Back to Journals » International Journal of Nanomedicine » Volume 16
Externally Triggered Novel Rapid-Release Sonosensitive Folate-Modified Liposomes for Gemcitabine: Development and Characteristics
Authors Omar MM ![]() , Hasan OA, Zaki RM, Eleraky NE
, Hasan OA, Zaki RM, Eleraky NE
Received 8 June 2020
Accepted for publication 10 December 2020
Published 28 January 2021 Volume 2021:16 Pages 683—700
DOI https://doi.org/10.2147/IJN.S266676
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Mahmoud M Omar,1,2 Omiya Ali Hasan,1,2 Randa Mohammed Zaki,3,4 Nermin E Eleraky5
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Deraya University, Minia, 61768, Egypt; 2Department of Pharmaceutics and Clinical Pharmacy, Faculty of Pharmacy,Sohag University, Sohag, 82524, Egypt; 3Department of Pharmaceutics, Faculty of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj, Saudi Arabia; 4Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt; 5Department of Pharmaceutics, Faculty of Pharmacy, Assiut University, Assiut, 71526, Egypt
Correspondence: Mahmoud M Omar
Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Deraya University, Deraya Square Street, Minia, 61768, Egypt
Tel +20 10 0933 2419
Email [email protected]
Purpose: To develop an externally triggered rapid-release targeted system for treating ovarian cancer, gemcitabine (GMC) was entrapped into sonosensitive (SoS) folate (Fo)-modified liposomes (LPs).
Methods: GMC-loaded LPs (GMC LPs), GMC-loaded Fo-targeted LPs (GMC-Fo LPs), and GMC-loaded Fo-targeted SoS LPs (GMC-SoS Fo LPs) were prepared utilizing a film-hydration technique and evaluated based on particle size, ζ-potential, and percentage entrapped drug. Cellular uptake of the fluorescent delivery systems in Fo-expressing ovarian cancer cells was quantified using flow cytometry. Finally, tumor-targeting ability, in vivo evaluation, and pharmacokinetic studies were performed.
Results: GMC LPs, GMC-Fo LPs, and GMC-SoS Fo LPs were successfully prepared, with sizes of < 120.3± 2.4 nm, 39.7 mV ζ-potential, and 86.3%± 1.84% entrapped drug. Cellular uptake of GMC-SoS Fo LPs improved 6.51-fold over GMC LPs (under ultrasonic irradiation — p< 0.05). However, cellular uptake of GMC-Fo LPs improved just 1.24-fold over GMC LPs (p> 0.05). Biodistribution study showed that of GMC concentration in tumors treated with GMC-SoS-Fo LPs (with ultrasound) improved 2.89-fold that of free GMC (p< 0.05). In vivo, GMC-SoS Fo LPs showed the highest antiproliferative and antitumor action on ovarian cancer.
Conclusion: These findings showed that externally triggered rapid-release SoS Fo-modified LPs are a promising system for delivering rapid-release drugs into tumors.
Keywords: sonosensitive liposome, gemcitabine, folate-modified liposomes, externally triggered, ovarian cancer
Introduction
Cancer is one of the most dangerous abnormalities threatening human life today. In Egyptian females, ovarian cancer is the fifth-commonest cause of death.1 Because of an inability to detect and diagnose ovarian cancer early, it spreads within the internal viscera. As such, most ovarian cancer patients cannot be diagnosed, leading to advanced stages (III or IV).2 Late diagnosis of ovarian cancer may lead to death within 5 years. Surgical removal of ovarian tumors and chemotherapy are basic approaches in standard treatment. Chemotherapy is a significant factor in treating cancer. There are many anticancer drugs, such as adriamycin, topotecan, and gemcitabine (GMC).3,4
Adriamycin may act by intercalating into DNA, leading to disruption of topoisomerase II–mediated DNA repair, or by producing free radicals, leading to damage of vital cellular components, such as cellular membranes, DNA, and proteins.5 The importance of adriamycin for treatment of ovarian cancer became limited, because of the rapid development of drug resistance. Topotecan is a derivative of camptothecin that interferes with DNA replication by binding to the topoisomerase I–DNA complex and preventing the religation of DNA, leading to cell death.6 It has effective action in metastatic ovarian cancer after failure of platinum-based therapy. It has many side effects, such as nausea, vomiting, bleeding, difficulty breathing, blood in urine, and extreme fatigue.7 Platinum-based chemotherapeutic drugs, eg, cisplatin, carboplatin, and oxaliplatin, are utilized for treating cancer. They mostly act on guanine N7, forming a 1,2-intrastrand cross-linking of DNA or DNA-protein cross-linking, inhibiting DNA repair and/or DNA synthesis in cancer cells. Cisplatin has been reported to be the superior analogue compared to carboplatin, based on its therapeutic efficacy across a spectrum of tumors and also its toxicity profile, especially the loss of hematologic toxicity.8 Paclitaxel is a microtubule-targeted chemotherapeutic drug for treating cancer. It causes stabilization of dynamic microtubule polymerization, leading to mitosis failure. Moreover, it changes other cellular functions that involve microtubules, such as intracellular signaling and organelle transport.9
GMC is an attractive choice in ovarian cancer management,10 because of its well-known efficacy in platinum/paclitaxel-resistant cancer, no cross-resistance with platinum compounds, and ease in combining it with other chemotherapeutics for treating recurrent ovarian cancer.11 It can be utilized for treating cancer of the ovaries, lungs, and pancreas.12 GMC penetrates the cell membrane and transforms into GMC diphosphate and GMC triphosphate through deoxycytidine kinase. Ribonucleotide reductase is inhibited by GMC diphosphate, leading to a halt in the synthesis of deoxynucleoside triphosphates required for DNA synthesis. At the same, GMC triphosphate is incorporated into DNA instead of endogenous deoxynucleoside triphosphates.13 The GMC-triphosphate metabolite is also utilized RNA synthesis, inhibiting it.14 However, the hydrophilicity, short plasma half-life, and rapid metabolism of GMC lead to increases doses required for effective plasma concentration for cancer-cell death, causing many side effects, such as hematologic neutropenia occurring more often than thrombocytopenia.15 GMC is a hydrophilic drug (soluble in water at 25 mg/mL) with an octanol–water partition coefficient (logP) of 1.123. Moreover, incorporation of GMC in liposomes (LPs) enhances its cytotoxic effectscompared to free drug.16
In an attempt to diminish the side effects of GMC, many delivery systems have been used, such as nanoparticles, transferosomes, and LPs.17–19 LPs are biocompatible, biodegradable, and safe and can be used to improve efficacy, decrease side effects, and enhance the stability of encapsulated drugs. Phospholipid bilayers of LPs may be used as membranes into which hydrophobic moieties can be inserted to target LPs toward the desired site.20,21 LPs are widely used as delivery systems because of relatively long circulation time, enhanced permeability-and-retention effect (EPR), and enhanced accumulation of drug into cancerous tissue, leading to improved therapeutic action and diminished toxic effects.22 Moreover, LPs have the capability of entrapping both hydrophobic (within the bilayer) and hydrophilic (within the core) drugs. They are atoxic and do not activate the immune system.21 Many attempts have been made to enhance the targeting ability of LPs by modifying, the surface such as PEGylated LPs and Fo-modified LPs.23,24 To improve cell-specific and intracellular delivery, LPs may be further conjugated with targeting ligands, such as Fo.25 Currently, Fo-modified LPs are utilized based on expression of Fo receptors, which is very high in most types of cancer, while expression is low in healthy tissue.26 These modified LPs have been characterized as having sustained release.27 However, there is great demand for developing a delivery systemwith an enhanced targeting index and an externally triggered rapid release of drug cargo within the cancerous tissue in ovaries.28
Because Fo-targeted LPs have proved to be highly effective in many medical applications, easily disrupted LPs have rarely been studied. Fo-conjugated LPs containing chlorin e6 (Ce6) could be studied for rapid-release capability after binding with cancerous cells.29 Encapsulation of drug in long-circulating PEGylated LPs has overcome limitations experienced with other non-PEGylated LPs.30 Moreover, proper PEGylation is also required, as a result of initial (and overall) tumor accumulation that still depends on passive extravasation.31 Active targeting (such as conjugation with Fo) is utilized to decrease disadvantages associated with passive targeting (such as decoration PEG conjugation and long circulation).32
The usse of most antitumor drugs for treatment of cancerous tissue is restricted, as a result of their low specificity.33 To counter this, antitumor drugs could be targeted to cancerous tissue. Moreover, high levels of antitumor drugs are desired within cancerous tissue, in order to kill abnormal cells. To achieve this aim, externally triggered rapid-release drug carriers should be developed.
The present study was carried out to achieve rapid release of the loaded drug within tumors. Formulated LPs should be sensitive to external stimuli, such as ultrasonic waves, and once applied, the drug should be released. The present study aimed to target a drug to the ovaries utilizing externally triggered novel rapid-release sonosensitive (SoS) Fo-modified LPs. SoS Fo-modified LPs were characterized by measuring particle size, ζ-potential, and encapsulation efficiency (EE). Moreover, ultrasound sensitivity, cellular association using flow cytometry, in vivo antitumor efficacy, and pharmacokinetics of the liposomal drug were examined.
Methods
Soybean phospholipids (L-α-phosphatidylcholine [PC, purity ≥99%), folic acid–polyethylene glycol–stearylamine (FA-PEG-SA) and cholesterol (purity ≥99%) were purchased from Xian Ruixi Biological Technology. Ce6 was purchased from Frontier Scientific (Salt Lake City, UT, USA). The CAOV3 cell line was obtained from the Chinese Academy of Sciences (Shanghai, China). RPMI 1640 medium and FBS were purchased from Sigma Chemical.
All reagents were of HPLC or analytical grade. Sprague Dawley rats were purchased from the animal house of the Faculty of Medicine, Assiut University, Egypt.
