Back to Journals » Infection and Drug Resistance » Volume 19
Genomic Characterization of Antibiotic-Resistant Enterococcus Spp. Strains in Clinical Settings of Yichang, China
Authors Yu H, Xiong P, Zhang H, Lu Z, Chen X, Yan R, Wang L, Yu L, Zhang Q, Zou L ![]() , Wang L
, Wang L
Received 2 December 2025
Accepted for publication 9 March 2026
Published 14 April 2026 Volume 2026:19 573082
DOI https://doi.org/10.2147/IDR.S573082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hazrat Bilal
Hong Yu,1– 3 Peng Xiong,2,3 HuiNa Zhang,2,3 Zhi Lu,2,3 XinLu Chen,2,3 RuHua Yan,2,3 Lei Wang,2,3 Long Yu,2,3 Qingyong Zhang,4,5 Lili Zou,2,3 Lu Wang2,3
1Department of Medical Ultrasound, Tongji Hospital, Tongji Medical College, and State Key Laboratory for Diagnosis and Treatment of Severe Zoonotic Infectious Diseases, Huazhong University of Science and Technology, Wuhan, Hubei, 430030, People’s Republic of China; 2Hubei Key Laboratory of Tumor Microenvironment and Immunotherapy, College of Basic Medical Sciences, China Three Gorges University, Yichang, Hubei, 443002, People’s Republic of China; 3Institute of Infection and Inflammation, College of Basic Medical Sciences, China Three Gorges University, Yichang, Hubei, 443002, People’s Republic of China; 4The First College of Clinical Medical Science, China Three Gorges University, Yichang, Hubei, 443002, People’s Republic of China; 5Department of Clinical Laboratory of Yichang Central People’s Hospital, Yichang, Hubei, 443002, People’s Republic of China
Correspondence: Lu Wang, Email [email protected]
Objective: Antimicrobial-resistant Enterococcus species are important causes of hospital-acquired infections, yet genomic surveillance data from Yichang, China, are lacking.
Methods: Between February 2023 and December 2024, 150 antibiotic-resistant Enterococcus isolates were collected from five hospitals in Yichang. Antimicrobial susceptibility testing was performed according to CLSI standards. Fifty-nine representative isolates underwent whole-genome sequencing for species identification, multilocus sequence typing (MLST), antimicrobial resistance (AMR) gene detection, virulence profiling, and phylogenetic analysis.
Results: E. faecalis (46.7%) and E. faecium (42.7%) were the predominant species, mainly isolated from urine specimens. High multidrug resistance rates were observed, particularly in E. faecium, which showed extensive resistance to β-lactams and fluoroquinolones. MLST analysis identified E. faecium ST78 and E. faecalis ST16 as the dominant clones, both corresponding to internationally reported epidemic lineages. Importantly, two novel E. faecalis sequence types were identified, each resulting from single-nucleotide substitutions in housekeeping genes, indicating ongoing local genetic evolution. Resistome analysis revealed enrichment of vancomycin, tetracycline, aminoglycoside, and lincosamide resistance genes in Yichang isolates compared with reference genomes. In addition, E. faecalis carried a broader repertoire of resistance and virulence genes than E. faecium.
Conclusion: Our findings demonstrate that whole-genome sequencing is not only a powerful tool for characterizing dominant epidemic clones but also an effective strategy for surveillance of atypical and emerging Enterococcus species. The detection of two novel sequence types and region-specific resistance patterns highlights the value of genomic monitoring in guiding infection-control policies, antimicrobial stewardship, and early warning of multidrug-resistant strain dissemination in hospital settings.
Keywords: antimicrobial-resistant Enterococcus, whole-genome sequencing, multilocus sequence typing, resistance gene, virulence gene
Introduction
Enterococcus spp. are Gram-positive cocci that are ubiquitous in the environmental-including soil, water, and the gastrointestinal tracts of humans and animals.1 These opportunistic pathogens are responsible for a wide spectrum of infections, such as urinary tract infections, intra-abdominal infections, and infective endocarditis.2–4 Enterococci currently account for approximately 10–15% of hospital-acquired infections worldwide and pose particular risks to elderly, immunocompromised, and critically ill patients, especially those undergoing invasive procedures or prolonged hospitalization.5,6 In these high-risk populations, infections are associated with increased morbidity, mortality, and healthcare burden.
Over recent decades, extensive use of broad-spectrum antibiotics and horizontal gene transfer have driven the emergence of multidrug-resistant (MDR) Enterococcus strains.7,8 Particularly concerning are resistances to glycopeptides and aminoglycosides, which considerably limit treatment options and increase mortality risk. Horizontal gene transfer further accelerates the evolution of multidrug-resistant (MDR) strains, complicating infection management in hospital environments.9 Recognizing the growing threat of antimicrobial resistance, the World Health Organization (WHO) classified vancomycin-resistant Enterococcus faecium (VRE) as a Priority 2: High pathogen in its Global Priority Pathogens List (initially released in 2017 and reaffirmed in 2024).10 This highlights the urgent need for improved surveillance and infection control strategies against VRE and other resistant Enterococcus lineages that are increasingly reported in clinical settings worldwide.11 However, focusing exclusively on clinically defined resistant subsets such as VRE may overlook the broader evolutionary landscape of resistance accumulation within Enterococcus populations.12 Resistance determinants often emerge progressively through clonal expansion and gene acquisition before reaching defined clinical thresholds.12 Therefore, comprehensive genomic surveillance of antibiotic-resistant Enterococcus spp., rather than restriction to VRE alone, is essential for early detection of high-risk clones and evolving resistance traits.
The advent of whole-genome sequencing (WGS) has revolutionized pathogen surveillance by enabling high-resolution insights into species diversity, virulence factors, and antimicrobial-resistance (AMR) determinants. Multilocus sequence typing (MLST), a foundational approach in molecular epidemiology, continues to provide a robust framework for strain classification.13 Comparative-genomic analyses have even revealed novel Enterococcus species, emphasizing the genus’s adaptability and evolutionary potential.
Although several regional studies in China have investigated the molecular epidemiology of Enterococcus,14,15 genomic data from Yichang remain scarce. As an important regional healthcare hub in central China, Yichang serves a large hospitalized and referred patient population. The lack of local genomic surveillance limits understanding of resistance evolution, clonal dissemination, and potential emergence of atypical or high-risk Enterococcus lineages in this region. Therefore, this study conducted comprehensive whole-genome sequencing of antimicrobial-resistant Enterococcus isolates collected from clinical settings in Yichang. By integrating phenotypic resistance profiles, MLST typing, and genomic analyses of AMR and virulence genes, we aimed to establish region-specific molecular epidemiological evidence to support antimicrobial stewardship and infection-control strategies.
