Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
Etiology-Driven Mouse Models of Hepatocellular Carcinoma: Paving the Way for Precision Oncology
Authors Cao W ![]() , Fan Y
, Fan Y ![]() , Chen H
, Chen H ![]() , Sun J, Jin X
, Sun J, Jin X ![]()
Received 29 April 2026
Accepted for publication 23 June 2026
Published 8 July 2026 Volume 2026:13 621043
DOI https://doi.org/10.2147/JHC.S621043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr David Gerber
Wuqing Cao,1,* Yiping Fan,1,* Heyi Chen,1,* Jichun Sun,1 Xiaoxin Jin2
1Department of Hepatobiliary and Pancreatic Surgery, The Third Xiangya Hospital of Central South University, Changsha, People’s Republic of China; 2Department of General Surgery, The Second Xiangya Hospital, Central South University, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jichun Sun, Email [email protected] Xiaoxin Jin, Email [email protected]
Background: Hepatocellular carcinoma (HCC) is biologically heterogeneous, and its genomic alterations, inflammatory context, and tumor immune microenvironment are strongly shaped by the underlying etiology, including chronic hepatitis B virus (HBV) infection, metabolic dysfunction-associated steatotic liver disease (MASLD), and alcohol exposure. This etiological diversity complicates the selection and interpretation of preclinical models. Genetically engineered mouse models (GEMMs), particularly when combined with dietary, chemical, viral, or alcohol-related insults, provide useful systems for dissecting how defined genetic drivers interact with disease-specific liver environments in an immunocompetent host.
Main Body: This review summarizes etiology-aligned GEMMs and related mouse models for HCC. In HBV-related models, we discuss how viral antigen exposure, HBV-associated genomic instability, Tert activation, and Trp53 loss support a multi-driver framework rather than a single-lesion model of carcinogenesis. In MASLD-associated HCC, we examine models involving Wnt/β-catenin activation, Acvr2a loss, Pten deficiency, and MUP-uPA driven steatohepatitis, emphasizing their value for studying immune exclusion, lactate-rich immunosuppression, IgA+ plasma-cell accumulation, and altered responses to immune checkpoint blockade. In alcohol-associated HCC, we review models centered on Aldh2 deficiency, ER stress-lysosomal lipid remodeling through the ATF4/LPLA2 axis, NF-κB-related inflammatory regulation, and neutrophil-driven tumor promotion. Across these etiologies, we compare model strengths and limitations, including tumor latency, penetrance, reproducibility, lack of cirrhotic remodeling, sex-dependent variability, microbiome-related environmental effects, and incomplete modeling of tumor-stroma co-evolution.
Short Conclusion: Etiology-aligned GEMMs can help match biological questions to appropriate preclinical platforms and generate testable hypotheses about therapy response. However, etiology alone should not be treated as a substitute for molecular profiling or clinical validation. Future models should integrate precise genetic engineering with fibrotic or cirrhotic backgrounds, microbiome-aware environmental modulation, and complementary human-relevant systems to better capture the complex evolution of human HCC.
Keywords: hepatocellular carcinoma, mouse models, HBV, MASLD, alcohol-associated hcc, precision medicine
Introduction
Primary liver cancer represents a major global health burden. According to GLOBOCAN statistics, it ranks as the sixth most common malignancy worldwide and the third leading cause of cancer-related mortality.1 Histologically, hepatocellular carcinoma (HCC) accounts for approximately 75% to 85% of all primary liver cancer cases.2 Although molecularly targeted therapies and immunotherapies have expanded the therapeutic landscape of HCC, the initiation and progression of this disease remain governed by complex interactions among genetic alterations, chronic liver injury, inflammation, fibrosis, and immune remodeling.3 A central feature of HCC is etiological heterogeneity: beyond traditional hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, metabolic dysfunction-associated steatotic liver disease (MASLD, formerly grouped under NAFLD) is rapidly emerging as a major contributor to HCC burden, while alcohol-associated HCC continues to rise globally.4,5
Animal models are indispensable for dissecting HCC pathogenesis and testing therapeutic hypotheses, but each model class has intrinsic limitations. Chemically induced models can reproduce selected injury and inflammatory processes but often lack genetic precision. Xenograft models are useful for tumor growth and drug-response studies but usually fail to recapitulate an intact immune system and the chronic liver-disease background in which most human HCCs arise. In contrast, genetically engineered mouse models (GEMMs), using systems such as Cre-loxP and CRISPR/Cas9, allow controlled manipulation of defined oncogenic drivers or tumor suppressors in an immunocompetent host.6,7 However, no single GEMM fully reproduces the complete inflammation–fibrosis–cirrhosis–HCC sequence observed in humans. Their interpretability depends on model-selection parameters such as tumor latency, penetrance, strain background, sex, environmental injury, microbiome status, immune competence, and the degree of fibrosis or cirrhosis achieved before tumor development.
Given the biological differences among HBV-related, MASLD-associated, and alcohol-associated HCC, etiology-aligned GEMMs provide a useful framework for matching specific mechanistic questions to appropriate preclinical platforms. For example, HBV-related models are useful for studying viral antigen exposure, HBx-mediated host-gene disruption, genomic instability, and cooperation with host drivers such as TERT activation or TP53 loss. MASLD-associated models can be used to interrogate Wnt/β-catenin activation, ACVR2A loss, metabolic stress, and immune exclusion. Alcohol-associated models provide insight into acetaldehyde metabolism, ER stress, lysosomal lipid remodeling, and inflammation-driven tumor promotion. Nevertheless, etiology should be regarded as a model-selection context rather than a validated surrogate for molecular profiling or clinical treatment selection.
This review summarizes the current landscape of etiology-aligned GEMMs and related mouse models for HCC, focusing on HBV-related, MASLD-associated, and alcohol-associated disease. HCV-related models are not treated as a central category because direct-acting antivirals have substantially reduced HCV-driven disease burden, but the residual HCC risk after sustained virologic response, particularly in patients with advanced fibrosis or cirrhosis, is addressed in the discussion of translational limitations. We emphasize model construction strategies, mechanistic insights, tumor latency and penetrance, reproducibility, fibrosis/cirrhosis status, and major limitations, with the goal of providing a critical framework for selecting appropriate preclinical systems rather than presenting GEMMs as direct substitutes for human molecular stratification.
HBV-Related HCC Models
Despite substantial progress in antiviral therapies, chronic Hepatitis B Virus (HBV) infection remains the primary driver of HCC globally, imposing a heavy clinical burden that is difficult to eradicate.8,9 In recent years, GEMMs based on the full HBV genome or specific viral proteins (eg., HBsAg, HBx) have successfully simulated the state of chronic viral antigen exposure in hosts. These models have played a key role in deciphering carcinogenic mechanisms, particularly confirming that the cytotoxicity of HBsAg and the transcriptional regulatory functions of HBx drive malignant transformation by inducing persistent hepatic inflammation, remodeling host gene expression profiles, and disrupting genomic stability.10–12
Beyond these mechanisms, HBV DNA integration into the host genome and the consequent genomic instability represent another core mechanism driving hepatocarcinogenesis.13 Whole-genome sequencing analyses have revealed characteristic high-frequency somatic mutations in HBV-related HCC, predominantly involving key genes such as TERT and TP53.14,15 To precisely mimic the synergistic oncogenic effects of viral proteins and host gene mutations, researchers frequently employ composite models combining HBx transgenic mice with gene recombination technologies. A paradigmatic example involves using the Cre-loxP system to specifically activate KrasG12D mutations in the livers of HBx transgenic mice, thereby confirming the synergistic effect of HBx and Kras mutations in accelerating hepatocarcinogenesis.16 Among the affected genes, Telomerase Reverse Transcriptase (TERT) holds special pathological significance: it is not only the most common target for HBV genomic integration,14 but its promoter mutations are also the most specific and highly prevalent genetic alterations in HBV-related HCC.17 Currently, intervention strategies targeting TERT and its downstream products—ranging from traditional inhibitors to emerging base-editing therapies for mutant promoters—have become a major direction in HCC therapeutic development.18
Regarding model construction, previous studies often utilized Tert knockout mice to analyze the roles of telomere shortening and genomic instability in liver cancer evolution.19 However, existing Tert overexpression models have largely focused on anti-aging and tissue regeneration. Authoritative studies indicate that in mice with normal tumor suppressor function (eg., wild-type p53), Tert overexpression alone primarily delays aging and extends lifespan, but is insufficient to induce spontaneous malignancies, particularly HCC.20,21 This limitation was further corroborated by recent findings from Mishima et al (2024). Using Cre-loxP technology to construct liver-specific Tert overexpression mice, they discovered that even under the backdrop of chemically induced chronic inflammation, TERT upregulation alone failed to cross the threshold for carcinogenesis; significant HCC development occurred only when combined with Trp53 deletion—a “second hit”.22 This suggests that the oncogenic process of Tert is strictly environment-dependent, often requiring synergy with genomic instability or specific oncogenic signals.