Preparation of Liposomal Samples
GMC LPs (GMC LPs) were prepared by the pH-gradient method using PC and cholesterol (PC:cholesterol 5.5:4.5).15,34 Briefly, LPs were prepared using thin-film hydration. To prepare non-Fo–modified LPs (500 mg lipid), a lipid composition of PC and cholesterol (55% and 45% molar ratio) were completely dissolved in a mixture of ether and chloroform (1:3 v:v). With a rotary evaporator (Büchi), the organic solution was evaporated under vacuum into a sterile conical flask, forming a dried thin membrane. Phosphate citrate buffer (50 mM, 10 mL, pH 2.2) was utilized to hydrate the membrane at 55°C (50 mg/mL), resulting in a colloidal solution, then the pH was raised to 7.2 using disodium hydrogen phosphate. The colloidal solution was extruded 12 times (mini-extruder, Avanti) through 100 nm polycarbonate filters, aiming to optimize the particle size, as shown in Figure S1 and S2 (supplementary section). GMC (concentration 0.9–1.6 mg/mL) was then incubated in the LPs at 65°C for 4 hours to get active loading. Separation of free GMC was carried out using extensive dialysis against buffer (20 mM HEPES, pH 7.4, containing 150 mM NaCl and 0.1 mM EDTA). GMC-Fo LPs were prepared by the same procedure, except FA-PEG-SA was added to the lipid mixture at 0.1% molar ratio. To prepare GMC-SoS Fo LPs, a lipid composition of PC and cholesterol (55% and 45% molar ratio) and FA-PEG-SA (0.1%) was completely dissolved in a mixture of ether and chloroform (1:3 v:v). The lipophilic sonosensitizer Ce6 ester (3%, 6% or 9%) was dissolved in the prepared organic phase. With the rotary evaporator, the organic solution was evaporated under vacuum into a sterile conical flask, forming a dried thin membrane. Then, all preparation steps used the same procedures mentioned previously. These procedures were performed under aseptic conditions and vertical laminar flow (vertical laminar flow hood, BZ series, Germfree).
Determination of Entrapment Efficiency and Drug Loading
GMC-EE and drug loading (DL) were calculated after purifying the LPs by ultrafiltration in centrifuge tubes with an Ultrafilter (Amicon Ultra, molecular weight cutoff 30 kDa, Millipore). Quantification was done spectrophotometrically.35 Briefly, LP formulations (0.5 mL) were placed in the upper part of the centrifuge tubes, then centrifuged at 6,000 rpm, 4°C for 30 minutes to separate free drug from the encapsulated LPs. The last step was repeated until obtaining all LP quantities. The amount of free drug in the filtrate was estimated at 267 nm spectrophotometrically (LC20A, Shimadzu). EE and DL were calculated using Equations 1 and 2:
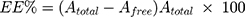
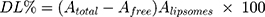
where Atotal, Afree, and Aliposomes represent GMC, free GMC, and the total LPs recovered, respectively.
Characterization of Liposomes
Morphology of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs was examined utilizing transmission electron microscopy. Briefly, aliquots were placed on hydrophobic copper grids and dried, then negatively stained with uranyl acetate (50 µL, 2.5% w:v). A Malvern Zetasizer Nano ZS Zen 3600 was utilized to investigate the colloidal dispersion to determine ζ-potential, particle size, and polydispersity index (PDI) values.36 Briefly, LP samples (200 μL) were diluted with deionized water (800 μL) to be analyzed based on dynamic light scattering using the Zetasizer.
Evaluation of Ultrasound Sensitivity
Acoustic Parameter Selection
GMC-SoS-Fo LPs were diluted eightfold with PBS (0.1 M, pH 7.4) and kept at 37°C for 10 minutes, then transferred into an ultrasound sonication system (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation with 10-second rests for a total of 0–150 seconds). Temperature of the LP preparations was fixed during ultrasonic irradiation utilizing a water bath containing a thermocouple. Drug release was determined after each ultrasonic irradiation and recorded. Colloidal dispersions were left for 10 minutes before they were purified by ultrafiltration in the Amicon centrifuge tubes.35 Quantification was done spectrophotometrically at 267 nm spectrophotometrically (Shimadzu LC20A), as per determination of EE.
Molar Ratio of Sonosensitizer
GMC release from GMC-SoS Fo LPs prepared withmolar ratios of 3%, 6%, or 9% Ce6 of total lipid was measured under the same procedures. However, the total period of ultrasonic irradiation was changed to 130 seconds.
In Vitro Drug Release
In vitro GMC release from GMC-loaded LPs was determined using dialysis bags.37 Briefly, 5 mL GMC-loaded LPs was placed into a cellophane bag (molecular weight cutoff 12–14 kDa), then inserted into 100 mL PBS (pH 7.4, 0.1 M, 37°C). Dissolution media were stirred gently and treated with ultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation with a 10-second rests for a total of 0–130 seconds) or not treated. Aliquots (2 mL) replaced with equal volume of fresh prepared PBS were withdrawn from the dissolution medium to be spectrophotometrically analyzed at at λmax=267 nm for measuring released GMC. Measurements were repeated three times, and are represented as means ± SD.
Storage Stability of Liposomes
To examine the stability of colloidal dispersions, LP dispersions were kept at different temperatures (4°C±2°C, 25°C±2°C, 37°C±2°C, 65% relative humidity) for 3 months. Aliquots were taken every month to investigate particle size, ζ-potential and EE.
Cell Culture
CAOV3 cells (adherent type) were incubated in RPMI 1640 media (50 mL) containing FBS (10%, 5 mL) 1%, streptomycin (0.1 mg/mL), and penicillin (200 IU, 1 mL) into flasks at 37±2°C/5% CO2. Cells were reseeded and suspended every 4 days and used for study at passages 5–20.
Quantitative Cellular Uptake of Liposomes Using Flow Cytometry
To determine the intercellular uptake of the GMC LPs, GMC-Fo LPs, and GMC-SoS Fo LPs, flow cytometry was utilized based on measuring mean fluorescence intensity.38 Briefly, cells were incubated into 48-well plates at a density 6×105 cells/well. Incubated cells were withdrawn from the medium after 24 hours, then washed with PBS and reseeded and suspended in fresh medium for 4 hours at 37°C±2°C/5% CO2 atmosphere. Cell suspensions were incubated for 48 hours with calcein LPs, calcein–Fo LPs, and calcein–SoS Fo LPs, respectively. Subsequently, they were treated with ultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation and with 10-second rests for a total of 0–130 seconds) or not treated, then incubated for 30 minutes at 37±2°C/5% CO2. Thereafter, culture media weres removed and sterile PBS used to rinse adherent monolayers. Trypsin enzyme (10 mL, 0.25% v:v) was utilized to liberate cells from the adhesion state. Cells were suspended in sterile PBS and analyzed with flow cytometry (Thermo Fisher Scientific) at excitation wavelength 495 nm and emission wavelength 516 nm. The LP dispersions were prepared to encapsulate calcein instead of GMC following the same aforementioned procedures.
Cell suspensions incubated with calcein-free LPs were treated withultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation and with 10-second rests for a total of 0–130 seconds) or not treated under the aforementioned conditions. The first sample (incubated with empty-LPs) was treated with ultrasonic irradiation, so was considered a control (control 1) for the tested samples treated with ultrasonic irradiation. The second sample (incubated with empty LPs) was not treated with ultrasonic irradiation, so was considered a control (control 2) for tested samples that did not receive-ultrasonic irradiation. These controls were used to subtract the effects of autofluorescence and ultrasonic irradiation.
After that, cells were washed with PBS twice and suspended in PBS before subjecting them to flow cytometry to calculate cellular binding.measurements were repeated three times.
Cytotoxicity
To examine the cell-killing effect of SoS Fo LPs, free GMC, GMC LPs, GMC-Fo LPs, GMC-SoS Fo LPs (no ultrasonic irradiation) and GMC-SoS Fo LPs (ultrasonic irradiation), cytotoxicity was assessed. Preparations were examined by MTS assays in CAOV3 cells. Briefly, cells were seeded in 96-well plates at 105 cells/well and incubated at 37°C in 5% CO2 for 48 hours. Thereafter, cells were treated with different concentrations (0–150 µM) of GMC from the formulations for 72 hours. Ultrasonic irradiation was applied for 150 seconds only to the last preparation after 48 hours’ incubation, then incubated for 24 hours (total incubation 72 hours). Subsequently, 20 µL MTS reagent (tetrazolium inner salt) per well was added and incubated for another 4 hours at 37°C in 5% CO2. Absorbance at 490 nm was measured using a multimode microplate reader to calculate cell viability:

where S[Test], S[Control], and S[Blank] represent the absorbance of the treatment group, untreated control group, and blank-culture medium group, respectively. Moreover,IC50 was calculated using an online calculator (https://www.aatbio.com/tools/ic50-calculator).
Establishment of Ovarian Cancer Model in Sprague Dawley Rats
A xenograft ovarian cancer model in rats obtained from the Faculty of Medicine, Assiut University was produced by surgical orthotopic implantation.39 Briefly, subcutaneous tumors were induced by injecting 106 CAOV3 cells into the right axillary regions of immunocompromised Sprague Dawley rats aged 30 days using 0.25 mL dexamethasone acetate (4 mg/mL) daily intramuscular injections. Immunosuppressive treatment was also started 5 days before the injection of CAOV3 cells and maintained throughout the experiment.
The resulting subcutaneous tumors were allowed to grow about 0.5 cm3, then isolated as small pieces (1 mm3). One piece was incubated surgically into the ovary. Development of ovarian cancer was monitored by ultrasound imaging.40 The study was conducted under license of the ethical approval committee of the Faculty of Medicine, Assiut University (Assiut 155–020, January 20, 2020), and in aqccordance with the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (eighth edition, revised 2011).
