Back to Journals » ImmunoTargets and Therapy » Volume 9
GM-CSF: A Promising Target in Inflammation and Autoimmunity
Authors Lee KMC ![]() , Achuthan AA
, Achuthan AA ![]() , Hamilton JA
, Hamilton JA
Received 3 September 2020
Accepted for publication 15 October 2020
Published 29 October 2020 Volume 2020:9 Pages 225—240
DOI https://doi.org/10.2147/ITT.S262566
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Kevin MC Lee,1 Adrian A Achuthan,1 John A Hamilton1,2
1Department of Medicine, Royal Melbourne Hospital, The University of Melbourne, Melbourne, VIC, 3050, Australia; 2Australian Institute for Musculoskeletal Science (AIMSS), The University of Melbourne and Western Health, Melbourne, VIC, Australia
Correspondence: John A Hamilton Email [email protected]
Abstract: The cytokine, granulocyte macrophage-colony stimulating factor (GM-CSF), was firstly identified as being able to induce in vitro the proliferation and differentiation of bone marrow progenitors into granulocytes and macrophages. Much preclinical data have indicated that GM-CSF has a wide range of functions across different tissues in its action on myeloid cells, and GM-CSF deletion/depletion approaches indicate its potential as an important therapeutic target in several inflammatory and autoimmune disorders, for example, rheumatoid arthritis. In this review, we discuss briefly the biology of GM-CSF, raise some current issues and questions pertaining to this biology, summarize the results from preclinical models of a range of inflammatory and autoimmune disorders and list the latest clinical trials evaluating GM-CSF blockade in such disorders.
Keywords: GM-CSF, inflammation, autoimmunity, therapeutic
Introduction
Granulocyte macrophage-colony stimulating factor (GM-CSF, CSF2) was originally defined as a hemopoietic growth factor due to its ability to form colonies of granulocytes and macrophages in vitro by proliferation and differentiation of bone marrow progenitor cells.1 A number of reports have revealed that GM-CSF and the GM-CSF receptor (GM-CSFR) levels are elevated and correlated with disease severity in many inflammatory/autoimmune diseases, for example, rheumatoid arthritis (RA).2 In addition, there is much preclinical data providing a strong rationale for the involvement of GM-CSF in such diseases. As a result, GM-CSF and GM-CSFR have both attracted great interest as potential therapeutic targets. This review provides a brief overview of the pleiotropic biology of GM-CSF and outlines some of the most recent preclinical findings and in particular the resultant clinical studies using GM-CSF- or GM-CSFR-targeting monoclonal antibodies (mAbs) in various diseases. It also summarizes some of the contentious issues and outstanding questions pertaining to GM-CSF biology. This review cannot obviously cover all aspect of the broad topic and further background information on GM-CSF biology and targeting can be found in earlier reviews (see, for example,2–6).
GM-CSF Biology
GM-CSF Receptor and Signaling
GM-CSF binds to the multimeric GM-CSFR, comprising a specific low-affinity ligand-binding α subunit (GM-CSFRα) and a signal-transducing β subunit (GM-CSFRβ), the latter shared with the interleukin-3 (IL-3) and IL-5 receptors. The activation of GM-CSFR triggers (i) phosphorylation of the GM-CSFRβ subunit, which commonly leads to the binding of signal transducer and activator of transcription 5 (STAT5) thereby initiating Janus Kinase (JAK) 2 signaling7–9 and (ii) activation of the MEK/ERK, phosphatidylinositol 3 kinase (PI3K) and NFκB pathways.10,11
The hemopoietic-specific transcription factor, interferon regulatory factor 4 (IRF4), is a key signaling molecule for the adoption of dendritic cell (DC)-like properties in GM-CSF-treated precursors, such as monocytes.12–16 Recently, it was reported that the GM-CSF stimulation of monocytes/macrophages in vitro leads to the formation of CCL17 via IRF4 as an important pathway, termed the GM-CSF/CCL17 axis (see below).17 Mechanistically, GM-CSF up-regulates IRF4 expression by enhancing JMJD3 demethylase activity.17 Additionally, GM-CSF-IRF4 signaling favours the polarization of pro-inflammatory macrophages and increased antigen presentation capability (ie increased MHC class II expression) during in vivo inflammation.18 In contrast, some literature indicates IRF5, but not IRF4, to be important for GM-CSF-induced macrophage polarization19,20 and IRF4 has been considered to have an anti-inflammatory role in macrophages (for example, enhanced interleukin (IL)-10 and reduced TNF production).21–23
GM-CSF and the Lung
While GM-CSF appears to be dispensable for steady state myeloid cell development in vivo,24 GM-CSF directly regulates the differentiation of liver-derived fetal monocytes into immature alveolar macrophages during embryonic development25 and also promotes the development of functional alveolar macrophages via PU.1.25,26 GM-CSF gene-deficient mice develop pulmonary alveolar proteinosis,24,27 which can also occur in humans due to genetic mutations or endogenous neutralizing antibody (53).28 This pathology results from compromised alveolar macrophage functions.29 In addition, GM-CSF deficient mice have been reported to be more susceptible to lung infections.30,31 Inhaled GM-CSF can protect mice from such infections30,32 via enhancing macrophage and DC function.30,31 These studies suggest that the availability of excess GM-CSF could be beneficial in certain circumstances.
Sources of GM-CSF in Inflammation
A wide range of cells can produce GM-CSF.2–6 During inflammation, major sources of GM-CSF are both hematopoietic, for example, T and B cells, and non-hematopoietic, for example, tissue resident cells.
T Cells
GM-CSF has been reported to be produced by TH1 and TH17 cells via STAT signaling33–35 and shown to be crucial for encephalitogenicity.33 A distinct subset of T cells, known as GM-CSF-producing TH cells, has recently been identified.35 GM-CSF-producing TH cells express undetectable T-bet, GATA-3, or RORγt and do not express a cytokine signature that other T cell subsets express.35,36 In humans, GM-CSF-producing TH cells can be identified as CCR10+ CCR4+ CXCR3− CCR6− cells.36 GM-CSF-producing TH cells have been implicated in autoimmune brain disease as fewer GM-CSF-producing TH cells correlated with less severe experimental autoimmune encephalomyelitis (EAE).35
Innate Lymphoid Cells
The innate lymphoid cell (ILC) family encompasses the classic cytotoxic natural killer (NK) cells and the non-cytotoxic ILCs.37,38 NK cells have been recently reported to produce GM-CSF when infiltrating into joints for the maintenance of inflammatory arthritis.39 Among the subsets of ILCs, type 3 ILCs have been shown to secrete GM-CSF in intestinal inflammation.40–42 In spondyloarthritis, type 3 ILCs are found to be enriched in the inflamed joint and are the predominate source of GM-CSF.43
B Cells
A subset of B cells, namely innate response activator (IRA) B cells, resides in nonlymphoid sites, such as the peritoneal and pleural cavities, and provides a first line of defense against infection.44 In a model of Toll-like receptor (TLR)-induced sepsis, IRA B cells, which produce GM-CSF and also reported to express the GM-CSFR, mediate a GM-CSF-dependent IgM protective mechanism against septic shock.44–46 Mixed chimeric mice with B cell-restricted GM-CSF deficiency had high bacterial titer morbidity after infection but did not develop alveolar proteinosis,44 indicating that B cell-derived GM-CSF is dispensable for surfactant clearance by alveolar macrophages. Memory B cells isolated from multiple sclerosis (MS) patients produce high levels of GM-CSF, termed GM-CSF+ B cells, and co-culture of these cells with macrophage-colony stimulating factor (M-CSF)-generated, human blood monocyte-derived macrophages initiates proinflammatory responses.47 Interestingly, dimethyl fumarate ameliorates MS and depletes GM-CSF-producing B cells in these patients48,49 consistent with a pathogenic role for these cells.