Although chronic HBV infection provides an environment of chronic inflammation and genomic damage, there is a lack of double transgenic models in the literature that combine HBV transgenes with Tert overexpression. In HBV-associated HCC, TP53 is a key driver gene with a mutation frequency second only to the TERT promoter.23 As a core tumor suppressor, p53 function is often directly inhibited by the HBV X protein (HBx), which blocks p53’s sequence-specific DNA binding and transcriptional activity, thereby deregulating the cell cycle and apoptosis.24 However, experiments have shown that Trp53 knockout alone in the context of HBV infection has low oncogenic efficiency and a long latency period, limiting its feasibility as a high-efficiency experimental model. Consequently, multi-gene editing strategies have been widely adopted. Literature confirms that simultaneous knockout of Trp53 and Pten in HBV transgenic mice using CRISPR/Cas9 significantly accelerates HCC onset and progression compared to Pten knockout alone.25 Importantly, the tumors induced by this model are not only pathologically confirmed as HCC but also mirror the molecular features of human HCC at the transcriptomic level.26 Thus, whether it is TERT activation or TP53 inactivation, HBV-driven carcinogenesis often requires a specific synergistic milieu—namely, virus-induced chronic inflammation and genomic instability. The findings by Mishima et al and the success of the Trp53/Pten dual-knockout model strongly validate the “Multi-hit” hypothesis in liver cancer modeling. Therefore, constructing dual/multi-driver models capable of simultaneously simulating the HBV persistent infection environment and key genetic variations (such as Tert or Trp53) represents a critical breakthrough for overcoming current limitations and precisely recapitulating the evolutionary laws of human HBV-HCC (Figure 1).
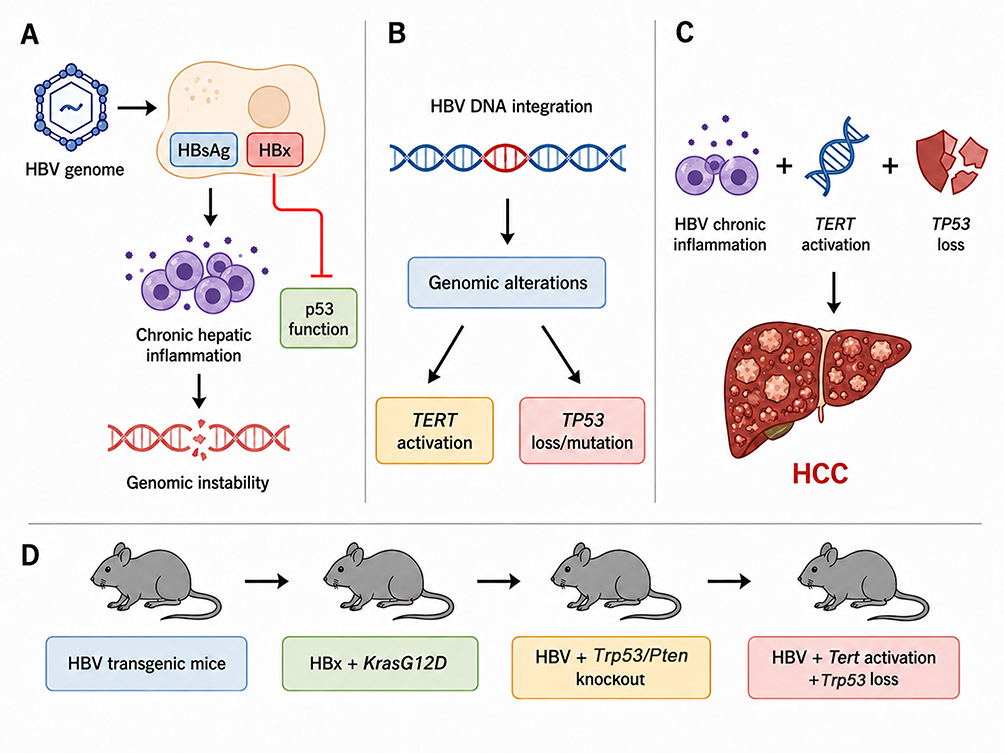 |
Figure 1 Mechanisms of HBV-associated hepatocarcinogenesis and the evolution of genetically engineered mouse models (GEMMs). (A) Chronic HBV antigen exposure promotes hepatocarcinogenesis through the expression of viral proteins such as HBsAg and HBx, which contribute to persistent hepatic inflammation, disruption of p53-related tumor-suppressive signaling, and genomic instability. (B) HBV DNA integration into the host genome generates genomic alterations that frequently converge on key driver events, including TERT activation and TP53 loss or mutation. (C) HBV-related HCC is better represented by an environment-dependent multi-hit model rather than by a single-driver mechanism. Chronic inflammatory injury, TERT activation, and TP53 inactivation may cooperate to cross the threshold for malignant transformation. (D) The conceptual evolution of HBV-related HCC mouse models progresses from simple HBV transgenic mice modeling chronic viral antigen exposure to composite models combining viral factors with oncogenic drivers (e.g., HBx plus Kras^G12D). High-efficiency models utilize CRISPR-mediated Trp53/Pten disruption in HBV transgenic backgrounds, and future multi-driver models aim to integrate HBV-associated injury with Tert activation and Trp53 loss. |
Metabolic-Associated HCC Models
Non-alcoholic fatty liver disease (NAFLD), recently redefined by international consensus as Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD), is currently the fastest-growing chronic liver disease globally. As the primary driver for the surge in HCC incidence,27 MASLD-associated HCC shows a trend toward younger patients and is projected to surpass viral hepatitis as the leading cause of liver cancer worldwide within the next decade.5 Furthermore, MASLD often acts as a critical comorbidity, synergizing with HBV, HCV, or alcohol consumption to accelerate malignant progression.28,29
At the molecular pathology level, metabolic-associated HCC exhibits a distinct landscape compared to viral HCC. While HBV-related HCC is enriched in TP53 mutations, metabolic HCC is driven by a unique mutational spectrum. Genomic analyses confirm that aberrant activation of the Wnt/β-catenin pathway (represented by CTNNB1 mutations) and inactivation of the TGF-β pathway (characterized by ACVR2A mutations) are the most prominent and characteristic molecular driver events in metabolic HCC.30 These unique features provide a clear genetic basis for utilizing gene-engineering technologies (eg., targeting Ctnnb1 or Acvr2a) to construct etiology-informed metabolic HCC mouse models.