Biodistribution Studies
To evaluate in vivo targeting efficiency of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs, a biodistribution and pharmacokinetic study was performed using 40 ovarian cancer xenograft models in the rats. Rats were randomly distributed into fife groups: the first (n=8) was intravenously injected with free GMC (2 mL, 4 mg/mL), and the second (n=8), third, fourth, and fifth were intravenously injected with LPs GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs (without ultrasound) and GMC-SoS-Fo LPs (with ultrasound) at 12.5 mg/kg. At 30 minutes after injection, the rats treated with GMC-SoS-Fo LPs were subjected to ultrasonic irradiation above the ovaries at both abdomen sides (sonication area of 2 cm2 and ultrasound of 2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation with 10-second rests for a total of 0–150 seconds). At 1 hour after injection, three rats from each group were humanely killed, and ovaries, kidneys, lungs, spleens, livers, and hearts collected. The drug was measured in the plasma as reported in the literature with some modification.41 Collected organs were kept at −20°C until further analysis. To detect GMC concentration, the organs collected were homogenized in fourfold volumes of double-distilled water. The homogenates (2 mL) spiked with 100 µL floxuridine (20 ng/mL) as internal standard were treated and vortexed with acetonitrile (1 mL) and centrifuged (2,000 rpm, 3 minutes) to precipitate the protein. The supernatants were evaporated under vacuum at 50°C. The dried residue was reconstituted with 2 mL mobile phase, which consisted of 40 mmol/L acetate ammonium buffer solution (pH 5.5):acetonitrile (97.5:2.5, v:v). Aliquots of reconstituted solution (50 µL) were injected into the column (C18, 250 mm×4.6 mm ID, 5 µm), and flow rate was set at 0.8 mL/minute for detection of the drug at λmax=268 nm.
Pharmacokinetic Studies
Twelve female Sprague Dawley rats weighing 250–280 g were utilized for the present pharmacokinetic studies and distributed randomly into four groups (n=3). The rats were kept under standard conditions at 25°C, relative humidity 55%±5% and free access to drinking water. Pharmacokinetic study of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs were conducted utilizing a single dose of 4 mg/kg GMC (intravenously via tail vain, diluted to 1 mL with isotonic saline solution 0.09% w:v). Blood samples (0.3 mL) were withdrawn at predetermined time intervals (5, 15, 30, 45, 60, 120, 180, 240, and 300 minutes) from the tail vein to separate the plasma. To conduct the analysis, 100 µL of floxuridine solution as internal standard (20 ng/mL) was added to each plasma sample. Plasma protein was precipitated in all samples by adding 1 mL acetonitrile to each, then vortexing for 10 minutes and centrifuging at 2,000 rpm for 4 minutes. Supernatants were withdrawn and evaporated under vacuum at 50°C. Dried residues were reconstituted with 2 mL mobile phase (40 mmol/L acetate ammonium buffer solution, pH 5.5):acetonitrile, 97.5:2.5, v:v). Analysis was performed as per the biodistribution study.
In Vivo Antitumor Efficacy
Ovarian cancer rat xenograft models were distributed into various groups, each with ten rats. The first was intravenously injected with GMC-free LPs (control group) and subjected to ultrasonic irradiation. The second, third, fourth and fifth were intravenously injected with 4 mg/kg free GMC solution, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs, respectively. The sixth group, injected with GMC-SoS-Fo LPs, was subjected to ultrasonic irradiation. The dose regimen was intravenous injecton 4 mg drug equivalent per kilogram on days 1, 7, 10, 13, 16, and 20. Tumors in each group were measured every 3 days up to 27 days using a commercial three-dimensional sonar (Artida, Toshiba Medical Systems).42 Survival time (from inoculation of tumors until death) was recorded for each group and plotted. Kaplan–Meier survival curves were plotted for each group. The body weight of each mouse was measured daily.
Statistical Analysis
All findings are presented as means ± SD. One way-ANOVAs were applied to calculate differences between any two groups. Significance was regarded as p<0.05, with Tukey–Kramer multiple assessments or two-sided Student t-tests for pairwise comparisons (GraphPad Prism 6.0, GraphPad, San Diego, CA, USA).
Results and Discussion
Targeting of drug-delivery systems into tumors is very important. There is currently huge demand to achieve rapid release of drugs after targeting tumor sites.
Characterization of Liposomes
Physical properties of the prepared LPs were investigated by measuring particle size, EE, and DL (Table 1). Mean particle sizes of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs were 120.3±2.4, 119.1±3.5, and 109.5±2.9 nm, respectively. Although a mini-extruder (100 nm polycarbonate filter) was used to optimize particle size, mean particle size was >100 nm. These results may be interpreted on the basis of elasticity of the LPs: they were squeezed through the polycarbonate filter under high pressure. After the LPs had passed the extruder, they attained their final size. Moreover, PDI values of all LPs was 0.209–0.272, suggesting high homogeneity. The EE of the prepared LPs was 82.8%–86.3%, confirming that conjugation of LPs with Fo moiety (GMC-Fo LPs) or Fo and Ce6 (GMC-SoS-Fo LPs) did not affect EE significantly (p>0.05). The ζ-potential of all LP formulations was more than >34.52±1.01 mV, as presented in Table 1, confirming stability of all formulations. Finally, DL of LPs was 3.45%±0.12%–3.73%±0.27%, showing that conjugation of the LPs had no effect on DL, as presented in Table 1. The small differences among GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs could be interpreted on the basis of the same lipid composition, while the other ingredients — Fo moiety (GMC-Fo LPs) or with Fo and Ce6 — were formulated with neglected ratio. Moreover, most Fo moieties were outside the phospholipid-bilayer membrane, so they may not have interfered with or had little effect on the phospholipid-bilayer membrane.
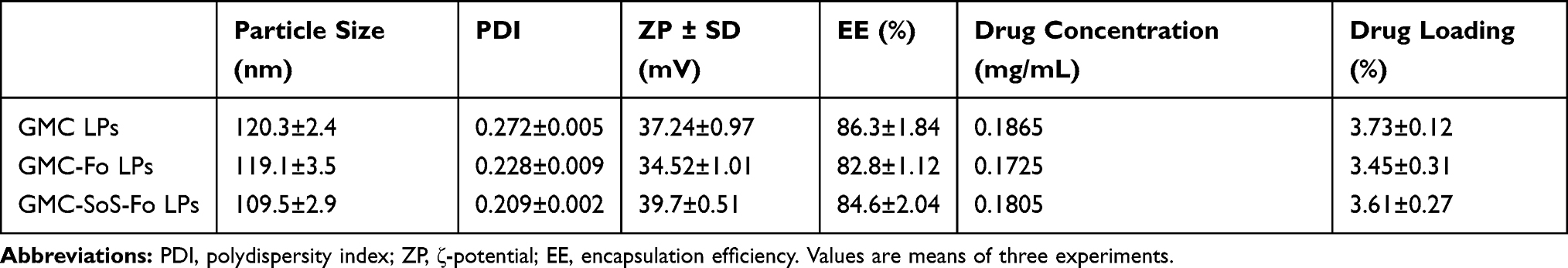 |
Table 1 Physical Properties of the Nanocarriers |
The ζ-potential of LPs may have great influence on drug accumulation within cells. Accumulation of cationic LPs is high after intravenous injection. Cationic macromolecules show higher glomerular permeability than anionic macromolecules of similar molecular weight. Larger cationic macromolecules accumulate in the kidney and liver as a result of many factors. The predominant charge of the cell surface is negative, leading to binding of cationic molecules to the cells. Interaction of negatively charged components of the blood with cationic macromolecules leads to embolization of aggregates. Moreover, cationic LPs are excellent as gene-delivery carriers and for liver, lung, and tumor targeting. Cationic LPs may be used as carriers to target cell nuclei. In addition, cationic LPs can cross the blood–brain barrier more easily than anionic LPs. Despite the many advantages of cationic LPs over viral vectors in gene delivery, the transfection efficiency of DNA-cationic LPs (lipoplexes) is too low and they have more toxicity than engineered viral vectors. Cationic LPs have been reported to target tumor vasculature selectively, due to an inherent yet unexplained mechanism.43 Moreover, a wide area of scientific research has focused on formulating cationic LPs to enhance their vascular targeting efficiency and diminish toxicity-related reactions.
Morphology of Liposomes
TEM of GMC-SoS-Fo LPs showed that spherical single-bilayer vesicles (unilamellar LPs) were formed, as shown in Figure 1, and their size concurrent with those obtained with the Zetasizer.
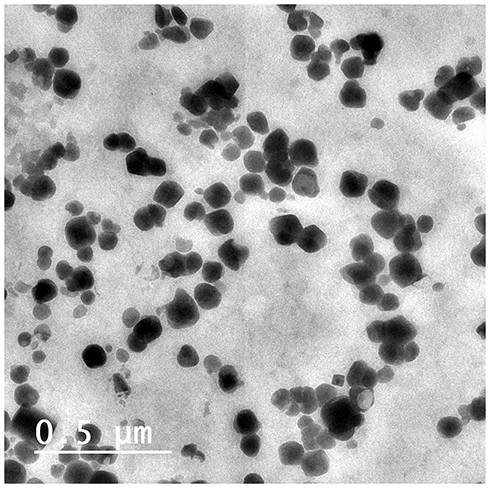 |
Figure 1 Transmission electron micrography of the GMC LPs when stained with uranyl acetate 10%. |
Evaluation of Ultrasound Sensitivity of Sonosensitive Liposomes
The release of GMC from GMC-SoS-Fo LPs was investigated under various periods of ultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation with 10-second rests for a total of 0–150 seconds), as shown in Figure 2. GMC release increased as a function of time, reaching 90.7%±3.7% at 130 seconds (10 seconds’ ultrasonic irradiation, 10-second pauses). However, an insignificant increase in GMC release was noted with an increase to 150 seconds, (92.4%±3.9%), so the period of ultrasonic irradiation was adjusted to 130 seconds in the following experiments.