Methods
Isolate Collection and Ethical Statement
Between February 2023 and December 2024, a total of 150 antibiotic-resistant Enterococcus isolates were collected from five hospitals in Yichang, Hubei Province, China—Yichang Central People’s Hospital, Yichang Second People’s Hospital, Yichang Youfu Hospital, Zigui County People’s Hospital, and Yidu First People’s Hospital. These institutions were selected to represent both plain and mountainous regions according to the city’s geomorphological distribution. Suspected Enterococcus colonies were preliminarily identified using the Autof MS 1000 MALDI-TOF MS system (Autobio Diagnostics, China), and confirmed isolates were stored at –80°C for subsequent analyses.
All isolates were obtained as residual anonymized materials from routine clinical diagnostic procedures, with no additional sampling or patient intervention conducted for this study. The use of these fully anonymized bacterial isolates and associated de‑identified clinical data (specimen type, ward, susceptibility profile) was reviewed and approved for exemption of ethical approval by the Ethics Committee of Yichang Central People’s Hospital (the lead institution). The exemption was granted in accordance with national regulations and international guidelines (eg., the Declaration of Helsinki) for retrospective studies that utilize irreversibly anonymized specimens and pose no risk to patient privacy or welfare.
Antimicrobial Susceptibility Testing
Antimicrobial susceptibility testing (AST) was performed using the VITEK 2 Compact automated system (bioMérieux, France), and minimum inhibitory concentrations (MICs) were interpreted according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (CLSI, 2023). The antimicrobial panel included routinely tested agents such as aminoglycosides, penicillins, vancomycin, and linezolid. Resistance phenotypes were determined to guide selection of representative isolates for genomic analysis.
Multilocus Sequence Typing (MLST)
MLST was carried out following the Enterococcus schemes available in the PubMLST database (https://pubmlst.org/data) using the MLST software package. Validated seven-locus schemes were applied for E. faecium (adk, atpA, ddl, gdh, gyd, pstS, and purK) and E. faecalis (aroE, gdh, gki, gyd, pstS, xpt, and yqiL). Allele assignment and sequence type (ST) designation were performed using custom Python scripts. Phylogenetic relationships were inferred from concatenated allele sequences. The distribution of STs was analyzed to identify dominant or high-risk clones and compared with previously reported global lineages.
AMR Gene and Virulence Gene Analysis
Fifty-nine representative isolates, selected based on their antimicrobial-resistance phenotypes, were subjected to WGS by Magigene Biotechnology (Guangdong, China). Genomic DNA was extracted using a standard phenol–chloroform method, and sequencing libraries were prepared according to the manufacturer’s protocols. Raw reads were quality-trimmed and assembled with SPAdes v3.15.5. Genome annotation was performed using Prokka v1.14.6.
Whole-Genome Sequencing and Assembly
Sequencing libraries were prepared using the Illumina DNA Prep kit and sequenced on an Illumina NovaSeq 6000 platform with 150-bp paired-end reads. Raw reads were quality-filtered using fastp (v0.23.2), removing adapters, and reads with Phred scores <20. Assemblies were generated using SPAdes (v3.15.5), and assembly quality was assessed by QUAST (v5.2.0). The average genome coverage depth was approximately 150×, with N50 values ranging from 29,159 to 1,485,635 bp.
Antimicrobial-resistance and virulence factors were identified via local BLAST searches against the CARD and VFDB databases, respectively. AMR genes were identified using the Comprehensive Antibiotic Resistance Database (CARD, version 3.2.7) with a minimum identity threshold of 90% and coverage ≥70%. Virulence factors were detected by BLASTp against the Virulence Factor Database (VFDB, version 5.0) under the same criteria. Mobile genetic elements were filtered out by cross-referencing with IslandViewer 4 predictions. The resulting genomic features were summarized and visualized.
Phylogenetic Reconstruction
Two complete reference genomes per species were retrieved from the NCBI database. Single-copy core genes shared among reference and study isolates were identified using OrthoFinder v2.5.5. Core-gene alignments were generated with MAFFT v7 (G-INS-i algorithm), concatenated, and used to infer a maximum-likelihood phylogeny in IQ-TREE v2.2.2 with 1,000 bootstrap replicates. Lactobacillus casei (GenBank accession: GCA_000309565.2) served as the outgroup.12 The distribution of resistance and virulence genes was mapped onto the phylogenetic tree, which was visualized in iTOL v6.
The best-fit substitution model was selected using ModelFinder implemented in IQ-TREE. Phylogenetic inference was performed using the GTR+F+I+G4 model with 1,000 ultrafast bootstrap replicates (-bb 1000). Core genes were defined as single-copy orthologs present in ≥95% of isolates. SNPs were extracted from concatenated alignments using custom Python scripts.
Statistical Analysis
Descriptive statistics, including means and standard deviations, were calculated using R software (v4.3.2). Principal component analysis (PCA) of resistance and virulence gene profiles was performed using the prcomp function in R. No formal hypothesis testing or inferential statistical comparisons were conducted.
Results
Epidemiological Distribution and Clinical Characteristics
A total of 150 antimicrobial-resistant Enterococcus isolates were recovered, including 64 E. faecium, 70 E. faecalis, 8 E. avium, 6 E. casseliflavus, and one each of E. gallinarum and E. cecorum. Among them, E. faecalis and E. faecium were the predominant species (Figure 1A). Most isolates were obtained during the winter-to-spring period (Figure 1B). Notably, 84% of patients had undergone invasive procedures, indicating that instrumentation was a major predisposing factor; however, infections also occurred in non-surgical cases (Figure 1C). The majority of isolates were recovered from urine and blood samples, reflecting the well-known tendency of Enterococcus to cause urinary tract and bloodstream infections and its ability to persist in hospital environments (Figure 1D). Correspondingly, most cases were concentrated in surgical wards, particularly urology, where urinary tract infections are common (Figure 1E).