CTNNB1 (β-catenin) mutations are the most characteristic molecular event in Metabolic Dysfunction-Associated Steatohepatitis (MASH)-HCC, occurring in approximately 20%–39% of cases, whereas they are rarer in HBV-related HCC.31 Mechanistically, mutations in Exon 3 of CTNNB1 are the most common. This region encodes serine/threonine residues crucial for β-catenin phosphorylation; mutations here prevent phosphorylation and ubiquitination, thereby allowing the protein to escape proteasomal degradation.32,33 This “stabilizing mutation” leads to cytoplasmic accumulation and nuclear translocation of β-catenin, constitutively activating Wnt signaling to drive cell proliferation, metabolic reprogramming, immune evasion, and metastasis.31,32 Early attempts to model this using Albumin (Alb) promoter-driven Cre recombinase to delete Exon 3 of Ctnnb1 were problematic; Harada et al found that this strategy caused severe hepatomegaly and lethality in mice within 20–30 days post-birth, without inducing overt liver tumors, suggesting that constitutive β-catenin activation alone during early development may be lethal or insufficient for independent carcinogenesis.34
To address this, researchers optimized modeling strategies. Professor Fangming Liu’s team utilized tail vein injection of Adenovirus-Cre to achieve spatiotemporally specific activation of Ctnnb1 in adult mouse livers, establishing a model that captures selected pathological and molecular features. Using this model, they further employed Cre-loxP technology to co-delete Trp53 or crossbreed with HBV transgenic mice, confirming that multi-gene synergy significantly accelerates tumor progression. They also revealed that oncogenic β-catenin-stimulated AKT2-CAD-mediated pyrimidine synthesis is a druggable vulnerability in liver cancer.35 Additionally, a team from Zhejiang University used in situ genome editing to systematically construct composite models combining CTNNB1 with the overexpression of oncogenes like MYC, RAS, AKT, or EGFR. These combined models achieved high tumor induction in selected combinations and modeled aspects of molecular heterogeneity.36 In the realm of immunotherapy, given that CTNNB1 mutations are often associated with T-cell exclusion (“immune-cold” tumors), recent studies using activated Ctnnb1 models have explored strategies to improve immune efficacy. It was reported that targeting Matrix Metalloproteinase 9 (MMP9) effectively restores the anti-tumor immune activity of CD8+ T cells in Ctnnb1-mutated HCC, thereby significantly enhancing the efficacy of anti-PD-1 therapy.37
ACVR2A, a key type II transmembrane serine/threonine kinase receptor in the TGF-β superfamily, plays a vital role in negative immune regulation and growth inhibition in maintaining liver homeostasis.38 Under physiological conditions, Activin signaling acts as a “molecular brake,” limiting excessive hepatocyte proliferation and inducing apoptosis in abnormal cells.39 However, loss of ACVR2A function leads to uncontrolled downstream signaling, relieving transcriptional repression of key glycolytic enzymes and resulting in significant upregulation of LDHA and MCT4, thus driving the Warburg effect in tumor cells. This metabolic reprogramming not only meets the energy demands of rapid tumor proliferation but, crucially, constructs a lactate-rich immunosuppressive microenvironment. High lactate concentrations specifically recruit regulatory T cells (Tregs) while directly inhibiting the cytotoxic function of CD8+ T cells, leading to immune escape.40,41 Large-scale genomic profiling confirms that ACVR2A mutations are indeed etiologically characteristic of MASH-HCC.31 Given its specificity and profound impact on the immune microenvironment, recent studies have primarily employed CRISPR-Cas9-edited syngeneic transplant models to elucidate its function. Yasukawa et al (2025) successfully replicated the high lactate metabolism and Treg infiltration features observed in human MASH-HCC by implanting Acvr2a-knockout mouse liver cancer cell lines into immunocompetent mice.40 Furthermore, liver-specific knockout mice based on the Acvr2a^flox/flox and Alb-Cre system provide genetic tools to further explore early molecular events in the progression from MASH to HCC.
Beyond these, Pten liver-specific knockout models and MUP-uPA transgenic models are typical GEMMs for simulating the pathological evolution of metabolic liver cancer. PTEN is a widely expressed tumor suppressor. Upon liver-specific Pten deletion via the Alb-Cre system, mice develop a steatosis–steatohepatitis–fibrosis–HCC sequence of “Steatosis → MASH → Fibrosis → HCC,” typically developing macroscopic tumor nodules by 12–15 months of age, with pathological morphology resembling selected features of human MASH-HCC.41,42 This model is widely used not only for studying HCC mechanisms but also for exploring MASH pathophysiology and drug interventions.43–45 The MUP-uPA mouse model is constructed by integrating the urokinase-type plasminogen activator (uPA) gene driven by the Major Urinary Protein (MUP) promoter into the genome. This model induces hepatocyte Endoplasmic Reticulum (ER) stress via uPA overexpression; when combined with a High-Fat Diet (HFD), mice rapidly progress to MASH within 10–18 weeks and develop tumors by 26–34 weeks.46
Using this model, Shalapour et al deeply analyzed the immune microenvironment of MASH-HCC, discovering that MASH-induced chronic inflammation promotes the accumulation of IgA+ plasma cells. These cells directly inhibit CD8+ T cell cytotoxicity by expressing PD-L1 and IL-10, creating an immunosuppressive landscape that shares features with human MASH-HCC.47 Building on this, a Nature study by Pfister et al further revealed that the MASH environment causes CD8+ T cells to adopt an “exhausted and auto-aggressive” state. These aberrant T cells lose their tumor surveillance function and instead promote HCC progression by exacerbating tissue damage, explaining why immune checkpoint inhibitors have limited efficacy in MASH-HCC.48 Regarding metabolic mechanisms, a recent Nature study identified Fructose-1,6-bisphosphatase 1 (FBP1), a key gluconeogenic enzyme, as having important tumor-suppressive functions. The aberrant loss of FBP1 was found to be a critical “molecular switch” driving the progression from MASH to HCC. Researchers utilized Cre-loxP technology to construct MUP-uPA/Fbp1 double-mutant and Trp53/Fbp1 double-mutant mice, confirming that Fbp1 knockout significantly accelerates MASH-HCC pathology and shortens tumor latency.49
Regarding clinical application, a review by Fornari summarized the efficacy evaluation of various immune and metabolic drugs in MASH-related HCC models but noted a lack of model trials targeting hepatocyte-specific delivery for precise intervention.50 In summary, unlike HBV-related HCC, which is primarily controlled by viral integration and TP53 mutations, MASH-HCC presents a unique genetic landscape characterized by CTNNB1 activation and ACVR2A inactivation. This essential difference dictates that research paradigms for viral liver cancer cannot simply be applied here. The “metabolic-immune” microenvironments replicated by models like Pten knockout or MUP-uPA confirm the existence of specific immune evasion mechanisms (eg., auto-aggressive CD8+ T cells) in metabolic HCC. Deepening the application of these specialized models will help us precisely address the growing challenge of metabolic HCC in the “post-viral hepatitis era” and provide theoretical support for developing combined metabolic-immune therapies distinct from traditional chemotherapy (Figure 2).
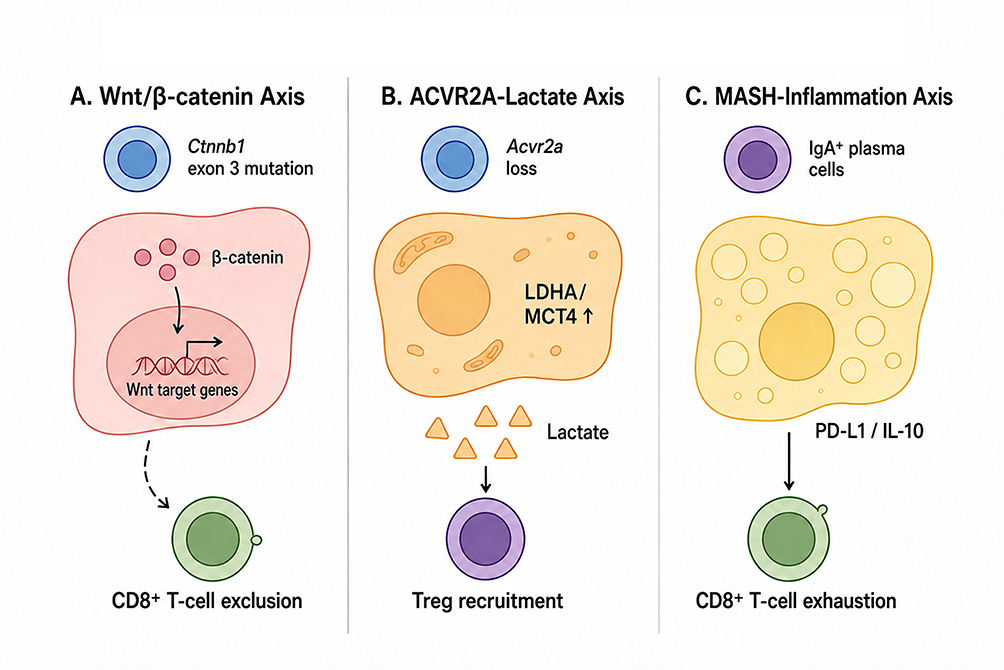 |
Figure 2 Distinct metabolic drivers and immune microenvironment remodeling in MASLD-associated HCC. (A) The Wnt/$\beta$-catenin axis represents a characteristic molecular pathway in metabolic HCC. Models with hepatocyte $\beta$-catenin activation, including Ctnnb1 exon 3 mutations or related genome-editing strategies, activate Wnt target-gene transcription and are associated with CD8+ T-cell exclusion, providing a mechanistic basis for immune-cold HCC phenotypes. (B) The Acvr2a–lactate axis illustrates how loss of Activin receptor signaling promotes metabolic reprogramming. Acvr2a deficiency increases LDHA/MCT4 expression and lactate production, generating a lactate-rich immunosuppressive microenvironment that favors regulatory T-cell (Treg) recruitment. (C) The MASH-inflammation axis highlights immune remodeling in steatohepatitis-associated HCC models. In models such as MUP-uPA combined with a high-fat diet, chronic metabolic inflammation promotes the accumulation of IgA+ plasma cells expressing PD-L1 and IL-10. This microenvironment promotes CD8+ T-cell exhaustion, suppresses cytotoxicity, and contributes to impaired antitumor immune surveillance and reduced responsiveness to immune checkpoint blockade. |
A critical appraisal of established MASLD-HCC models is therefore essential. Liver-specific Pten deletion recapitulates selected aspects of steatohepatitis-associated hepatocarcinogenesis, but its long latency, pathway-specific genetic background, and variable penetrance limit its generalizability as a universal MASLD-HCC model.41–45 Similarly, MUP-uPA combined with high-fat diet accelerates steatohepatitis and tumor development, but the model is strongly shaped by uPA-induced endoplasmic reticulum stress and dietary injury, which may introduce metabolic and inflammatory confounders.46,47,49 These models should therefore be selected according to the specific biological question, with latency, penetrance, strain background, sex, fibrosis stage, and reproducibility reported whenever available.