Tissue Resident Cells
Tissue resident cells can also be a potential source of GM-CSF during inflammation. For example, it has been shown that fibroblast-like synoviocytes are important for the initiation of experimental autoimmune arthritis via their GM-CSF production.50 GM-CSF has been reported to be expressed by cardiac fibroblasts in models of Kawasaki disease and myocarditis.51,52 Epithelial cells can produce GM-CSF in response to allergenic stimuli53,54 and such production can restore alveolar barrier function.55 In addition, studies also reported that endothelial cells can produce GM-CSF in response to pro-inflammatory cytokine stimulation.56–58
GM-CSF-Responsive Cells
In vitro, GM-CSF can regulate proliferation and/or activation of myeloid cells, namely monocytes, macrophages, DCs, neutrophils and eosinophils. At sites of inflammation, GM-CSF can be proinflammatory through recruitment of myeloid cells and/or by enhancing their survival and activation.6,59 However, it has been reported that prolonged exposure to GM-CSF can lead to the generation of monocyte-derived suppressor cells.60
In addition to being able to control the development of monocytes and macrophages from bone marrow precursors in vitro, GM-CSF can regulate multiple functions in the differentiated cells, including cell survival, proliferation and maturation, via transcription factors, such as PU.1 and IRF4.17,26,61 During infection, GM-CSF has been shown to boost macrophage antimicrobial functions, such as enhanced phagocytosis62 and increased production of reactive oxygen species.63,64 GM-CSF stimulates monocytes/macrophages to secrete some pro-inflammatory cytokines (for example, IL-6, IL-23 and CCL17)17,65–67 and these cells are often hyperinflammatory (“primed”) when they encounter a second stimulus (for example, lipopolysaccharide (LPS)).68–70 Macrophages are remarkably plastic cells and have been classified into various so-called “polarization states” (for example, M1 vs M2) in different diseased tissues. As regards the expression of certain pro-inflammatory cytokines, GM-CSF has been considered to shift the phenotype of macrophages into a M1-like, pro-inflammatory polarization state;71 however, such cells have also been considered to have dual M1/M2 characteristics,53,67 and GM-CSF-activated monocytes have been reported to alleviate experimental colitis.67 As a result, it has been recommended that the M1/M2 polarization terminology not be applied to GM-CSF action.2,17,61,72
GM-CSF also promotes the development of migratory CD103+ CD11b+ DCs,40,73 while negatively regulating the development of resident CD8+ DCs.74 GM-CSF, often in combination with IL-4, is widely used to generate in vitro murine and human DC populations from bone marrow precursors and blood monocytes, respectively.11,75–77 Heterogeneity in GM-CSF-induced bone marrow DCs has been reported, comprising at least two populations, namely the Fms-like tyrosine kinase 3 (FLT-3)+ DC or the CD11c+ MHCII+ CD115+ monocyte-derived DC (MoDC).78 There has been debate in the literature regarding the role of GM-CSF in the in vivo generation of MoDCs.79–85 Some studies have shown GM-CSF to be dispensable for the differentiation of MoDCs;82 in contrast, it has been demonstrated that NFκB-dependent GM-CSF production in CD4+ T cells is required for the generation of MoDCs.81,83,84 Very recently, to add fuel to this debate GM-CSF has been proposed86 to differentiate a population of DC3s independently of conventional DCs (cDCs) or monocyte-restricted progenitors. Besides the regulation of DC numbers, there is evidence that GM-CSF also regulates their function, including antigen presentation (as indicated by increased MHCII expression78) and inflammatory responses (as indicated by increased IL-6 and IL-23 secretion87).
Neutrophils
GM-CSF can enhance the survival, adhesion and trafficking of neutrophils.88,89 During infection, GM-CSF also upregulates the antimicrobial functions of neutrophils, such as phagocytosis90 and formation of extracellular traps.91 One study reported that the expression of PU.1 in neutrophils of pulmonary alveolar proteinosis patients was normal, indicating that GM-CSF is not involved in steady state neutrophil development.89
GM-CSF/CCL17 Axis
The chemokine, CCL17 (formerly called thymus and activation-regulated chemokine [TARC]) was originally implicated in the preferential attraction of TH2 cells;92 however, it can also attract regulatory T cells.93 Its most recognized receptor is CCR494 although the atypical chemokine receptor 2 (ACR2) has also been reported to be a CCL17 receptor.95 As mentioned above, GM-CSF in vitro upregulates CCL17 formation via an IRF4/JMJD3-dependent mechanism in human and mouse monocytes/macrophages,17 as well as in inflammation models.18,80 It is to be noted that GM-CSF is not the only mediator that regulates CCL17 production in these populations; for example, IL-4 also upregulates the CCL17 expression in human monocytes and mouse macrophages via a similar mechanism.96 This GM-CSF/CCL17 axis has been found to be important in controlling inflammatory arthritic and osteoarthritic pain in pre-clinical models;17,80,97–99 importantly, this axis appears to be active in humans, since a neutralizing GM-CSFR mAb in RA patients leads to a sustained reduction in circulating CCL17 levels.100 Preclinical studies suggest that CCL17 may not necessarily be acting as a T cell chemokine in its control of inflammation and its associated pain.17,80,97–99 Mechanistically, whether CCL17 has a direct effect on neurons for pain induction remains an open question101 – there are conflicting reports as to whether CCR4, the CCL17 receptor, is expressed on neurons.101–104 However, whatever the mechanism, it is clear that neutralizing CCL17 peripherally with a mAb ameliorates inflammatory arthritic99 and osteoarthritic97,98 pain. Additionally, other studies have also reported a non-chemotactic role for CCL17, for example, a role(s) in regulating inflammation by restricting regulatory T cell expansion.105,106
GM-CSF Biology: Current Issues and Questions
There are still a number of issues and questions pertaining to GM-CSF biology, which need to be addressed as they have potential implications for the clinical targeting of GM-CSF in inflammation and autoimmunity. It should be borne in mind that this biology may vary depending on the processes and tissues involved in the particular clinical indication in question. Which responding cell type(s) is relevant and whether GM-CSF regulates their number and/or activation/differentiation status are important considerations ― the latter issue may have implications for therapeutic delivery and dosing since the extent of GM-CSF neutralization/depletion is likely to be critical.6 There is still plenty of controversy around the role of GM-CSF in DC development in vivo.79–85 It would also be worth knowing how significant is GM-CSF-dependent IRF4 signaling in monocytes/macrophages, including the so-called GM-CSF/CCL17 axis discussed above. Depending on the particular model of inflammation/autoimmunity being studied, the relevant cell type(s) producing GM-CSF varies, again with possible implications for therapeutic strategies. GM-CSF administration systemically can have pro-inflammatory and anti-inflammatory effects ― as discussed previously, these responses to exogenous GM-CSF may or may not be predictive of the findings when endogenous (locally acting?) GM-CSF is neutralized/depleted.2
GM-CSF has been documented for its role in peripheral pain.17,99,107,108 However, whether in this capacity GM-CSF is acting directly on neurons (nociceptors), including acting centrally, remains unclear as conflicting reports have been published.101,109–115 Additionally, GM-CSF has also been reported to have neuroprotective effects following nerve injury.116,117 Further research is obviously needed to clarify how GM-CSF interacts with the nervous system.
GM-CSF in Disease
Inflammatory Arthritis
Early studies measuring cytokines in synovial fluid and blood from patients with RA showed increased GM-CSF levels, as well as increased expression of GM-CSFR, in inflamed synovial tissue.118,119 Administration of GM-CSF to RA patients led to disease flares.120 A genome-wide association study revealed that mutations in CSF2 (the gene that encodes GM-CSF) contribute to genetic susceptibility in RA.121 Based in part on the priming of blood monocytes with GM-CSF, it was recently suggested that GM-CSF neutralization be considered as a potential therapeutic approach for the treatment of ankylosing spondylitis.122
The contribution of GM-CSF to the pathogenesis of experimental inflammatory arthritis is well documented in the literature. GM-CSF-deficient mice fail to develop arthritis and associated pain in several inflammatory arthritis models, including collagen-induced arthritis (CIA), antigen-induced arthritis (AIA), zymosan-induced arthritis (ZIA) and K/BxN serum-transfer arthritis (STA).17,39,108,123 The administration of neutralizing GM-CSF mAbs ameliorated existing disease in these models.99,124 Regarding the relevance of GM-CSF to arthritic pain, as mentioned earlier GM-CSF is implicated in regulating inflammatory and arthritic pain via downstream CCL17.17,98,99 Interestingly, high levels of circulating GM-CSF have been shown to correlate with the responsiveness of RA patients to anti-TNF agents.125 Consistent with the concept of the GM-CSF/CCL17 axis (see above), RA patients treated with anti-GM-CSFR mAb (mavrilimumab) have reduced circulating CCL17 levels, suggesting that CCL17 could be a biomarker for anti-GM-CSF or anti-GM-CSFR treatment.100
Osteoarthritis
Osteoarthritis (OA) was once considered a non-inflammatory arthropathy; however, it is now well-recognized that there can be a significant inflammatory component contributing to OA clinical symptoms, for example, chronic pain. The expression of GM-CSF and its receptor have been found in OA synovial tissue126,127 and reported to be negatively correlated with pain.126 In contrast, in a collagenase-induced, joint instability OA model, GM-CSF-deficient mice were protected from associated pain and osteophyte development.107 Consistent with this data, neutralizing anti-GM-CSF mAb effectively ameliorated pain in the same model.97,107 This pain amelioration was observed when the neutralizing mAb was administered early or late in this model but early administration was needed for it to be effective on joint damage.97 This data has led to clinical trials in OA for targeting GM-CSF (see below) or CCL17 (https://clinicaltrials.gov/show/NCT03485365). Synovial inflammation, characterized by macrophage infiltration, is often more prominent in early OA lesions, while advanced OA is more commonly associated with structural changes (for example, cartilage degeneration and/or osteophyte formation).128–130 Given that GM-CSF regulates a wide range of macrophage functions (see above), it could be that optimal clinical improvement might be seen in patients with early OA as opposed to patients with advanced OA disease.
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic autoimmune/inflammatory disease of the central nervous system (CNS) and is characterized by demyelination and subsequent axonal degeneration. While it is widely believed that TH17 cells are the main encephalitogenic population in EAE,131 the most widely used MS model, it was reported that their key secreted cytokine, IL-17, is dispensable for the development of EAE.132,133 Instead, it was later shown that GM-CSF secreted by TH17 cells is the main cytokine contributing to encephalitogenicity134 via the activation of microglia within the CNS.135 GM-CSF-activated microglia adopt a M1-like (inflammatory) phenotype136 and produce highly neurotoxic molecules such as tumor necrosis factor (TNF), IL-1 and IL-6.137 It has been proposed that GM-CSF promotes the breakdown of the blood brain barrier enabling entry of circulating Ly6Chi monocytes and stimulates the differentiation of monocyte-derived antigen-presenting cells.138,139 These differentiated cells share a similar phenotype to macrophages found in active MS lesions.140,141 Additional mouse studies have demonstrated that GM-CSF deletion results in fewer monocyte-derived cells in the CNS parenchyma following EAE induction,142 and GM-CSF administration leads to more cells migrating into the CNS parenchyma.143 Elevated GM-CSF levels have been reported in the cerebrospinal fluid of patients with active MS.144,145 Glatiramer acetate, a FDA-approved drug to treat MS, has been shown to upregulate regulatory T cells and reduce GM-CSF levels in mice with EAE.146 These reports demonstrate that GM-CSF plays a central role in EAE and indicate that GM-CSF might be a therapeutic target in MS.
