Back to Journals » Journal of Inflammation Research » Volume 19
Hypoxia-Inducible Factors in Rheumatoid Arthritis: Central Pathogenic Roles and Therapeutic Opportunities
Authors Luo TT, He LX, Yin Q, Wang ZF, Zuo J ![]()
Received 14 February 2026
Accepted for publication 26 April 2026
Published 23 May 2026 Volume 2026:19 600186
DOI https://doi.org/10.2147/JIR.S600186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Peng Wang
Ting-Ting Luo,1,2,* Li-Xia He,1,* Qin Yin,1 Zhong-Fang Wang,1 Jian Zuo2,3
1Department of Pharmacy, The Second Affiliated Hospital of Wannan Medical College, Wuhu, People’s Republic of China; 2Xin’an Medical Research Center, The First Affiliated Hospital of Wannan Medical College (Yijishan Hospital), Wuhu, People’s Republic of China; 3Pharmacy Department, The First Affiliated Hospital of Wannan Medical College, Wuhu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Zuo, Email [email protected]
Abstract: Rheumatoid arthritis (RA) joints are characterized by a persistently hypoxic microenvironment that drives activation of hypoxia-inducible factors (HIFs). In RA synovium, both HIF-1α and HIF-2α accumulate across key cellular compartments (fibroblast-like synoviocytes, macrophages, endothelial cells, and chondrocytes) and correlate with disease activity, positioning HIF signaling as a central node linking hypoxic stress to inflammation, metabolic rewiring, angiogenesis, and joint destruction. Notably, HIF-1α primarily regulates inflammatory responses and metabolic adaptation, whereas HIF-2α plays a more prominent role in cartilage degradation and structural joint damage, highlighting isoform-specific functions in RA pathogenesis. This review summarizes current evidence on the role of HIF in RA, focusing on its regulatory effects on immune cell function, immunometabolism, angiogenesis, and cartilage and bone destruction. We also discuss emerging therapeutic strategies targeting HIF signaling, including small-molecule inhibitors, gene-silencing approaches, and natural products. Finally, we address translational challenges, particularly isoform selectivity, local drug delivery, and biomarker-guided patient stratification. This review aims to provide an integrated perspective on the pathogenic and therapeutic roles of HIF in RA and to highlight future directions for HIF-targeted therapies.
Keywords: rheumatoid arthritis, hypoxia-inducible factor, angiogenesis, immunometabolism, cartilage destruction, therapeutic targeting
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic, erosive arthritis, with a global prevalence of approximately 0.5%–1%.1 Recent epidemiological studies further indicate that RA continues to impose a substantial global health burden, contributing significantly to long-term disability, reduced quality of life, and healthcare utilization across different populations.2,3 It primarily targets synovial joints, leading to chronic inflammation of the synovium (synovitis), formation of pannus tissue rich in new blood vessels, and progressive destruction of articular cartilage and bone. Clinically, RA causes joint pain, swelling, and stiffness and can result in deformity and functional disability if inadequately controlled.4 Over the past decades, significant advances have been made in understanding RA immunopathogenesis–involving complex interactions among infiltrating immune cells (T cells, B cells, macrophages, neutrophils, etc.) and resident fibroblast-like synoviocytes (FLS)–which drive the production of pro-inflammatory cytokines (eg tumor necrosis factor-α, interleukin-1β) and proteases that mediate tissue damage.5,6 These insights have led to targeted therapies (such as cytokine inhibitors) that have improved outcomes for many patients.7 Nonetheless, a substantial subset of RA patients do not achieve sustained remission with current treatments, indicating the need for new therapeutic targets that address underexplored aspects of RA pathology. In this context, targeting HIF signaling is of particular interest because it addresses a relatively underexplored upstream mechanism that links hypoxia to inflammation, metabolic reprogramming, angiogenesis, and joint destruction in RA.
One distinctive and under-targeted feature of the RA joint microenvironment is hypoxia (an inadequate oxygen supply) relative to demand. The inflamed RA synovium is highly cellular and metabolically active, yet suffers from disordered and insufficient microvasculature, resulting in markedly low oxygen tension.8,9 As early as 1970, it was reported that oxygen partial pressure in RA synovial fluid is roughly half that of normal joints.10 In fact, thickened RA synovial tissues can exhibit oxygen levels as low as 1–4% O2 (10–30 mmHg), correlating inversely with local inflammation severity.11 Several factors contribute to this chronic hypoxia:12,13 (i) hyperplastic synovial lining and infiltrating leukocytes consume oxygen at an accelerated rate, outpacing the supply from immature new vessels; (ii) elevated intra-articular pressure from joint effusions compresses blood vessels and reduces perfusion; and (iii) vasoconstrictive mediators (eg. angiotensin-converting enzyme) are upregulated in RA, further diminishing blood flow. Thus, hypoxia is a fundamental pathophysiological element of RA joints – not merely a consequence of inflammation but also an active driver of disease processes. Hypoxic conditions in the synovium can perpetuate inflammation and aberrant angiogenesis in a vicious cycle: inflammation increases oxygen demand and disrupts blood flow, leading to hypoxia, which in turn triggers molecular pathways that exacerbate inflammation and sprout more dysfunctional vessels.
The key transcriptional regulators of cellular adaptation to low oxygen are the hypoxia-inducible factors (HIFs).14 HIFs are heterodimeric transcription factors composed of an oxygen-regulated HIF-α subunit (HIF-1α, HIF-2α, or HIF-3α) and a constitutively expressed HIF-β subunit (also known as ARNT). Under normoxia, HIF-α proteins are rapidly hydroxylated by prolyl hydroxylase domain enzymes (PHDs) and targeted for ubiquitination and proteasomal degradation via the von Hippel–Lindau (VHL) E3 ligase complex. Hypoxia inhibits the oxygen-dependent PHDs, allowing HIF-α to escape degradation, accumulate, and translocate into the nucleus where it dimerizes with HIF-β. The active HIF complex binds to hypoxia-response elements (HREs) in target gene promoters and recruits co-activators to drive transcription. HIF target genes number in the hundreds and collectively facilitate adaptive responses to hypoxia, including angiogenesis (eg. vascular endothelial growth factor, VEGF), metabolic shifts to anaerobic glycolysis, erythropoiesis, pH regulation, and cell survival pathways.15,16 In healthy physiology, HIF-mediated responses help maintain oxygen homeostasis during situations like vigorous exercise or high altitude.17 In pathological settings such as cancer and chronic inflammation, however, HIF signaling is often aberrantly activated, leading to profound downstream effects on disease progression.18
RA synovial tissue is now recognized as a quintessential example of a hypoxic inflammatory milieu with overactive HIF signaling. Studies have shown robust accumulation of both HIF-1α and HIF-2α proteins in RA synovium, detected in the nuclei of synovial lining cells (FLS and macrophage-like cells), endothelial cells, and chondrocytes.19–21 In contrast, normal synovium shows little or no HIF expression. Notably, the level of HIF-1α/2α in RA correlates with local disease activity, implying HIF activation is not just a marker of hypoxia but a driver of pathogenic mechanisms.22 For example, HIF downstream genes such as VEGF, glucose transporter-1 (GLUT1), and matrix metalloproteinases (MMPs) are highly expressed in RA joints, contributing to vascular proliferation, intense glycolytic activity, and tissue destruction, respectively.23 Functional studies further demonstrate HIF’s causal role: HIF-1α deletion or inhibition can attenuate experimental arthritis severity, whereas overexpression of HIF-2α in mouse joints leads to spontaneous arthritic damage.21,24,25 HIF signaling also interacts with other pivotal inflammatory pathways (such as NF-κB and Notch), compounding its impact on gene expression in RA lesions.21,26 Collectively, these findings position HIF as a critical node in the RA pathogenesis network–integrating hypoxic stress with immune and stromal cell dysfunction to drive chronic inflammation and joint damage.
Given the centrality of HIF in RA and the fact that current therapies do not directly address hypoxic adaptation, there is growing interest in therapeutically targeting the HIF pathway. Lessons from oncology have yielded various HIF inhibitors and related agents, some of which are now being repurposed or tested in preclinical RA models.27,28 The therapeutic strategies discussed in this review were selected because they target distinct levels of the HIF axis, including direct HIF inhibition, modulation of upstream regulatory mechanisms, and interference with downstream pathogenic effectors. Targeting HIF in RA is conceptually attractive because HIF controls multiple downstream processes – inflammation, angiogenesis, metabolism, and osteoarticular destruction – that are all aberrant in RA. A HIF-focused intervention could therefore target an upstream regulatory pathway that influences multiple pathogenic cascades in RA, potentially breaking the cycle of inflammation and hypoxia that sustains disease. However, because HIF pathways also play essential roles in normal physiology (eg. immunity against infections, wound healing, blood cell production), any systemic HIF inhibition carries risks that must be carefully managed.
In recent years, increasing attention has been paid to the potential role of hypoxia-related biomarkers in rheumatoid arthritis. Several studies have reported that synovial expression levels of HIF-1α and HIF-2α correlate with disease activity, inflammatory cytokine levels, and radiographic progression, suggesting that HIF signaling may serve not only as a pathogenic driver but also as a potential biomarker for disease severity and therapeutic response.29–31 In addition, circulating hypoxia-related markers, including VEGF, lactate, and glycolysis-related enzymes, have been proposed as indicators of synovial hypoxia and metabolic reprogramming in RA.32,33 These findings support the concept that hypoxia and HIF activation are closely associated with disease activity and may provide a basis for biomarker-guided precision therapy in RA.
In this review, we examine the multifaceted role of HIF in RA pathogenesis to highlight advances in understanding how HIF impacts immune cell function, synovial metabolism, pathological angiogenesis, and cartilage/bone homeostasis. We then discuss the emerging therapeutic strategies targeting HIF in RA, including small-molecule inhibitors, natural product derivatives, and gene therapy approaches, and evaluate their preclinical efficacy. Finally, we address the current challenges and future perspectives for translating HIF-targeted therapies into clinical use, as well as the potential for HIF-related biomarkers to guide personalized RA treatment. By synthesizing these insights, we aim to assess whether HIF can be leveraged as a viable new target to improve outcomes in RA, especially for patients who remain refractory to existing therapies.
The Regulatory Effect of HIF on Immune Cells
Chronic synovitis in RA is sustained by a complex interplay of innate and adaptive immune cells within the hypoxic joint environment.21,34 HIF-1α in particular is known to modulate the development, survival, and function of various immune cells, often promoting pro-inflammatory phenotypes. Meanwhile, HIF-2α may contribute to longer-term inflammatory responses in certain cell types.35,36 This section discusses how HIF influences major immune cell populations in RA (summarized schematically in Figure 1) and thereby amplifies the immune-mediated pathology.