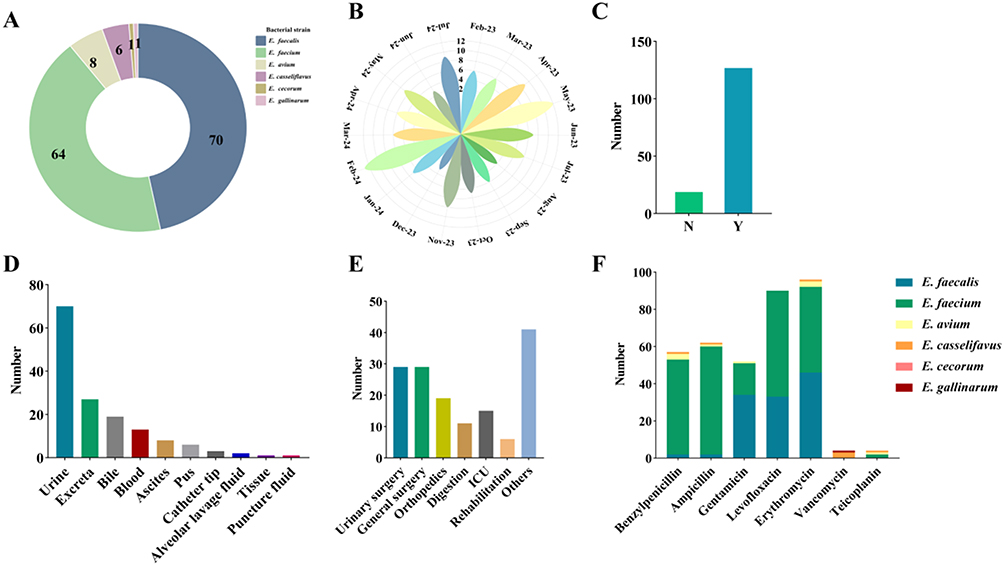 |
Figure 1 Summary of Clinical Isolate Information. (A) Species composition of antimicrobial-resistant Enterococcus isolates; (B) Temporal distribution of sample collection; (C) History of invasive procedures including surgery; (D) Specimen sources of isolates; (E) Distribution of isolates across hospital wards; (F) Antimicrobial resistance profiles of the 150 clinical isolates representing six Enterococcus species. |
Antimicrobial susceptibility testing revealed widespread multidrug resistance among the 150 clinical isolates. High resistance rates were observed for aminoglycosides, tetracyclines, and macrolides (Figure 1F). Both E. faecalis and E. faecium exhibited strong resistance to macrolides and fluoroquinolones, but their overall resistance profiles differed. E. faecalis showed notably high resistance to aminoglycosides—73.0% to erythromycin, 50.0% to levofloxacin, and 49.3% to gentamicin—whereas E. faecium demonstrated predominant resistance to β-lactams, with rates of 91.1% for penicillin G and 92.1% for ampicillin, along with 82.1% resistance to erythromycin, 24.6% to gentamicin, and 91.9% to levofloxacin. Collectively, these findings indicate that E. faecium exhibited a broader and more robust antimicrobial resistance phenotype than E. faecalis (Supplementary Table S1).
Genomic Characteristics and Assembly Quality of the Sequenced Isolates
A total of 59 Enterococcus isolates underwent whole-genome sequencing, including 28 E. faecium, 22 E. faecalis, 4 E. avium, 3 E. casseliflavus, and one isolate each of E. gallinarum and E. cecorum. Genome sizes ranged from 2.43 to 4.55 Mb, with GC contents between 36.58% and 42.55%. The average sequencing depth was approximately 150×. Assembly N50 values ranged from 29,159 to 1,485,635 bp. Detailed assembly statistics are provided in Supplementary Table S2, and all genome data have been deposited in the NCBI database under the accession numbers listed in Supplementary Table S2.
Species-Level Genomic Features
Species-specific genomic summaries are presented in Supplementary Table S3. The mean genome sizes of E. faecium and E. faecalis were 2.95 ± 0.12 Mb and 3.00 ± 0.12 Mb, respectively, with GC contents of 37.70 ± 0.19% and 37.27 ± 0.18%. E. avium exhibited a larger average genome size (4.45 ± 0.10 Mb), whereas E. casseliflavus and E. gallinarum displayed higher GC contents (>40%). The single E. cecorum genome measured 2.43 Mb with a GC content of 36.58%. The numbers of predicted genes, rRNA operons, and tRNAs varied among species.
MLST Typing Characteristics
Based on the 59 clinical Enterococcus genomes sequenced in this study and two complete reference genomes per species for E. faecium, E. faecalis, E. avium, E. casseliflavus, E. cecorum, and E. gallinarum retrieved from NCBI (12 references in total), together with Lactobacillus casei as an outgroup, we identified 509 452 SNPs across 679 single-copy core genes and constructed a maximum-likelihood phylogenetic tree. STs for E. faecium and E. faecalis were assigned via the PubMLST scheme, and the resulting ST information was merged with epidemiological metadata to generate a transmission network. In Figure 2A, isolates sharing the same ST cluster tightly within the same phylogenetic branch, confirming their close genetic relatedness. Several regional patterns emerge. E. faecium exhibited a distinct epidemiological imprint, with most isolates derived from urine, and ST78 predominates (16/28, 57.1%). In contrast, E. faecalis was more genetically heterogeneous, displaying wider ward and specimen distributions and a dispersed ST profile, although ST16 remains the leading type (10/22, 45.5%). Notably, isolates within the same phylogenetic branch did not consistently originate from the same department or specimen category, implying multiple independent transmission routes within the study hospitals. The dominance of ST78 in E. faecium and the expanding assortment of E. faecalis lineages—particularly the ascendant ST16—highlight the dynamic population structure of Enterococcus in this region.
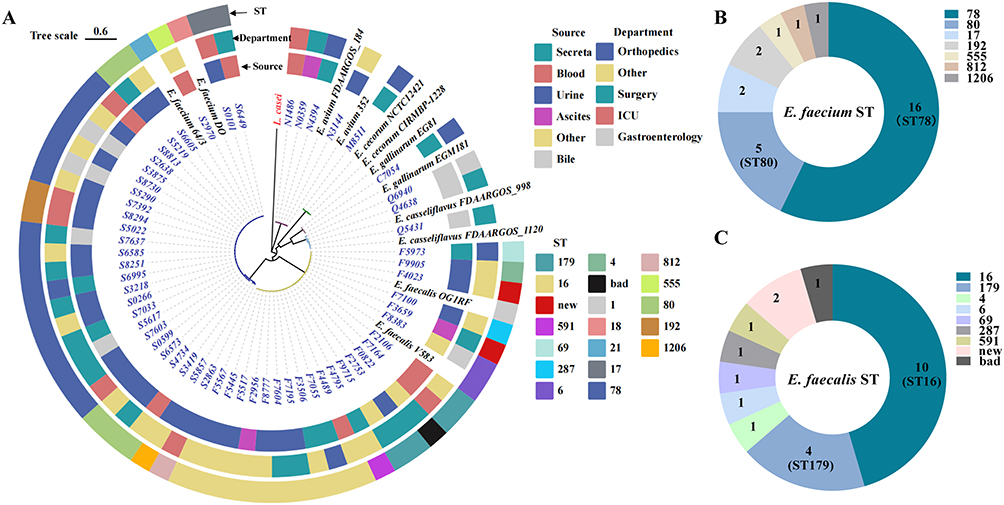 |
Figure 2 Phylogeny and MLST profiles of Enterococcus isolates. (A) Maximum-likelihood phylogenetic tree of clinical isolates constructed from core-genome SNPs; (B) Sequence-type distribution of sequenced E. faecium isolates (n = 28), ST78 is the predominant type (16 isolates); (C) Sequence-type distribution of sequenced E. faecalis isolates (n = 22), ST16 is the predominant type (10 isolates). |
Among the 28 sequenced E. faecium isolates, seven STs were identified (Figure 2B): ST78 (16 isolates, 57.1%), ST80 (5, 17.9%), ST17 (2, 7.1%), ST192 (2, 7.1%), and the rarer ST555, ST812, and ST1206 (one isolate each, 3.6%). A survey of 23,616 publicly available E. faecium genomes in NCBI showed that these STs account for 3.51%, 17.77%, 7.03%, 1.88%, 0.89%, 0.03%, and 0.01% of typed genomes, respectively, confirming ST78 and ST80 as globally prevalent lineages. The 22 E. faecalis isolates likewise yielded seven STs (Figure 2C): ST16 (10 isolates, 45.5%), ST179 (4, 18.2%), and single representatives of ST4, ST6, ST69, ST287, and ST591 (each 4.5%). Among 9071 E. faecalis genomes in NCBI, these STs occur at frequencies of 8.11%, 6.05%, 1.06%, 8.02%, 0.10%, 0.60%, and 0.15%, respectively, highlighting ST16 as both the local and an international epidemic clone.