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a chronic immune-mediated disease affecting the gastrointestinal tract consisting of two main subtypes: Crohn’s disease (CD) and ulcerative colitis (UC).147 Impaired innate immunity plays a critical pathogenic role in IBD.148 GM-CSF has been identified as a key mediator of chronic inflammation in models of colitis.41,149,150 Other studies using dextran sodium sulfate (DSS)-induced colitis reported that GM-CSF-deficient mice developed more severe colitis;151,152 mechanistically, it has been claimed that type 3 ILC-derived GM-CSF modulates the macrophage phenotype to prevent intestinal fibrosis.42 In line with a potential beneficial role of GM-CSF in IBD, GM-CSF administration can improve IBD experimentally153 and in some patients;152,154 also, high levels of circulating anti-GM-CSF autoantibodies have been found to correlate with worse CD prognosis.155,156
Interstitial Lung Disease
Interstitial lung disease (ILD) comprise heterogenous inflammatory lung parenchyma disorders that can lead to alveolitis and ultimately fibrosis.157 It is also a serious complication associated with systemic rheumatic diseases.157 Experimentally, the SKG mouse with a mutation in the Zap-70 gene158,159 develops spontaneous arthritis, ILD and IBD.160 The lungs of SKG mice develop fibrosis associated with intense infiltrates of GM-CSF+ IL-17A+ neutrophils, pathological features that are reminiscent of human ILD.161 GM-CSF blockade reduces these features, including the degree of fibrosis, in SKG mice, with IL-17 blockade being less effective.162 Interestingly, CCL17-expressing macrophages have been implicated in mediating peritoneal fibrosis.163 Given these findings, together with the data in lung inflammation models wherein GM-CSF blockade impaired CCL17 expression in alveolar macrophages,80 in our view further studies examining the role of the GM-CSF/CCL17 axis in lung fibrosis are warranted.
Aortic Aneurysm
Dissecting aortic aneurysm is an important and often life-threatening condition. It was reported that mice deficient in Kif6, the gene encoding the transcription factor, Krueppel-like factor 6, developed worse aortic aneurysm.164 In the same study, GM-CSF was identified to be an effector molecule downstream of Krueppel-like factor 6 and the administration of GM-CSF exacerbated aortic aneurysm formation, with GM-CSF antagonism having the opposite effect.164 In the aortic root GM-CSF induces CD11b+ Gr-1+ Ly6Chi inflammatory monocyte accumulation, with anti-GM-CSF mAb administration resulting in reduced inflammation and dilation.165 These findings suggest that GM-CSF blockade might be an effective therapeutic approach in aortic aneurysm.
Allergic Disease
GM-CSF has been reported to be involved in the TH2 response in allergic airway inflammation via activation of DCs.53,166,167 In a mouse model of asthma, allergen-exposed epithelial cells secrete GM-CSF, which activates DCs and also prolongs eosinophil survival.53,168 Administration of a GM-CSF neutralizing mAb also led to reduced allergic hyperresponsiveness.167,168 As a result, an anti-GM-CSF mAb has been tested in a Phase II trial for severe asthma (see below). Interestingly, it has been reported that alveolar DC-derived CCL17 is critical for airway inflammation169,170 and CCL17 airway expression correlates with asthmatic disease severity.171,172 In our view these data warrant a detailed study examining the role of the GM-CSF/CCL17 axis in asthma, with CCL17 being a potential target for treating allergic disease and/or a biomarker for patient selection.
Obesity and Its Associated Meta-Inflammation
Obesity is now widely considered as a low-grade, chronic inflammatory disease that contributes to metabolic dysfunction, ectopic lipid deposition and insulin resistance.173,174 With progressive obesity, adipose tissue macrophages (ATMs) have been considered to be a key cell type contributing to metabolic inflammation, insulin resistance and the impairment of adipocyte function.175–177 In response to diet-induced obesity (DIO) in mice, elevated GM-CSF levels can be detected in serum,178 peritoneal fluid,179 and adipose tissue;180 moreover, GM-CSF is required for DIO-induced adipose tissue inflammation, as GM-CSF gene-deficient mice had reduced number of infiltrating ATMs and crown-like features in adipose tissue,180 in spite of increased adiposity and body weight.181,182 It was also reported that GM-CSF gene-deficient mice exhibited improved metabolic status, namely insulin sensitivity to glucose, compared with their wild-type counterparts,180 and that GM-CSF-responsive myeloid cells play a key role in this improvement.181 As a result, GM-CSF has been proposed to be a key mediator whose actions might explain the difference between obese individuals with normal glucose tolerance (metabolically “healthy”) and those with type 2 diabetes (metabolically “unhealthy”).181 In addition to type 2 diabetes, GM-CSF has also been implicated in other obesity-exacerbated diseases, for example, in the obesity-mediated enhancement of breast cancer metastasis.183 How GM-CSF plays a role during obesity-induced meta-inflammation remains to be explored.
Covid-19
In coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), there is a characteristic hyperactive immune response that leads to an overwhelming infiltration of inflammatory myeloid cells (particularly monocytes, macrophages and neutrophils) into the lungs.184–190 More recent studies have questioned the validity of referring to the COVID-19 hyperactive immune response as the “cytokine storm” seen in chimeric antigen receptor (CAR) T cell-related cytokine release syndrome (CRS).191,192 The COVID-19-related hyperactive immune response resembles a phenotype of secondary haemophagocytic lymphohistiocytosis, often referred to as “macrophage activation syndrome”.184–189 In COVID-19 patients, increased percentages of GM-CSF-expressing leukocytes have been found in the blood.193 Inhibition of GM-CSF activity in models of hyperinflammatory conditions that share similar pathology to late stages of COVID-19, such as CAR T cell-related CRS and neurotoxicity,194 graft versus host disease-associated CRS195 and inflammatory lung diseases,162,196–198 was shown to be beneficial. The relevance of GM-CSF to COVID-19 and its potential as a therapeutic target have been reviewed recently.199,200
Clinical Studies with the Blockade of GM-CSF and Its Receptor
A number of clinical trials neutralizing GM-CSF (see Table 1) or GM-CSFR (see Table 2) using mAbs have been/are being carried out. Encouragingly, no serious adverse events have been noted so far, for example infections and compromised lung function, with the data from a long-term open label extension (OLE) study in RA patients being particularly promising in this regard.100
 |
Table 1 Past and Current Status of GM-CSF-Based Therapies |
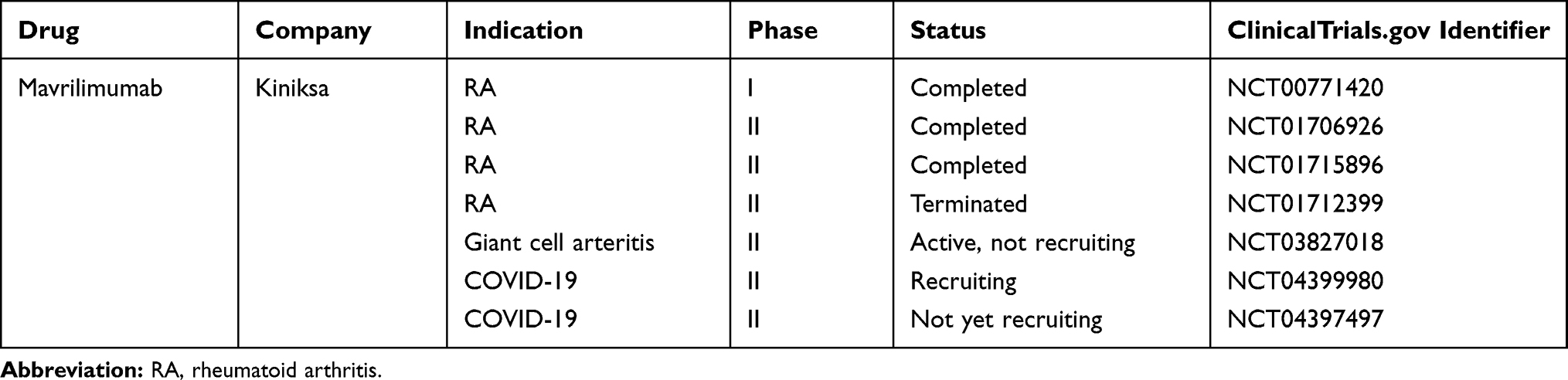 |
Table 2 Past and Current Status of GM-CSFR-Based Therapies |
Otilimab
Otilimab (formerly known as MOR-103 and GSK3196165) is an IgG1 mAb, developed by MorphoSys AG, that binds to GM-CSF and prevents its interaction with GM-CSFRα; it is currently being produced by GSK for use in several randomized controlled trials (RCTs).