Alcohol-Associated HCC Models
Epidemiological Background and Current Models
Recent epidemiological data indicate a continuous upward trend in global alcohol exposure, with approximately 30% of new liver cancer cases annually attributable to long-term alcohol consumption.51 The pathological evolution of Alcohol-Associated HCC (AL-HCC) spans the entire spectrum from Alcoholic Fatty Liver (AFL) and Alcoholic Steatohepatitis (ASH) to cirrhosis and HCC. This malignant transformation involves multidimensional molecular remodeling, in which ALDH2 (alcohol metabolism), LPLA2 (lipid remodeling and stress), NF-κB1 (inflammatory regulation), and c-MYC (oncogene activation) exhibit distinct etiology-dependent functions in the pathogenic pathway of alcohol.52,53 Traditional AL-HCC mouse models mainly rely on combinatorial chemical induction (eg., Ethanol combined with CCl4 or Lieber-DeCarli diet).54 To overcome the issues of low tumorigenicity and long cycles in traditional models, Alarcón-Sánchez et al (2024) developed a novel chemical model based on “Multiple Hepatotoxics.” By combining ethanol, Lipopolysaccharide (LPS), sucrose, and low-dose Diethylnitrosamine (DEN), they successfully reproduced the full spectrum of human ALD pathology—from steatosis and inflammatory fibrosis to eventual HCC—in mice.55 Although such improved chemical models have made breakthroughs in simulating multifactorial synergy, their complex inducing factors make it difficult to precisely dissect the specific contributions of single genes or pathways in an alcohol context. Therefore, GEMMs based on specific targets remain irreplaceable for elucidating AL-HCC molecular mechanisms and screening targeted drugs.
Key GEMMs and Mechanisms
ALDH2 Deficiency Model: A Specific Target for Metabolic-Immune Interaction
Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) is the key enzyme for clearing acetaldehyde and exhibits high etiological specificity. ALDH2 mutations are rare in non-alcoholic HCC (<5%) but significantly promote carcinogenesis under alcohol exposure via the accumulation of acetaldehyde-DNA adducts; notably, this mutation is highly prevalent in East Asian populations.56,57 To simulate the high-risk clinical scenario of “cirrhosis background + alcohol exposure,” Seo et al (2019) constructed a liver-specific Aldh2 knockout mouse model. This model achieved a tumorigenesis rate of 80%-90% at 28 weeks, effectively addressing the deficiencies of traditional models.58 Recent studies further revealed that acetate accumulation resulting from ALDH2 deficiency serves as “metabolic fuel” for tumor-associated macrophages, driving HCC metastasis, thus establishing it as a critical target for metabolic-immune therapy.59
ER Stress and Lysosomal Lipid Remodeling Model (ATF4/LPLA2 Axis)
Beyond direct acetaldehyde toxicity, alcohol-induced organelle dysfunction is another core mechanism of AL-HCC. A recent study by Zhou et al (2025) revealed a novel oncogenic axis linking Endoplasmic Reticulum (ER) stress to lysosomal lipid remodeling.60 The study found that chronic alcohol exposure-induced ER stress upregulates the transcription factor ATF4, which directly binds to and activates the promoter of PLA2G15 (LPLA2). Abnormally high expression of LPLA2 leads to the significant accumulation of BMP (bis(monoacylglycero)phosphate), a lipid unique to lysosomes. This aberrant lipid microenvironment further activates the MAPK/ERK signaling pathway, driving tumor proliferation. Notably, this mechanism was validated in an HBx transgenic mouse (HBx-Tg) model fed with alcohol: alcohol significantly accelerated HBx-mediated hepatocarcinogenesis via the ATF4/LPLA2 axis. This suggests that constructing liver-specific Pla2g15 overexpression mice or Pla2g15/Cln5 gene-modified mice holds promise as an ideal new tool for studying the synergistic carcinogenesis of alcohol-induced metabolic remodeling and viral infection.60
NF-κB1 Deficiency Model: Neutrophil-Driven Inflammatory Mechanisms
The NF-κB pathway permeates the entire process of alcohol-induced chronic inflammation initiation, malignant transformation, and drug resistance.61,62 Research by Wilson et al confirmed that Nfkb1 is a key tumor suppressor gene inhibiting neutrophil-driven HCC. Nfkb1 knockout mice constructed via ES cell homologous recombination exhibit chronic liver disease characteristics, with approximately 50% progressing spontaneously to HCC by 20 months of age, and show increased sensitivity to chemical carcinogens.63 Furthermore, the regulatory effect of NF-κB1 on PD-L1 transcription and subsequent tumor immune evasion—achieved through direct binding to the PD-L1 promoter—has been validated in general hepatocellular carcinoma models (not restricted to alcohol-associated disease).64 This regulatory mechanism is equally applicable to alcohol-driven hepatocarcinogenesis. A current limitation is that this model typically involves whole-body knockout; there is an urgent need to develop liver-specific NF-κB pathway modification models to exclude the interference of systemic inflammation.65
P16 (CDKN2A)-Related Mechanisms: Epigenetic Remodeling and Dual Roles
P16 is a classic tumor suppressor, and its inactivation in AL-HCC exhibits unique metabolic dependence. Recent clinical research (Gut, 2025) revealed the dynamic dual role of P16 in alcoholic liver disease: in the early stages, alcohol-induced DNA damage activates P16 expression, inducing cellular senescence and the secretion of SASP factors, thereby exacerbating liver inflammation;66 in advanced stages, long-term alcohol metabolism disrupts the SAMe (methyl donor) cycle, leading to hypermethylation of the CDKN2A promoter and expression silencing, ultimately promoting the malignant transformation from cirrhosis to HCC.67 Although p16 editing has been explored in large-animal platforms such as Oncopigs, no established mouse model currently reproduces the dynamic sequence of early CDKN2A activation followed by late epigenetic silencing during alcohol-associated hepatocarcinogenesis.68 Therefore, CDKN2A should be viewed as a clinically relevant epigenetic mechanism and a defined modeling gap, rather than as an established AL-HCC mouse model. Future work will require mouse systems capable of separating transient senescence-associated Cdkn2a activation from stable promoter methylation-mediated silencing in advanced alcohol-associated liver disease.
Myc Overexpression Model: Etiology-Oriented Molecular Subtyping
The c-Myc protein is a core molecule maintaining stem cell pluripotency,69 and the MYC gene is highly expressed in up to 70% of patients with HBV- and alcohol-associated hepatocellular carcinoma.70 Genomic analysis shows that the tumor transcriptomic profile of Myc overexpression mice most closely matches human alcohol-associated liver cancer, embodying the “Etiology-Oriented” feature of the model.71 Liver-specific Myc overexpression mice constructed using Cre-loxP technology progress rapidly, with male mice exhibiting early lesions at 47 days and advancing to late-stage liver cancer within 3 months.72 As this model not only reproduces the aggressive phenotype of AL-HCC but also presents specific immune barriers, it is now widely used to evaluate the efficacy of immunotherapies.73
In summary, existing GEMMs have evolved from single oncogene validation to deep mechanistic dissection of alcohol-specific metabolic toxicity (eg., ALDH2, PLA2G15 pathways) and microenvironmental remodeling (eg., NF-κB, P16). The latest findings by Zhou et al (2025) regarding the ER stress-lipid metabolism axis offer a completely new perspective for understanding the synergistic carcinogenic mechanisms of alcohol and viruses (HBV). However, single gene modifications still struggle to fully replicate the complex etiological features of human AL-HCC. Future modeling strategies will trend toward “multi-gene modification combined with environmental induction” to better simulate the tumor immune microenvironment of clinical patients, thereby providing a more reliable preclinical platform for screening precision therapeutics for alcoholic liver cancer (Figure 3).