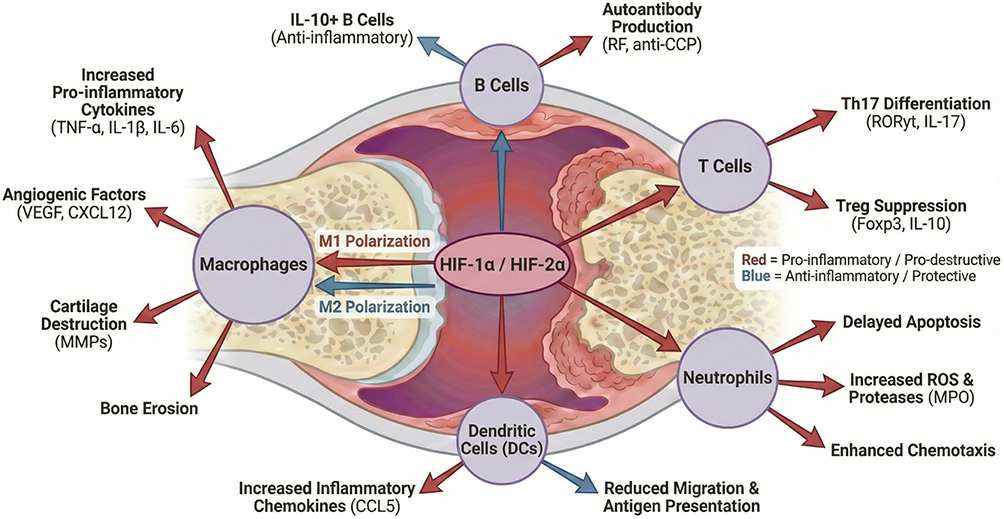 |
Figure 1 In the hypoxic rheumatoid synovium, stabilization and activation of HIF-1α and HIF-2α reprogram multiple immune cell compartments toward pathogenic responses. HIF signaling promotes macrophage M1 polarization and enhances the production of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) as well as angiogenic mediators (VEGF, CXCL12), contributing to synovitis, neovascularization, cartilage degradation (via MMPs), and bone erosion. In lymphocytes, HIF activity supports B-cell-mediated autoantibody production (RF, anti-CCP) and skews T-cell responses toward Th17 differentiation (RORγt, IL-17), while impairing regulatory T-cell (Treg) function (Foxp3, IL-10). HIF signaling also enhances neutrophil persistence and effector functions, including delayed apoptosis, increased reactive oxygen species (ROS) and protease activity (eg, MPO), and augmented chemotaxis. In dendritic cells, HIF-dependent programs regulate inflammatory chemokine production (eg, CCL5) and may reduce migration and antigen-presenting capacity. Red arrows indicate pro-inflammatory and tissue-destructive effects, whereas blue arrows represent anti-inflammatory or regulatory effects. Arrows denote functional regulation or association rather than direct causal interactions. The majority of mechanisms illustrated are supported by evidence from animal models and in vitro studies, while selected features—such as increased synovial HIF expression, angiogenesis, and cytokine production—have also been validated in human RA tissues. |
Macrophages
Macrophages are abundant in RA synovium and are key producers of pro-inflammatory cytokines (TNF, IL-1β, IL-6) and tissue-degrading enzymes (MMPs). HIF-1α is essential for the full inflammatory activity of macrophages. In landmark experiments, deletion of HIF-1α in myeloid cells (macrophages and neutrophils) dramatically reduced the severity of inflammatory diseases, including arthritis, in mice. Cramer et al showed that mice lacking HIF-1α in macrophages had attenuated joint inflammation in arthritis models, albeit at the cost of impaired bactericidal function of those macrophages.24 Consistently, RA patient synovium exhibits high HIF-1α expression in CD68+ macrophages in the lining layer, highlighting that these cells experience and respond to hypoxia in vivo.8 HIF-1α endows macrophages with several pathogenic advantages in RA. Under hypoxic conditions, HIF-1α drives macrophages to produce greater amounts of IL-1β, TNF-α, IL-6 and chemokines, amplifying local inflammation. At the same time, HIF-1 induces expression of VEGF, CXCL12 (SDF-1) and other angiogenic factors, promoting the vascularization of pannus.30 Through these secreted factors, HIF-1–activated macrophages both intensify synovial inflammation and support the expanding microcirculation that sustains the pannus. Indeed, stabilizing HIF-1α in macrophages (for instance, with an iron chelator) delays their apoptosis and boosts cytokine production. Conversely, silencing HIF-1α shifts macrophages toward a more “resolving” phenotype.30 Macrophage polarization is also strongly influenced by HIF-1α. Classically activated M1 macrophages (pro-inflammatory) rely predominantly on glycolytic metabolism and significantly upregulate HIF-1α in hypoxia, whereas alternatively activated M2 macrophages (anti-inflammatory/tissue-reparative) have lower HIF-1α levels and depend more on oxidative metabolism.37 HIF-1α is a known driver of the metabolic switch to glycolysis that characterizes M1 macrophages. Experimental ablation of HIF-1α causes macrophages to skew from an M1 toward an M2 phenotype, with reduced inflammatory cytokine production and increased expression of repair mediators.37 In hypoxic RA synovia, the activation of HIF-1α in macrophages likely reinforces the M1-polarized, disease-promoting state–these hypoxia-conditioned macrophages secrete abundant TNF-α, IL-1β and other factors that drive a positive feedback loop of synovitis.11,38 There is some evidence that HIF-2α in macrophages may also contribute, possibly sustaining chronic inflammatory functions of macrophages over longer term.39 Overall, HIF-1α serves as an important amplifier of acute inflammatory responses in RA macrophages, integrating with pathways like NF-κB. In fact, HIF-1α and NF-κB act cooperatively: under normoxia, PHD enzymes hydroxylate and inhibit IKK, stabilizing IκB and restraining NF-κB, but hypoxia relieves this block, enabling NF-κB activation alongside HIF-1α. The result is a vicious cycle of inflammation and hypoxia–cytokines fuel HIF activation and vice versa.40 In addition to hypoxia itself, inflammatory and infectious stimuli may further enhance HIF-1α activation in rheumatoid arthritis. Exogenous infectious agents, including bacterial and viral components, can activate Toll-like receptor (TLR) signaling pathways in immune cells such as macrophages and dendritic cells.41 TLR activation promotes NF-κB signaling, which increases HIF-1α transcription and stabilization even under non-severe hypoxic conditions.42 In the hypoxic synovial microenvironment, this synergistic interaction between infection-related inflammatory signaling and hypoxia may further amplify HIF-1α activity, leading to increased production of pro-inflammatory cytokines, angiogenic factors, and metabolic mediators.43 This mechanism may help explain why mucosal or systemic infections can trigger disease flares in patients with rheumatoid arthritis. Accordingly, targeting HIF-1α in macrophages may represent a rational strategy to disrupt this hypoxia-inflammation amplification loop and thereby attenuate synovial inflammation in RA.
T Cells
HIF profoundly affects RA T cell numbers, subsets, and functions. Hypoxic conditions favor T cell survival by stabilizing HIF-1α, which is relevant in the nutrient- and oxygen-poor synovium where effector T cells accumulate.44 In RA, low joint oxygen tension and downstream HIF signals allow activated T cells to survive and persist despite a hostile microenvironment.11,44 Furthermore, HIF-1α has been identified as a decisive factor in CD4+ T cell lineage differentiation. It tilts the balance between pro-inflammatory T helper 17 (Th17) cells and anti-inflammatory regulatory T cells (Treg).45 Under hypoxia, HIF-1α promotes Th17 development while inhibiting Treg formation. Mechanistically, HIF-1α acts as a metabolic sensor and transcriptional regulator: it enhances the expression of RORγt, the master transcription factor for Th17 cells, thereby increasing IL-17 production, and simultaneously it interferes with Foxp3, the lineage-defining factor of Tregs.45 In a seminal study, Dang et al demonstrated that hypoxia-induced HIF-1α drives naïve CD4+ T cells to differentiate into Th17 at the expense of Tregs.45 HIF-1α was found to physically interact with Foxp3, targeting it for proteasomal degradation, which counteracts any direct hypoxia-driven increase in Foxp3 expression. The net effect is a skewed Th17/Treg ratio favoring inflammation.46 In RA joints, Th17 cells are indeed abundant and produce IL-17 that exacerbates inflammation and bone erosion, whereas Treg numbers and function are insufficient to control the inflammation.47 The hypoxic synovial microenvironment likely contributes to this imbalance via HIF-1α.45 In T cells, HIF-1α-driven metabolic reprogramming, particularly enhanced glycolysis, favors Th17 cells differentiation and supports inflammatory effector programs.46 Th17 cells are highly glycolytic, and HIF-1α (downstream of mTOR signaling) ensures T cells preferentially use glycolysis to meet the demands of proliferation and cytokine production. Studies in RA models confirm HIF-1α’s role in T cell pathogenicity.46,48 For example, overexpression of HIF-1α in RA FLS was shown to augment the expansion of co-cultured Th1 and Th17 cells, suggesting that HIF-1α in synoviocytes can provide a microenvironment that boosts pro-inflammatory T cells. Additionally, inhibiting HIF-1α can reduce Th17 responses and/or bolster Treg responses.49 Taken together, HIF-1α activation is a major contributor to the loss of immune tolerance in RA, favoring Th17-driven inflammation. By inhibiting HIF-1α, it may be possible to partially restore the Th17/Treg balance and ameliorate the excessive immune activation in RA.
Neutrophils
Neutrophils are present in RA joints (particularly in synovial fluid) and are short-lived cells that normally undergo apoptosis to resolve inflammation. Hypoxia and HIF-1 prolong neutrophil longevity and activity in RA, which can exacerbate tissue damage. In vitro, hypoxia inhibits neutrophil apoptosis via a HIF-1α–dependent mechanism involving delayed caspase activation.50 Studies have shown that chemically stabilizing HIF-1α (using an iron chelator) or incubating neutrophils in low oxygen prevents their programmed cell death, partly by upregulating the NF-κB pathway and increasing secretion of macrophage inflammatory protein-1β (MIP-1β) – a chemokine that provides survival signals to neutrophils.44,50 High levels of reactive oxygen species and proteases from neutrophils were detected in the synovial fluid of rheumatoid arthritis (RA), which is related to their overactivation mediated by HIF-1α.30 In addition, myeloperoxidase and other substances released by neutrophils under hypoxic conditions can also cause tissue damage and exacerbate inflammation. It is worth mentioning that neutrophils also activate the HIF-1α pathway after taking up antigen-antibody complexes, suggesting that immune complex deposition may prolong neutrophil lifespan through the hypoxia/HIF mechanism and participate in the persistent inflammation of RA synovium.50 In summary, HIF-1α makes the neutrophil response in the acute inflammatory phase of RA more intense and persistent by delaying neutrophil apoptosis and enhancing their chemotaxis and pro-inflammatory functions. In the treatment of RA, drug intervention of HIF-1α or its downstream signaling may promote timely neutrophil apoptosis and accelerate inflammation resolution.
Dendritic Cells
Dendritic cells (DCs) are antigen-presenting cells found in the RA synovium that bridge innate and adaptive immunity. Hypoxia and HIF-1 influence DC differentiation and function in ways that can amplify RA inflammation.51 Notably, the transition of monocytes to dendritic cells is profoundly affected by low oxygen – with over 2000 genes showing altered expression when this process occurs under hypoxia.52,53 HIF-1α skews DCs towards a more inflammatory phenotype: under hypoxic conditions, DCs show enhanced production of inflammatory chemokines like CCL5, which can recruit neutrophils and other leukocytes, but they exhibit reduced migratory capacity and diminished antigen-presenting function.53,54 Mancino et al reported that hypoxia-exposed DCs produce higher levels of pro-inflammatory cytokines but have impaired ability to stimulate T cells due to decreased expression of co-stimulatory molecules and chemokine receptors needed for migration to lymph nodes.51 In RA, synovial DCs express high levels of the HIF-inducible triggering receptor TREM-1, indicating activation of hypoxic response pathways in these cells.55 The net effect of HIF on DCs in RA may be to keep them stationed in the joint, secreting cytokines that perpetuate local inflammation rather than efficiently maturing and exiting to induce immune tolerance. This could contribute to the sustained inflammatory milieu in RA synovium.