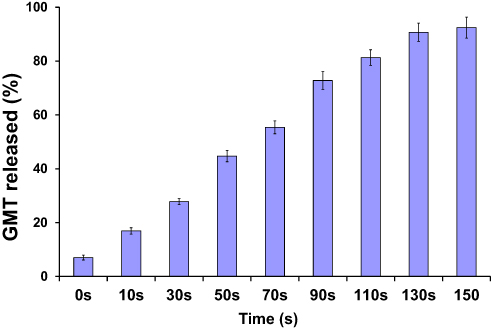 |
Figure 2 In vitro release of GMC from GMC-SoS-Fo LPs (6% Ce6) with various ultrasonic irradiation periods in PBS (0.1 M, pH 7.4) at 37°C. Results depicted as means ± SD (n=3). |
To confirm the role of Ce6 as activator of SoS GMC LPs, GMC-Fo LPs, and SoS LPs with various concentrations of Ce6 (3%, 6%, and 9%) incubated at 37°C for 20 minutes were examined under the specific ultrasonic irradiation condition of 2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation and 10-second rests for a total of 0–130 seconds), as shown in Figure 3. Minimal GMC release was recorded for all LPs untreated with ultrasonic irradiation. GMC release from conventional liposomes (GMC LPs) and GMC-Fo LPs was also unchanged under the same condition, as shown in Figure 3. However, GMC release from GMC-SoS-Fo LPs with 3% Ce6, GMC-SoS-Fo LPs with 6% Ce6, and GMC-SoS-Fo LPs with 9% Ce6 treated with ultrasonic irradiation was 48.7%±3.9%, 89.7%±4.2%, and 90.1%±2.7%, respectively. The results demonstrated that Ce6 content was a fundamental factor in releasing GMC from relevant LPs, while increased GMC release resulted from increasing the Ce6 content to 9% compared to the release from GMC-SoS-Fo LPs with 6% Ce6 (p<0.05). Ultrasonic irradiation conditions (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation and 10-second rests for a total of 0–130 seconds) may be insufficient to completely activate SoS LPs GMC-SoS-Fo LPs with 9% Ce6. However, 6% Ce6 was the optimum concentration under the same ultrasonic irradiation conditions to produce sonodynamic activity, resulting in burst release of GMC from GMC-SoS-Fo LPs. These results may be attributed to the ability of the SoS activator (Ce6) to form pores or irreparably damage the LP bilayer.44,45 Sonosensitivity of DSPE-containing LPs has been attributed to DSPE possibly forming an inverted hexagonal frame within the liposome bilayer as a response to high temperature or pressure.46 As a result of ultrasonic irradiation, DSPE in the lamellar liquid crystalline converted to a hexagonal phase, forming cavities because the polar head groups occupied less space than theapolar moieties.47
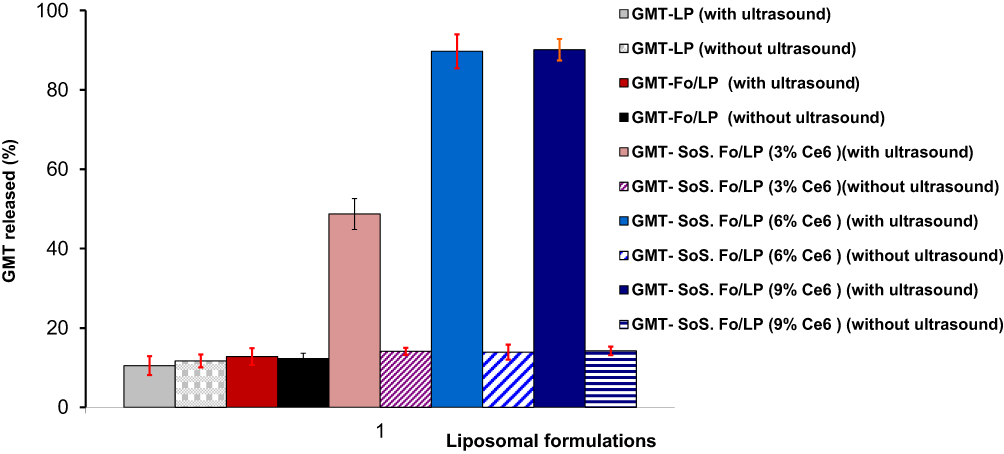 |
Figure 3 In vitro release of GMC from conventional LPs and Fo-targeted LPs with different percentage of SoS activator (Ce6 0, 3%, 6%, and 9%) treated or untreated with ultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation with a 10-second rest, for a total of 0–130 seconds), in PBS (0.1 M, pH 7.4) at 37°C. Results depicted as means ± SD (n=3). |
In Vitro Release of Liposomes
In vitro release of GMC from GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs treated with sonication or not is represented in Figure 4. GMC release from GMC-SoS-Fo LPs showed ultrasounddependence: as GMC-SoS-Fo LPs were exposed to ultrasonic irradiation, they were disrupted, releasing GMC promptly. However, GMC release from GMC-SoS-Fo LPs not treated with ultrasonic irradiation was 9.5%±1.5% after incubation of 50 minutes. Moreover, GMC LPs and GMC-Fo LPs represented ultrasound independence and minimal release change was observed, as shown in Figure 4.
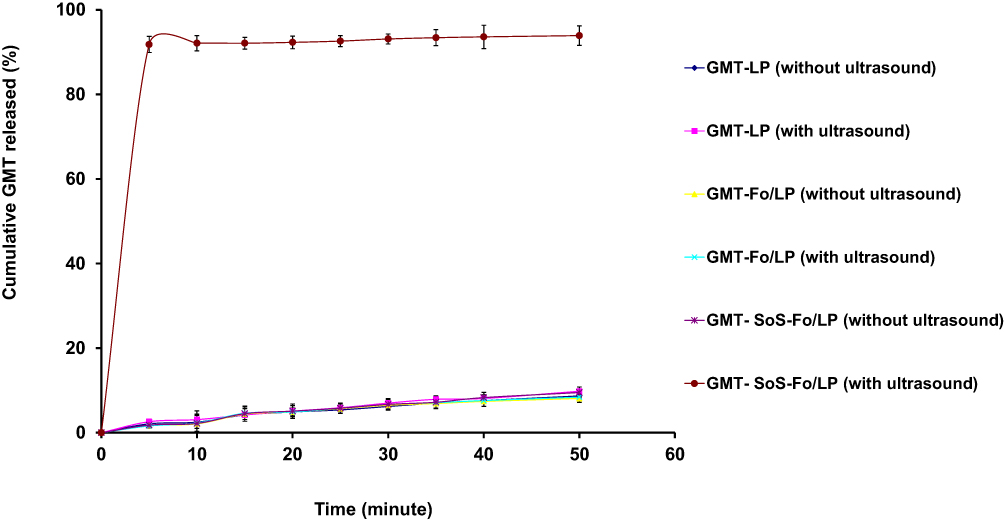 |
Figure 4 In vitro release of GMC from GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs untreated or treated with ultrasonic irradiation in isotonic PBS) pH 7.4, 37°C). Results depicted as means ±SD (n=3). |
GMC release from GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs incubated for 24 hours in isotonic PBS (pH 7.4, 37°C) was minimal (<23%). However, subjecting GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs to ultrasonicirradiation at 25 hours led to burst release (93.6%±3.9) only with GMC-SoS-Fo LPs, while insignificant release changes were observed with GMC LPs and GMC-Fo LPs, as shown in Figure 5. The results confirmed that release from GMC-SoS-Fo LPs was low until LPs were in contact with ultrasonic irradiation: burst release was achieved, suggesting little drug released in blood until the LPs reached the desired site (ovarian tumors) and were subjected to ultrasound. GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs showed a strong tendency to be stable upon no ultrasonic wave exposure. There were no changes in drug release from GMC LPs or GMC-Fo LPs in the absence or presence of ultrasonic irradiation. These findings could be explained by insensitivity of GMC LPs and GMC-Fo LPs to ultrasonic irradiation. However, GMC-SoS-Fo LPs were disrupted, releasing the encapsulated drug once they were stimulated with acoustic energy. The sensitizer Ce6 was a key component, whereas disruption of the LP membrane may have occurred as a result of vibration of Ce6 to an extent that was sufficient to destroy the bilayer membrane or form sufficient pores for escaping the encapsulated drug. SoS LPs for ultrasound-triggered release have been developed.48 As ultrasonic irradiation was applied, gas bubbles oscillated and collapsed in media, so introducing high mechanical pressure increases release substantially. This could be explained by the formation and collapse of small gas nuclei in the hydrophobic region of the lipid bilayer during exposure to ultrasonic irradiation, thereby inducing the formation of transient pores through which drugs are released.44
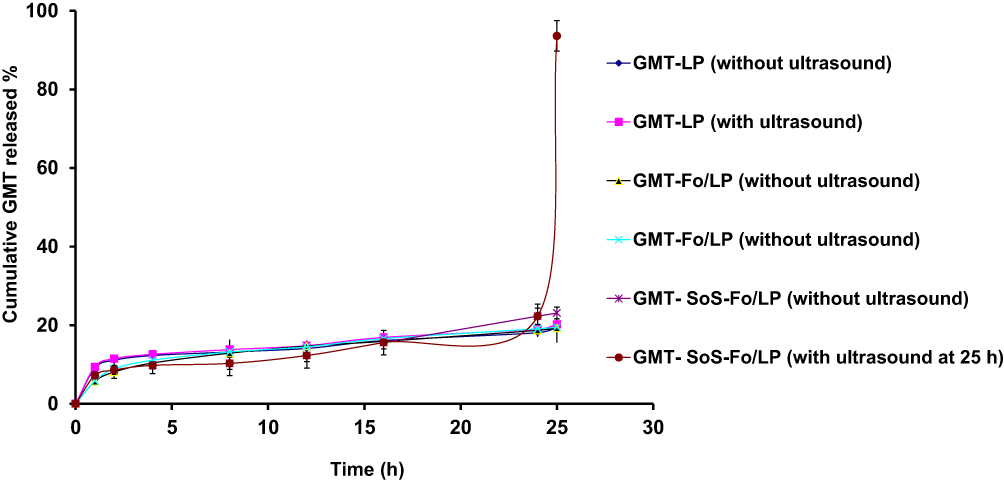 |
Figure 5 In vitro release of GMC from GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPsuntreated or treated with ultrasonic irradiation after 24 hours’ incubation in isotonic PBS (pH 7.4, 37°C). Results depicted as means ± SD (n=3). |
Stability
GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs were physically and chemically stable in the conditions examined (4 C, up to 3 months), as presented in Table 2. EE change forGMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs) was insignificant. Particle size growth, ζ-potential reduction, and PDI values of the LPs showed insignificant changes, proving that monodispersion of LPs was stable. Finally, ultrasonic sensitivity of GMC-SoS-Fo LPs continued without change during storage.