Yichang’s dominant resistant clones, E. faecium ST78 and E. faecalis ST16, correspond to globally prevalent lineages, whereas rare types such as E. faecium ST1206 and ST812 were also present locally. During ST assignment, three E. faecalis isolates (F0822, F3659, and F4023) did not match any known profiles: F0822 lacked one allele due to a sequencing gap, while F3659 and F4023 each harboured a single-nucleotide change that generated novel STs (Figure 3 and Supplementary Table S4). The emergence of these new STs indicates ongoing local evolution and dissemination, potentially driven by adaptation, strain importation, or the risk of future outbreaks.
 |
Figure 3 Identification of two novel E. faecalis sequence types. (A) Isolate F3659 carries a single-nucleotide substitution in the housekeeping gene pstS (allele pstS_17; g.277 C→T), as indicated by the red arrow; (B) Isolate F4023 carries a single-nucleotide substitution in the housekeeping gene aroE (allele aroE_1; g.127 C→A), as indicated by the red arrow. Asterisks (*) indicate conserved positions in the alignment. |
Resistance - Gene Profile Analysis
CARD screening detected 11 classes of AMR genes among the clinical Enterococcus isolates, and their distribution differed markedly by species (Figure 4). Among the rarer taxa, E. avium (n = 4) carried between one and four AMR genes per genome, whereas E. casseliflavus (n = 3) harboured none of the surveyed determinants. The single E. gallinarum isolate contained six AMR genes, including five vancomycin-resistance loci, while the lone E. cecorum genome encoded 14 genes that mainly conferred resistance to aminoglycosides, lincosamides, and tetracyclines. In the predominant species, E. faecium (n = 28) carried 2–12 AMR genes, principally targeting aminoglycosides, β-lactams, macrolides, tetracyclines, and fluoroquinolones; notably, six isolates possessed vancomycin-resistance genes. E. faecalis (n = 22) encoded 5–20 AMR genes, predominantly harbors resistance genes against macrolides, quinolones, tetracyclines, and aminoglycosides, and also carries smaller numbers of resistance genes for diaminopyrimidines, lincosamides, nucleosides, and cephalosporins. Thus, although the rarer species generally harboured fewer resistance genes, the E. gallinarum isolate exhibited extensive vancomycin resistance and the E. cecorum isolate possessed a broad profile of aminoglycosides, lincosamides, and tetracyclines. The large and varied resistomes of E. faecium and E. faecalis underscore their clinical importance, especially given the presence of vancomycin-resistant E. faecium strains circulating in the Yichang region.
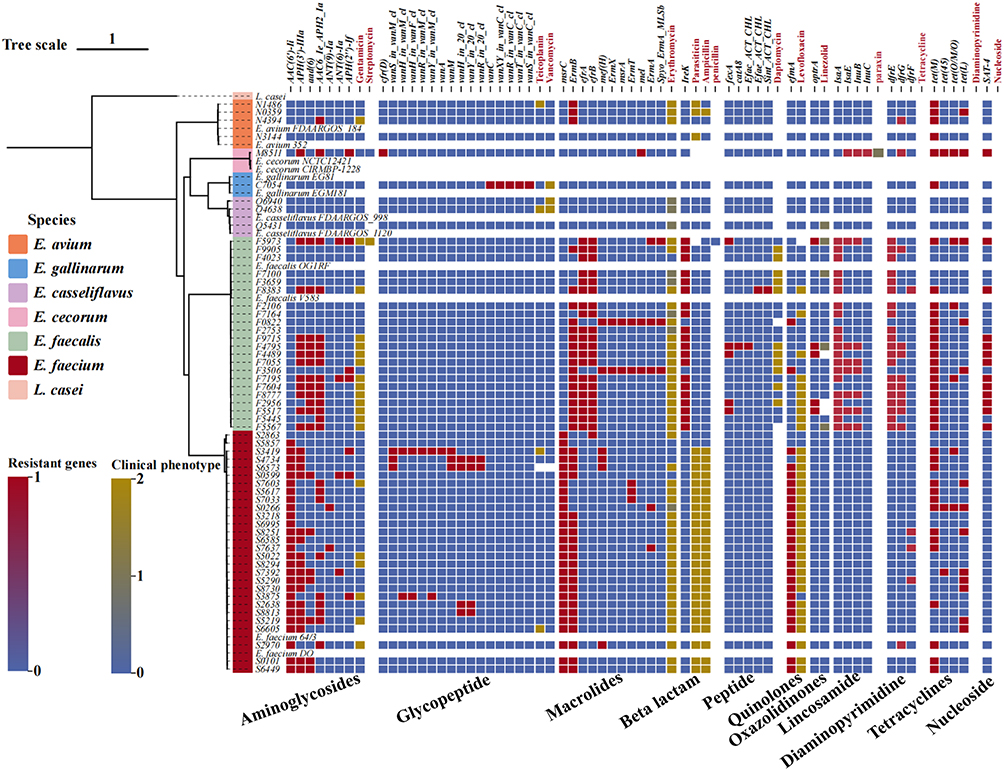 |
Figure 4 Correlation between resistome and phenotype in Enterococcus isolates. Eleven classes of antimicrobial-resistance genes were identified across the study isolates. On the x-axis, black labels denote individual resistance genes, while red labels indicate the corresponding antibiotic classes tested phenotypically. Blank cells signify either the absence of that gene class or the lack of susceptibility data for the matching drug. Resistant genes: 0=gene absent; 1=gene present. Clinical phenotype: 0=susceptible; 1=intermediate; 2=resistant. |
Analysis of the 59 sequenced Enterococcus isolates revealed heterogeneous distributions of antimicrobial resistance genes across species (Figure 5A). The most prevalent resistance determinants included ermB (macrolide resistance), tetM/tetL (tetracycline resistance), and aac(6’)-aph(2”) (high-level aminoglycoside resistance). Six E. faecium isolates and the single E. gallinarum isolate carried vancomycin-resistance genes (van cluster). Notably, although >90% of E. faecium isolates were phenotypically resistant to ampicillin and penicillin G, no acquired β-lactamase genes were detected, consistent with intrinsic PBP-mediated resistance. Principal component analysis further demonstrated interspecies clustering based on resistome composition (Figure 5B). Compared with NCBI reference genomes, Yichang isolates were enriched in vancomycin-, tetracycline-, aminoglycoside-, and lincosamide-resistance genes (Figure 5C).