B Cells
B lymphocytes are central to RA pathogenesis through autoantibody production (eg, rheumatoid factor and anti-CCP) and antigen presentation.56 Direct effects of HIF on B cells in RA are less well characterized than for other immune cells, but emerging evidence indicates that HIF-1α is critical for a subset of immunoregulatory IL-10–producing B cells, also referred to as B10 cells.57 Meng et al reported that B cell–specific deletion of HIF-1α in mice markedly reduced the frequency of peripheral IL-10+ regulatory B cells, while increasing pro-inflammatory Th17 cells, thereby exacerbating collagen-induced arthritis (CIA).57 These findings suggest that, in B cells, HIF-1α promotes IL-10 production and constrains inflammation. Mechanistically, antigen receptor and Toll-like receptor (TLR) stimulation induces HIF-1α expression in B cells, and HIF-1α can directly enhance IL-10 gene transcription, thereby conferring an anti-inflammatory phenotype on a fraction of B cells.57 In patients with rheumatoid arthritis (RA), IL-10–producing B cells are thought to suppress autoimmune responses, and functional impairment of this population may aggravate disease.58 Although direct evidence validating the HIF–B cell axis in human RA remains limited, these animal studies collectively support a context-dependent, bidirectional role of HIF-1α in B-cell immunity—contributing to autoantibody-driven pathology while exerting anti-inflammatory effects within specific B-cell subsets.57,59 Further dissection of HIF signaling across distinct B-cell lineages may help elucidate previously underappreciated mechanisms underlying autoantibody generation and dysregulated immunoregulation in RA.59,60 In addition to regulatory B cells, hypoxia may also influence plasma cell differentiation and autoantibody production in RA.61 Germinal centers within secondary lymphoid organs and ectopic lymphoid structures in RA synovium are often characterized by hypoxic conditions, which may affect B-cell maturation and antibody affinity maturation.61 HIF-1α has been reported to regulate B-cell metabolism and survival during germinal center reactions, promoting glycolysis and supporting plasma cell differentiation. Hypoxia-driven HIF signaling may therefore contribute to the persistence of autoreactive plasma cells and sustained production of autoantibodies such as rheumatoid factor (RF) and anti-citrullinated protein antibodies (anti-CCP), which are key mediators of RA pathogenesis.62,63 Furthermore, hypoxia may influence class-switch recombination and antibody affinity maturation through metabolic reprogramming and altered cytokine signaling within germinal centers.61 Although direct evidence in RA remains limited, these findings suggest that hypoxia and HIF signaling may play a role in shaping the autoantibody response in RA, representing another potential mechanism linking hypoxia to autoimmune pathology.
Overall, HIFs—particularly HIF-1α—are broadly expressed across multiple immune cell types in RA and exert pleiotropic immunomodulatory functions. On the one hand, HIF-1α confers a survival and functional “advantage” to innate immune cells in hypoxic microenvironments by enhancing the viability and microbicidal activity of macrophages and neutrophils, and by promoting the production of pro-inflammatory mediators by dendritic cells (DCs) and B cells. On the other hand, HIF signaling disrupts the balance of adaptive immunity, amplifying pathogenic Th17 responses while weakening the anti-inflammatory functions of regulatory T cells (Tregs) and regulatory IL-10–producing B cells.35 Figure 1 (schematic) summarizes the HIF-1α–centered regulatory network across major immune compartments in RA: in macrophages, HIF-1α promotes M1-associated mediators and pro-angiogenic factors; in T cells, it drives Th17 differentiation and suppresses Treg programs; in B cells, it supports the maintenance of IL-10+ B-cell responses; and in neutrophils and DCs, it prolongs effector functions and inflammatory activity. Collectively, these actions shift synovial inflammation toward a self-sustaining, dysregulated state. Accordingly, the HIF pathway has emerged as a key entry point for understanding RA immunopathology and a promising target for therapeutic intervention.
The Influence of HIF on Energy Metabolism
Cellular metabolism is tightly linked to immune function, and in RA the hypoxic environment forces synovial cells to alter their metabolic pathways. RA has increasingly been recognized as a disease with disordered metabolism in inflamed joints – characterized by high rates of glycolysis (glucose breakdown to lactate) even in the presence of oxygen, akin to the “Warburg effect” seen in tumors.64,65 HIF-1α is a central mediator of this metabolic reprogramming, as it induces many glycolytic enzymes and transporters that enable cells to generate ATP under low oxygen conditions.66 In the RA synovium, evidence of accelerated glycolysis is clear: analyses of synovial fluid and tissue show increased activity of glycolytic enzymes, depletion of glucose, and accumulation of lactate, indicating a shift to anaerobic metabolism in the inflamed joint.67 For example, one study using high-field NMR spectroscopy found significantly elevated lactate levels in RA synovial fluid compared to normal joints, correlating with local hypoxia.68 Under hypoxic conditions, HIF-1α upregulates a broad array of metabolic genes, including glucose transporters (GLUT1/GLUT3) to boost glucose uptake, and key glycolytic enzymes such as hexokinase, phosphofructokinase, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and lactate dehydrogenase (LDH).69 This HIF-driven program enables synovial cells to maintain ATP production via glycolysis when mitochondrial oxidative phosphorylation is limited by oxygen scarcity.66 Indeed, RA synovial fibroblasts and macrophages rely heavily on glycolysis for energy, and inhibiting glycolytic flux has been shown to impair their aggressive behavior (eg. pro-inflammatory cytokine secretion and invasiveness).38,65
An important consequence of HIF-1 induced glycolysis in RA is the generation of metabolic byproducts that can further perpetuate autoimmunity and tissue damage. Notably, HIF-1α increases the expression of enzymes like glucose phosphate isomerase (GPI), α-enolase, and aldolase–which, beyond their metabolic roles, can act as autoantigens in RA.70,71 GPI and enolase are known targets of autoantibodies in RA, and their upregulation in the hypoxic joint may break tolerance and drive autoantibody production. For instance, α-enolase expression is elevated in RA synovial fibroblasts under hypoxia, and anti-citrullinated enolase antibodies have been detected in RA patients, linking metabolic stress to autoimmune response.72 Thus, HIF-1’s attempt to help cells adapt (by ramping up glycolysis) inadvertently creates neoantigens that stimulate RA’s autoimmune cycle. Additionally, the end-product of glycolysis, lactate, accumulates in RA joints and can acidify the microenvironment, potentially activating acid-sensing pathways that promote pain and cartilage degradation.73 Lactate has also been shown to stabilize HIF-1α further, by inhibiting prolyl hydroxylases, thereby reinforcing HIF activation in an autocrine manner.74 This feed-forward loop can sustain a high-glycolysis, high-inflammation state in the synovium.
Macrophages in RA illustrate a tight, HIF-1α-centered coupling between metabolism and inflammation. Synovial macrophages adopt a hypermetabolic state, with increased glucose uptake, enhanced glycolysis, and accumulation of lactate and succinate, alongside upregulation of glycolytic enzymes such as α-enolase. Succinate, a key immunometabolite, can stabilize HIF-1α and thereby promote IL-1β production, linking metabolic rewiring to inflammatory output.38,75 Consistently, RA patient serum drives healthy human macrophages toward aerobic glycolysis, elevates HIF-1α, and markedly increases IL-1β; this response correlates with HK2 and glycolytic activity and is attenuated by glycolysis inhibitors (2-DG or 3-bromopyruvate) or HIF-1α knockdown.38 The induced IL-1β strongly correlated with hexokinase-2 expression and glycolytic activity, and importantly, pharmacologic blockade of glycolysis (using 2-deoxyglucose or 3-bromopyruvate) or knockdown of HIF-1α drastically reduced IL-1β production in these macrophages.38 Moreover, inflammatory cytokines in RA serum, including IL-1β and TNF-α, further amplify HIF-1α and glycolytic gene expression, forming a feed-forward loop that sustains IL-1β production and chronic synovial inflammation.38
Synovial fibroblasts, too, are subject to HIF-1α’s metabolic control. RA fibroblast-like synoviocytes (RA-FLS) notoriously exhibit a shift toward glycolysis and an increase in mitochondrial dysfunction, supporting their aggressive growth and invasive behavior in the hypoxic joint.65 HIF-1α in RA-FLS promotes glycolytic enzyme expression and is required for their hypertrophic and invasive phenotype. Studies have shown that silencing HIF-1α or inhibiting its transcriptional activity in RA-FLS leads to reduced glycolysis, decreased production of VEGF and MMPs, and even triggers apoptosis in these cells, indicating they are “addicted” to HIF-driven metabolic pathways for survival.76–78 It is reported that HIF-1α knockdown in RA-FLS under normoxic conditions could mimic the effect of hypoxia resolution–the fibroblasts shifted away from glycolysis and underwent cell death, pointing to HIF-1α as a potential metabolic Achilles’ heel of the pannus.76 These findings provide a strong rationale for targeting metabolic pathways or HIF-1α itself as a means to dampen synovial expansion in RA.