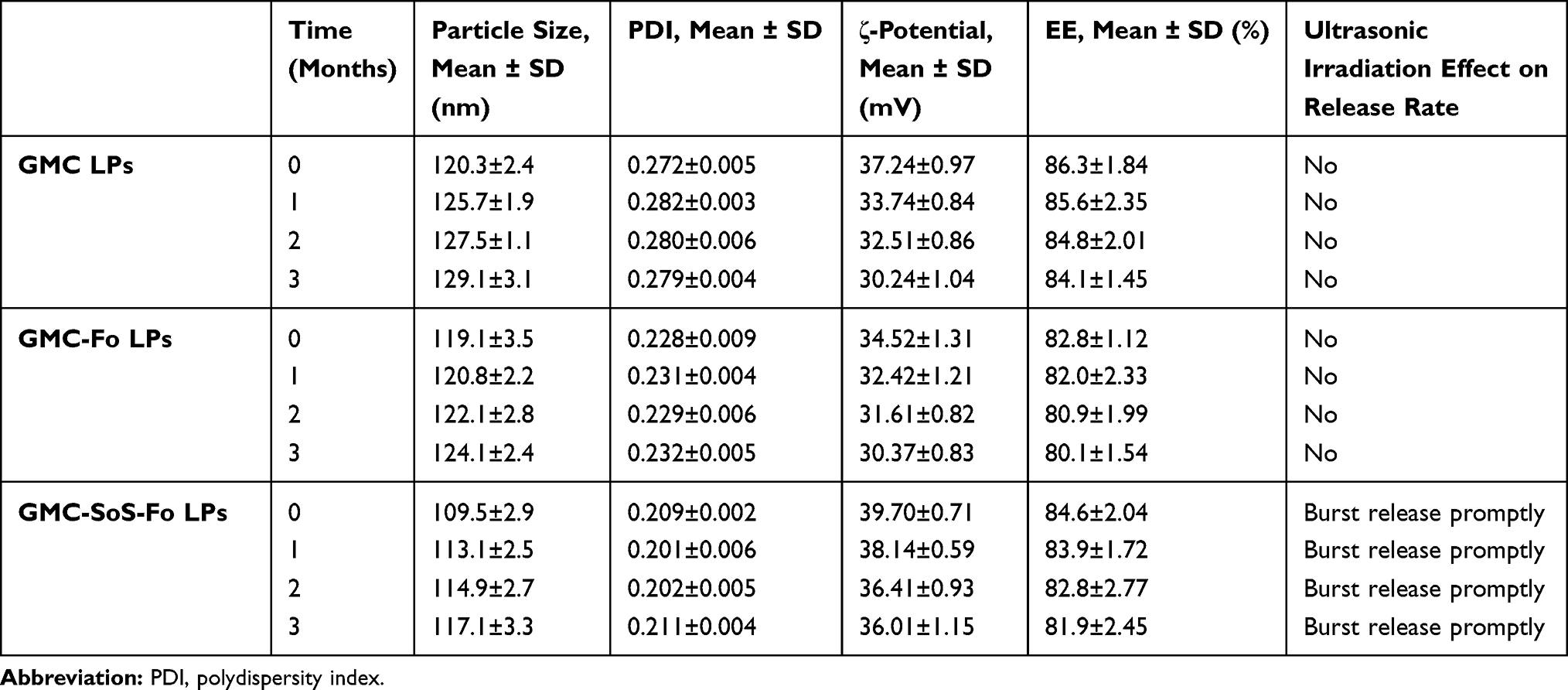 |
Table 2 Particle Size, PDI, Encapsulation Efficiency, and Effect of Ultrasonic Irradiation on Release of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs) Over 3 Months |
Cellular Uptake of Liposomes on Flow Cytometry
Flow-cytometry analysis was utilized to quantify cellular uptake of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs with or without ultrasonic irradiation. The cell populations that bound and absorbed released calcein were identified by green fluorescence. Calcein acetoxymethyl molecules have no green fluorescence, but were converted to green fluorescent calceinacetoxymethyl by intracellular metabolism. Cellular uptake of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs incubated with the cell line and untreated with ultrasonic irradiation was 12.5%±5.7%, 15.85%±3.9%, and 16.12%±4.9%, respectively. Additionally, cellular uptake of GMC LPs and GMC-Fo LPs incubated with the cell line and treated with ultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation, and 10-second rests for a total of 0–130 seconds), was 13.4%±4.5% and 15.45%±5.2%, respectively. However, cellular uptake of SoS targeted LPs (GMC-SoS-Fo LPs) incubated with the cell line treated with ultrasonic irradiation (2 MHz, 1.5 W/cm2 power intensity, 10 seconds’ ultrasonic irradiation, and 10-second rests for a total of 0–130 seconds), was 81.4%±8.5%, as shown in Figure 6. Compared to the cellular uptake achieved using GMC LPs not treated with ultrasonic irradiation, that achieved using GMC-SoS-Fo LPs treated with ultrasonic irradiation was improved 6.51-fold. These findings can be explained on the basis of intercellular uptake based on the Fo ligand followed by burst release as a response to ultrasonic irradiation. Ce6 molecules inserted into SoS targeted LPs were vibrated under the effect of ultrasonic irradiation, creating pores or rupturing the nanocarriers. Moreover, the extracellular liberated calcein acetoxymethyl can be easily engulfed by cells, converting it to have greenfluorescence. These results prove that SoS targeted LPs can bind strongly with Fo-expressing cells and release cargo promptly as a result of ultrasonic irradiation.
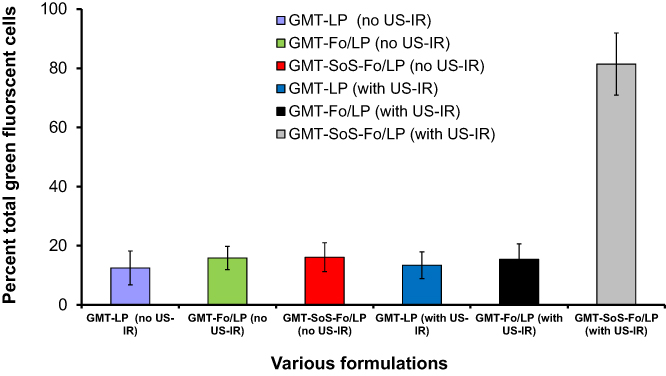 |
Figure 6 Flow-cytometry findings for GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs) when incubated with the CAOV3 cell line and treated or untreated with US-IR. Abbreviation: US-IR, ultrasonic irradiation. Notes: Measurements estimated after 30 minutes’ incubation, and are averages of three results ± SD. |
In Vitro Cytotoxicity Assays
The cytotoxic effects of SoS Fo LPs, free GMC, GMC LPs, GMC-Fo LPs, GMC-SoS Fo LPs (no ultrasonic irradiation), and GMC-SoS Fo LPs (ultrasonic irradiation) against the CAOV3 cells are illustrated in Figure 7. The CAOV3 cell line was chosen for this study because its viability is unaffected by the chemotherapeutic agent cisplatin at concentrations >200 µM and GMC >1,000 nM.49 The drug-free carrier system (SoS Fo LPs) alone showed no effect on cell viability, confirming the biocompatibility of SoS Fo LPs. CAOV3 cells were treated with various concentrations of GMC. Free GMC, GMC LPs, GMC-Fo LPs, and GMC-SoS Fo LPs (untreated) reduced cell viability in a dose-dependent manner up to 80 µM. However, GMC-SoS Fo LPs (treated with ultrasonic irradiation) reduced cell viability in a dose-dependent manner up to 40 µM. The highest cellular cytotoxic effect was reported with GMC-SoS Fo LPs (treated with ultrasonic irradiation, 40 µM drug equivalent). Our results showed that GMC-SoS Fo LPs (treated with ultrasonic irradiation, 80 µM drug equivalent) had an insignificant increase in cellular cytotoxic effect compared to GMC-SoS Fo LPs (treated with ultrasonic irradiation, 40 µM drug equivalent).
 |
Figure 7 Cytotoxic effect of GMC. CAOV3 cells were grown and treated with increasing concentrations of GMC in different liposomal formulations. Cell viability was evaluated by MTS reagent(see Methods). Values expressed asmeans ± SEM of at least three different experiments in triplicate. |
Pharmacokinetic and Biodistribution Studies
Pharmacokinetic and biodistribution evaluations of the formulations were performed to examine in vivo ovarian cancer–targeting efficiency. GMC concentration in plasma after intravenous injection in LPs was measured, as presented in Figure 8. Limits of determination detection were 0.1617 and 0.4903 µg/mL, respectively, calculated using calibration curves as per International Council for Harmonisation guidelines. Accuracy and precision of analytical assays were achieved across their linear range.
GMC plasma concentration reached its peak (24.1±1.2 µg/mL) in five minutes after a single GMC injection. It quickly decreased to be around 8.78% of the peak value within 1 hour, suggesting rapidly eliminationt. The rapid plasma elimination of GMC following intravenous injection of the pure form of the drug could be explained by the rapid metabolism of GMC and conversion into its inactive metabolites. These metabolites are more susceptible to elimination, and this was confirmed by increased concentration of GMC in kidneys, as shown in Figure 8.
GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs showed insignificant differences in plasma concentration. These results confirmed that the Ce6 component did not interfere with long circulation in LPs (Table 3).