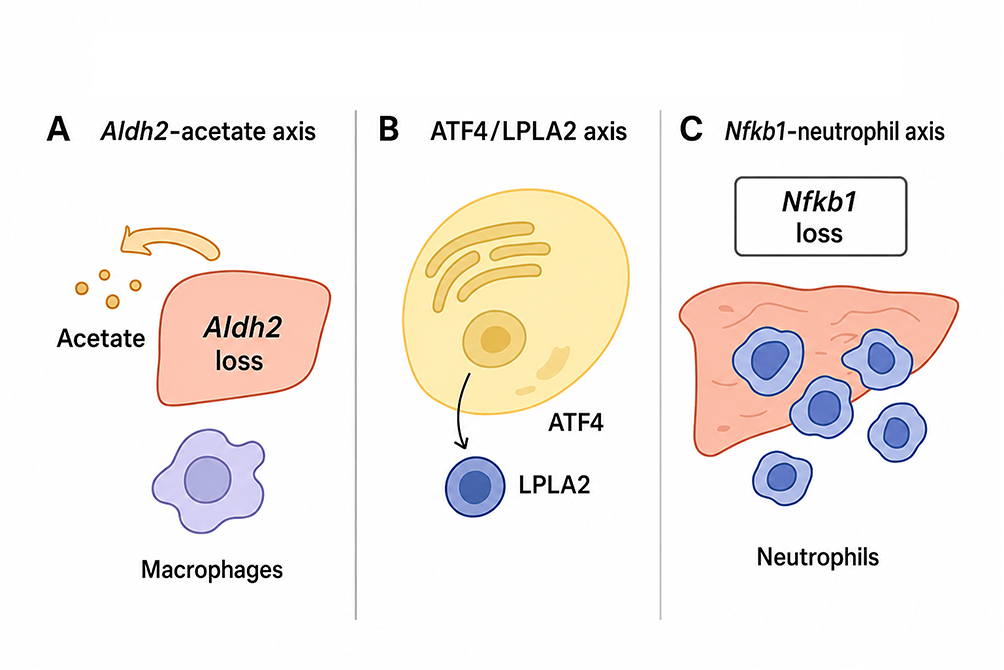 |
Figure 3 Unique metabolic and inflammatory mechanisms in Alcohol-Associated HCC models (A) The Aldh2–acetate axis illustrates how impaired aldehyde metabolism can promote alcohol-associated hepatocarcinogenesis. Liver-specific Aldh2 deficiency enhances tumor development under alcohol exposure, while acetate accumulation may provide metabolic support for tumor-associated macrophages (TAMs) and facilitate metastatic progression. (B) The ATF4/LPLA2 axis links alcohol-induced endoplasmic reticulum (ER) stress to lysosomal lipid remodeling. Chronic alcohol exposure activates ATF4, which upregulates LPLA2/PLA2G15 and promotes the accumulation of lysosome-enriched BMP lipids, thereby enhancing oncogenic signaling and hepatocarcinogenesis. (C) The Nfkb1–neutrophil axis represents an inflammation-driven mechanism of liver tumor promotion. Nfkb1 deficiency enhances chronic hepatic inflammation and neutrophil-associated tumor development. However, because established Nfkb1 models commonly involve systemic gene disruption, future liver- or cell-type-specific models are needed to separate hepatocyte-intrinsic effects from systemic inflammatory contributions. |
Beyond intrinsic cellular signaling and classical environmental insults, the gut-liver axis is an important modifier of etiology-associated hepatocarcinogenesis. Microbiota-derived metabolites can reshape hepatic inflammation, bile acid signaling, metabolic stress, and tumor immunity in both MASLD- and alcohol-associated liver diseases.74 Future GEMM strategies should therefore incorporate microbiome-aware approaches, including co-housing, antibiotic depletion, germ-free or gnotobiotic housing, fecal microbiota transplantation, defined dietary protocols, or colonization with patient-derived microbial communities. These approaches may help distinguish hepatocyte-intrinsic genetic effects from microbiome-dependent inflammatory signals and improve the reproducibility of etiology-aligned preclinical models.
Conclusion and Perspectives
This article has systematically reviewed key advances in Genetically Engineered Mouse Models (GEMMs) for recapitulating Hepatocellular Carcinoma (HCC) induced by diverse etiologies. From simulating HBV integration-driven carcinogenesis (eg., Tert promoter mutations,17 HBx and Trp53 synergy25,26), to elucidating immune microenvironment remodeling in metabolic HCC (eg., the discovery of IgA+ plasma cells in MUP-uPA models47 and the critical role of Fbp1 loss49), and exploring metabolic defects in alcohol-driven carcinogenesis (Aldh2 mutations58 and NF-κB pathways63), GEMMs have successfully established an etiology-based research atlas covering multiple molecular subtypes (Table 1).
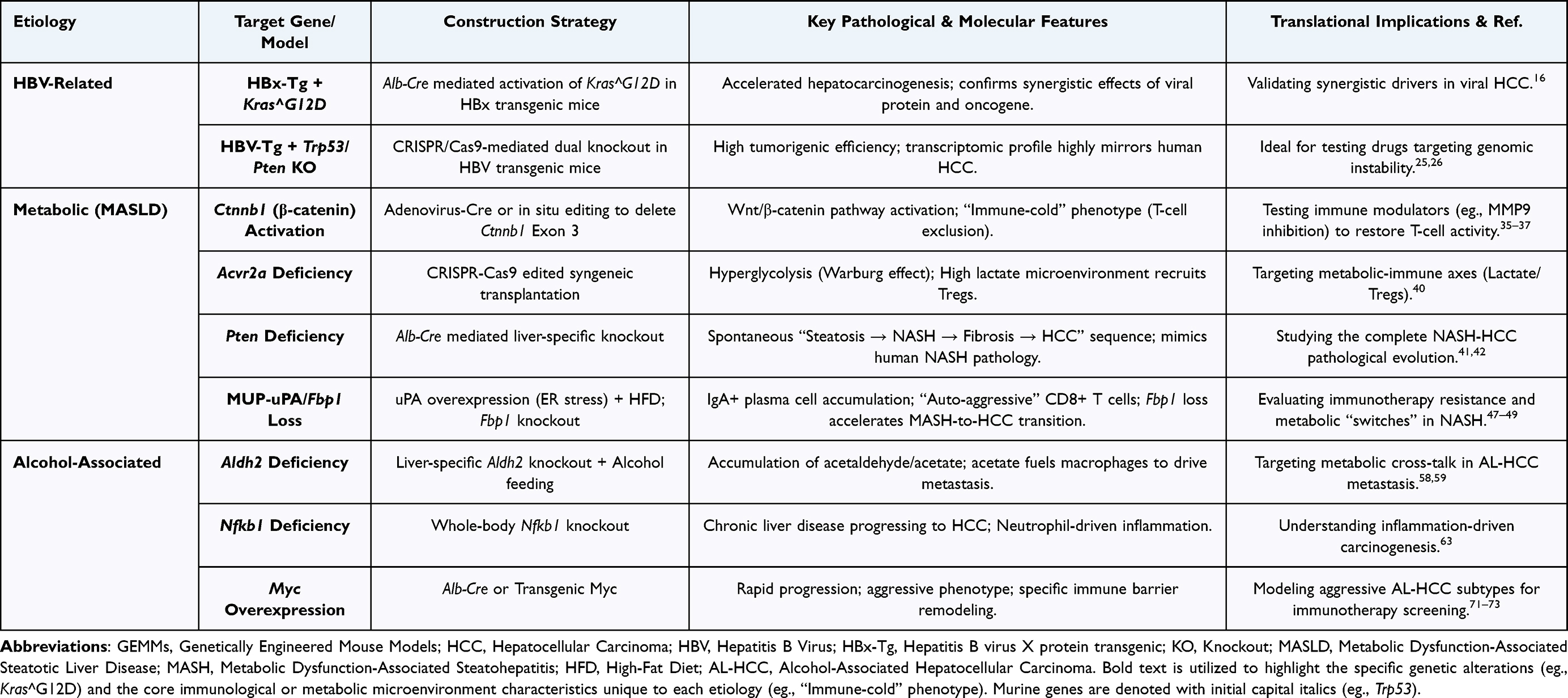 |
Table 1 Key Genetically Engineered Mouse Models (GEMMs) for Etiology-Specific Hepatocellular Carcinoma |
Therefore, etiology-based GEMM research may help prioritize biologically relevant model systems and generate testable hypotheses regarding therapeutic vulnerability across HCC subtypes.3,71 However, etiology alone should not be treated as a validated surrogate for molecular profiling or as a current basis for clinical treatment selection. Its predictive value for therapy response requires rigorous validation in human cohorts and prospective translational studies. In addition, although HCV-related models are not the central focus of this review because direct-acting antivirals have substantially reduced HCV-driven disease burden,75,76 sustained virologic response does not completely eliminate HCC risk, particularly in patients with pre-existing advanced fibrosis or cirrhosis. HCV-associated models therefore remain relevant for selected questions, including post-SVR carcinogenesis, persistent epigenetic or inflammatory alterations, fibrotic niche remodeling, and surveillance strategies in post-cure populations.77