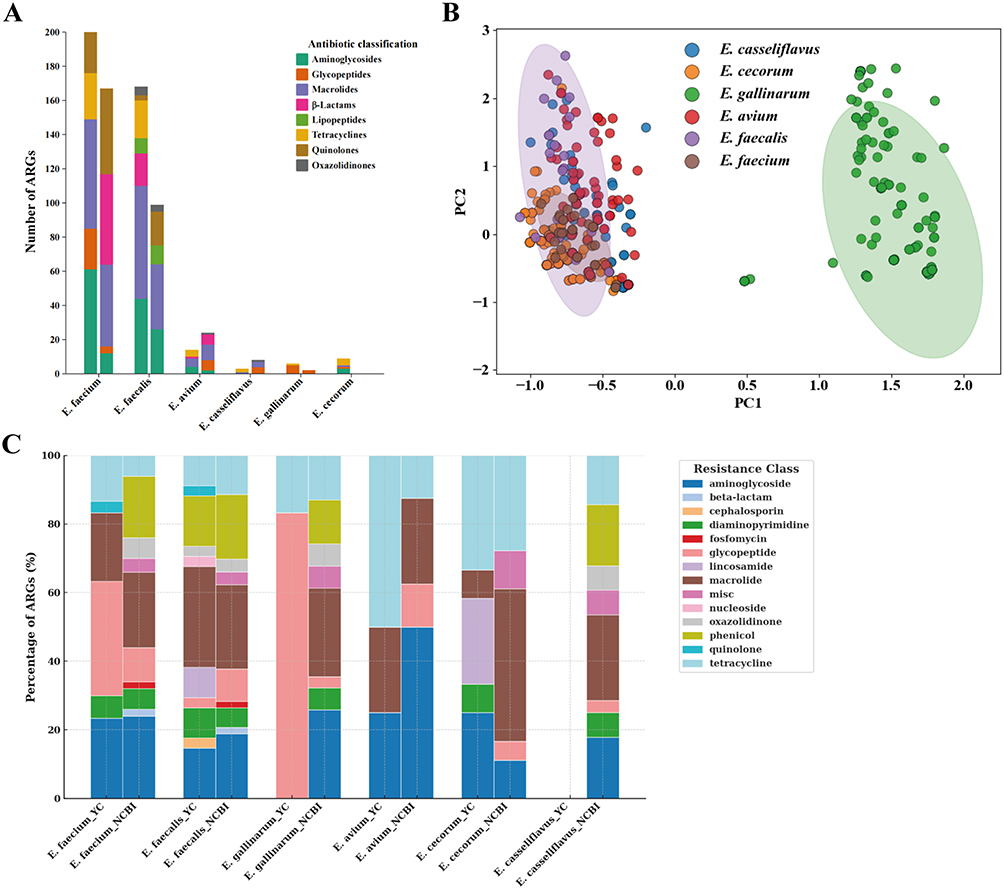 |
Figure 5 Analysis of Antimicrobial Phenotypes and Resistance Genes. (A) Stacked bar plot showing the distribution and counts of antibiotic resistance genes (ARGs) across six Enterococcus species; (B) Principal component analysis (PCA) of ARG profiles among six Enterococcus species; (C) Comparative distribution of ARG classes between Yichang isolates and reference genomes from the NCBI database. |
Virulence Gene Profiles
Comparative analysis using the VFDB revealed widespread distribution of key virulence determinants (Figure 6). Prominent adhesins included esp, ace, and efaA, supporting strong colonization potential. Several isolates, particularly E. faecalis, harbored cytolysin-associated genes (eg., cylA) and gelatinase (gelE), along with genes involved in immune evasion and effector secretion systems. Overall, E. faecalis exhibited a broader and denser virulence-gene repertoire than E. faecium, consistent with enhanced host adaptation and pathogenic potential.
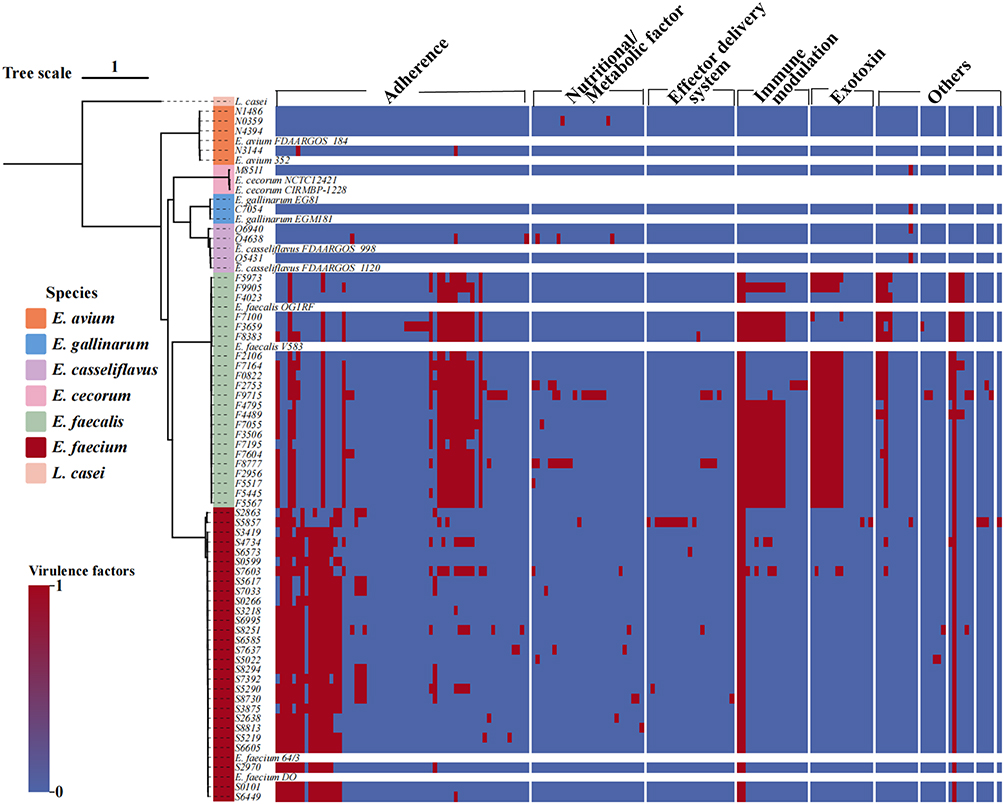 |
Figure 6 Virulence gene profiles of sequenced Enterococcus isolates. Nine categories of virulence genes were detected, with factors from E. faecium and E. faecalis predominating. Black squares indicate the presence of a gene (1), and white squares indicate absence (0). |
A quantitative comparison of virulence determinants showed that E. faecalis harboured a broader set of virulence factors than E. faecium (Figure 7A). Principal component analysis further highlighted clear interspecies separation based on virulence-gene repertoires (Figure 7B). Compared with six reference Enterococcus species from the NCBI database, Yichang isolates exhibited reduced prevalence of adherence genes but enrichment in exoenzymes, nutrient/metabolism factors, and effector secretion systems, yielding a more diverse virulence profile (Figure 7C).