A short-term, dose-escalation phase Ib/IIa trial in randomized RA patients (n=96, NCT01023256) showed improved efficacy in all outcomes (ACR and European League Against Rheumatism (EULAR) response) compared with placebo with no pulmonary function test abnormalities being reported.201 The subsequent double-blind, placebo-controlled phase IIa (NCT02799472) and IIb (NCT02504671) trials consistently showed clinical improvement in RA patients receiving otilimab and it was well tolerated. Circulating CCL17 levels declined only in the otilimab group, supporting the existence of the GM-CSF/CCL17 axis in humans. GSK has announced the start of a clinical development program (ContRAst) embracing three Phase III trials aiming to evaluate the efficacy and safety of otilimab in RA patients with inadequate response to i) conventional synthetic/biologic disease modifying anti-rheumatic drugs (DMARDs) (NCT03970837), ii) methotrexate (NCT03980483) and (iii) biologic DMARDs and/or JAK inhibitors (NCT04134728). A long-term safety and efficacy study with otilimab has also commenced (NCT04333147).
The results of an exploratory, 12-week, phase IIa study of otilimab in patients with hand OA (n=44, NCT02683785) have been reported and, while not statistically significant in this small study, reduction in pain, accompanied by improvement in functional impairment, was noted.202 Patients have been recruited to determine the efficacy of otilimab in COVID-19 (NCT04376684).199
Lenzilumab
Lenzilumab (formerly known as KB003) is an IgG1-neutralizing anti-GM-CSF mAb and has been tested successfully in a randomized phase II trial in RA (NCT00995449). A Phase I trial using lenzilumab in patients with chronic myelomonocytic leukemia (CMML) has been completed (NCT02546284); 33% of patients showed durable clinical benefit, which appears to be better in a distinct subtype of CMML patients, warranting further studies to identify CMML subtypes more likely to respond.203 A Phase II, randomized, double-blind, placebo-controlled, 24-week study in asthma patients (n=311; NCT01603277) has been performed; overall, there were no effects on asthma control although there appeared to be improvements in patients with eosinophilic asthma.204 Recently, lenzilumab was administered to a small cohort of patients (n=12) with COVID-19 pneumonia and found to associate with improved clinical outcome with no mortality observed;205 a subsequent phase III study has commenced patient recruitment to evaluate efficacy and safety of lenzilumab (NCT04351152).
TJM2
TJM2 is an IgG1-neutralizing anti-GM-CSF mAb. Phase I trials in healthy subjects (NCT03794180) and in patients with severe COVID-19 (NCT04341116) have commenced.
Namilumab
Namilumab (formerly known as MT203), an IgG1-neutralizing anti-GM-CSF mAb, has been investigated in double-blind, placebo controlled, randomized trials in healthy individuals (NCT02354599)206 and in RA patients (NCT01317797),207 which established that namilumab has an acceptable tolerability profile.206,207 Phase 1b (NCT01317797) and phase II studies (NCT02379091) in RA demonstrated efficacy, with the latter study further reporting dose-response effects.207,208 A phase II trial investigating the efficacy of namilumab in plaque psoriasis was also completed (NCT02129777) with no significant difference being recorded for this end point between placebo-treated and namilumab-treated individuals.209 Patients are being recruited for a phase IIa trial using namilumab in axial spondyloarthritis (NCT03622658).
Gimsilumab
Gimsilumab (also known as KIN-1901), a fully human IgG1 mAb, has been investigated in a double-blind, placebo controlled, randomized trial in healthy subjects and subjects with ankylosing spondylitis (NCT04205851). Patients are being recruited for a phase II trial in COVID-19 (NCT04351243).
Mavrilimumab
Mavrilimumab (CAM-3001) is a humanized IgG4 mAb with high affinity to the GM-CSFRα chain.210 The efficacy and safety profiles of mavrilimumab have been investigated in a phase I trial in 32 RA patients (NCT00771420) and in the subsequent EARTH clinical development program, including two phase IIa-IIb RCTs (EARTH EXPLORER 1 and EARTH EXPLORER 2).211,212 In the phase IIb, placebo-controlled EARTH EXPLORER 1 study (NCT01706926), moderate-to-severe active RA patients (n=236) with ongoing methotrexate treatment, received three different doses of mavrilimumab (150, 100 and 30 mg). The DAS28-CRP score among these RA patients was significantly decreased in all mavrilimumab subgroups compared with placebo, with the optimal response being 150mg mavrilimumab.212 The EARTH EXPLORER 2 study (NCT01715896), which was a phase II, double-blind, randomized trial, evaluated the benefits of using mavrilimumab (100mg every other week, n=70) in long-standing, active RA patients (mean disease duration 6.7 years, and mean baseline DAS28-ESR 6.5), who had not responded to a conventional synthetic DMARD or TNF inhibitor.211 This study included a parallel treatment with an anti-TNF mAb (golimumab) (50 mg every 4 weeks, n=68). No statistical difference was seen between RA patients treated with mavrilimumab or golimumab, which could be due to the fact that a suboptimal mavrilimumab dose (100 mg every other week) was used in this trial as opposed to the most effective dose (150mg every other week), confirmed by the EARTH EXPLORER 1 study.212 Peripheral biomarkers and pathophysiological pathways modulated by mavrilimumab and golimumab were also assessed in the study. While a number of mediators were suppressed by both mAbs, mavrilimumab, but not golimumab, was able to suppress serum levels of CCL17 and CCL22 and to induce sustained differential suppression of peripheral disease markers in anti-TNF inadequate responders.211
The long-term efficacy and safety profile of mavrilimumab were also explored in an OLE study (NCT01712399). All patients (n=422), who completed the double-blind phase of EARTH EXPLORER 1 and 2 trials (study 1109; NCT01712399), had the opportunity to enter the study and receive mavrilimumab 100 mg every other week plus methotrexate for a 3-year follow-up period.100 At week 122, 65.0% and 40.6% patients achieved a DAS28-CRP score of <3.2 and <2.6, respectively,100 demonstrating a sustained benefit in measures of RA disease outcomes. The overall safely profile of mavrilimumab appears to be promising, particularly regarding pulmonary alveolar proteinosis. In this OLE study, biomarker analyses support the hypothesis that GM-CSF regulates CCL17 and CCL22 as sustained suppression of CCL17 and CCL22 was seen in mavrilimumab-treated patients over a longer follow-up period.
New clinical trials are underway to evaluate the benefits of mavrilimumab in patients with giant cell arteritis (NCT03827018) and in COVID-19 patients (NCT04399980 and NCT04397497).199 It has recently been reported that mavrilimumab treatment was associated with improved clinical outcomes compared with standard care with patients with severe COVID-19.213
Concluding Remarks and Future Perspectives
The preclinical rationale for targeting GM-CSF in inflammation/autoimmunity is solid, and the results from early phase clinical trials of GM-CSF or GM-CSFR blockade in RA patients, and possibly asthma and COVID-19, are encouraging. However, careful ongoing evaluation of adverse effects, particularly in the lungs and gut, is clearly paramount but, as mentioned above, it seems so far that the anti-GM-CSF and anti-GM-CSFR mAbs used in clinical trials are without major safety concerns. It is hoped that there are other indications (see Table 1) where GM-CSF targeting will turn out to provide potential benefit. The key role of GM-CSF in inflammatory pain was highlighted above. In this connection, the rapid and dramatic effect of mavrilimumab on RA pain has been highlighted211,212 and, intriguingly, it has been speculated that the dramatic effects of the JAK1/2 inhibitor, baricitinib, also on RA pain may be due to its inhibition of GM-CSF signaling.214 Circulating biomarkers, such as CCL17, may aid in the selection of an indication for GM-CSF-based therapeutics and even of patients within such an indication, thus hopefully leading to better clinical outcomes. It is hoped that targeting GM-CSF is successful in patients who are non-responders to biologics, for example, those targeting other inflammatory mediators, such as TNF and IL-6.
Disclosure
The employer of K.M.-C. L., A.A.A and J.A.H., the University of Melbourne, has licensed patented technology relating to therapeutically targeting GM-CSF to MorphoSys AG, Germany.
References
1. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. 1980;56(6):947–958. doi:10.1182/blood.V56.6.947.947
2. Hamilton JA, Cook AD, Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. 2016;16(1):53–70.
3. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–544. doi:10.1038/nri2356
4. Wicks IP, Roberts AW, Targeting GM. CSF in inflammatory diseases. Nat Rev Rheumatol. 2016;12(1):37–48. doi:10.1038/nrrheum.2015.161
5. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity. 2016;45(5):963–973. doi:10.1016/j.immuni.2016.10.026
6. Hamilton JA. GM-CSF in inflammation. J Exp Med. 2020;217(1):e20190945.
7. Hansen G, Hercus TR, McClure BJ, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134(3):496–507. doi:10.1016/j.cell.2008.05.053
8. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol. 2002;71(3):511–519.
9. Perugini M, Brown AL, Salerno DG, et al. Alternative modes of GM-CSF receptor activation revealed using activated mutants of the common beta-subunit. Blood. 2010;115(16):3346–3353. doi:10.1182/blood-2009-08-235846
10. Achuthan A, Aslam ASM, Nguyen Q, et al. Glucocorticoids promote apoptosis of proinflammatory monocytes by inhibiting ERK activity. Cell Death Dis. 2018;9(3):267. doi:10.1038/s41419-018-0332-4
11. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;119(15):3383–3393.