Despite promising prospects, current GEMMs still suffer from two fatal shortcomings:
First, the lack of a cirrhotic background. Clinical HCC predominantly arises on a foundation of cirrhosis, whereas existing models (especially Alb-Cre mediated specific gene knockouts) often induce tumors directly, skipping the classic “inflammation-fibrosis-cirrhosis” evolutionary stage (Table 2). Mechanistically, the absence of cirrhosis is not merely a histological limitation. Cirrhosis alters extracellular matrix stiffness, sinusoidal architecture, hypoxia, vascular remodeling, immune-cell localization, hepatocyte regenerative pressure, liver functional reserve, and drug metabolism. As a result, non-cirrhotic GEMMs may overestimate therapeutic efficacy or fail to capture toxicity and immune-response patterns that emerge in cirrhotic human HCC.6,50 Sex should also be incorporated as a biological variable in GEMM-based HCC research. Human HCC shows marked male predominance, and several mouse models exhibit sex-dependent differences in tumor latency, penetrance, inflammatory signaling, and hormone-regulated immune responses. Future studies should report animal sex, analyze male and female mice separately when feasible, and avoid extrapolating male-only data to all biological contexts.78
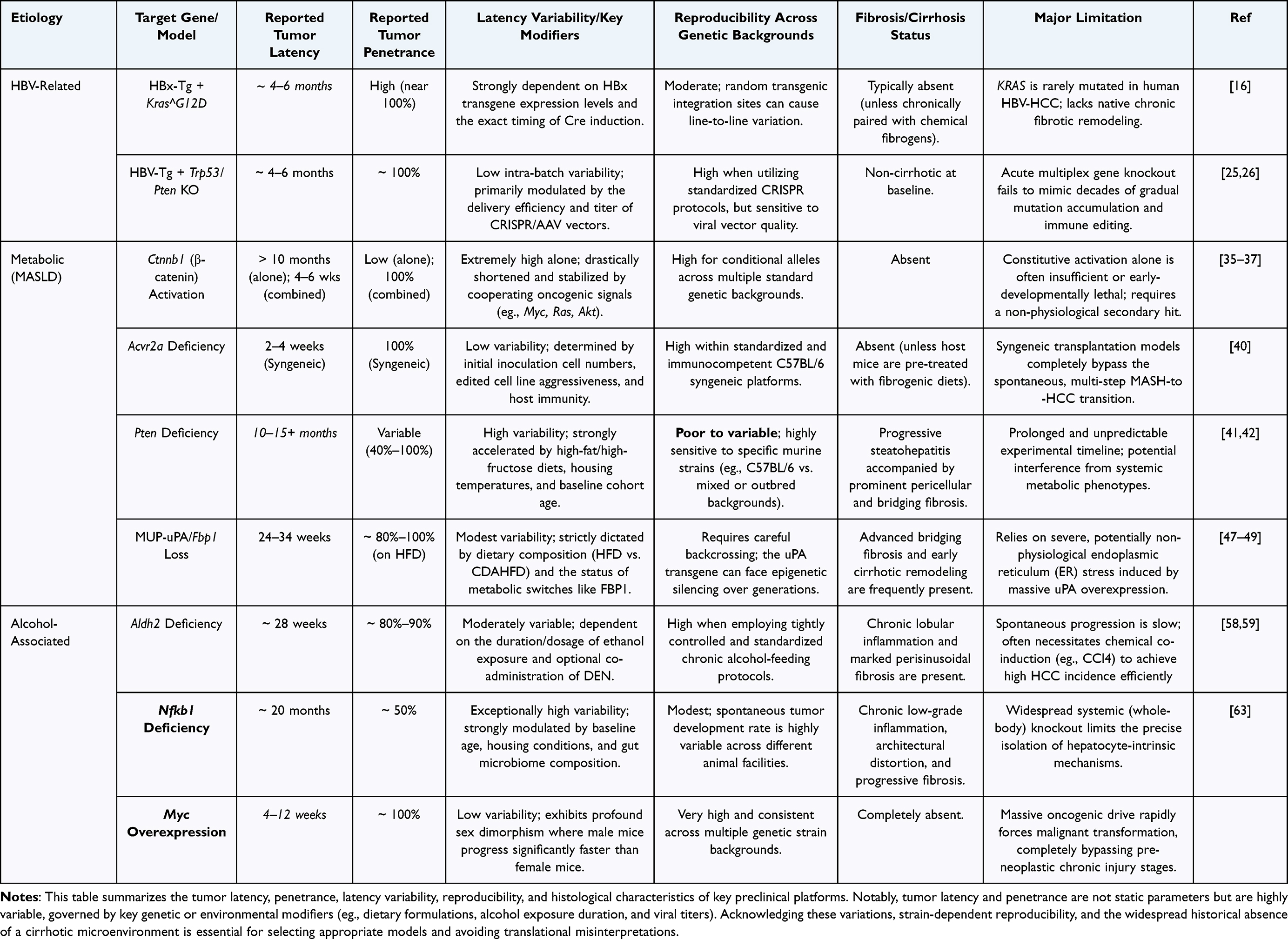 |
Table 2 Critical Appraisal of Quantitative Parameters and Limitations in Etiology-Specific HCC GEMMs |
Second, insufficient tissue specificity and challenges of cellular heterogeneity. The liver is a complex organ composed of highly heterogeneous cell populations; beyond hepatocytes responsible for metabolic functions, it contains various non-parenchymal cells such as Kupffer cells, stellate cells, and endothelial cells. However, some existing models (such as those involving systemic gene modifications of NF-κB or Cdkn2a) lack strict targeting of hepatocytes, making it difficult to completely rule out interference from systemic inflammation or extrahepatic organs.10,63 Given the distinct functions of different cell subpopulations in tumorigenesis, future model construction must emphasize “Hepatocyte-specific Targeting” to precisely distinguish intrinsic oncogenic driver events in hepatocytes from confounding effects of non-parenchymal cells or systemic backgrounds, thereby avoiding mechanistic misinterpretations caused by cellular heterogeneity.35,36
To overcome these limitations, future GEMM designs will advance toward multi-gene orthogonal editing systems based on Cre-loxP, Flp-FRT, and Dre-rox. Current liver cancer GEMMs rely heavily on a single Cre-loxP system for hepatocyte-specific modification (eg., Alb-Cre mediated Ctnnb1 or Pten knockout), making it difficult to dissect dynamic interactions between tumor cells and the Tumor Microenvironment (TME). By integrating three non-interfering recombinase systems—Cre-loxP, Flp-FRT, and Dre-rox—researchers can achieve precise, “divide-and-conquer” regulation within the same mouse. For instance, one could use the Cre system to specifically induce oncogenic mutations (eg., Trp53 loss) in hepatocytes, while simultaneously using the Dre system to manipulate immune checkpoints or fibrotic genes in Kupffer cells or stellate cells, and the Flp system to control the temporal window of gene expression. This strategy not only ensures absolute specificity of gene editing in specific cell subpopulations, avoiding off-target effects, but also authentically simulates the co-evolutionary mechanisms of tumor parenchyma and stroma in HCC development, providing the most sophisticated genetic tools for dissecting the complex tumor microenvironment.