The Influence of HIF on Angiogenesis
Pathologic angiogenesis is a well-established feature of RA, and HIF signaling is a primary trigger of the aberrant blood vessel formation in inflamed joints.79 Angiogenesis in RA is thought to be, at least in part, a response to synovial hypoxia: new blood vessels sprout to increase oxygen delivery, but they often form in a disorganized manner and fail to resolve the hypoxic stress, thereby perpetuating a cycle of neovascularization and inflammation.80 HIF-1α, as the oxygen sensor of cells, upregulates a host of angiogenic factors in RA, linking low oxygen tension to robust vascular proliferation in the pannus. The most prominent HIF target is vascular endothelial growth factor (VEGF), a potent endothelial cell mitogen and chemokine.81 RA patients have markedly elevated VEGF levels in synovial fluid and tissue, and VEGF correlates with disease activity and radiographic progression.82 Hypoxia-induced HIF-1α is a major driver of this VEGF overexpression: HIF-1α binds hypoxia-response elements in the VEGF gene promoter, leading to transcriptional upregulation of VEGF in RA synovial fibroblasts, macrophages, and T cells within the hypoxic joint.83,84 More broadly, synovial hypoxia is thought to amplify pathological angiogenesis via HIF-dependent programs (including VEGF), and the resulting immature neovasculature can promote ongoing leukocyte recruitment, sustaining synovitis and perpetuating the inflammatory–angiogenic cycle. In addition to VEGF, HIF-1α induces other pro-angiogenic mediators such as placental growth factor (PlGF), angiopoietin 2, and stromal cell–derived factor 1 (SDF-1/CXCL12), all of which have been implicated in RA angiogenesis.85,86 SDF-1 in particular is upregulated by hypoxia in synovial fibroblasts and promotes the trafficking of endothelial progenitors and leukocytes to the joint, contributing both to angiogenesis and inflammation.85 HIF-driven angiogenesis in RA creates a self-reinforcing loop. Newly formed vessels deliver additional oxygen and nutrients, which might transiently relieve hypoxia.87 Moreover, RA synovial microvessels are frequently dysfunctional: endothelial cells display a “leaky” inflammatory phenotype with increased permeability, and a substantial fraction of neovessels are immature and lack adequate periendothelial (pericyte/mural cell) coverage—features that undermine effective perfusion and leave regions of persistently low oxygen tension.80,88 The persistent hypoxia keeps HIF-1α active, which in turn generates more VEGF and pro-angiogenic signals, fueling further vessel growth. In parallel, inflammatory cytokines abundant in RA (eg, TNF-α, IL-6, IL-1β) crosstalk with hypoxia/HIF pathways to magnify angiogenesis.89,90 Mechanistically, TNF-α can promote HIF-1α accumulation through NF-κB–dependent signaling, providing a route by which inflammation strengthens HIF-driven transcriptional programs.91 In RA synovial fibroblasts, cytokines such as TNF and IL-1β can directly stimulate VEGF production, and hypoxia further augments this VEGF output—together linking inflammatory cues and low oxygen tension to heightened angiogenic drive.85 Similarly, Toll-like receptor (TLR)–driven innate activation can synergize with hypoxia/HIF-1α to boost VEGF and other inflammatory mediators in RA synovial fibroblasts, and TLR-driven macrophage cytokines combined with local hypoxia can activate synovial cells to secrete key angiogenic growth factors including VEGF and basic fibroblast growth factor (bFGF).92 The net effect is an “angiogenic switch” in RA tissue, shifting the balance toward sustained neovascularization. Histologic and immunohistochemical studies further support this integrated model: HIF-1α is prominently expressed in RA synovium (notably lining and sublining stromal compartments), and HIF/HIF-related angiogenic activation is linked with increased microvessel density and VEGF pathway activation.20 Importantly, synovial HIF-1α burden correlates with the degree of vascularity as well as inflammatory cell infiltration and synovitis severity, underscoring HIF’s central role at the intersection of angiogenesis and inflammation in RA.
Given the crucial contribution of angiogenesis to RA pathology, there has been interest in anti-angiogenic approaches to therapy. The involvement of HIF-1α in RA angiogenesis makes it an appealing target. In animal models, inhibiting HIF-1α (eg, intra-articular siRNA) reduces VEGF, improves synovial vessel structure, and alleviates arthritis severity.76 Likewise, blocking VEGF or its receptors suppresses synovial angiogenesis and inflammation;93 these approaches tend to work best when given early, while effects are weaker once disease is established, likely due to already-expanded vasculature and redundant pro-angiogenic pathways. Clinical trials of anti-angiogenic agents in RA remain limited and have shown only modest benefits.92 Nonetheless, the concept is supported indirectly by clinical experience: effective anti-inflammatory treatments such as TNF inhibitors can lower synovial VEGF and partially normalize vessel morphology in responders, consistent with reduced HIF-driven angiogenic stimuli helping to resolve synovitis.32,88
The Influence of HIF on Cartilage and Bone Destruction
The tissue destruction in RA is mainly manifested as erosion of articular cartilage and localized bone damage, ultimately leading to joint deformity and loss of function. Driven by inflammation and hypoxia, pannus tissue invades beneath the articular cartilage, where activated fibroblasts and osteoclasts act in concert at this interface to degrade both cartilage and bone.94,95 The HIF pathway also plays an important role in this destructive process, but its effects are dual and stage-specific.67
In articular cartilage, HIF-1α plays a protective role in chondrocyte survival and extracellular matrix homeostasis. Given the intrinsically hypoxic nature of articular cartilage, HIF-1α is essential for chondrocyte adaptation to low oxygen tension and for maintaining viability.96,97 Under physiological conditions, HIF-1α sustains the synthesis of key matrix components, including aggrecan and type II collagen, while restraining excessive chondrocyte hypertrophy and calcification. Accordingly, mice with HIF-1α deficiency display impaired cartilage development, underscoring its importance in maintaining cartilage integrity.97–99 In the inflammatory milieu of RA, however, the function of HIF-1α within cartilage may be suppressed or overridden by HIF-2α. HIF-2α (EPAS1) has been described as a “catabolic factor” in cartilage, and a substantial body of evidence indicates that HIF-2α upregulates multiple cartilage-degrading enzymes, such as MMP-13 and the aggrecanases ADAMTS-4/5, thereby promoting matrix breakdown.25,100 Transgenic chondrocyte-specific overexpression of HIF-2α has been shown to be sufficient to induce spontaneous arthritic changes, characterized by severe cartilage degradation and accompanying bone erosion. In line with this, in collagen-induced arthritis models, genetic deletion of HIF-2α significantly attenuates joint swelling and limits cartilage injury even when HIF-1α remains present, supporting an HIF-1α–independent and central role for HIF-2α in driving RA-associated cartilage destruction.21,25 Mechanistically, HIF-2α directly induces the expression of cartilage matrix–degrading enzymes (eg, MMP-1 and MMP-13) and inflammatory mediators (eg, IL-6) in chondrocytes and synovial fibroblasts, and it enhances RANKL expression to facilitate osteoclast activation.21,25 In addition, HIF-2α promotes chondrocyte production of chemokines (eg, CXCL1 and CCL20), which recruit synovial fibroblasts and inflammatory cells beneath the cartilage, thereby fostering an invasive destructive complex at the cartilage–synovium interface.101 Moreover, chemokines induced by HIF-2α have been shown to facilitate directed migration of fibroblast-like synoviocytes and chondrocytes toward the cartilage–bone junction, which in turn accelerates cartilage erosion.101 By contrast, HIF-1α appears to counteract cartilage catabolism. Evidence suggests that HIF-1α can suppress NF-κB–dependent induction of HIF-2α, thereby reducing MMP-13 production. Thus, HIF-1α partially antagonizes HIF-2α–driven cartilage destruction and functions as a key regulator of chondrocyte homeostasis.99 Nevertheless, under persistent inflammation in RA, HIF-1α activity may be chronically downregulated or insufficient, allowing HIF-2α signaling to predominate. Collectively, HIF-2α emerges as a major driver of cartilage matrix degradation and chondrocyte loss in RA, whereas HIF-1α is protective but may be functionally overwhelmed in pathological settings. Therefore, therapeutic inhibition of HIF-2α has been proposed as a promising strategy to preserve articular cartilage.25 Cartilage destruction in RA is also mediated by multiple degradative enzymes and inflammatory factors, many of which are influenced by HIF signaling. Synovial fibroblasts and chondrocytes produce key matrix metalloproteinases (MMP-1, MMP-3, and MMP-13) that degrade collagen and proteoglycans and thereby directly drive cartilage breakdown. Hypoxia can amplify MMP expression through cooperative activation of HIF-1α and NF-κB;78,102 for example, IL-1β induces MMP-1 and MMP-13 in chondrocytes under normoxia, and hypoxia further enhances this response. Similarly, under hypoxia, IL-17 and TNF-α synergistically promote FLS release of MMP-2 and MMP-9, strengthening their invasive phenotype, whereas HIF-1α inhibition attenuates these effects.103,104 Furthermore, Hypoxia/HIF signaling also modulates chondrocyte survival programs. HIF-1α can induce autophagy-related genes and improve tolerance to nutrient deprivation and low oxygen, potentially delaying cell death.97,105,106 However, in RA, oxidative stress and inflammatory mediators may override these protective effects, leading to increased apoptosis and abnormal differentiation.107 In addition, excessive HIF-2α activity is associated with chondrocyte hypertrophy and cartilage calcification/ossification, including type X collagen induction and osteophyte-related changes, which may also emerge in end-stage RA.108
With respect to bone destruction, HIF signaling primarily promotes osteoclastogenesis and osteoclast activity. In RA, pathological bone erosion is largely driven by excessive osteoclast differentiation downstream of the RANKL/RANK axis. Both HIF-1α and HIF-2α can increase RANKL expression in synovial fibroblasts and T cells, thereby indirectly facilitating osteoclast formation. In addition, osteoclasts exhibit enhanced functional activity under hypoxic conditions, which is closely linked to HIF activation.21,109,110 In RA synovial tissue, a substantial proportion of osteoclasts show nuclear HIF-1α expression, indicating a highly HIF-activated state compared with osteoclasts in osteoarthritis or normal synovium. These HIF-1α–positive osteoclasts robustly express the HIF target gene ANGPTL4, whereas ANGPTL4 expression is minimal in osteoclasts from non-RA synovial tissues.111 ANGPTL4 has been shown to enhance osteoclast-mediated bone resorption, and neutralizing ANGPTL4 can attenuate bone erosion in experimental arthritis.112,113 Thus, hypoxia-driven HIF-1α may promote osteoclast-derived pro-resorptive mediators such as ANGPTL4, amplifying osteoclast activity and creating a positive feedback loop within the inflamed joint.
Beyond transcriptional effects, HIF-1α also regulates metabolic programming during osteoclast differentiation. Because osteoclastogenesis is highly energy-demanding, hypoxia-induced HIF-1α shifts osteoclast precursors toward glycolytic metabolism to support proliferation and functional maturation.114 Hypoxia can further enhance osteoclast resistance to apoptosis, allowing these cells to persist longer in inflamed joints and thereby worsen bone destruction.115 Although the role of HIF-2α in osteoclasts is less well defined, its capacity to promote RANKL expression suggests an overall pro-osteoclastogenic effect. Consistently, HIF-2α deficiency in RA models is associated with markedly reduced bone erosion, partly due to decreased osteoclast formation.21 Overall, HIF signaling drives RA-associated bone destruction by reinforcing RANKL/OPG imbalance and by activating osteoclast metabolic and effector programs, ultimately intensifying pathological bone resorption.