12. Gao Y, Nish SA, Jiang R, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity. 2013;39(4):722–732. doi:10.1016/j.immuni.2013.08.028
13. Inaba K, Yashiro T, Hiroki I, Watanabe R, Kasakura K, Nishiyama C. Dual roles of PU.1 in the expression of PD-L2: direct transactivation with IRF4 and indirect epigenetic regulation. J Immunol. 2020;205(3):822–829.
14. Lehtonen A, Veckman V, Nikula T, et al. Differential expression of IFN regulatory factor 4 gene in human monocyte-derived dendritic cells and macrophages. J Immunol. 2005;175(10):6570–6579. doi:10.4049/jimmunol.175.10.6570
15. Williams JW, Tjota MY, Clay BS, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. 2013;4(1):2990. doi:10.1038/ncomms3990
16. Yashiro T, Yamaguchi M, Watanuki Y, Kasakura K, Nishiyama C. The transcription factors PU.1 and IRF4 determine dendritic cell-specific expression of RALDH2. J Immunol. 2018;201(12):3677–3682. doi:10.4049/jimmunol.1800492
17. Achuthan A, Cook AD, Lee MC, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. 2016;126(9):3453–3466. doi:10.1172/JCI87828
18. Lee MC, Lacey DC, Fleetwood AJ, Achuthan A, Hamilton JA, Cook AD. GM-CSF- and IRF4-dependent signaling can regulate myeloid cell numbers and the macrophage phenotype during inflammation. J Immunol. 2019;202(10):3033–3040. doi:10.4049/jimmunol.1801549
19. Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231–238. doi:10.1038/ni.1990
20. Weiss M, Blazek K, Byrne AJ, Perocheau DP, Udalova IA. IRF5 is a specific marker of inflammatory macrophages in vivo. Mediators Inflamm. 2013;2013:245804. doi:10.1155/2013/245804
21. Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol. 2009;183(3):1598–1606. doi:10.4049/jimmunol.0803302
22. Satoh T, Takeuchi O, Vandenbon A, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11(10):936–944. doi:10.1038/ni.1920
23. Honma K, Udono H, Kohno T, et al. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci U S A. 2005;102(44):16001–16006. doi:10.1073/pnas.0504226102
24. Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91(12):5592–5596. doi:10.1073/pnas.91.12.5592
25. Guilliams M, De Kleer I, Henri S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210(10):1977–1992. doi:10.1084/jem.20131199
26. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15(4):557–567. doi:10.1016/S1074-7613(01)00218-7
27. Reed JA, Ikegami M, Cianciolo ER, et al. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol. 1999;276(4):L556–563.
28. Trapnell BC, Nakata K, Bonella F, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers. 2019;5(1):16. doi:10.1038/s41572-019-0066-3
29. Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349(26):2527–2539.
30. Chen GH, Teitz-Tennenbaum S, Neal LM, et al. Local GM-CSF-dependent differentiation and activation of pulmonary dendritic cells and macrophages protect against progressive cryptococcal lung infection in mice. J Immunol. 2016;196(4):1810–1821. doi:10.4049/jimmunol.1501512
31. Huang FF, Barnes PF, Feng Y, et al. GM-CSF in the lung protects against lethal influenza infection. Am J Respir Crit Care Med. 2011;184(2):259–268. doi:10.1164/rccm.201012-2036OC
32. Todd EM, Ramani R, Szasz TP, Morley SC. Inhaled GM-CSF in neonatal mice provides durable protection against bacterial pneumonia. Sci Adv. 2019;5(8):eaax3387. doi:10.1126/sciadv.aax3387
33. McWilliams IL, Rajbhandari R, Nozell S, Benveniste E, Harrington LE. STAT4 controls GM-CSF production by both Th1 and Th17 cells during EAE. J Neuroinflammation. 2015;12(1):128. doi:10.1186/s12974-015-0351-3
34. O’Malley JT, Eri RD, Stritesky GL, et al. STAT4 isoforms differentially regulate Th1 cytokine production and the severity of inflammatory bowel disease. J Immunol. 2008;181(7):5062–5070. doi:10.4049/jimmunol.181.7.5062
35. Sheng W, Yang F, Zhou Y, et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014;24(12):1387–1402. doi:10.1038/cr.2014.154
36. Noster R, Riedel R, Mashreghi MF, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. 2014;6(241):241ra280. doi:10.1126/scitranslmed.3008706
37. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348(6237):aaa6566. doi:10.1126/science.aaa6566
38. Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. doi:10.1038/nature14189
39. Louis C, Souza-Fonseca-Guimaraes F, Yang Y, et al. NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J Exp Med. 2020;217:5.
40. Mortha A, Chudnovskiy A, Hashimoto D, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343(6178):1249288. doi:10.1126/science.1249288
41. Pearson C, Thornton EE, McKenzie B, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. 2016;5:e10066.
42. Castro-Dopico T, Fleming A, Dennison TW, et al. GM-CSF calibrates macrophage defense and wound healing programs during intestinal infection and inflammation. Cell Rep. 2020;32(1):107857. doi:10.1016/j.celrep.2020.107857
43. Al-Mossawi MH, Chen L, Fang H, et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. 2017;8(1):1510. doi:10.1038/s41467-017-01771-2
44. Weber GF, Chousterman BG, Hilgendorf I, et al. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J Exp Med. 2014;211(6):1243–1256. doi:10.1084/jem.20131471
45. Rauch PJ, Chudnovskiy A, Robbins CS, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335(6068):597–601. doi:10.1126/science.1215173
46. Snapper CM, Moorman MA, Rosas FR, Kehry MR, Maliszewski CR, Mond JJ. IL-3 and granulocyte-macrophage colony-stimulating factor strongly induce Ig secretion by sort-purified murine B cell activated through the membrane Ig, but not the CD40, signaling pathway. J Immunol. 1995;154(11):5842–5850.
47. Li R, Rezk A, Miyazaki Y, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. 2015;7(310):310ra166. doi:10.1126/scitranslmed.aab4176
48. Li R, Rezk A, Ghadiri M, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in b cell subsets of patients with multiple sclerosis. J Immunol. 2017;198(2):691–698. doi:10.4049/jimmunol.1601649
49. Smith MD, Martin KA, Calabresi PA, Bhargava P. Dimethyl fumarate alters B-cell memory and cytokine production in MS patients. Ann Clin Transl Neurol. 2017;4(5):351–355. doi:10.1002/acn3.411
50. Hirota K, Hashimoto M, Ito Y, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity. 2018;48(6):1220–1232e1225. doi:10.1016/j.immuni.2018.04.009
51. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks IP. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. 2016;213(10):1983–1998. doi:10.1084/jem.20151853
52. Chen G, Bracamonte-Baran W, Diny NL, et al. Sca-1(+) cardiac fibroblasts promote development of heart failure. Eur J Immunol. 2018;48(9):1522–1538. doi:10.1002/eji.201847583
53. Willart MA, Deswarte K, Pouliot P, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. 2012;209(8):1505–1517. doi:10.1084/jem.20112691
54. Sheih A, Parks WC, Ziegler SF. GM-CSF produced by the airway epithelium is required for sensitization to cockroach allergen. Mucosal Immunol. 2017;10(3):705–715.