To make orthogonal recombinase systems more actionable, they should be integrated with controlled environmental injury models rather than discussed only as abstract genetic tools. For example, hepatocyte-specific oncogenic events induced by Cre-loxP could be combined with Flp-FRT- or Dre-rox-based manipulation of hepatic stellate cells, Kupffer cells, endothelial cells, or immune compartments. These genetic systems may be further paired with fibrogenic or metabolic injury protocols, such as CCl4, thioacetamide, CDAHFD, ethanol-containing regimens, or carefully titrated multiple-hepatotoxic protocols.55 However, models containing low-dose DEN require cautious interpretation because DEN introduces mutagenic pressure that may obscure driver-specific mechanisms. Such hybrid strategies should therefore be validated by lineage tracing, cell-type-resolved transcriptomics, fibrosis staging, and standardized reporting of tumor latency and penetrance.
Recent studies on RNA epigenetic regulation and three-dimensional MASLD modeling further highlight the need to integrate GEMMs with complementary molecular and human-relevant systems. For example, NAT10-mediated ac4C modification has been implicated in SMAD3 mRNA stability and HCC progression,79 while three-dimensional dynamic coculture systems have been developed to model progressive stages of MASLD.80 These platforms do not replace GEMMs, but they may help validate candidate mechanisms and improve cross-platform translational inference.
Data Sharing Statement
Not applicable. Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval and Consent to Participate
Not applicable. This manuscript is a review article and does not involve primary research on humans or animals conducted by the authors. All data discussed are from previously published studies.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by Hunan Provincial Natural Science Foundation of China (2026JJ30210) (2023JJ30841) (2023JJ30752).
Disclosure
The authors declare that they have no competing interests.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–15. doi:10.3322/caac.21834
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
3. Rebouissou S, Nault J-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol. 2020;72(2):215–229. doi:10.1016/j.jhep.2019.08.017
4. Rumgay H, Arnold M, Ferlay J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77(6):1598–1606. doi:10.1016/j.jhep.2022.08.021
5. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–238. doi:10.1038/s41575-020-00381-6
6. Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15(9):536–554. doi:10.1038/s41575-018-0033-6
7. Maddalo D, Manchado E, Concepcion CP, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516(7531):423–427. doi:10.1038/nature13902
8. Jeng W-J, Papatheodoridis GV, Lok ASF. Hepatitis B. Lancet. 2023;401(10381):1039–1052. doi:10.1016/S0140-6736(22)01468-4
9. Choi W-M, Yip TC-F, Wong GL-H, et al. Hepatocellular carcinoma risk in patients with chronic hepatitis B receiving tenofovir- vs. entecavir-based regimens: individual patient data meta-analysis. J Hepatol. 2023;78(3):534–542. doi:10.1016/j.jhep.2022.12.007
10. Chisari FV, Klopchin K, Moriyama T, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59(6):1145–1156. doi:10.1016/0092-8674(89)90770-8
11. Kim C-M, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351(6324):
12. Slagle BL, Bouchard MJ. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb Perspect Med. 2016;6(3):a021402. doi:10.1101/cshperspect.a021402
13. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64(1):S84–S101. doi:10.1016/j.jhep.2016.02.021
14. Amaddeo G, Cao Q, Ladeiro Y, et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2015;64(5):820–829. doi:10.1136/gutjnl-2013-306228
15. Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24(3):245–250. doi:10.1038/73448
16. Ye H, Zhang C, Wang BJ, et al. Synergistic function of Kras mutation and HBx in initiation and progression of hepatocellular carcinoma in mice. Oncogene. 2014;33(43):5133–5138. doi:10.1038/onc.2013.468
17. Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi:10.1038/ncomms3218
18. Zhao G, Ma Q, Yang H, et al. Base editing of the mutated TERT promoter inhibits liver tumor growth. Hepatology. 2024;79(6):1310–1323. doi:10.1097/HEP.0000000000000700
19. Nault JC, Ningarhari M, Rebouissou S, Zucman-Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol. 2019;16(9):544–558. doi:10.1038/s41575-019-0165-3
20. Tomás-Loba A, Flores I, Fernández-Marcos PJ, et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell. 2008;135(4):609–622. doi:10.1016/j.cell.2008.09.034
21. de Jesus B B, Vera E, Schneeberger K, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012;4(8):691–704. doi:10.1002/emmm.201200245
22. Mishima M, Takai A, Takeda H, et al. TERT upregulation promotes cell proliferation via degradation of p21 and increases carcinogenic potential. J Pathol. 2024;264(3):318–331. doi:10.1002/path.6351
23. Schulze K, Imbeaud S, Letouzé E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–511. doi:10.1038/ng.3252
24. Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A. 1994;91(6):2230–2234. doi:10.1073/pnas.91.6.2230
25. Liu Y, Qi X, Zeng Z, et al. CRISPR/Cas9-mediated p53 and Pten dual mutation accelerates hepatocarcinogenesis in adult hepatitis B virus transgenic mice. Sci Rep. 2017;7(1):2796. doi:10.1038/s41598-017-03070-8
26. Chen Z, Yang J, Song Y, et al. HCC model induced by p53 and pten knockout in hbv-transgenic mice mirrors human hcc at the transcriptome level. J Med Virol. 2024;96(12):e70120. doi:10.1002/jmv.70120
27. Rinella ME, Lazarus JV, Ratziu V, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;78(6):1966–1986. doi:10.1097/HEP.0000000000000520
28. Park S, Davis AM, Prevention PAA. Diagnosis, and treatment of hepatocellular carcinoma. JAMA. 2024;332(12):1013–1014. doi:10.1001/jama.2024.14101
29. Wong YJ, Nguyen VH, Yang HI, et al. Impact of fatty liver on long-term outcomes in chronic hepatitis B: a systematic review and matched analysis of individual patient data meta-analysis. Clin Mol Hepatol. 2023;29(3):705–720. doi:10.3350/cmh.2023.0004
30. Pinyol R, Torrecilla S, Wang H, et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol. 2021;75(4):865–878. doi:10.1016/j.jhep.2021.04.049
31. Dantzer C, Dif L, Vaché J, Basbous S, Billottet C, Moreau V. Specific features of ß-catenin-mutated hepatocellular carcinomas. Br J Cancer. 2024;131(12):1871–1880. doi:10.1038/s41416-024-02849-7
32. Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18(21):5931–5942. doi:10.1093/emboj/18.21.5931
33. Xu C, Xu Z, Zhang Y, Evert M, Calvisi DF, Chen X. β-Catenin signaling in hepatocellular carcinoma. J Clin Invest. 2022;132(4):e154515. doi:10.1172/JCI154515
34. Harada N, Miyoshi H, Murai N, et al. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer Res. 2002;62(7):1971–1977.