Therapeutic Strategies Targeting HIF
The central involvement of HIF in RA pathogenesis makes it an enticing therapeutic target. By modulating HIF activity, one could theoretically hit multiple disease pathways at once-inflammation, angiogenesis, and tissue destruction – offering a disease-modifying approach for refractory RA. Over the past decade, researchers have explored various strategies to interfere with HIF signaling in RA, drawing from advances in cancer and ischemia therapies where HIF is also pivotal.67,116 These strategies include direct HIF inhibitors, gene therapy approaches, hypoxia-activated prodrugs, and targeting downstream effectors of HIF. While most are in preclinical or early development stages, they collectively highlight the promise and challenges of anti-HIF therapy in RA. (Table 1)
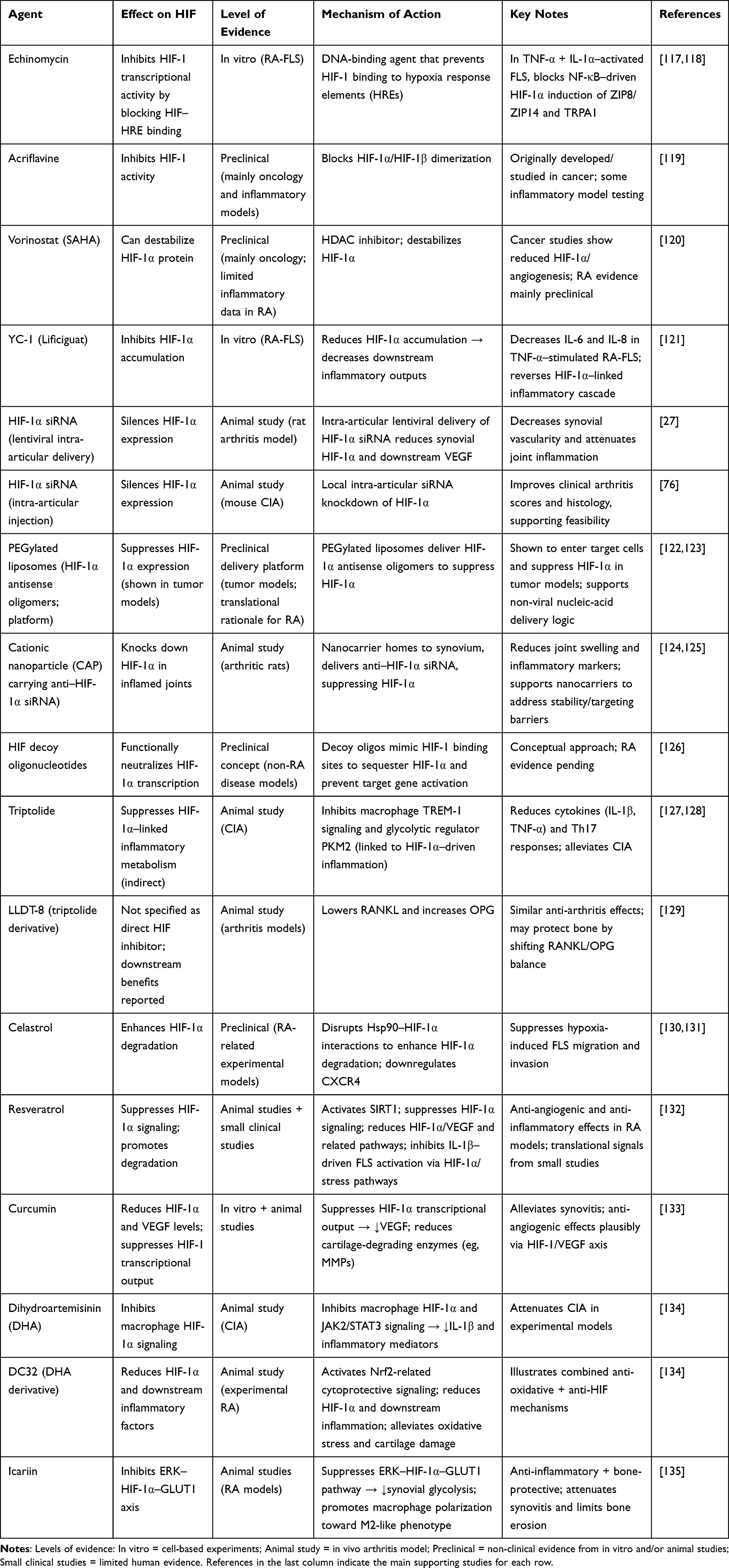 |
Table 1 Therapeutic Candidates and Delivery Strategies Targeting the HIF Pathway in Rheumatoid Arthritis |
Small-Molecule Inhibitors
A number of small molecules have been identified that inhibit HIF-1α transcriptional activity or stability. These were largely first studied in oncology, but some have been tested in inflammatory models. Examples include echinomycin (a DNA-binding agent that prevents HIF-1 binding to hypoxia response elements), acriflavine (which blocks HIF-1α/HIF-1β dimerization) and vorinostat (SAHA) (a histone deacetylase inhibitor that can destabilize HIF-1α protein).117,119,120 In cancer models, such agents showed efficacy in reducing HIF-1α levels and angiogenesis, and some progressed to clinical trials with mixed results. In the context of RA, these inhibitors have so far been tested mainly in vitro or in animal studies. For instance, the compound YC-1 (lificiguat), which inhibits HIF-1α accumulation, was used on RA synovial fibroblasts and found to decrease their production of IL-6 and IL-8, especially when they were stimulated with TNF-α – essentially reversing the HIF-1α–mediated inflammatory cascade in these cells.121 Similarly, when FLSs were activated with TNF-α and IL-1α, NF-κB–driven HIF-1α signaling promoted the expression of ZIP8/ZIP14 and TRPA1, whereas echinomycin pretreatment prevented these increases, supporting a direct role for HIF-1–dependent transcriptional programs in synoviocyte activation.118 However, a key limitation of these agents is limited specificity, as many do not clearly discriminate between HIF-1α and HIF-2α and may exert substantial off-target effects.136,137 In addition, systemic inhibition of HIF signaling can be difficult to tolerate because HIF pathways are essential for normal physiology, including hematopoiesis and wound repair.138,139 Consistent with this, clinical studies of HIF-1–targeting compounds have reported dose-dependent toxicities such as cytopenias, elevations in hepatic transaminases, and gastrointestinal adverse events.140 This emphasizes that while pan-HIF blockade can ameliorate arthritis in experimental settings, careful calibration or targeting will be required for human use.
Gene Silencing and Molecular Therapy
An alternative to small molecules is to directly silence HIF-1α gene expression in affected tissues. In experimental arthritis, HIF-1α–targeted siRNA has produced encouraging therapeutic benefits. For example, intra-articular delivery of HIF-1α siRNA via a lentiviral vector reduced synovial HIF-1α expression and downstream VEGF levels, leading to decreased synovial vascularity and attenuated joint inflammation in a rat arthritis model.27 Similarly, intra-articular injection of HIF-1α siRNA in mice with CIA improved clinical arthritis scores and histology, confirming the feasibility of this approach.76 Viral vectors can achieve efficient gene transfer, but concerns regarding immunogenicity and insertional mutagenesis have constrained their clinical translation in RA.141 Consequently, interest has increasingly shifted toward non-viral delivery platforms for HIF-1α siRNA or antisense oligonucleotides. Nanoparticle-based systems are particularly attractive; for instance, PEGylated liposomes loaded with HIF-1α antisense oligomers have been shown to enter target cells and suppress HIF-1α expression in tumor models, thereby improving the efficacy of co-administered therapeutics.122,123 In RA, a recent study formulated a cationic nanoparticle (CAP) carrying an anti–HIF-1α siRNA specifically designed to target inflamed joints-when administered systemically in arthritic rats, these nanoparticles homed to the synovium, knocked down HIF-1α, and resulted in decreased joint swelling and inflammatory markers.124 This innovative approach suggests that nanocarriers can overcome the stability and targeting issues associated with siRNA, making HIF silencing a more practical strategy.125 Another molecular approach is to use decoy oligonucleotides that mimic HIF-1 binding sites, sequestering HIF-1α and preventing it from activating gene transcription. Such decoys have been tested in other diseases (eg, a HIF decoy was tried in a cancer trial), but not yet reported in RA specifically.126
Natural Products and Bioactive Compounds from Traditional Medicines
Triptolide shows strong anti-inflammatory effects in RA models and can alleviate CIA by reducing cytokines (eg, IL-1β, TNF-α) and Th17 responses, partly through inhibition of macrophage TREM-1 signaling and the glycolytic regulator PKM2, which is linked to HIF-1α–driven inflammation.127,128 However, because triptolide is limited by toxicity, the derivative LLDT-8 provides similar anti-arthritis effects in animals and may protect bone by lowering RANKL and increasing OPG.129 Celastrol has broad anti-inflammatory actions and can enhance HIF-1α degradation by disrupting Hsp90–HIF-1α interactions. It also suppresses hypoxia-induced FLS migration and invasion, in part by downregulating CXCR4.130,131 Resveratrol, a grape-skin polyphenol, has antioxidant and anti-inflammatory effects in RA models and small clinical studies. It may act by activating SIRT1 and suppressing HIF-1α signaling, thereby reducing HIF-1α activity and promoting its degradation. In RA rats, resveratrol lowers synovial HIF-1α/VEGF and related pathways (eg, STAT3), with reduced angiogenesis and inflammatory cell infiltration. It also inhibits IL-1β–driven FLS activation by downregulating HIF-1α and stress pathways such as MAPK/c-JUN.132 Poor bioavailability remains a major limitation, prompting efforts to develop analogs and improved delivery systems. Curcumin can reduce HIF-1α and VEGF levels by suppressing HIF-1α transcriptional output in vitro. In RA models, it alleviates synovitis and reduces cartilage-degrading enzymes (eg, MMPs), with anti-angiogenic effects plausibly linked to inhibition of the HIF-1/VEGF axis.133 Dihydroartemisinin (DHA) has been reported to attenuate CIA by inhibiting macrophage HIF-1α and JAK2/STAT3 signaling, thereby reducing IL-1β and other inflammatory mediators. A derivative (DC32) was further shown to activate Nrf2-related cytoprotective signaling, reduce HIF-1α and downstream inflammatory factors, and alleviate synovial oxidative stress and cartilage damage in experimental RA.134 Icariin, a flavonoid, shows both bone-protective and anti-inflammatory effects in RA models. Mechanistically, it is associated with inhibition of the ERK–HIF-1α–GLUT1 axis, leading to reduced synovial glycolysis, and it also promotes macrophage polarization toward an M2-like phenotype. Together, these actions help attenuate synovitis and limit bone erosion.135 Overall, many natural products converge on HIF-associated pathways while simultaneously modulating oxidative stress, immunity, and metabolism, which may be advantageous in a multifactorial disease such as RA. Future work should clarify their direct molecular targets within the HIF network (eg, specific HIF-1α interaction sites and upstream regulators such as PHD enzymes) to guide rational optimization and translation.
Conclusion
The hypoxic microenvironment in rheumatoid arthritis (RA) joints plays a pivotal role in disease progression by activating the hypoxia-inducible factor (HIF) pathway. Acting as a central regulator, HIF integrates and amplifies key pathological processes, including inflammation, metabolic dysregulation, and angiogenesis. Elevated expression of HIF-1α and HIF-2α enhances the production of proinflammatory cytokines and angiogenic factors from immune effector cells, aggravating synovitis and pannus formation. Concurrently, HIF-driven metabolic reprogramming in synovial cells leads to excess lactate and autoantigen production, exacerbating autoimmunity. HIF-2α also directly contributes to cartilage and bone destruction by inducing catabolic factors like MMPs and RANKL. As such, HIF serves as a pathological “hub”, perpetuating a positive feedback loop in RA.
Although current biologic therapies targeting TNF or IL-6 have improved RA outcomes, a significant subset of patients remains unresponsive. Targeting the HIF pathway offers a promising multipronged strategy, given its upstream integration of hypoxia, oxidative stress, and inflammation, and its downstream impact on metabolism, immunity, angiogenesis, and tissue destruction. However, the challenge lies in minimizing systemic side effects. Future strategies should aim at selective inhibition—such as HIF-2α-specific small molecules to protect joint tissues while preserving HIF-1α functions—or localized delivery systems to concentrate drugs within synovial tissue. Moreover, hypoxia/HIF biomarkers could guide treatment timing and monitor therapeutic response, especially when integrated with imaging and metabolomics. Combination approaches with conventional DMARDs may also help disrupt the inflammation–hypoxia vicious cycle more effectively.
