. Xiao M, Liu D, Xu Y, et al. Role of PFKFB3-driven glycolysis in sepsis. Ann Med. 2023;55(1):1278–1289. doi:10.1080/07853890.2023.2191217
44. Wang G, Jiang C, Fang J, et al. Pentraxin-3 as a predictive marker of mortality in sepsis: an updated systematic review and meta-analysis. Crit Care. 2022;26(1):167. doi:10.1186/s13054-022-04032-x
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identifying Potential Effective Diagnostic and Prognostic Biomarkers in Sepsis by Bioinformatics Analysis and Validation
Huang X, Tan J, Chen X, Zhao L
International Journal of General Medicine 2022, 15:6055-6071
Published Date: 6 July 2022
Analysis and Verification of Glycosylation Characteristics of Septic Acute Kidney Injury
Chen S, Gan H
Journal of Inflammation Research 2023, 16:5881-5897
Published Date: 5 December 2023
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024
Unveiling Novel Protein Biomarkers for Psoriasis Through Integrated Analysis of Human Plasma Proteomics and Mendelian Randomization
Mao R, Zhang T, Yang Z, Li J
Psoriasis: Targets and Therapy 2024, 14:179-193
Published Date: 7 December 2024
Exploring the Causal Relationship and Molecular Mechanisms Between Fasting Insulin and Androgenetic Alopecia: A Mendelian Randomization Study with Bioinformatics Analysis
Ding X, Bai Z, Wang W, Gu Y, Li Z, Fan Y
Clinical, Cosmetic and Investigational Dermatology 2025, 18:355-365
Published Date: 7 February 2025