55. Cakarova L, Marsh LM, Wilhelm J, et al. Macrophage tumor necrosis factor-alpha induces epithelial expression of granulocyte-macrophage colony-stimulating factor: impact on alveolar epithelial repair. Am J Respir Crit Care Med. 2009;180(6):521–532. doi:10.1164/rccm.200812-1837OC
56. Schuett J, Schuett H, Oberoi R, et al. NADPH oxidase NOX2 mediates TLR2/6-dependent release of GM-CSF from endothelial cells. FASEB J. 2017;31(6):2612–2624. doi:10.1096/fj.201600729R
57. Pare A, Mailhot B, Levesque SA, et al. IL-1beta enables CNS access to CCR2(hi) monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc Natl Acad Sci U S A. 2018;115(6):E1194–E1203. doi:10.1073/pnas.1714948115
58. Montanari E, Stojkovic S, Kaun C, et al. Interleukin-33 stimulates GM-CSF and M-CSF production by human endothelial cells. Thromb Haemost. 2016;116(2):317–327. doi:10.1160/TH15-12-0917
59. Hamilton JA, Anderson GP. GM-CSF biology. Growth Factors. 2004;22(4):225–231. doi:10.1080/08977190412331279881
60. Ribechini E, Hutchinson JA, Hergovits S, et al. Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv. 2017;1(14):947–960. doi:10.1182/bloodadvances.2017006858
61. Lacey DC, Achuthan A, Fleetwood AJ, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol. 2012;188(11):5752–5765. doi:10.4049/jimmunol.1103426
62. Collins HL, Bancroft GJ. Cytokine enhancement of complement-dependent phagocytosis by macrophages: synergy of tumor necrosis factor-alpha and granulocyte-macrophage colony-stimulating factor for phagocytosis of cryptococcus neoformans. Eur J Immunol. 1992;22(6):1447–1454. doi:10.1002/eji.1830220617
63. LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J Clin Invest. 1999;103(4):563–569. doi:10.1172/JCI5212
64. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS
65. McDermott AJ, Frank CR, Falkowski NR, McDonald RA, Young VB, Huffnagle GB. Role of GM-CSF in the inflammatory cytokine network that regulates neutrophil influx into the colonic mucosa during clostridium difficile infection in mice. Gut Microbes. 2014;5(4):476–484. doi:10.4161/gmic.29964
66. Darrieutort-Laffite C, Boutet MA, Chatelais M, et al. IL-1beta and TNFalpha promote monocyte viability through the induction of GM-CSF expression by rheumatoid arthritis synovial fibroblasts. Mediators Inflamm. 2014;2014:241840. doi:10.1155/2014/241840
67. Dabritz J, Weinhage T, Varga G, et al. Reprogramming of monocytes by GM-CSF contributes to regulatory immune functions during intestinal inflammation. J Immunol. 2015;194(5):2424–2438. doi:10.4049/jimmunol.1401482
68. Sorgi CA, Rose S, Court N, et al. GM-CSF priming drives bone marrow-derived macrophages to a pro-inflammatory pattern and downmodulates PGE2 in response to TLR2 ligands. PLoS One. 2012;7(7):e40523. doi:10.1371/journal.pone.0040523
69. Lendemans S, Rani M, Selbach C, Kreuzfelder E, Schade FU, Flohe S. GM-CSF priming of human monocytes is dependent on ERK1/2 activation. J Endotoxin Res. 2006;12(1):10–20. doi:10.1177/09680519060120010201
70. Jablonska E, Kiluk M, Markiewicz W, Jablonski J. Priming effects of GM-CSF, IFN-gamma and TNF-alpha on human neutrophil inflammatory cytokine production. Melanoma Res. 2002;12(2):123–128. doi:10.1097/00008390-200204000-00004
71. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178(8):5245–5252. doi:10.4049/jimmunol.178.8.5245
72. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
73. King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med. 2010;207(5):953–961. doi:10.1084/jem.20091844
74. Daro E, Pulendran B, Brasel K, et al. Polyethylene glycol-modified GM-CSF expands CD11b(high)CD11c(high) but notCD11b(low)CD11c(high) murine dendritic cells in vivo: a comparative analysis with Flt3 ligand. J Immunol. 2000;165(1):49–58. doi:10.4049/jimmunol.165.1.49
75. Suzuki H, Katayama N, Ikuta Y, et al. Activities of granulocyte-macrophage colony-stimulating factor and interleukin-3 on monocytes. Am J Hematol. 2004;75(4):179–189. doi:10.1002/ajh.20010
76. Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. doi:10.1084/jem.176.6.1693
77. Conti L, Gessani S. GM-CSF in the generation of dendritic cells from human blood monocyte precursors: recent advances. Immunobiology. 2008;213(9–10):859–870. doi:10.1016/j.imbio.2008.07.017
78. Helft J, Bottcher J, Chakravarty P, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. 2015;42(6):1197–1211. doi:10.1016/j.immuni.2015.05.018
79. Louis C, Cook AD, Lacey D, et al. Specific contributions of CSF-1 and GM-CSF to the dynamics of the mononuclear phagocyte system. J Immunol. 2015;195(1):134–144. doi:10.4049/jimmunol.1500369
80. Lee KM, Jarnicki A, Achuthan A, et al. CCL17 in inflammation and pain. J Immunol. 2020;205(1):213–222. doi:10.4049/jimmunol.2000315
81. Ko HJ, Brady JL, Ryg-Cornejo V, et al. GM-CSF-responsive monocyte-derived dendritic cells are pivotal in Th17 pathogenesis. J Immunol. 2014;192(5):2202–2209. doi:10.4049/jimmunol.1302040
82. Greter M, Helft J, Chow A, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36(6):1031–1046. doi:10.1016/j.immuni.2012.03.027
83. Reynolds G, Gibbon JR, Pratt AG, et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. 2016;75(5):899–907. doi:10.1136/annrheumdis-2014-206578
84. Campbell IK, van Nieuwenhuijze A, Segura E, et al. Differentiation of inflammatory dendritic cells is mediated by NF-kappaB1-dependent GM-CSF production in CD4 T cells. J Immunol. 2011;186(9):5468–5477. doi:10.4049/jimmunol.1002923
85. Hamilton JA. GM-CSF-dependent inflammatory pathways. Front Immunol. 2019;10:2055. doi:10.3389/fimmu.2019.02055
86. Bourdely P, Anselmi G, Vaivode K, et al. Transcriptional and functional analysis of CD1c(+) human dendritic cells identifies a CD163(+) subset priming CD8(+)CD103(+) T cells. Immunity. 2020;53(2):335–352e338. doi:10.1016/j.immuni.2020.06.002
87. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205(10):2281–2294. doi:10.1084/jem.20071119
88. Yong KL, Rowles PM, Patterson KG, Linch DC. Granulocyte-macrophage colony-stimulating factor induces neutrophil adhesion to pulmonary vascular endothelium in vivo: role of beta 2 integrins. Blood. 1992;80(6):1565–1575. doi:10.1182/blood.V80.6.1565.1565
89. Sakagami T, Uchida K, Suzuki T, et al. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N Engl J Med. 2009;361(27):2679–2681. doi:10.1056/NEJMc0904077
90. Wright HL, Thomas HB, Moots RJ, Edwards SW. RNA-seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS One. 2013;8(3):e58598. doi:10.1371/journal.pone.0058598
91. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16(11):1438–1444. doi:10.1038/cdd.2009.96
92. Alferink J, Lieberam I, Reindl W, et al. Compartmentalized production of CCL17 in vivo: strong inducibility in peripheral dendritic cells contrasts selective absence from the spleen. J Exp Med. 2003;197(5):585–599. doi:10.1084/jem.20021859
93. Iellem A, Mariani M, Lang R, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194(6):847–853. doi:10.1084/jem.194.6.847
94. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol. 2015;27(1):11–20. doi:10.1093/intimm/dxu079
95. Russo RC, Savino B, Mirolo M, et al. The atypical chemokine receptor ACKR2 drives pulmonary fibrosis by tuning influx of CCR2(+) and CCR5(+) IFNgamma-producing gammadeltaT cells in mice. Am J Physiol Lung Cell Mol Physiol. 2018;314(6):L1010–L1025. doi:10.1152/ajplung.00233.2017
96. Hsu AT, Lupancu TJ, Lee MC, et al. Epigenetic and transcriptional regulation of IL4-induced CCL17 production in human monocytes and murine macrophages. J Biol Chem. 2018;293(29):11415–11423. doi:10.1074/jbc.RA118.002416
97. Lee KM, Prasad V, Achuthan A, Fleetwood AJ, Hamilton JA, Cook AD. Targeting GM-CSF for collagenase-induced osteoarthritis pain and disease in mice. Osteoarthritis Cartilage. 2020;28(4):486–491. doi:10.1016/j.joca.2020.01.012
98. Lee MC, Saleh R, Achuthan A, et al. CCL17 blockade as a therapy for osteoarthritis pain and disease. Arthritis Res Ther. 2018;20(1):62. doi:10.1186/s13075-018-1560-9
99. Cook AD, Lee MC, Saleh R, et al. TNF and granulocyte macrophage-colony stimulating factor interdependence mediates inflammation via CCL17. JCI Insight. 2018;3(6):6. doi:10.1172/jci.insight.99249
100. Burmester GR, McInnes IB, Kremer JM, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor alpha monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70(5):679–689.
101. Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol. 2018;39(3):240–255. doi:10.1016/j.it.2017.12.003
102. Thakur M, Crow M, Richards N, et al. Defining the nociceptor transcriptome. Front Mol Neurosci. 2014;7:87. doi:10.3389/fnmol.2014.00087
103. Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21(14):5027–5035. doi:10.1523/JNEUROSCI.21-14-05027.2001
104. Li CL, Li KC, Wu D, et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res. 2016;26(1):83–102. doi:10.1038/cr.2015.149
105. Weber C, Meiler S, Doring Y, et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. 2011;121(7):2898–2910. doi:10.1172/JCI44925
106. Heiseke AF, Faul AC, Lehr HA, et al. CCL17 promotes intestinal inflammation in mice and counteracts regulatory T cell-mediated protection from colitis. Gastroenterology. 2012;142(2):335–345. doi:10.1053/j.gastro.2011.10.027
107. Cook AD, Pobjoy J, Steidl S, et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther. 2012;14(5):R199. doi:10.1186/ar4037
108. Cook AD, Pobjoy J, Sarros S, et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in inflammatory and arthritic pain. Ann Rheum Dis. 2013;72(2):265–270.
109. Stosser S, Schweizerhof M, Kuner R. Hematopoietic colony-stimulating factors: new players in tumor-nerve interactions. J Mol Med (Berl). 2011;89(4):321–329.