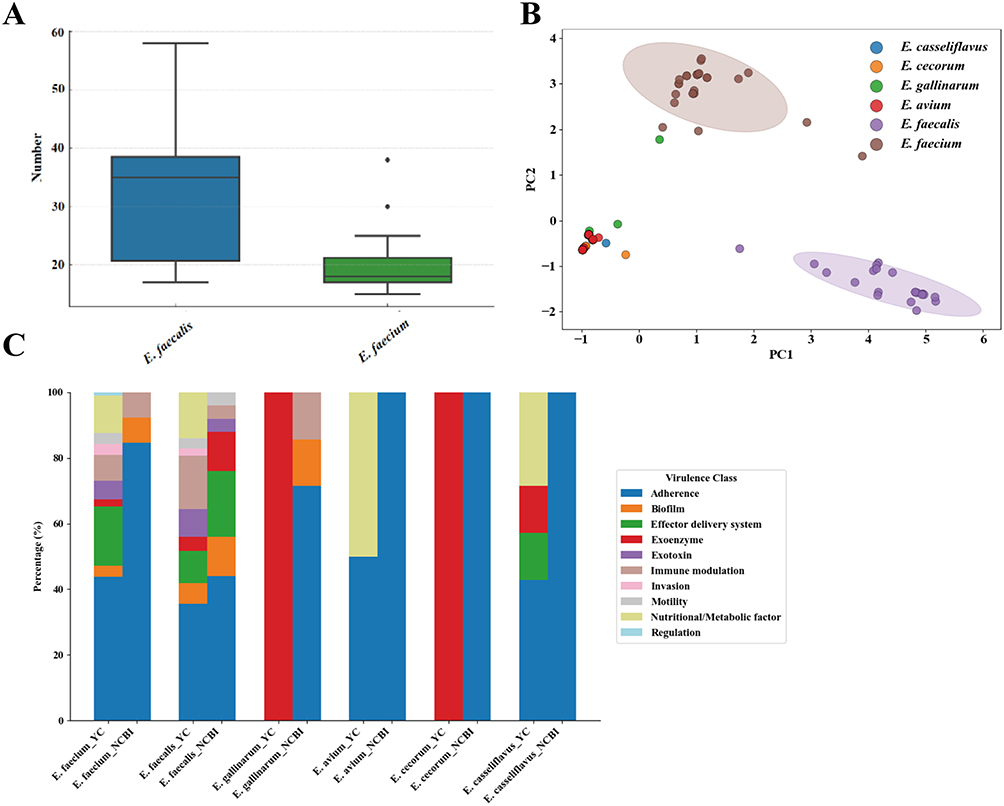 |
Figure 7 Integrated Analysis of Virulence Genes, ST Types, and Resistance Genes in Sequenced Isolates. (A) Comparison of total virulence-factor counts between E. faecium and E. faecalis; (B) Principal-component analysis of virulence-gene repertoires across six Enterococcus species; (C) Comparative analysis of virulence genes in antibiotic-resistant Enterococcus isolates from the Yichang region against those in the NCBI database. |
Resistance traits correlate with both the number of acquired AMR genes and the burden of virulence factors (Figure 8). Isolates belonging to high-prevalence sequence types—E. faecalis ST16 and ST179, and E. faecium ST78, ST192, and ST80—exhibit the densest clustering of virulence and resistance genes, reflecting their tight evolutionary relatedness. Although E. avium and E. casseliflavus carry comparatively few virulence or resistance genes, they are nonetheless isolated with appreciable frequency in clinical practice. Conversely, the rare species E. gallinarum and E. cecorum display extensive resistomes yet fewer virulence determinants. Notably, E. cecorum—mostly reported in poultry outbreaks—was recovered from a wound exudate in a trauma patient in this study. Given the zoonotic nature of enterococcal infections, clinicians should remain alert to the outbreak potential of such uncommon species.
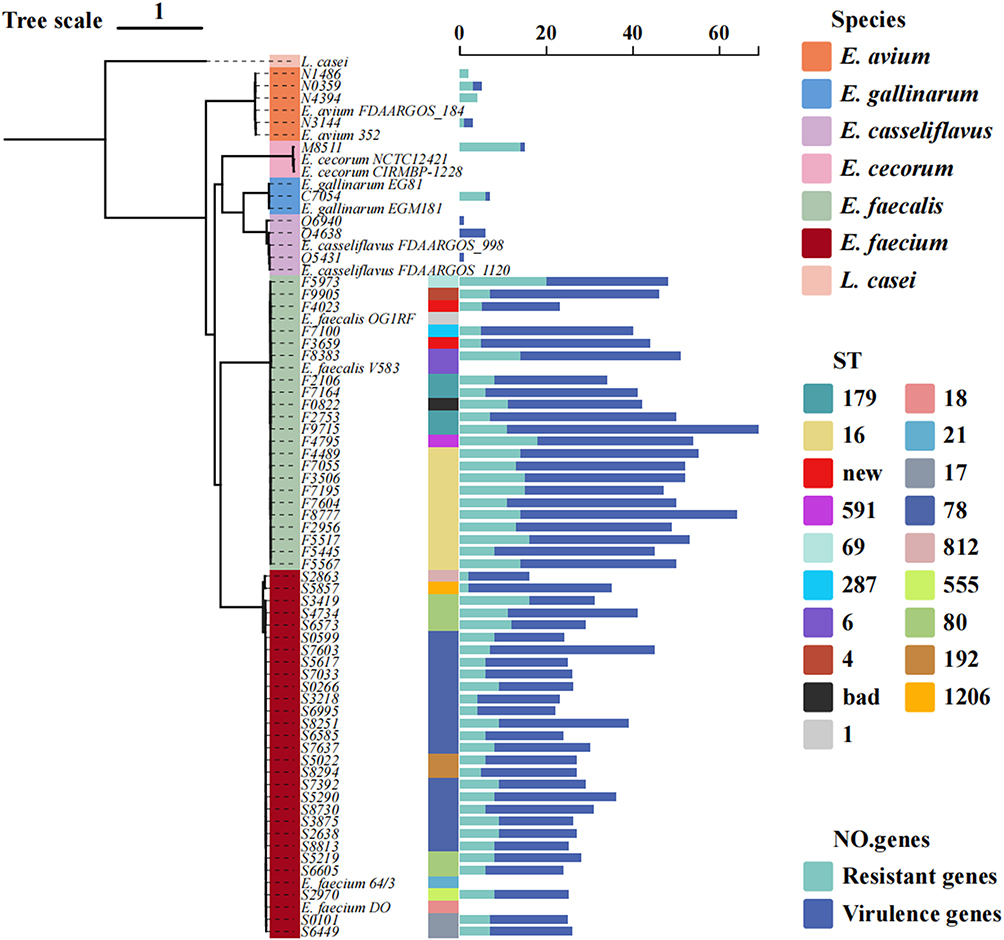 |
Figure 8 Comprehensive analysis of Enterococcus genomic data in conjunction with phylogenetic tree analysis. Bar chart summarising, for each ST, the numbers of resistance and virulence genes detected in the sequenced isolates. |
Discussion
Enterococcus species are ubiquitous opportunistic pathogens and have become major contributors to hospital-acquired infections worldwide. Their ability to acquire multidrug resistance through horizontal gene transfer and adaptive genome evolution has resulted in escalating therapeutic challenges.16,17 The increasing incidence of enterococcal infections—particularly among elderly, immunocompromised, and critically ill patients—reflects both selective antibiotic pressure and the ecological resilience of these organisms.18–21 Their resistance spectrum is broad, encompassing β-lactams, aminoglycosides, macrolides, tetracyclines, and other classes.22,23
Through WGS, our study provides the first comprehensive genomic characterization of antibiotic-resistant Enterococcus isolates from Yichang. Consistent with national and global epidemiological patterns, E. faecalis and E. faecium were the predominant species, accounting for the majority of infections, most frequently originating from urine specimens in surgical and urology wards. This distribution aligns with previous reports highlighting the urinary tract as a common source of enterococcal infection in hospitalized patients.24 MLST analysis identified E. faecium ST78 and E. faecalis ST16 as dominant lineages, both of which correspond to internationally disseminated epidemic clones. The detection of two novel E. faecalis sequence types, each arising from single-nucleotide substitutions in housekeeping genes, provides evidence of ongoing local microevolution. Although further functional investigation is warranted, the emergence of these novel STs suggests adaptive diversification within regional Enterococcus populations under antimicrobial selection pressure. The coexistence of epidemic clones and newly emerging lineages highlights the dynamic nature of resistance evolution in this setting.
Comparative genomic profiling revealed that Yichang isolates display a distinct antimicrobial resistance landscape, characterized by enrichment of vancomycin- and tetracycline-resistance genes and relative reduction in phenicol and oxazolidinone determinants compared with publicly available reference genomes. Notably, six E. faecium isolates and one E. gallinarum isolate harbored vancomycin-resistance genes, underscoring the importance of vancomycin-variable Enterococcus as a potential reservoir for resistance dissemination.25 In parallel, virulence profiling demonstrated that E. faecalis possessed a broader repertoire of virulence determinants than E. faecium, particularly in effector secretion and immune-evasion modules, suggesting enhanced host adaptation potential.26 Although E. gallinarum and E. cecorum were infrequently isolated, their relatively extensive resistomes indicate that atypical species may still pose clinical risk. The recovery of E. cecorum, a species commonly associated with poultry outbreaks, from a human wound infection further supports the potential zoonotic interface of enterococcal infections.27