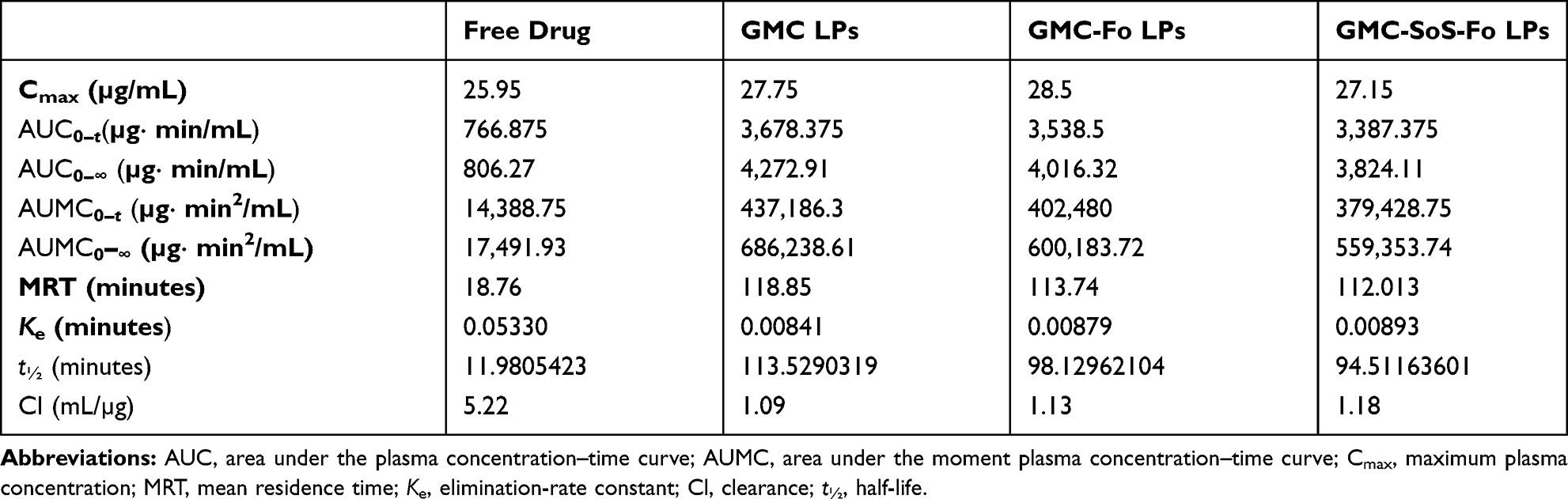 |
Table 3 Pharmacokinetic Parameters of GMC After IV Injection in Different Formulations (Free Drug, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs) |
However, pharmacokinetic parameters of free GMC — t½, AUC0–t, AUC0–∞, mean residence time, and clearance — were significantly different from GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs (p<0.05, Table 3).
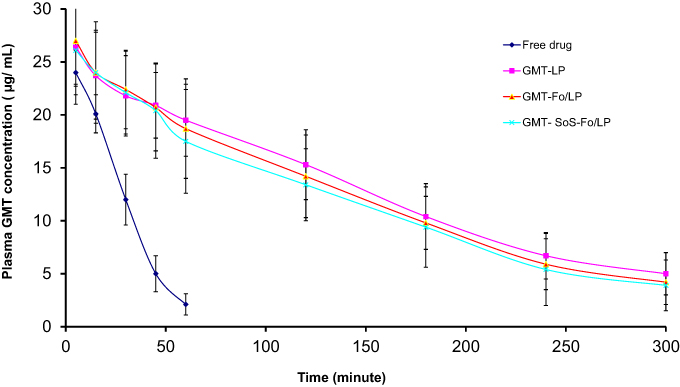 |
Figure 8 Plasma concentration of GMC versus time after IV injection of free drug, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs in rats (n=3). Results are depicted as means ± SD (n=3). |
Selective targeting of drug to tumors was confirmed by the concentration of GMC in tumors following intravenous injection of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs without ultrasound and GMC-SoS-Fo LPs with ultrasound, as presented in Figure 9. Concentration of GMC in tumors treated with GMC-SoS-Fo LPs with ultrasound was 2.89-fold that of free GMC, while concentration of GMC in tumors treated with GMC-SoS-Fo LPs with ultrasound was 1.98-fold that of GMC LPs, 1.44-fold that of GMC-Fo LPs, and 1.42-foldthat of GMC-SoS-Fo LPs with no ultrasound. The highest concentration of GMC was achieved in tumors treated with GMC-SoS-Fo LPs with ultrasound, leading not only to improved specificity of the drug and killing of cancerous cells but also to decreased side effects. These benefits of using GMC-SoS-Fo LPs with ultrasound encourage further application of SoS delivery systems for treating tumors.
These findings could be explained by the targeting efficiency of GMC-SoS-Fo LPs and their externally triggered rapid release, producing high concentrations within tumor tissue. The dual mechanism of GMC-SoS-Fo LPs may potentially support anticancer efficiency of GMC in vivo. To confirm these results clinically, evaluation of antitumor activity of various LPs was performed in vivo.
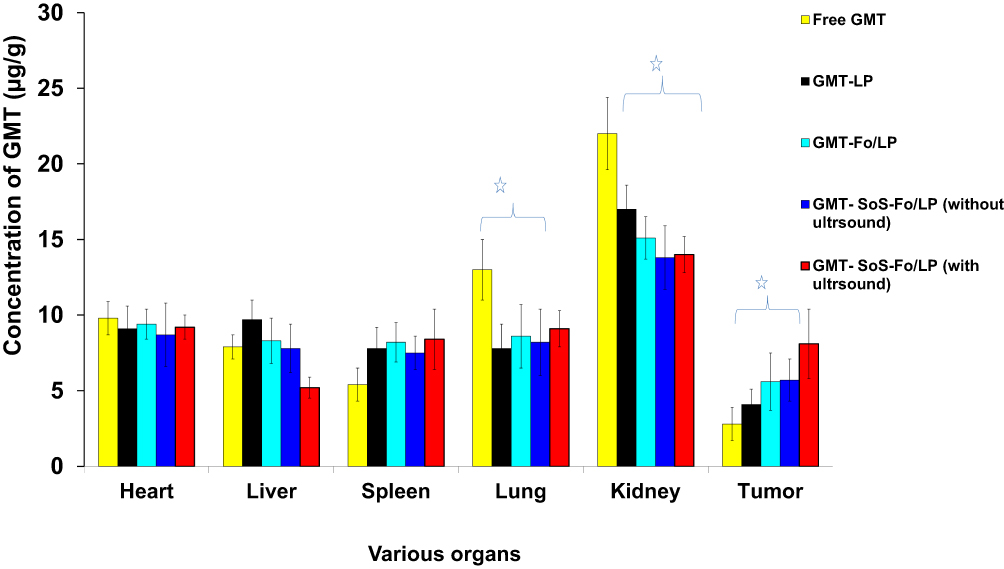 |
Figure 9 Biodistribution of GMC in heart, liver, spleen, lung, and kidney and ovarian cancer 30minutes after IV injection of free GMC, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs without ultrasonic irradiation and GMC-Fo LPs and GMC-SoS-Fo LPs with ultrasonic irradiation. Results depicted as means ± SD (n=10). *p<0.05. |
In Vivo Antitumor Activity
Repression of tumor growth following intravenous injection of free GMC, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs with or without ultrasonic irradiation was evaluated, as shown in Figure 10. To determine tumor-repression effects, the ratio of ovary volume at the end of the experiment to ovary volume at the beginning of the experiment was measured. The higher the ovary-growth ratio is, the lower the tumor-repression effect. Ovary-growth ratio for free GMC, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs with or without ultrasonic irradiation were 3.46, 2.76, 2.25, 1.75, 1.71, and 1.21, respectively. As predicted, the tumor-growth ratio forGMC-SoS-Fo LPs with ultrasonic irradiation was lowest — 1.21. This can be attributed to the targeting efficiency of the drug-delivery system, relatively long circulation time of the nanocarriers, and external triggering by ultrasonic irradiation, resulting in rapid release of the drug cargo.
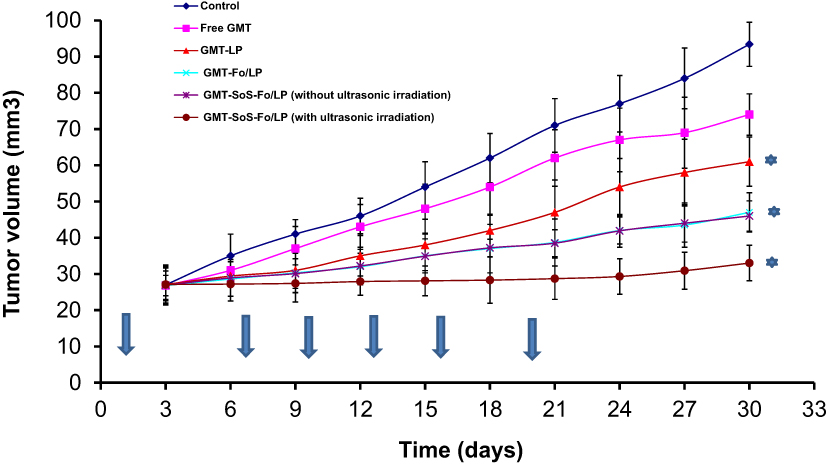 |
Figure 10 Antitumor activity of GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs without ultrasonic irradiation and GMC-SoS-Fo LPs with ultrasonic irradiation against isotonic saline and free drug in xenograft ovarian cancer rat models. Results depicted as means ± SD (n=10). Arrows show the days when treatments were given. *p<0.5. |
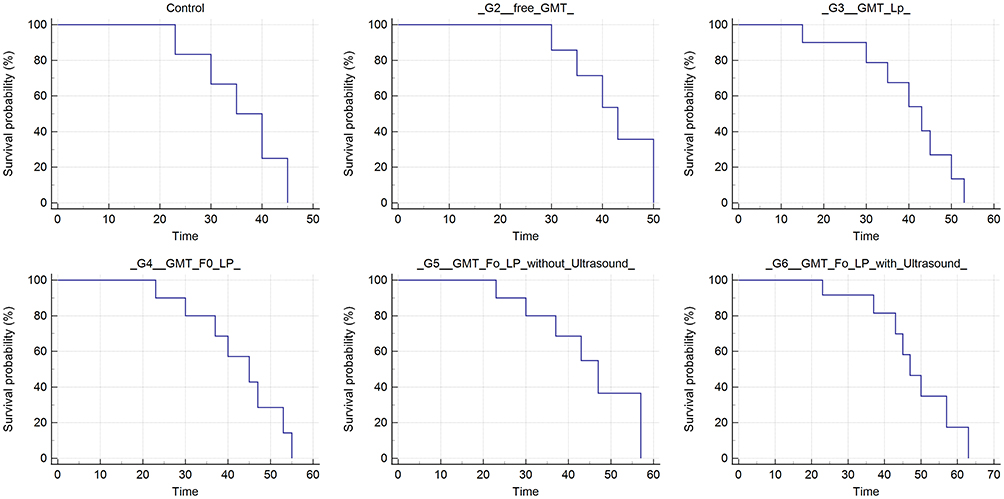 |
Figure 11 Kaplan–Meier survival curves for rats treated intravenously with GMC-free LPs (control group), 4 mg/kg of free GMC solution (G1), GMC LPs (G2), GMC-Fo LPs (G3), GMC-SoS-Fo LPs (G4), and GMC-SoS-Fo LPs (G5) subjected to ultrasonic irradiation. Drug dose administered was 4 mg/kg (n=3 for all groups). |
As for the Kaplan–Meier survival curves, the median survival of GMC-SoS-Fo LPs (G5) with ultrasonic irradiation–treated rats was compared to all other groups. Significant improvement in median survival and thus slowdown of tumor recurrence was observed in groups treated with GMC-SoS-Fo LPs (G5) with ultrasonic irradiation compared to untreated and other groups (Figure 11).