In conclusion, HIF signaling serves as a central link between hypoxia and multiple pathogenic processes in RA, including immune dysregulation, metabolic reprogramming, angiogenesis, and cartilage and bone destruction. Among the HIF isoforms, HIF-1α and HIF-2α appear to play distinct but complementary roles in driving synovial inflammation and structural damage. These findings support the concept that targeting HIF signaling may represent a promising therapeutic strategy in RA. However, future studies should focus on improving isoform selectivity, optimizing local delivery approaches, validating hypoxia-related biomarkers, and determining whether HIF-targeted strategies can be effectively integrated with conventional DMARD-based therapies. Compared with HIF-1α and HIF-2α, the role of HIF-3α in rheumatoid arthritis remains largely unexplored, and further studies are needed to determine whether it exerts distinct regulatory or protective functions in the hypoxic synovial microenvironment.
Abbreviations
ASIC1a, acid-sensing ion channel 1a; CAP, cationic nanoparticle; CIA, collagen-induced arthritis; CXCR4, C–X–C chemokine receptor 4; DHA, dihydroartemisinin; ERK, extracellular signal–regulated kinase; FLS, fibroblast-like synoviocytes; GLUT1, glucose transporter 1; HDAC, histone deacetylase; HIF, hypoxia-inducible factor; HIF-1α/HIF-1β/HIF-2α, hypoxia-inducible factor-1 alpha/1 beta/2 alpha; HRE, hypoxia response element; Hsp90, heat shock protein 90; IL, interleukin; JAK2, Janus kinase 2; MMPs, matrix metalloproteinases; NF-κB, nuclear factor kappa B; Nrf2, nuclear factor erythroid 2–related factor 2; OPG, osteoprotegerin; PEG, polyethylene glycol; PKM2, pyruvate kinase M2; RA, rheumatoid arthritis; RA-FLS, rheumatoid arthritis fibroblast-like synoviocytes; RANKL, receptor activator of nuclear factor κB ligand; RIPK3, receptor-interacting protein kinase 3; SAHA, suberoylanilide hydroxamic acid (vorinostat); siRNA, small interfering RNA; SIRT1, sirtuin 1; STAT3, signal transducer and activator of transcription 3; TNF-α, tumor necrosis factor alpha; TRPA1, transient receptor potential ankyrin 1; TREM-1, triggering receptor expressed on myeloid cells 1; VEGF, vascular endothelial growth factor; ZIP8/ZIP14, Zrt/Irt-like proteins 8/14.
Data Sharing Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.
Author Contributions
Ting-Ting Luo: Conceptualization, Investigation, Writing – original draft, Visualization.
Li-Xia He: Investigation, Data curation, Writing – review and editing.
Qin Yin: Data curation, Project administration, Software, Writing – review and editing.
Zhong-Fang Wang: Investigation, Writing – review and editing.
Jian Zuo: Conceptualization, Supervision, Project administration, Writing – review and editing.
All authors made a significant contribution to the work reported, whether in the conception, study design, literature review, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Key Project of Natural Science Foundation of Universities in Anhui Province (2023AH051739), Wannan Medical College Youth Research Fund (WK2024ZQNZ79), the Quality Engineering Project of Wannan Medical College (2025jyxm096), the Horizontal Research Project of Wannan Medical College (HXKT2022010), and the “Three New” Project of the Second Affiliated Hospital of Wannan Medical College (Y25017).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–19. doi:10.1016/S0140-6736(16)30173-8
2. Ma Y, Chen H, Lv W, et al. Global, regional and national burden of rheumatoid arthritis from 1990 to 2021, with projections of incidence to 2050: a systematic and comprehensive analysis of the global burden of disease study 2021. Biomarker Res. 2025;13(1):47. doi:10.1186/s40364-025-00760-8
3. Song Y, Chen Y, Wen L, et al. Health-related quality of life profiles in patients with rheumatoid arthritis: a latent profile analysis. Front Public Health. 2024;12:1478376. doi:10.3389/fpubh.2024.1478376
4. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16(6):316–333.
5. Firestein GS, McInnes IB. Immunopathogenesis of Rheumatoid Arthritis. Immunity. 2017;46(2):183–196. doi:10.1016/j.immuni.2017.02.006
6. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol. 2021;22(1):10–18.
7. Liu S, Ma H, Zhang H, et al. Recent advances on signaling pathways and their inhibitors in rheumatoid arthritis. Clin Immunol. 2021;230:108793. doi:10.1016/j.clim.2021.108793
8. Konisti S, Kiriakidis S, Paleolog EM. Hypoxia--a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol. 2012;8(3):153–162. doi:10.1038/nrrheum.2011.205
9. McGarry T, Biniecka M, Veale DJ, et al. Hypoxia, oxidative stress and inflammation. Free Radic Biol Med. 2018;125:15–24. doi:10.1016/j.freeradbiomed.2018.03.042
10. Lund‐Olesen K. Oxygen tension in synovial fluids. Arthritis Rheum. 1970;13(6):769–776. doi:10.1002/art.1780130606
11. Ng C, Biniecka M, Kennedy A, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheumatic Dis. 2010;69(7):1389–1395. doi:10.1136/ard.2009.119776
12. Fearon U, Hanlon MM, Floudas A, et al. Cellular metabolic adaptations in rheumatoid arthritis and their therapeutic implications. Nat Rev Rheumatol. 2022;18(7):398–414. doi:10.1038/s41584-022-00771-x
13. Wang X, Garrabou G, Barcos T, et al. The role of reactive oxygen species in the rheumatoid arthritis-associated synovial microenvironment. Antioxidants. 2022;11(6):1129. doi:10.3390/antiox11061129
14. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270(3):1230–1237. doi:10.1074/jbc.270.3.1230
15. Taylor CT, Scholz CC. The effect of HIF on metabolism and immunity. Nat Rev Nephrol. 2022;18(9):573–587. doi:10.1038/s41581-022-00587-8
16. Cowman SJ, Koh MY. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer. 2022;8(1):28–42. doi:10.1016/j.trecan.2021.10.004
17. Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semi Cancer Biol. 2009;19(1):12–16. doi:10.1016/j.semcancer.2008.11.009
18. Maxwell PH. The HIF pathway in cancer. Semin Cell Dev Biol. 2005;16(4–5):523–530. doi:10.1016/j.semcdb.2005.03.001
19. Guo X, Chen G. Hypoxia-inducible factor is critical for pathogenesis and regulation of immune cell functions in rheumatoid arthritis. Front Immunol. 2020;11:1668. doi:10.3389/fimmu.2020.01668
20. Giatromanolaki A, Sivridis E, Maltezos E, et al. Upregulated hypoxia inducible factor-1α and-2α pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2003;5(4):R193. doi:10.1186/ar756
21. Ryu J-H, Chae C-S, Kwak J-S, et al. Hypoxia-inducible factor-2α is an essential catabolic regulator of inflammatory rheumatoid arthritis. PLoS Biol. 2014;12(6):e1001881. doi:10.1371/journal.pbio.1001881
22. Hanlon MM, Canavan M, Barker BE, et al. Metabolites as drivers and targets in rheumatoid arthritis. Clin Exp Immunol. 2022;208(2):167–180. doi:10.1093/cei/uxab021
23. Horváth E, Sólyom Á, Székely J, Nagy EE, Popoviciu H. Inflammatory and metabolic signaling interfaces of the hypertrophic and senescent chondrocyte phenotypes associated with osteoarthritis. Int J Mol Sci. 2023;24(22):16468.
24. Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi:10.1016/S0092-8674(03)00154-5
25. Yang S, Kim J, Ryu J-H, et al. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16(6):687–693. doi:10.1038/nm.2153
26. Chen J, Cheng W, Li J, et al. Notch‐1 and Notch‐3 mediate hypoxia‐induced activation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheumatol. 2021;73(10):1810–1819. doi:10.1002/art.41748
27. Hu Y, Zhang T, Chen J, et al. Downregulation of hypoxia-inducible factor-1α by RNA interference alleviates the development of collagen-induced arthritis in rats. Mol Ther Nucleic Acids. 2020;19:1330–1342.
28. Hua S, Dias TH. Hypoxia-inducible factor (HIF) as a target for novel therapies in rheumatoid arthritis. Front Pharmacol. 2016;7:184.
29. Brouwer E, Gouw AS, Posthumus MD, et al. Hypoxia inducible factor-1-alpha (HIF-1 alpha) is related to both angiogenesis and inflammation in rheumatoid arthritis. Clin Experim Rheumatol. 2009;27(6):945–951.
30. Hu F, Shi L, Mu R, et al. Hypoxia-inducible factor-1α and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PLoS One. 2013;8(8):e72650. doi:10.1371/journal.pone.0072650
31. Vordenbäumen S, Sewerin P, Lögters T, et al. Inflammation and vascularisation markers of arthroscopically-guided finger joint synovial biospies reflect global disease activity in rheumatoid arthritis. Clin Experim Rheumatol. 2014;32(1):117–120.
32. Taylor PC. VEGF and imaging of vessels in rheumatoid arthritis. Arthritis Res Ther. 2002;4(Suppl 3):S99. doi:10.1186/ar582
33. Zhou K-L, Zhu Z-H, Zhou J-P, et al. Increased hexokinase-2 as a novel biomarker for the diagnosis and correlating with disease severity in rheumatoid arthritis. Medicine. 2021;100(25):e26504. doi:10.1097/MD.0000000000026504
34. Muz B, Khan MN, Kiriakidis S, et al. Hypoxia. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res Therapy. 2009;11(1):201. doi:10.1186/ar2568
35. McGettrick AF, O’Neill LA. The role of HIF in immunity and inflammation. Cell Metab. 2020;32(4):524–536.
36. Imtiyaz HZ, Williams EP, Hickey MM, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120(8):2699–2714.
37. Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37(9):364–372. doi:10.1016/j.tibs.2012.06.004
38. Jia Y, Li R, Huang L, et al. The Glycolysis-HIF-1α axis induces IL-1β of macrophages in rheumatoid arthritis. Arthritis Res Ther. 2025;27(1):180. doi:10.1186/s13075-025-03647-z
39. Jeelani I, Moon J-S, da Cunha FF, et al. HIF-2α drives hepatic Kupffer cell death and proinflammatory recruited macrophage activation in nonalcoholic steatohepatitis. Sci Trans Med. 2024;16(764):eadi0284. doi:10.1126/scitranslmed.adi0284
40. Korbecki J, Simińska D, Gąssowska-Dobrowolska M, et al. Chronic and cycling hypoxia: drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: a review of the molecular mechanisms. Int J Mol Sci. 2021;22(19):10701.