Collectively, these findings delineate a region-specific adaptive pattern among Enterococcus isolates in Yichang. The convergence of multidrug resistance, virulence diversification, and emerging sequence types underscores the necessity of sustained genomic surveillance to monitor atypical species and prevent dissemination of high-risk clones in clinical settings.
Conclusions
Whole-genome sequencing of 59 Enterococcus isolates from Yichang revealed that E. faecalis and E. faecium remain the dominant species, with ST16 and ST78 as prevailing epidemic lineages. Two novel E. faecalis sequence types indicate ongoing microevolution under local antibiotic pressure. The regional resistome is marked by enrichment of vancomycin, tetracycline, aminoglycoside, and lincosamide genes, whereas virulence profiles shift from adhesion factors toward exoenzymes and secretion systems, reflecting adaptive diversification.
These findings support the implementation of sustained genomic surveillance, strengthened infection-control measures, and targeted monitoring of atypical Enterococcus species to limit dissemination of multidrug-resistant and emerging high-risk clones in clinical settings.
Data Sharing Statement
The genome datasets presented in this study can be found in online NCBI database (https://www.ncbi.nlm.nih.gov/). Detailed information on clinical strains can be found in the Supplementary Material.
Acknowledgments
Special thanks to the teachers in the clinical laboratory of relevant hospitals in Yichang City for providing the strains required for the experiment (Yichang Second People’s Hospital, Yichang Youfu Hospital, Zigui County People’s Hospital, and Yidu First People’s Hospital).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the National Natural Science Foundation of China (32201986), the Natural Science Foundation of Hubei Province (2022CFB743, 2024AFD145), Open Fund of Hubei Provincial Clinical Research Center for Precise Prevention and Treatment of Elderly Gastrointestinal Cancer (2024EGC-01), Open Foundation of Hubei Province Key Laboratory of Tumor Microenvironment and Immunotherapy (2024ZLKF2-51).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ramos S, Silva V, Dapkevicius MDLE, Igrejas G, Poeta P. Enterococci, from harmless bacteria to a pathogen. Microorganisms. 2020;8(8):1118. doi:10.3390/microorganisms8081118
2. McDonald JR, Olaison L, Anderson DJ, et al. Enterococcal endocarditis: 107 cases from the international collaboration on endocarditis merged database. Am J Med. 2005;118(7):759–13. doi:10.1016/j.amjmed.2005.02.020
3. Zhu Z, Du W, Yang Y, et al. Enterococci independently increase the risk for initial antibiotic treatment failure and prolonged hospitalization in adult patients with complicated urinary tract infection: a retrospective cohort study. Infection. 2025;53(1):307–315. doi:10.1007/s15010-024-02372-0
4. Prosty C, Sorin M, Katergi K, et al. Revisiting the evidence base that informs the use of adjunctive therapy for Enterococcus faecalis endocarditis: a systematic review and meta-analysis. Clin Infect Dis. 2024;79(5):1162–1171. doi:10.1093/cid/ciae379
5. Lu Z, Mclnnes RS, Allen F, Gadar K, van Schaik W. Resistance to last-resort antibiotics in enterococci. FEMS Microbiol Rev. 2025;49:fuaf057. doi:10.1093/femsre/fuaf057
6. Cattoir V. The multifaceted lifestyle of enterococci: genetic diversity, ecology and risks for public health. Curr Opin Microbiol. 2022;65:73–80. doi:10.1016/j.mib.2021.10.013
7. Wei L, Li M, Xia F, et al. Phosphate transport system mediates the resistance of Enterococcus faecalis to multidrug. Microbiol Res. 2021;249:126772. doi:10.1016/j.micres.2021.126772
8. Werner G, Neumann B, Weber RE, et al. Thirty years of VRE in Germany - “expect the unexpected”: the view from the national reference centre for staphylococci and Enterococci. Drug Resistance Updates. 2020;53:100732. doi:10.1016/j.drup.2020.100732
9. Yoshino Y. Enterococcus casseliflavus infection: a review of clinical features and treatment. Infect Drug Resist. 2023;16:363–368. doi:10.2147/IDR.S398739
10. Yu X, Yu Z, Huang L, et al. Global epidemiology and genomic perspectives on vancomycin-resistant Enterococcus faecium: rising antimicrobial resistance and transmission risks within the one health framework. Sci total environ. 2025;1002:180595. doi:10.1016/j.scitotenv.2025.180595
11. El Haddad L, Hanson BM, Arias CA, et al. Emergence and transmission of daptomycin and vancomycin-resistant Enterococci between patients and hospital rooms. Clin Infect Dis. 2021;73(12):2306–2313. doi:10.1093/cid/ciab001