35. Liu F, Gai X, Wu Y, et al. Oncogenic β-catenin stimulation of AKT2-CAD-mediated pyrimidine synthesis is targetable vulnerability in liver cancer. Proc Natl Acad Sci U S A. 2022;119(39):e2202157119. doi:10.1073/pnas.2202157119
36. Tang M, Zhao Y, Zhao J, et al. Liver cancer heterogeneity modeled by in situ genome editing of hepatocytes. Sci Adv. 2022;8(25):eabn5683. doi:10.1126/sciadv.abn5683
37. Cai N, Cheng K, Ma Y, et al. Targeting MMP9 in CTNNB1 mutant hepatocellular carcinoma restores CD8+ T cell-mediated antitumour immunity and improves anti-PD-1 efficacy. Gut. 2024;73(6):985–999. doi:10.1136/gutjnl-2023-331342
38. Olsen OE, Wader KF, Hella H, et al. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun Signal. 2015;13:27. doi:10.1186/s12964-015-0104-z
39. Du R, Wen L, Niu M, et al. Activin receptors in human cancer: functions, mechanisms, and potential clinical applications. Biochem Pharmacol. 2024;222:116061. doi:10.1016/j.bcp.2024.116061
40. Yasukawa K, Shimada S, Akiyama Y, et al. ACVR2A attenuation impacts lactate production and hyperglycolytic conditions attracting regulatory T cells in hepatocellular carcinoma. Cell Rep Med. 2025;6(4):102038. doi:10.1016/j.xcrm.2025.102038
41. Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. 2018;19(9):547–562. doi:10.1038/s41580-018-0015-0
42. Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12):1774–1783. doi:10.1172/JCI20513
43. Kodama T, Yi J, Newberg JY, et al. Molecular profiling of nonalcoholic fatty liver disease-associated hepatocellular carcinoma using SB transposon mutagenesis. Proc Natl Acad Sci U S A. 2018;115(44):E10417–E10426. doi:10.1073/pnas.1808968115
44. Gjorgjieva M, Calo N, Sobolewski C, et al. Hepatic IR and IGF1R signaling govern distinct metabolic and carcinogenic processes upon PTEN deficiency in the liver. JHEP Rep. 2024;7(4):101305. doi:10.1016/j.jhepr.2024.101305
45. Kawamura S, Matsushita Y, Kurosaki S, et al. Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. J Clin Invest. 2022;132(11):e151895. doi:10.1172/JCI151895
46. Nakagawa H, Umemura A, Taniguchi K, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26(3):331–343. doi:10.1016/j.ccr.2014.07.001
47. Shalapour S, Lin XJ, Bastian IN, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017;551(7680):340–345. doi:10.1038/nature24302
48. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592(7854):450–456. doi:10.1038/s41586-021-03362-0
49. Gu L, Zhu Y, Nandi SP, et al. FBP1 controls liver cancer evolution from senescent MASH hepatocytes. Nature. 2025;637(8045):461–469. doi:10.1038/s41586-024-08317-9
50. Fornari F, Giovannini C, Piscaglia F, Gramantieri L. Animal models of hepatocellular carcinoma: current applications in clinical research. J Hepatocell Carcinoma. 2022;9:1263–1278. doi:10.2147/JHC.S347946
51. Manthey J, Shield KD, Rylett M, Hasan OSM, Probst C, Rehm J. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: a modelling study. Lancet. 2019;393(10190):2493–2502. doi:10.1016/S0140-6736(18)32744-2
52. Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68(3):526–549. doi:10.1016/j.jhep.2017.09.016
53. Yeo YH, Abdelmalek M, Khan S, et al. Current and emerging strategies for the prevention of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2025;22(3):173–190. doi:10.1038/s41575-024-01021-z
54. Zhang HE, Henderson JM, Gorrell MD. Animal models for hepatocellular carcinoma. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):993–1002. doi:10.1016/j.bbadis.2018.08.009
55. Alarcón-Sánchez BR, Idelfonso-García OG, Guerrero-Escalera D, et al. A model of alcoholic liver disease based on different hepatotoxics leading to liver cancer. Biochem Pharmacol. 2024;228:116209. doi:10.1016/j.bcp.2024.116209
56. Chang MH, You SL, Chen CJ, et al. Long-term effects of hepatitis b immunization of infants in preventing liver cancer. Gastroenterology. 2016;151(3):472–480.e1. doi:10.1053/j.gastro.2016.05.048
57. Jin S, Chen J, Chen L, et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc Natl Acad Sci U S A. 2015;112(29):9088–9093. doi:10.1073/pnas.1510757112
58. Seo W, Gao Y, He Y, et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol. 2019;71(5):1000–1011. doi:10.1016/j.jhep.2019.06.018
59. Shen L, Wang S, Gao C, et al. Tumour-associated macrophages serve as an acetate reservoir to drive hepatocellular carcinoma metastasis. Nat Metab. 2025;7(11):2268–2283. doi:10.1038/s42255-025-01393-9
60. Zhou H, Ba J, Xiao C, et al. Alcohol activates ATF4/LPLA2-mediated BMP metabolism to enhance HBV-induced hepatocellular carcinogenesis. J Hepatol. doi:10.1016/j.jhep.2025.08.022
61. Wang F, Yang JL, Yu KK, et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol Cancer. 2015;14(1):10. doi:10.1186/s12943-014-0274-0
62. Lukas K, Nguyen J, Necas C, Dave K, Venketaraman V. Targeting the NF-κB Pathway in Cancer: mechanisms, Resistance, and Therapeutic Potential Across Tumor Types. Pharmaceuticals. 2025;18(11):1764. doi:10.3390/ph18111764
63. Wilson CL, Jurk D, Fullard N, et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat Commun. 2015;6:6818. doi:10.1038/ncomms7818
64. Wu Y, Tao Q, Xie J, et al. Indole-3-carbinol inhibits PD-L1-mediated immune evasion in hepatocellular carcinoma via suppressing NF-κB p105 Ubiquitination. Phytomedicine. 2025;141:156692. doi:10.1016/j.phymed.2025.156692
65. Ji Y, Ni C, Shen Y, et al. ESRP1-mediated biogenesis of circPTPN12 inhibits hepatocellular carcinoma progression by PDLIM2/NF-κB pathway. Mol Cancer. 2024;23(1):143. doi:10.1186/s12943-024-02056-1
66. Rodrigo-Torres D, Kilpatrick AM, Ferreira-Gonzalez S, et al. Longitudinal paired liver biopsies and transcriptome profiling in alcohol-associated hepatitis reveal dynamic changes in cellular senescence. Gut. 2025;74(9):1500–1513. doi:10.1136/gutjnl-2024-334094
67. French SW. Epigenetic events in liver cancer resulting from alcoholic liver disease. Alcohol Res. 2013;35(1):57–67.
68. Elkhadragy L, Carlino MJ, Jordan LR, et al. Development of a genetically tailored implantation hepatocellular carcinoma model in Oncopigs by somatic cell CRISPR editing. Dis Model Mech. 2025;18(1):dmm052079. doi:10.1242/dmm.052079
69. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi:10.1016/j.cell.2007.11.019
70. Schlaeger C, Longerich T, Schiller C, et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology. 2008;47(2):511–520. doi:10.1002/hep.22033
71. Friemel J, Frick L, Unger K, et al. Characterization of HCC Mouse Models: towards an Etiology-Oriented Subtyping Approach. Mol Cancer Res. 2019;17(7):1493–1502. doi:10.1158/1541-7786.MCR-18-1045
72. Mei Y, Zhou C, Liang CY, et al. A method to establish a c-Myc transgenic mouse model of hepatocellular carcinoma. MethodsX. 2020;7:100921. doi:10.1016/j.mex.2020.100921
73. Liu Y, Xun Z, Ma K, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78(4):770–782. doi:10.1016/j.jhep.2023.01.011
74. Ma C, Han M, Heinrich B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391):eaan5931. doi:10.1126/science.aan5931
75. Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973–1982. doi:10.1056/NEJMoa1402869
76. Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol. doi:10.1016/j.jhep.2017.08.030
77. Hamdane N, Jühling F, Crouchet E, et al. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology. 2019;156(8):2313–2329.e7. doi:10.1053/j.gastro.2019.02.038
78. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi:10.1126/science.1140485
79. Zhang Y, Dong Y, Chen S, et al. Targeting NAT10 inhibits hepatocarcinogenesis via ac4c-mediated smad3 mrna stability. Exploration. 2025;5(6):20250075. doi:10.1002/EXP.20250075
80. Huang Z, Li L, Dudley K, et al. Three-dimensional dynamic cell models for metabolic dysfunction-associated steatotic liver disease progression. BME Front. 2025;6:0181. doi:10.34133/bmef.0181
81. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–1341.e23. doi:10.1016/j.cell.2017.05.046
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Awareness and Predictors of the Use of Bioinformatics in Genome Research in Saudi Arabia
Alomair L, Abolfotouh MA
International Journal of General Medicine 2023, 16:3413-3425
Published Date: 11 August 2023
Establishment and Clinical Application of the Nomogram Related to Risk or Prognosis of Hepatocellular Carcinoma: A Review
Wang X, Zhao M, Zhang C, Chen H, Liu X, An Y, Zhang L, Guo X
Journal of Hepatocellular Carcinoma 2023, 10:1389-1398
Published Date: 22 August 2023
Advancing Hepatocellular Carcinoma Management Through Peritumoral Radiomics: Enhancing Diagnosis, Treatment, and Prognosis
Huang Y, Qian H
Journal of Hepatocellular Carcinoma 2024, 11:2159-2168
Published Date: 4 November 2024
Hydrogel-Based Immunomodulation of Tumor Immune Microenvironment in Hepatocellular Carcinoma: Current Strategies and Future Directions
Zhang S, Ding L, Hou T, Lin D, Qu Y
International Journal of Nanomedicine 2026, 21:576091
Published Date: 4 March 2026
Integrating Combination Therapy and Smart Embolic Materials to Overcome the Therapeutic Ceiling of Transarterial Chemoembolization in Hepatocellular Carcinoma
Fu Y, Bai C, Ling Y, Liu Y, Li W, Li G
Journal of Hepatocellular Carcinoma 2026, 13:620956
Published Date: 19 June 2026