110. Schweizerhof M, Stosser S, Kurejova M, et al. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med. 2009;15(7):802–807. doi:10.1038/nm.1976
111. Ridwan S, Bauer H, Frauenknecht K, von Pein H, Sommer CJ. Distribution of granulocyte-monocyte colony-stimulating factor and its receptor alpha-subunit in the adult human brain with specific reference to Alzheimer’s disease. J Neural Transm (Vienna). 2012;119(11):1389–1406. doi:10.1007/s00702-012-0794-y
112. Bali KK, Venkataramani V, Satagopam VP, Gupta P, Schneider R, Kuner R. Transcriptional mechanisms underlying sensitization of peripheral sensory neurons by granulocyte-/granulocyte-macrophage colony stimulating factors. Mol Pain. 2013;9:48. doi:10.1186/1744-8069-9-48
113. Nicol LSC, Thornton P, Hatcher JP, et al. Central inhibition of granulocyte-macrophage colony-stimulating factor is analgesic in experimental neuropathic pain. Pain. 2018;159(3):550–559. doi:10.1097/j.pain.0000000000001130
114. Zhang F, Wang Y, Liu Y, et al. Transcriptional regulation of voltage-gated sodium channels contributes to GM-CSF-induced pain. J Neurosci. 2019;39(26):5222–5233. doi:10.1523/JNEUROSCI.2204-18.2019
115. Tewari D, Cook AD, Lee MC, et al. Granulocyte-macrophage colony stimulating factor as an indirect mediator of nociceptor activation and pain. J Neurosci. 2020;40(11):2189–2199. doi:10.1523/JNEUROSCI.2268-19.2020
116. Schabitz WR, Kruger C, Pitzer C, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J Cereb Blood Flow Metab. 2008;28(1):29–43. doi:10.1038/sj.jcbfm.9600496
117. Kelso ML, Elliott BR, Haverland NA, Mosley RL, Gendelman HE. Granulocyte-macrophage colony stimulating factor exerts protective and immunomodulatory effects in cortical trauma. J Neuroimmunol. 2015;278:162–173. doi:10.1016/j.jneuroim.2014.11.002
118. Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II. Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest. 1989;83(3):876–882. doi:10.1172/JCI113971
119. Fiehn C, Wermann M, Pezzutto A, Hufner M, Heilig B. Plasma GM-CSF concentrations in rheumatoid arthritis, systemic lupus erythematosus and spondyloarthropathy. Z Rheumatol. 1992;51(3):121–126.
120. Hazenberg BP, Van Leeuwen MA, Van Rijswijk MH, Stern AC, Vellenga E. Correction of granulocytopenia in Felty’s syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood. 1989;74(8):2769–2770. doi:10.1182/blood.V74.8.2769.2769
121. Okada Y, Terao C, Ikari K, et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet. 2012;44(5):511–516. doi:10.1038/ng.2231
122. Shi H, Chen L, Ridley A, et al. GM-CSF primes proinflammatory monocyte responses in ankylosing spondylitis. Front Immunol. 2020;11:1520. doi:10.3389/fimmu.2020.01520
123. Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1998;161(7):3639–3644.
124. Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001;3(5):293–298. doi:10.1186/ar318
125. Bystrom J, Clanchy FI, Taher TE, et al. Response to treatment with TNFalpha inhibitors in rheumatoid arthritis is associated with high levels of GM-CSF and GM-CSF(+) T lymphocytes. Clin Rev Allergy Immunol. 2017;53(2):265–276. doi:10.1007/s12016-017-8610-y
126. van Helvoort EM, Eijkelkamp N, Lafeber F, Mastbergen SC. Expression of granulocyte macrophage-colony stimulating factor and its receptor in the synovium of osteoarthritis patients is negatively correlated with pain. Rheumatology (Oxford). 2020. doi:10.1093/rheumatology/keaa199
127. Berenbaum F, Rajzbaum G, Amor B, Toubert A. Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur Cytokine Netw. 1994;5(1):43–46.
128. Moradi B, Rosshirt N, Tripel E, et al. Unicompartmental and bicompartmental knee osteoarthritis show different patterns of mononuclear cell infiltration and cytokine release in the affected joints. Clin Exp Immunol. 2015;180(1):143–154. doi:10.1111/cei.12486
129. Klein-Wieringa IR, de Lange-brokaar BJ, Yusuf E, et al. Inflammatory cells in patients with endstage knee osteoarthritis: a comparison between the synovium and the infrapatellar fat pad. J Rheumatol. 2016;43(4):771–778. doi:10.3899/jrheum.151068
130. Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–1267. doi:10.1136/ard.2004.025270
131. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol. 2019;41(3):283–297.
132. Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119(1):61–69.
133. Codarri L, Gyulveszi G, Tosevski V, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–567. doi:10.1038/ni.2027
134. El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–575. doi:10.1038/ni.2031
135. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178(1):39–48. doi:10.4049/jimmunol.178.1.39
136. Gonzalez H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation. 2014;11(1):201. doi:10.1186/s12974-014-0201-8
137. Parajuli B, Sonobe Y, Kawanokuchi J, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation. 2012;9:268.
138. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113(14):3190–3197. doi:10.1182/blood-2008-07-168575
139. Vogel DY, Kooij G, Heijnen PD, et al. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur J Immunol. 2015;45(6):1808–1819. doi:10.1002/eji.201444960
140. Vogel DY, Vereyken EJ, Glim JE, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation. 2013;10(1):35. doi:10.1186/1742-2094-10-35
141. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–717. doi:10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
142. Duncker PC, Stoolman JS, Huber AK, Segal BM. GM-CSF promotes chronic disability in experimental autoimmune encephalomyelitis by altering the composition of central nervous system-infiltrating cells, but is dispensable for disease induction. J Immunol. 2018;200(3):966–973.
143. McQualter JL, Darwiche R, Ewing C, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194(7):873–882. doi:10.1084/jem.194.7.873
144. Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998;20(3):373–382. doi:10.3109/08923979809034820
145. Perrella O, Carrieri PB, De Mercato R, Buscaino GA. Markers of activated T lymphocytes and T cell receptor gamma/delta+ in patients with multiple sclerosis. Eur Neurol. 1993;33(2):152–155. doi:10.1159/000116923
146. Aharoni R, Eilam R, Schottlender N, et al. Glatiramer acetate increases T- and B -regulatory cells and decreases granulocyte-macrophage colony-stimulating factor (GM-CSF) in an animal model of multiple sclerosis. J Neuroimmunol. 2020;345:577281. doi:10.1016/j.jneuroim.2020.577281
147. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12(4):205–217. doi:10.1038/nrgastro.2015.34
148. Marks DJ, Rahman FZ, Sewell GW, Segal AW. Crohn’s disease: an immune deficiency state. Clin Rev Allergy Immunol. 2010;38(1):20–31. doi:10.1007/s12016-009-8133-2
149. Griseri T, McKenzie BS, Schiering C, Powrie F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity. 2012;37(6):1116–1129. doi:10.1016/j.immuni.2012.08.025
150. Griseri T, Arnold IC, Pearson C, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity. 2015;43(1):187–199. doi:10.1016/j.immuni.2015.07.008
151. Xu Y, Hunt NH, Bao S. The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res. 2008;18(12):1220–1229. doi:10.1038/cr.2008.310
152. Dabritz J. Granulocyte macrophage colony-stimulating factor and the intestinal innate immune cell homeostasis in Crohn’s disease. Am J Physiol Gastrointest Liver Physiol. 2014;306(6):G455–465. doi:10.1152/ajpgi.00409.2013
153. Bernasconi E, Favre L, Maillard MH, et al. Granulocyte-macrophage colony-stimulating factor elicits bone marrow-derived cells that promote efficient colonic mucosal healing. Inflamm Bowel Dis. 2010;16(3):428–441. doi:10.1002/ibd.21072
154. Dieckgraefe BK, Korzenik JR. Treatment of active Crohn’s disease with recombinant human granulocyte-macrophage colony-stimulating factor. Lancet. 2002;360(9344):1478–1480. doi:10.1016/S0140-6736(02)11437-1
155. Gathungu G, Zhang Y, Tian X, et al. Impaired granulocyte-macrophage colony-stimulating factor bioactivity accelerates surgical recurrence in ileal Crohn’s disease. World J Gastroenterol. 2018;24(5):623–630. doi:10.3748/wjg.v24.i5.623
156. Denson LA, Jurickova I, Karns R, et al. Genetic and transcriptomic variation linked to neutrophil granulocyte-macrophage colony-stimulating factor signaling in pediatric crohn’s disease. Inflamm Bowel Dis. 2019;25(3):547–560. doi:10.1093/ibd/izy265
157. Atzeni F, Gerardi MC, Barilaro G, Masala IF, Benucci M, Sarzi-Puttini P. Interstitial lung disease in systemic autoimmune rheumatic diseases: a comprehensive review. Expert Rev Clin Immunol. 2018;14(1):69–82. doi:10.1080/1744666X.2018.1411190
158. Sakaguchi N, Takahashi T, Hata H, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426(6965):454–460. doi:10.1038/nature02119
159. Guerard S, Boieri M, Hultqvist M, Holmdahl R, Wing K. The SKG mutation in ZAP-70 also confers arthritis susceptibility in C57 black mouse strains. Scand J Immunol. 2016;84(1):3–11. doi:10.1111/sji.12438
160. Benham H, Rehaume LM, Hasnain SZ, et al. Interleukin-23 mediates the intestinal response to microbial beta-1,3-glucan and the development of spondyloarthritis pathology in SKG mice. Arthritis Rheumatol. 2014;66(7):1755–1767. doi:10.1002/art.38638