Finally, body weight in the groups was measured to evaluate the safety of the dosages. Insignificant body-weight changes were recorded for groups injected with GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs with or without ultrasonic irradiation, as shown in Figure 12. However, the control group and that intravenously injected with free GMC were characterized by acute body-weight reduction because of cancer cachexia and the toxicity of untargeted dosage forms. Repression of tumor growth inhibited weight loss.
In cancer-suffering living bodies, loss of body weight with specific loss of skeletal muscle and adipose tissue is recorded, which is called cachexia. This is highly associated with cancers of the pancreas, ovaries, stomach, lungs, and bowels. Weight loss may be due to processes related to metabolic changes mediated by excess release of proinflammatory cytokines and increased activity of the sympathetic nervous system. Both catecholamines and proinflammatory cytokines promote catabolic processes.36 Proinflammatory cytokines, in addition to decreasing the efficacy of growth hormones, work in the central nervous system as mediators of inflammation and act as catabolic factors stimulating proteolytic pathways, leading to muscle atrophy and increased adipose-tissue breakdown.50 Proinflammatory cytokines, including IL6, IL10, IL1β, and TNFα, stimulate muscle-protein degradation, cause contractile dysfunction, and inhibit myogenesis, in addition to promoting adipose-tissue waste, inhibition of adipocyte differentiation, lipolysis stimulation, and increased apoptosis in adipocytes.51
 |
Figure 12 Weight loss recorded for groups treated with control (isotonic saline solution), free GMC, GMC LPs, GMC-Fo LPs, and GMC-SoS-Fo LPs without ultrasonic irradiation and GMC-SoS-Fo LPs with ultrasonic irradiation) in xenograft ovarian cancer rat models. Results depicted as the means ± SD (n=10). |
Finally, these formulations succeeded in managing ovarian cancer because of their accumulation within tumors via passive (GMC LPs) or active (GMC-Fo LPs and GMC-SoS-Fo LPs) targeting. Accumulation of nanocarriers in tumors has been reported as due to the EPR effect,52 because of leaky vasculature and lymphatic drainage within cancerous tissue. Physicochemical properties of nanocarriers, such as particle size, charge of surface, and circulation time(longevity), can influence their accumulation within tumors. Particle size of nanocarriers is an important parameter affecting the EPR effect of nanocarriers.53 To achieve extravasation of drug in cancerous tissue, the mean size of drug carriers should be about 100 nm. At the same time, EE should be also be sufficient. These technological requirements are important for effective in vivo–delivered drugs.54,55
The negative charge of the luminal surface of blood vessels, due to the presence of carboxylate sugars and the sulfate group, interact with positively charged nanocarriers, preventing them from recirculation.56 However, a high positive charge may lead to opsonization and clearance of nanocarriers from the circulation57 and may be the reason for insignificant difference in tumor accumulation found between GMT LPs and GMT-Fo LPs.
Longevity can influence both passive and active accumulation of nanocarriers, because of the large number of passages achieved via the target. To prevent the capture and clearance of nanocarriers, steric protection should be achieved by grafting polymers, such as PEG, on the surface of nanocarriers. Steric protection can be explained on the basis of shielding surface charges, improved hydrophilicity, and formation of a polymeric layer covering the nanocarriers’ surfaces, leading to repulsion between blood components and nanocarriers.58
Active targeting can be achieved via two techniques. The first is decorating the nanocarrier’s surface with ligands, such as Fo, that interact with specific receptors. Fo receptors are extensively expressed in tumor tissue. The choice of the targeting ligand represents a significant factor in the engineering of targeted nanocarriers.59 The second technique is achieved via formulating stimulus-sensitive nanocarriers that respond to internal stimuli, such as changes in the pathological area or external stimuli ultrasonic irradiation.
Ultrasonic waves are mechanical waves with periodic continuous vibrations at frequencies of 20 kHz or more. They are safe; however, they have exceptional tissue-penetrating ability without major attenuation of energy.60 The effect of ultrasonic waves can be localized by irradiating specific areas and choosing an SoS drug-delivery system with cancer affinity.61 In the present study, the sensitizer was a key component. Distribution and cellular uptake of Ce6 was the most important factor in its therapeutic action, due to short life and very short diffusion distance of some radical products, such as peroxides, derived from the sensitizer and produced after its cellular uptake.62 Li et al reported that Ce6 enhanced cytotoxicity and apoptotic response as a result of Ce6 accumulation within mitochondria, leading to damage and apoptosis.63 Subsequently, the damaged mitochondria released many chemical factors, eg, cytochrome C. The factors released stimulate activation of the caspase 9 proenzyme to its active form, causing cell apoptosis.63
The present study used ultrasonic irradiation to enhance delivery of GMC to ovarian cancer upon GMC-SoS-Fo LPs. This technique used ultrasonic irradiation to increase the permeabilization of phospholipid membranes, inducing GMC release from nanocarriers at cancerous ovaries and increasing drug uptake into cancer cells.
Conclusion
Targeted SoS LPs (GMC-SoS-Fo LPs) have a spherical shape and are nanosized with optimum ζ-potential. Compared to untargeted LPs (GMC LPs) and targeted nonultrasonic sensitive LPs (GMC-Fo LPs), the targeted SoS LPs succeeded in releasing the drug cargo as a response to ultrasonic irradiation as external stimuli. Moreover, targeted SoS LPs enhanced intracellular uptake, leading to significant inhibition of tumor growth.
The study data showed that GMC-SoS-Fo LPs are a promising anticancer drug- delivery system. To enhance the targeting efficiency of SoS LPs, Fo ligands were inserted into the the prepared LPs (active targeting) and SoS LPs were prepared in the nanosize range (passive targeting).
In future, we need to develop SoS theranostic LPs to explore the usage of these drug-delivery systems not only for oncotherapy but also for diagnosis.
Abbreviations
GMC, gemcitabine; GMC LPs, GMC-loaded LPs; GMC-Fo LPs, GMC-loaded Fo-targeted LPs; GMC-SoS Fo LPs, GMC-loaded Fo-targeted SoS LPs; PC, L-α-phosphatidylcholine; EE, entrapment efficiency); DL, drug loading; PDI, polydispersity index; EPR, enhanced permeability and retention; Ce6, chlorin e6.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ibrahim A, et al. Cancer incidence in Egypt: results of the national population-based cancer registry program. J Cancer Epidemiol. 2014;2014:18. doi:10.1155/2014/437971
2. Goff BA, et al. Ovarian carcinoma diagnosis. Cancer. 2000;89(10):2068–2075. doi:10.1002/1097-0142(20001115)89:10<2068::AID-CNCR6>3.0.CO;2-Z
3. Ten Bokkel Huinink W, et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J Clin Oncol. 1997;15(6):2183–2193. doi:10.1200/JCO.1997.15.6.2183
4. Gershenson DM, et al. Single-agent cis-platinum therapy for advanced ovarian cancer. Obstet Gynecol. 1981;58(4):487–496.
5. Ozols RF, et al. Phase I and pharmacological studies of adriamycin administered intraperitoneally to patients with ovarian cancer. Cancer Res. 1982;42(10):4265–4269.