41. Jiménez-Dalmaroni MJ, Gerswhin ME, Adamopoulos IE. The critical role of toll-like receptors—from microbial recognition to autoimmunity: a comprehensive review. Autoimmunity Rev. 2016;15(1):1–8. doi:10.1016/j.autrev.2015.08.009
42. Rius J, Guma M, Schachtrup C, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008;453(7196):807–811. doi:10.1038/nature06905
43. Hu F, Mu R, Zhu J, et al. Hypoxia and hypoxia-inducible factor-1α provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheumatic Dis. 2014;73(5):928–936. doi:10.1136/annrheumdis-2012-202444
44. Makino Y, Nakamura H, Ikeda E, et al. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol. 2003;171(12):6534–6540. doi:10.4049/jimmunol.171.12.6534
45. Dang EV, Barbi J, Yang H-Y, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–784. doi:10.1016/j.cell.2011.07.033
46. Shi LZ, Wang R, Huang G, et al. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–1376.
47. Niu Q, Cai B, Huang ZC, Shi YY, Wang LL. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int. 2012;32(9):2731–2736.
48. Zhang Q, Wang L, Jiang J, et al. Critical role of AdipoR1 in regulating Th17 cell differentiation through modulation of HIF-1α-dependent glycolysis. Front Immunol. 2020;11:2040. doi:10.3389/fimmu.2020.02040
49. Hu F, Liu H, Xu L, et al. Hypoxia‐inducible factor‐1α perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. European J Immunol. 2016;46(3):742–751. doi:10.1002/eji.201545784
50. Walmsley SR, Print C, Farahi N, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J Exp Med. 2005;201(1):105–115. doi:10.1084/jem.20040624
51. Mancino A, Schioppa T, Larghi P, et al. Divergent effects of hypoxia on dendritic cell functions. Blood J Am Soc Hematol. 2008;112(9):3723–3734.
52. Bosco MC, Varesio L. Dendritic cell reprogramming by the hypoxic environment. Immunobiology. 2012;217(12):1241–1249. doi:10.1016/j.imbio.2012.07.023
53. Ricciardi A, Elia AR, Cappello P, et al. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res. 2008;6(2):175–185. doi:10.1158/1541-7786.MCR-07-0391
54. Blengio F, Raggi F, Pierobon D, et al. The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology. 2013;218(1):76–89. doi:10.1016/j.imbio.2012.02.002
55. Canavan M, Walsh AM, Bhargava V, et al. Enriched Cd141+ DCs in the joint are transcriptionally distinct, activated, and contribute to joint pathogenesis. JCI Insight. 2018;3(23):e95228. doi:10.1172/jci.insight.95228
56. Toes RE. The autoantibody response in rheumatoid arthritis: what makes it unique? Semin Arthritis Rheumatism. 2025;65:152356.
57. Meng X, Grötsch B, Luo Y, et al. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun. 2018;9(1):251. doi:10.1038/s41467-017-02683-x
58. Daien CI, Gailhac S, Mura T, et al. Regulatory B10 cells are decreased in patients with rheumatoid arthritis and are inversely correlated with disease activity. Arthritis Rheumatol. 2014;66(8):2037–2046. doi:10.1002/art.38666
59. Li L, Feng C, Qin J, et al. Regulation of humoral immune response by HIF-1α-dependent metabolic reprogramming of the germinal center reaction. Cell Immunol. 2021;367:104409. doi:10.1016/j.cellimm.2021.104409
60. Zhang J, Wu X, Ma J, Long K, Sun J, Li M, Ge L. Hypoxia and hypoxia-inducible factor signals regulate the development, metabolism, and function of B cells. Front Immunol. 2022;13:967576.
61. Abbott RK, Thayer M, Labuda J, et al. Germinal center hypoxia potentiates immunoglobulin class switch recombination. J Immunol. 2016;197(10):4014–4020.
62. Meng X, Asadi-Asadabad S, Cao S, et al. Metabolic rewiring controlled by HIF-1α tunes IgA-producing B-cell differentiation and intestinal inflammation. Cell Mol Immunol. 2025;22(1):54–67.
63. Mueller A-L, Payandeh Z, Mohammadkhani N, et al. Recent advances in understanding the pathogenesis of rheumatoid arthritis: new treatment strategies. Cells. 2021;10(11):3017.
64. Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol. 2017;13(5):291–301. doi:10.1038/nrrheum.2017.49
65. Biniecka M, Canavan M, McGarry T, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheumatic Dis. 2016;75(12):2192–2200. doi:10.1136/annrheumdis-2015-208476
66. Kierans S, Taylor C. Regulation of glycolysis by the hypoxia‐inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599(1):23–37. doi:10.1113/JP280572
67. Quiñonez-Flores CM, González-Chávez SA, Pacheco-Tena C. Hypoxia and its implications in rheumatoid arthritis. J Biomed Sci. 2016;23(1):62. doi:10.1186/s12929-016-0281-0
68. Naughton D, Whelan M, Smith EC, et al. An investigation of the abnormal metabolic status of synovial fluid from patients with rheumatoid arthritis by high field proton nuclear magnetic resonance spectroscopy. FEBS Lett. 1993;317(1–2):135–138. doi:10.1016/0014-5793(93)81508-W
69. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–214. doi:10.1016/j.tips.2012.01.005
70. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671. doi:10.1172/JCI67230
71. Adamus G. Impact of autoantibodies against glycolytic enzymes on pathogenicity of autoimmune retinopathy and other autoimmune disorders. Front Immunol. 2017;8:505. doi:10.3389/fimmu.2017.00505
72. Chang X, Wei C. Glycolysis and rheumatoid arthritis. Int J Rheumatic Dis. 2011;14(3):217–222. doi:10.1111/j.1756-185X.2011.01598.x
73. De saedeleer CJ, Copetti T, Porporato PE, et al. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One. 2012;7(10):e46571. doi:10.1371/journal.pone.0046571
74. Hu W, Wang K, Dong Y, et al. RIPK3 promotes ASIC1a-mediated fibroblast-like synoviocyte migration and invasion via malate shuttle-driven mitochondrial respiration in rheumatoid arthritis. Theranostics. 2025;15(17):8719. doi:10.7150/thno.113974
75. Tannahill GÁ, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
76. Del Rey MJ, Valín Á, Usategui A, et al. Hif-1α knockdown reduces glycolytic metabolism and induces cell death of human synovial fibroblasts under normoxic conditions. Sci Rep. 2017;7(1):3644. doi:10.1038/s41598-017-03921-4
77. Del Rey MJ, Izquierdo E, Caja S, et al. Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia‐inducible transcription factor 1α/vascular endothelial growth factor–mediated pathway in immunodeficient mice. Arthritis Rheum. 2009;60(10):2926–2934. doi:10.1002/art.24844
78. Ahn J, Koh E-M, Cha H-S, et al. Role of hypoxia-inducible factor-1α in hypoxia-induced expressions of IL-8, MMP-1 and MMP-3 in rheumatoid fibroblast-like synoviocytes. Rheumatology. 2008;47(6):834–839. doi:10.1093/rheumatology/ken086
79. Zheng Q-H, Zhai Y, Wang Y-H, et al. The role of hypoxic microenvironment in rheumatoid arthritis. Front Immunol. 2025;16:1633406. doi:10.3389/fimmu.2025.1633406
80. Middleton J, Americh L, Gayon R, et al. Endothelial cell phenotypes in the rheumatoid synovium: activated, angiogenic, apoptotic and leaky. Arthritis Res Ther. 2004;6(2):60. doi:10.1186/ar1156
81. Park SY, Lee SW, Kim HY, et al. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF‐1α activation. European J Immunol. 2015;45(4):1216–1227. doi:10.1002/eji.201444908
82. Ballara S, Taylor PC, Reusch P, et al. Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheum. 2001;44(9):2055–2064. doi:10.1002/1529-0131(200109)44:9<2055::AID-ART355>3.0.CO;2-2
83. Fava RA, Olsen NJ, Spencer-Green G, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180(1):341–346. doi:10.1084/jem.180.1.341
84. Westra J, Brouwer E, van Roosmalen IA, et al. Expression and regulation of HIF-1alpha in macrophages under inflammatory conditions; significant reduction of VEGF by CaMKII inhibitor. BMC Musculoskeletal Disord. 2010;11(1):61. doi:10.1186/1471-2474-11-61
85. Hitchon C, Wong K, Ma G, et al. Hypoxia‐induced production of stromal cell–derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46(10):2587–2597. doi:10.1002/art.10520
86. Yoo S-A, Park J-H, Hwang S-H, et al. Placental growth factor-1 and-2 induce hyperplasia and invasiveness of primary rheumatoid synoviocytes. J Immunol. 2015;194(6):2513–2521. doi:10.4049/jimmunol.1402900
87. Stevens C, Blake DR, Merry P, et al. A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum. 1991;34(12):1508–1513. doi:10.1002/art.1780341206
88. Izquierdo E, Cañete JD, Celis R, et al. Immature blood vessels in rheumatoid synovium are selectively depleted in response to anti-TNF therapy. PLoS One. 2009;4(12):e8131. doi:10.1371/journal.pone.0008131
89. Paleolog EM. The vasculature in rheumatoid arthritis: cause or consequence? Int J Experim Pathol. 2009;90(3):249–261. doi:10.1111/j.1365-2613.2009.00640.x
90. Paleolog EM, Young S, Stark AC, et al. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor α and interleukin‐1 in rheumatoid arthritis. Arthritis Rheum. 1998;41(7):1258–1265. doi:10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1
91. Zhou J, Schmid T, Brune B. Tumor necrosis factor-α causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. ?mol Biol Cell. 2003;14(6):2216–2225. doi:10.1091/mbc.e02-09-0598
92. Elshabrawy HA, Chen Z, Volin MV, et al. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis. 2015;18(4):433–448. doi:10.1007/s10456-015-9477-2
93. De Bandt M, Ben Mahdi MH, Ollivier V, et al. Blockade of vascular endothelial growth factor receptor I (VEGF-RI), but not VEGF-RII, suppresses joint destruction in the K/BxN model of rheumatoid arthritis. J Immunol. 2003;171(9):4853–4859. doi:10.4049/jimmunol.171.9.4853
94. Ainola M, Mandelin JA, Liljeström MP, et al. Pannus invasion and cartilage degradation in rheumatoid arthritis: involvement of MMP-3 and interleukin-1b. Clin Exp Rheumatol. 2005;23(5):644–650.
95. Bartok B, Firestein GS. Fibroblast‐like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–255. doi:10.1111/j.0105-2896.2009.00859.x
96. Duval E, Leclercq S, Elissalde JM, et al. Hypoxia‐inducible factor 1α inhibits the fibroblast‐like markers type I and type III collagen during hypoxia‐induced chondrocyte redifferentiation: hypoxia not only induces type II collagen and aggrecan, but it also inhibits type I and type III collagen in the hypoxia‐inducible factor 1α–dependent redifferentiation of chondrocytes. Arthritis Rheum. 2009;60(10):3038–3048.