161. Kwon OC, Lee EJ, Chang EJ, et al. IL-17A(+)GM-CSF(+) neutrophils are the major infiltrating cells in interstitial lung disease in an autoimmune arthritis model. Front Immunol. 2018;9:1544. doi:10.3389/fimmu.2018.01544
162. Shiomi A, Usui T, Ishikawa Y, Shimizu M, Murakami K, Mimori T. GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J Immunol. 2014;193(2):849–859. doi:10.4049/jimmunol.1303255
163. Chen YT, Hsu H, Lin CC, et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J Pathol. 2020;250(1):55–66. doi:10.1002/path.5350
164. Son BK, Sawaki D, Tomida S, et al. Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat Commun. 2015;6(1):6994. doi:10.1038/ncomms7994
165. Ye P, Chen W, Wu J, et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest. 2013;123(5):2317–2331. doi:10.1172/JCI67356
166. Nouri-Aria KT, Masuyama K, Jacobson MR, et al. Granulocyte/macrophage-colony stimulating factor in allergen-induced rhinitis: cellular localization, relation to tissue eosinophilia and influence of topical corticosteroid. Int Arch Allergy Immunol. 1998;117(4):248–254. doi:10.1159/000024019
167. Cates EC, Fattouh R, Wattie J, et al. Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF-mediated mechanism. J Immunol. 2004;173(10):6384–6392. doi:10.4049/jimmunol.173.10.6384
168. Yamashita N, Tashimo H, Ishida H, et al. Attenuation of airway hyperresponsiveness in a murine asthma model by neutralization of granulocyte-macrophage colony-stimulating factor (GM-CSF). Cell Immunol. 2002;219(2):92–97. doi:10.1016/S0008-8749(02)00565-8
169. Ait Yahia S, Azzaoui I, Everaere L, et al. CCL17 production by dendritic cells is required for NOD1-mediated exacerbation of allergic asthma. Am J Respir Crit Care Med. 2014;189(8):899–908. doi:10.1164/rccm.201310-1827OC
170. Perros F, Hoogsteden HC, Coyle AJ, Lambrecht BN, Hammad H. Blockade of CCR4 in a humanized model of asthma reveals a critical role for DC-derived CCL17 and CCL22 in attracting Th2 cells and inducing airway inflammation. Allergy. 2009;64(7):995–1002. doi:10.1111/j.1398-9995.2009.02095.x
171. Yuan L, Zhang X, Yang M, et al. Airway epithelial integrin beta4 suppresses allergic inflammation by decreasing CCL17 production. Clin Sci (Lond). 2020;134(13):1735–1749. doi:10.1042/CS20191188
172. Chen YL, Chiang BL. Targeting TSLP with shRNA alleviates airway inflammation and decreases epithelial CCL17 in a murine model of asthma. Mol Ther Nucleic Acids. 2016;5:e316. doi:10.1038/mtna.2016.29
173. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. doi:10.1038/nature21363
174. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi:10.1038/nature05485
175. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi:10.1172/JCI200319451
176. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW
177. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi:10.1172/JCI29881
178. Boi SK, Buchta CM, Pearson NA, et al. Obesity alters immune and metabolic profiles: new insight from obese-resistant mice on high-fat diet. Obesity (Silver Spring). 2016;24(10):2140–2149. doi:10.1002/oby.21620
179. Shaw OM, Pool B, Dalbeth N, Harper JL. The effect of diet-induced obesity on the inflammatory phenotype of non-adipose-resident macrophages in an in vivo model of gout. Rheumatology (Oxford). 2014;53(10):1901–1905. doi:10.1093/rheumatology/keu174
180. Kim DH, Sandoval D, Reed JA, et al. The role of GM-CSF in adipose tissue inflammation. Am J Physiol Endocrinol Metab. 2008;295(5):E1038–1046. doi:10.1152/ajpendo.00061.2008
181. Plubell DL, Fenton AM, Wilmarth PA, et al. GM-CSF driven myeloid cells in adipose tissue link weight gain and insulin resistance via formation of 2-aminoadipate. Sci Rep. 2018;8(1):11485. doi:10.1038/s41598-018-29250-8
182. Pamir N, Liu NC, Irwin A, et al. Granulocyte/macrophage colony-stimulating factor-dependent dendritic cells restrain lean adipose tissue expansion. J Biol Chem. 2015;290(23):14656–14667. doi:10.1074/jbc.M115.645820
183. Quail DF, Olson OC, Bhardwaj P, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol. 2017;19(8):974–987. doi:10.1038/ncb3578
184. Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020;39(5):405–407. doi:10.1016/j.healun.2020.03.012
185. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368(6490):473–474. doi:10.1126/science.abb8925
186. Mehta P, McAuley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi:10.1016/S0140-6736(20)30628-0
187. Liao M, Liu Y, Yuan J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020;26(6):842–844. doi:10.1038/s41591-020-0901-9
188. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000e1003. doi:10.1016/j.chom.2020.04.009
189. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217(6):6. doi:10.1084/jem.20200652
190. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–970. doi:10.2217/imt-2016-0020
191. Sinha P, Matthay MA, Calfee CS. Is a “cytokine storm” relevant to COVID-19? JAMA Intern Med. 2020;180(9):1152. doi:10.1001/jamainternmed.2020.3313
192. Del Valle DM, Kim-Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26(10):1636–1643. doi:10.1038/s41591-020-1051-9
193. Zhou Y, Fu B, Zheng X, et al. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl Sci Rev. 2020;7(6):998–1002. doi:10.1093/nsr/nwaa041
194. Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709. doi:10.1182/blood-2018-10-881722
195. Tugues S, Amorim A, Spath S, et al. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci Transl Med. 2018;10(469):469. doi:10.1126/scitranslmed.aat8410
196. Puljic R, Benediktus E, Plater-Zyberk C, et al. Lipopolysaccharide-induced lung inflammation is inhibited by neutralization of GM-CSF. Eur J Pharmacol. 2007;557(2–3):230–235. doi:10.1016/j.ejphar.2006.11.023
197. De Alessandris S, Ferguson GJ, Dodd AJ, et al. Neutrophil GM-CSF receptor dynamics in acute lung injury. J Leukoc Biol. 2019;105(6):1183–1194. doi:10.1002/JLB.3MA0918-347R
198. Bozinovski S, Jones JE, Vlahos R, Hamilton JA, Anderson GP. Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J Biol Chem. 2002;277(45):42808–42814. doi:10.1074/jbc.M207840200
199. Lang FM, Lee KM-C, Teijaro JR, Becher B, Hamilton JA. GM-CSF-based treatments in COVID-19: reconciling opposing therapeutic approaches. Nat Rev Immunol. 2020;20(8):507–514. doi:10.1038/s41577-020-0357-7
200. Bonaventura A, Vecchie A, Wang TS, et al. Targeting GM-CSF in COVID-19 pneumonia: rationale and strategies. Front Immunol. 2020;11:1625. doi:10.3389/fimmu.2020.01625
201. Behrens F, Tak PP, Ostergaard M, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74(6):1058–1064. doi:10.1136/annrheumdis-2013-204816
202. Schett GBC, Berkowitz M, Davy K, et al. A Phase iia study of anti-GM-CSF antibody GSK3196165 in subjects with inflammatory hand osteoarthritis. Arthritis Rheumatol. 2018;70.
203. Patnaik MM, Sallman DA, Mangaonkar AA, et al. Phase 1 study of lenzilumab, a recombinant anti-human GM-CSF antibody, for chronic myelomonocytic leukemia. Blood. 2020;136(7):909–913. doi:10.1182/blood.2019004352
204. Molfino NA, Kuna P, Leff JA, et al. Phase 2, randomised placebo-controlled trial to evaluate the efficacy and safety of an anti-GM-CSF antibody (KB003) in patients with inadequately controlled asthma. BMJ Open. 2016;6(1):e007709. doi:10.1136/bmjopen-2015-007709
205. Temesgen Z, Assi M, Vergidis P, et al. First clinical use of lenzilumab to neutralize GM-CSF in patients with severe COVID-19 pneumonia. medRxiv. 2020.
206. Tanaka S, Harada S, Hiramatsu N, Nakaya R, Kawamura M. Randomized, double-blind, placebo-controlled, phase I study of the safety and pharmacokinetics of namilumab in healthy Japanese and Caucasian men. Int J Clin Pharmacol Ther. 2018;56(11):507–517. doi:10.5414/CP203235
207. Huizinga TW, Batalov A, Stoilov R, et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):53. doi:10.1186/s13075-017-1267-3
208. Taylor PC, Saurigny D, Vencovsky J, et al. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: a randomized, controlled trial. Arthritis Res Ther. 2019;21(1):101.
209. Papp KA, Gooderham M, Jenkins R, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) as a therapeutic target in psoriasis: randomized, controlled investigation using namilumab, a specific human anti-GM-CSF monoclonal antibody. Br J Dermatol. 2019;180(6):1352–1360. doi:10.1111/bjd.17195
210. Nair JR, Edwards SW, Moots RJ. Mavrilimumab, a human monoclonal GM-CSF receptor-alpha antibody for the management of rheumatoid arthritis: a novel approach to therapy. Expert Opin Biol Ther. 2012;12(12):1661–1668. doi:10.1517/14712598.2012.732062
211. Weinblatt ME, McInnes IB, Kremer JM, et al. A randomized phase IIb study of mavrilimumab and golimumab in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(1):49–59. doi:10.1002/art.40323
212. Burmester GR, McInnes IB, Kremer J, et al. A randomised phase IIb study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1020–1030. doi:10.1136/annrheumdis-2016-210624
213. De Luca G, Cavalli G, Campochiaro C, et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: a single-centre, prospective cohort study. Lancet Rheumatol. 2020;2(8):E465–E473. doi:10.1016/S2665-9913(20)30170-3
214. McInnes IB, Byers NL, Higgs RE, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. 2019;21(1):183. doi:10.1186/s13075-019-1964-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.