6. Coleman RL. Emerging role of topotecan in front-line treatment of carcinoma of the ovary. oncologist. 2002;7(90005):46–55. doi:10.1634/theoncologist.7-suppl_5-46
7. Rave-Fränk M, et al. Combined effect of topotecan and irradiation on the survival and the induction of chromosome aberrations in vitro. Strahlentherapie und Onkologie. 2002;178(9):497–503. doi:10.1007/s00066-002-0959-y
8. Lokich J. What is the “best” platinum: cisplatin, carboplatin, or oxaliplatin. Cancer Invest. 2001;19(7):756–760. doi:10.1081/CNV-100106152
9. Alexandre JRM, et al. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 2007;67(8):3512–3517. doi:10.1158/0008-5472.CAN-06-3914
10. Ozols RF. The Current Role of Gemcitabine in Ovarian Cancer. In Seminars in Oncology. Elsevier; 2001.
11. Le TN, et al. A retrospective evaluation of activity of gemcitabine/platinum regimens in the treatment of recurrent ovarian cancer. Gynecologic Oncology Research Practice. 2017;4(1):16. doi:10.1186/s40661-017-0053-x
12. Tomita Y, et al. The safety and efficacy of cisplatin plus gemcitabine in recurrent ovarian cancer. Int J Clin Oncol. 2014;19(4):662–666. doi:10.1007/s10147-013-0599-5
13. Alvarellos M, et al. PharmGKB summary: gemcitabine pathway. Pharmacogenet Genomics. 24(11):564–574. doi:10.1097/FPC.0000000000000086
14. Mackey JR, Mani RS, Selner M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58(19):4349–4357.
15. Immordino ML, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Controlled Release. 2004;100(3):331–346. doi:10.1016/j.jconrel.2004.09.001
16. Celano M, et al. Cytotoxic effects of gemcitabine-loaded liposomes in human anaplastic thyroid carcinoma cells. BMC Cancer. 2004;4(1):1–5. doi:10.1186/1471-2407-4-63
17. Mody VV, et al. Magnetic nanoparticle drug delivery systems for targeting tumor. Applied Nanoscience. 2014;4(4):385–392. doi:10.1007/s13204-013-0216-y
18. Kim A, et al. In vitro and in vivo transfection efficiency of a novel ultradeformable cationic liposome. Biomaterials. 2004;25(2):305–313. doi:10.1016/S0142-9612(03)00534-9
19. Tahover E, Patil YP, Gabizon AA. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: focus on liposomes. Anticancer Drugs. 2015;26(3):241–258. doi:10.1097/CAD.0000000000000182
20. Perche F. Recent trends in multifunctional liposomal nanocarriers for enhanced tumor targeting. J Drug Deliv. 2013;2013:705265. doi:10.1155/2013/705265
21. Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine. 2013;8(9):1509–1528. doi:10.2217/nnm.13.118
22. Corvo ML, et al. Liposomes as delivery system of a Sn (IV) complex for cancer therapy. Pharm Res. 2016;33(6):1351–1358. doi:10.1007/s11095-016-1876-6
23. Allen TM. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol Sci. 1994;15(7):215–220. doi:10.1016/0165-6147(94)90314-X
24. Li X, et al. In vitro and in vivo evaluation of folate receptor-targeting amphiphilic copolymer-modified liposomes loaded with docetaxel. Int J Nanomedicine. 2011;6:1167.
25. Chen Y, et al. Baicalin loaded in folate-PEG modified liposomes for enhanced stability and tumor targeting. Colloids Surf B Biointerfaces. 2016;140:74–82. doi:10.1016/j.colsurfb.2015.11.018
26. Fasehee H, et al. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: in vitro and in vivo investigations. J Nanobiotechnology. 2016;14(1):32. doi:10.1186/s12951-016-0183-z
27. Lukyanov AN, et al. Tumor-targeted liposomes: doxorubicin-loaded long-circulating liposomes modified with anti-cancer antibody. J Controlled Release. 2004;100(1):135–144. doi:10.1016/j.jconrel.2004.08.007
28. Kumar P, Huo P, Liu B. Formulation Strategies for Folate-Targeted Liposomes and Their Biomedical Applications. Pharmaceutics. 2019;11(8):381. doi:10.3390/pharmaceutics11080381
29. Zhang P, et al. Iron oxide nanoparticles as nanocarriers to improve chlorin e6-based sonosensitivity in sonodynamic therapy. Drug Des Devel Ther. 2018;12:4207. doi:10.2147/DDDT.S184679
30. Newman MS, et al. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother Pharmacol. 1999;43(1):1–7. doi:10.1007/s002800050855
31. Yamada A, et al. Design of folate-linked liposomal doxorubicin to its antitumor effect in mice. Clinical Cancer Research. 2008;14(24):8161–8168. doi:10.1158/1078-0432.CCR-08-0159
32. Torchilin VP. Passive and Active Drug Targeting: Drug Delivery to Tumors as an Example, in Drug Delivery. Springer; 2010:3–53.
33. Monteiro LOF, et al. Paclitaxel-loaded folate-coated long circulating and pH-sensitive liposomes as a potential drug delivery system: A biodistribution study. Biomedicine Pharmacotherapy. 2018;97:489–495. doi:10.1016/j.biopha.2017.10.135
34. Gulati M, et al. Lipophilic drug derivatives in liposomes. Int J Pharm. 1998;165(2):129–168. doi:10.1016/S0378-5173(98)00006-4
35. Liu R, et al. Liquid crystalline nanoparticles as an ophthalmic delivery system for tetrandrine: development, characterization, and in vitro and in vivo evaluation. Nanoscale Res Lett. 2016;11(1):254. doi:10.1186/s11671-016-1471-0
36. Egelhaaf SU, et al. Determination of the size distribution of lecithin liposomes: a comparative study using freeze fracture, cryoelectron microscopy and dynamic light scattering. J Microsc. 1996;184(3):214–228. doi:10.1046/j.1365-2818.1996.1280687.x
37. Omar MM, Hasan OA, El Sisi AM. Preparation and optimization of lidocaine transferosomal gel containing permeation enhancers: a promising approach for enhancement of skin permeation. Int J Nanomedicine. 2019;14:3705–3727. doi:10.2147/IJN.S201356
38. Salem HF, et al. Targeting brain cells with glutathione-modulated nanoliposomes: in vitro and in vivo study. Drug Des Devel Ther. 2015;9:3705. doi:10.2147/DDDT.S85302
39. X Fu, R M Hoffman. Human ovarian carcinoma metastatic models constructed in nude mice by orthotopic transplantation of histologically-intact patient specimens. Anticancer Res. 1993;13(2):283–6. Available from: https://pubmed.ncbi.nlm.nih.gov/8517640/
40. Zuo L, Roberts WJ, Evans KD. Diagnostic ultrasound imaging of mouse diaphragm function. J Vis Exp. 2014;86:51290.
41. Lin NM, et al. Determination of gemcitabine and its metabolite in human plasma using high-pressure liquid chromatography coupled with a diode array detector. Acta Pharmacol Sin. 2004;25:1584–1589.
42. Pflanzer R, et al. Advanced 3D-sonographic imaging as a precise technique to evaluate tumor volume. Transl Oncol. 2014;7(6):681–686. doi:10.1016/j.tranon.2014.09.013
43. Lila ASA, Ishida T, Kiwada H. Targeting anticancer drugs to tumor vasculature using cationic liposomes. Pharm Res. 2010;27(7):1171–1183. doi:10.1007/s11095-010-0110-1
44. Schroeder A, Kost J, Barenholz Y. Ultrasound, liposomes, and drug delivery: principles for using ultrasound to control the release of drugs from liposomes. Chem Phys Lipids. 2009;162(1–2):1–16. doi:10.1016/j.chemphyslip.2009.08.003
45. Suzuki R, et al. Progress in the development of ultrasound-mediated gene delivery systems utilizing nano-and microbubbles. J Control Release. 2011;149(1):36–41. doi:10.1016/j.jconrel.2010.05.009
46. Evjen TJ, et al. Distearoylphosphatidylethanolamine-based liposomes for ultrasound-mediated drug delivery. European J Pharmaceutics Biopharmaceutics. 2011;75(3):327–333. doi:10.1016/j.ejpb.2010.04.012
47. Kusube M, Matsuki H, Kaneshina S. Thermotropic and barotropic phase transitions of N-methylated dipalmitoylphosphatidylethanolamine bilayers. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 2005;1668(1):25–32. doi:10.1016/j.bbamem.2004.11.002
48. Huang S-L, MacDonald RC. Acoustically active liposomes for drug encapsulation and ultrasound-triggered release. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 2004;1665(1–2):134–141. doi:10.1016/j.bbamem.2004.07.003
49. Kawaguchi H, et al. Gemcitabine as a molecular targeting agent that blocks the Akt cascade in platinum-resistant ovarian cancer. J Ovarian Res. 2014;7(1):1–11. doi:10.1186/1757-2215-7-38
50. Steinman J, DeBoer MD. Treatment of Cachexia: Melanocortin and Ghrelin Interventions, in Vitamins & Hormones. Elsevier; 2013:197–242.
51. Rania S Abdel-Rashid, Doaa A Helal, Mahmoud M Omar,and Amani M El Sisi; Nanogel loaded with surfactant based nanovesicles for enhanced ocular delivery of acetazolamide. Int J Nanomedicine. 2019;14:2973–2983.
52. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Part 1):6387–6392.
53. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. doi:10.1016/j.addr.2012.10.002
54. Cosco D, et al. In vivo activity of gemcitabine-loaded PEGylated small unilamellar liposomes against pancreatic cancer. Cancer Chemother Pharmacol. 2009;64(5):1009–1020. doi:10.1007/s00280-009-0957-1
55. Celia C, et al. Gemcitabine-loaded innovative nanocarriers vs GEMZAR: biodistribution, pharmacokinetic features and in vivo antitumor activity. Expert Opin Drug Deliv. 2011;8(12):1609–1629. doi:10.1517/17425247.2011.632630
56. Ding H-M. Controlling cellular uptake of nanoparticles with pH-sensitive polymers. Sci Rep. 2013;3:2804. doi:10.1038/srep02804
57. Xiao K, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. doi:10.1016/j.biomaterials.2011.01.021
58. Gabizon A, Papahadjopoulos D. The role of surface charge and hydrophilic groups on liposome clearance in vivo. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 1992;1103(1):94–100. doi:10.1016/0005-2736(92)90061-P
59. Bertrand N, Leroux J-C. The journey of a drug-carrier in the body: an anatomo-physiological perspective. J Controlled Release. 2012;161(2):152–163. doi:10.1016/j.jconrel.2011.09.098
60. Jensen JRA. A model for the propagation and scattering of ultrasound in tissue. J Acoust Soc Am. 1991;89(1):182–190. doi:10.1121/1.400497
61. FERIL, Jr. LB, Kondo T. Biological effects of low intensity ultrasound: the mechanism involved, and its implications on therapy and on biosafety of ultrasound. J Radiat Res. 2004;45(4):479–489. doi:10.1269/jrr.45.479
62. Yumita N. Membrane lipid peroxidation as a mechanism of sonodynamically induced erythrocyte lysis. Int J Radiat Biol. 1996;69(3):397–404. doi:10.1080/095530096145959
63. Li Q, et al. Efficacy of chlorin e6-mediated sono-photodynamic therapy on 4T1 cells. Cancer Biother Radiopharm. 2014;29(1):42–52. doi:10.1089/cbr.2013.1526
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.