97. Schipani E, Ryan HE, Didrickson S, et al. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15(21):2865–2876. doi:10.1101/gad.934301
98. Pfander D, Cramer T, Schipani E, et al. HIF-1α controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116(9):1819–1826. doi:10.1242/jcs.00385
99. Okada K, Mori D, Makii Y, et al. Hypoxia-inducible factor-1 alpha maintains mouse articular cartilage through suppression of NF-κB signaling. Sci Rep. 2020;10(1):5425. doi:10.1038/s41598-020-62463-4
100. Sheu S-Y, Ho S-R, Sun J-S, et al. Arthropod steroid hormone (20-Hydroxyecdysone) suppresses IL-1β-induced catabolic gene expression in cartilage. BMC Complementary Alternative Med. 2015;15(1):1. doi:10.1186/s12906-015-0520-z
101. Huh YH, Lee G, Lee K-B, et al. HIF-2α-induced chemokines stimulate motility of fibroblast-like synoviocytes and chondrocytes into the cartilage-pannus interface in experimental rheumatoid arthritis mouse models. Arthritis Res Therapy. 2015;17(1):302. doi:10.1186/s13075-015-0816-x
102. Cha HS, Ahn KS, Jeon CH, et al. Influence of hypoxia on the expression of matrix metalloproteinase-1,-3 and tissue inhibitor of metalloproteinase-1 in rheumatoid synovial fibroblasts. Clin Experim Rheumatol. 2003;21(5):593–598.
103. Lee Y-A, Choi HM, Lee S-H, et al. Hypoxia differentially affects IL-1β-stimulated MMP-1 and MMP-13 expression of fibroblast-like synoviocytes in an HIF-1α-dependent manner. Rheumatology. 2012;51(3):443–450. doi:10.1093/rheumatology/ker327
104. Li G, Zhang Y, Qian Y, et al. Interleukin-17A promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing MMP2 and MMP9 expression through NF-κB/HIF-1α pathway. Mol Immunol. 2013;53(3):227–236. doi:10.1016/j.molimm.2012.08.018
105. Bohensky J, Leshinsky S, Srinivas V, et al. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol. 2010;25(4):633–642. doi:10.1007/s00467-009-1310-y
106. Lu J, Peng Y, Zou J, et al. Hypoxia inducible factor-1α is a regulator of autophagy in osteoarthritic chondrocytes. Cartilage. 2021;13(2_suppl):1030S–1040S. doi:10.1177/19476035211035434
107. Kondo N, Kanai T, Okada M. Rheumatoid arthritis and reactive oxygen species: a review. Curr Issues Mol Biol. 2023;45(4):3000–3015. doi:10.3390/cimb45040197
108. Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochimica et Biophysica Acta (BBA)-Proteins Proteomics. 2012;1824(1):133–145. doi:10.1016/j.bbapap.2011.06.020
109. Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. 2012;8(11):656–664. doi:10.1038/nrrheum.2012.153
110. Park H-J, Baek KH, Lee H-L, et al. Hypoxia inducible factor-1α directly induces the expression of receptor activator of nuclear factor-κB ligand in periodontal ligament fibroblasts. Mol Cells. 2011;31(6):573–578. doi:10.1007/s10059-011-1055-x
111. Swales C, Athanasou NA, Knowles HJ. Angiopoietin-like 4 is over-expressed in rheumatoid arthritis patients: association with pathological bone resorption. PLoS One. 2014;9(10):e109524. doi:10.1371/journal.pone.0109524
112. Knowles HJ, Cleton-Jansen A-M, Korsching E, et al. Hypoxia-inducible factor regulates osteoclast-mediated bone resorption: role of angiopoietin-like 4. FASEB J. 2010;24(12):4648–4659. doi:10.1096/fj.10-162230
113. Ke L, He Q, Qu J, et al. Bone-protective effects of neutralizing angiopoietin-like protein 4 monoclonal antibody in rheumatoid arthritis. Mol Ther. 2024;32(12):4497–4513. doi:10.1016/j.ymthe.2024.09.031
114. Morten KJ, Badder L, Knowles HJ. Differential regulation of HIF‐mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts. J Pathol. 2013;229(5):755–764. doi:10.1002/path.4159
115. Knowles HJ, Athanasou NA. Acute hypoxia and osteoclast activity: a balance between enhanced resorption and increased apoptosis. J Pathol. 2009;218(2):256–264. doi:10.1002/path.2534
116. Li H, Wu Q-Y, Teng X-H, et al. The pathogenesis and regulatory role of HIF-1 in rheumatoid arthritis. Cent Eur J Immunol. 2023;48(4):338–345. doi:10.5114/ceji.2023.134217
117. Kong D, Park EJ, Stephen AG, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65(19):9047–9055. doi:10.1158/0008-5472.CAN-05-1235
118. Hatano N, Matsubara M, Suzuki H, et al. HIF-1α dependent upregulation of ZIP8, ZIP14, and TRPA1 modify intracellular Zn2+ accumulation in inflammatory synoviocytes. Int J Mol Sci. 2021;22(12):6349. doi:10.3390/ijms22126349
119. Mangraviti A, Raghavan T, Volpin F, et al. HIF-1α-targeting acriflavine provides long term survival and radiological tumor response in brain cancer therapy. Sci Rep. 2017;7(1):14978. doi:10.1038/s41598-017-14990-w
120. Kong X, Lin Z, Liang D, et al. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1α. Mol Cell Biol. 2006;26(6):2019–2028. doi:10.1128/MCB.26.6.2019-2028.2006
121. Wang G, Wang J, Li X, et al. Hypoxia and TNF-α synergistically induce expression of IL-6 and IL-8 in human fibroblast-like synoviocytes via enhancing TAK1/NF-κB/HIF-1α signaling. Inflammation. 2023;46(3):912–924. doi:10.1007/s10753-022-01779-x
122. Wang Y, Minko T. A novel cancer therapy: combined liposomal hypoxia inducible factor 1 alpha antisense oligonucleotides and an anticancer drug. Biochem Pharmacol. 2004;68(10):2031–2042. doi:10.1016/j.bcp.2004.07.017
123. Pakunlu RI, Wang Y, Saad M, et al. In vitro and in vivo intracellular liposomal delivery of antisense oligonucleotides and anticancer drug. J Control Release. 2006;114(2):153–162. doi:10.1016/j.jconrel.2006.06.010
124. Liu X, Guo R, Huo S, et al. CaP-based anti-inflammatory HIF-1α siRNA-encapsulating nanoparticle for rheumatoid arthritis therapy. J Control Release. 2022;343:314–325. doi:10.1016/j.jconrel.2022.01.029
125. Bai L, Su P. Nanocarrier-based delivery of siRNA therapeutics in rheumatoid arthritis: immune mechanisms and translational perspectives. Front Immunol. 2025;16:1718256. doi:10.3389/fimmu.2025.1718256
126. Kumari A, Kaur A, Aggarwal G. The emerging potential of siRNA nanotherapeutics in treatment of arthritis. Asian J Pharm Sci. 2023;18(5):100845. doi:10.1016/j.ajps.2023.100845
127. Shen M-Y, Wang X, Di Y-X, et al. Triptolide inhibits Th17 differentiation via controlling PKM2-mediated glycolysis in rheumatoid arthritis. Immuno Immunotoxicol. 2022;44(6):838–849. doi:10.1080/08923973.2022.2086139
128. Palsson-McDermott EM, Curtis A, Goel G, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21(1):65–80. doi:10.1016/j.cmet.2014.12.005
129. Zeng J-Z, Ma L-F, Meng H, et al. (5R)-5-hydroxytriptolide (LLDT-8) prevents collagen-induced arthritis through OPG/RANK/RANKL signaling in a rat model of rheumatoid arthritis. Exp Ther Med. 2016;12(5):3101–3106. doi:10.3892/etm.2016.3739
130. Li G-Q, Liu D, Zhang Y, et al. Anti-invasive effects of celastrol in hypoxia-induced fibroblast-like synoviocyte through suppressing of HIF-1α/CXCR4 signaling pathway. Int Immunopharmacol. 2013;17(4):1028–1036. doi:10.1016/j.intimp.2013.10.006
131. Huang L, Zhang Z, Zhang S, et al. Inhibitory action of Celastrol on hypoxia-mediated angiogenesis and metastasis via the HIF-1α pathway. Int J Mol Med. 2011;27(3):407–415. doi:10.3892/ijmm.2011.600
132. Yang G, Chang -C-C, Yang Y, et al. Resveratrol alleviates rheumatoid arthritis via reducing ROS and inflammation, inhibiting MAPK signaling pathways, and suppressing angiogenesis. J Agric Food Chemistry. 2018;66(49):12953–12960. doi:10.1021/acs.jafc.8b05047
133. Li RL, He L-Y, Zhang Q, et al. HIF-1α is a potential molecular target for herbal medicine to treat diseases. Drug Des Devel Ther. 2020;14:4915–4949. doi:10.2147/DDDT.S274980
134. Zhang M, Wu D, Xu J, et al. Suppression of NLRP3 inflammasome by dihydroarteannuin via the HIF‐1α and JAK3/STAT3 signaling pathway contributes to attenuation of collagen-induced arthritis in mice. Front Pharmacol. 2022;13:884881. doi:10.3389/fphar.2022.884881
135. Chen M, Wang Z, Chen H, et al. Biomimetic nanoparticles inhibit the HIF-1α/iNOS/NLRP3 pathway to alleviate rheumatoid arthritis. Nano Lett. 2025;25(10):3807–3816. doi:10.1021/acs.nanolett.4c05782
136. Yu T, Tang B, Sun X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for cancer therapy. Yonsei Med J. 2017;58(3):489–496. doi:10.3349/ymj.2017.58.3.489
137. Vanderborght B, De Muynck K, Lefere S, et al. Effect of isoform-specific HIF-1α and HIF-2α antisense oligonucleotides on tumorigenesis, inflammation and fibrosis in a hepatocellular carcinoma mouse model. Oncotarget. 2020;11(48):4504–4520. doi:10.18632/oncotarget.27830
138. Hong WX, Hu MS, Esquivel M, et al. The role of hypoxia-inducible factor in wound healing. Adv Wound Care. 2014;3(5):390–399. doi:10.1089/wound.2013.0520
139. Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi:10.1016/j.stem.2010.06.020
140. Jeong W, Rapisarda A, Park SR, et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother Pharmacol. 2014;73(2):343–348. doi:10.1007/s00280-013-2362-z
141. Deviatkin AA, Vakulenko YA, Akhmadishina LV, et al. Emerging concepts and challenges in rheumatoid arthritis gene therapy. Biomedicines. 2020;8(1):9. doi:10.3390/biomedicines8010009
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
From Methotrexate Resistance to Biologic and Targeted Pharmacotherapy: A Decade of Phase 4 Clinical Trials Evidence in Rheumatoid Arthritis
Alsharif ST
Drug Design, Development and Therapy 2026, 20:581822
Published Date: 8 January 2026
APOE and CCR2: Potential Macrophage-Specific Biomarkers in the Rheumatoid Arthritis Synovial Microenvironment Identified by Bioinformatics and Experimental Verification in Murine Models
Wang D, Lu L, Zhang Y, Shang W
Journal of Inflammation Research 2026, 19:587712
Published Date: 25 March 2026
Mitochondrial Hub Genes in Rheumatoid Arthritis Identified by Machine Learning and Mendelian Randomization with Experimental Validation
Luo X, Dong Z, Song Y, Hu J, Liao S, Ding X
Journal of Inflammation Research 2026, 19:576901
Published Date: 27 March 2026
Nanobiosensors for Rheumatoid Arthritis Biomarkers: Advances and Clinical Translational Potential
Zhang X, Song X, Chen M, Lei Y, Li J
International Journal of Nanomedicine 2026, 21:620504
Published Date: 8 July 2026