12. Akter T, Haque MN, Ehsan R, et al. Virulence and antibiotic-resistance genes in Enterococcus faecalis associated with streptococcosis disease in fish. Sci Rep. 2023;13(1):1551. doi:10.1038/s41598-022-25968-8
13. Yan S, Jiang Z, Zhang W, et al. Genomes-based MLST, cgMLST, wgMLST and SNP analysis of salmonella typhimurium from animals and humans. Comp Immunol Microbiol Infect Dis. 2023;96:101973. doi:10.1016/j.cimid.2023.101973
14. Chen W, Wang Q, Wu H, et al. Molecular epidemiology, phenotypic and genomic characterization of antibiotic-resistant enterococcal isolates from diverse farm animals in Xinjiang, China. Sci total environ. 2024;912:168683. doi:10.1016/j.scitotenv.2023.168683
15. Zhao J, Liu R, Sun Y, Yang X, Yao J. Tracing enterococci persistence along a pork production chain from feed to food in China. Anim Nutr. 2022;9:223–232. doi:10.1016/j.aninu.2022.01.005
16. Cunha S, Miranda C, Martins Â, et al. Analysis of antibiotic-resistant and virulence genes of Enterococcus detected in calf colostrum—one health perspective. Animals. 2023;13(12):1900. doi:10.3390/ani13121900
17. Shahveh M, Tajbakhsh E, Momtaz H, Ranjbar R. Molecular characterization of Enterococcus faecalis and Enterococcus faecium isolated from a meat source in Shahrekord local markets, Iran. Arch Razi Inst. 2023;78(4):1387–1396. doi:10.32592/ARI.2023.78.4.1387
18. Das S, Bombaywala S, Srivastava S, et al. Genome plasticity as a paradigm of antibiotic resistance spread in ESKAPE pathogens. Environ Sci Pollut Res Int. 2022;29(27):40507–40519. doi:10.1007/s11356-022-19840-5
19. Goh HMS, Yong MHA, Chong KKL, Kline KA. Model systems for the study of Enterococcal colonization and infection. Virulence. 2017;8(8):1525–1562. doi:10.1080/21505594.2017.1279766
20. Song H, Yoo JS, Unno T. Discerning the dissemination mechanisms of antibiotic resistance genes through whole genome sequencing of extended-spectrum beta-lactamase (ESBL)-producing E. coli isolated from veterinary clinics and farms in South Korea. Sci total environ. 2024;926:172068. doi:10.1016/j.scitotenv.2024.172068
21. Torres C, Alonso CA, Ruiz-Ripa L, et al. Antimicrobial resistance in Enterococcus spp. of animal origin. Microbiol Spectr. 2018;6(4). doi:10.1128/microbiolspec.arba-0032-2018
22. Khan A, Miller WR, Axell-House D, Munita JM, Arias CA, Humphries RM. Antimicrobial susceptibility testing for enterococci. J Clin Microbiol. 2022;60(9):e00843–21. doi:10.1128/jcm.00843-21
23. Wang J, Cassone M, Gibson K, et al. Gut microbiota features on nursing home admission are associated with subsequent acquisition of antibiotic-resistant organism colonization. Clin Infect Dis. 2020;71(12):3244–3247. doi:10.1093/cid/ciaa662
24. Farsi S, Salama I, Escalante-Alderete E, Cervantes J. Multidrug-resistant enterococcal infection in surgical patients, what surgeons need to know. Microorganisms. 2023;11(2):238. doi:10.3390/microorganisms11020238
25. Sun L, Chen Y, Hua X, et al. Tandem amplification of the vanM gene cluster drives vancomycin resistance in vancomycin-variable enterococci. J Antimicrob Chemother. 2020;75(2):283–291. doi:10.1093/jac/dkz461
26. Ko KKK, Chng KR, Nagarajan N. Metagenomics-enabled microbial surveillance. Nat Microbiol. 2022;7(4):486–496. doi:10.1038/s41564-022-01089-w
27. Rhoads DD, Pummill J, Alrubaye AAK. Molecular genomic analyses of Enterococcus cecorum from sepsis outbreaks in broilers. Microorganisms. 2024;12(2):250. doi:10.3390/microorganisms12020250
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Characteristics, Virulence Profile, and Molecular Epidemiology of Klebsiella pneumoniae Infections in Kidney Transplant Recipients
Fan R, Zuo Y, Wang B, Shi D, Wang X, Wang Z, Huang Y
Infection and Drug Resistance 2025, 18:1303-1311
Published Date: 6 March 2025
Surveillance of Antibiotic Resistance and Molecular Epidemiology of Staphylococcus aureus in Baotou, Inner Mongolia, China
Zhang W, Liu F, Li J, Zhang L, Hu T
Infection and Drug Resistance 2025, 18:4887-4900
Published Date: 13 September 2025
Serotype Distribution, Antibiotic Resistance Characteristics, and Virulence Factor Analysis of Group B Streptococcus Isolates in Guangzhou Based on Whole-Genome Sequencing
Ling Y, Deng Y, Zhang P, Yuan K, Ye L, Zhang X, Chen X, Cui S, Liu J, Zhao Y
Infection and Drug Resistance 2026, 19:549193
Published Date: 15 January 2026
Molecular Epidemiology and Antimicrobial Resistance of Group B Streptococcus in Hainan, China: Genomic Insights from Perinatal and Adult Clinical Isolates
Zhao HM, Song XD, Li J, Miao BB, Zhang JY, Gong XY, He Y, Wang YJ, Fu YS, Wu H
Infection and Drug Resistance 2026, 19:617393
Published Date: 15 July 2026