")
Back to Journals » International Journal of Nanomedicine » Volume 16
Dual Targeting of Endoplasmic Reticulum by Redox-Deubiquitination Regulation for Cancer Therapy
Authors Cai B , Hou M , Zhang S, Xin Z, Huang J, Yang J, Wang Y, Cai X, Xie S , Zhang C, Huang Y
Received 26 May 2021
Accepted for publication 22 July 2021
Published 30 July 2021 Volume 2021:16 Pages 5193—5209
DOI https://doi.org/10.2147/IJN.S321612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Biao Cai,1,* Mengfei Hou,2,* Shijun Zhang,3,* Zhixiang Xin,1 Jiwei Huang,1 Jingxing Yang,2 Yueming Wang,1 Xingyun Cai,1 Shaowei Xie,3 Chunfu Zhang,2 Yiran Huang1
1Department of Urology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, People’s Republic of China; 2School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, 200030, People’s Republic of China; 3Department of Ultrasound, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yiran Huang
Department of Urology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 160 Pujian Road, Shanghai, 200127, People’s Republic of China
Tel +86 21-68383396
Email [email protected]
Shaowei Xie
Department of Ultrasound, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 160 Pujian Road, Shanghai, 200127, People’s Republic of China
Tel +86 21-68383396
Email [email protected]
Background: Recently, nanocatalyst-induced endoplasmic reticulum (ER) stress for cancer therapy has been attracting considerable attention. However, cancer cells are often able to overcome ER stress-induced death by activating the unfolded protein response (UPR), making nanocatalytic monotherapy a poor defense against cancer progression.
Purpose: In this study, to improve the nanocatalytic treatment efficacy, a phase change material (PCM) was used to encapsulate the upstream ER stress initiator, iron oxide nanoparticles (Fe3O4 NPs), and the downstream UPR modulator, PR-619. Subsequently, the tumor-homing peptide tLyP-1 was coupled to it to form tLyP-1/PR-619/Fe3O4@PCM (tPF@PCM) theranostic platform.
Materials and Methods: tPF@PCM was synthesized using nanoprecipitation and resolidification methods followed by the EDC/NHS cross-linking method. The targeting capacity of tPF@PCM was evaluated in vitro and in vivo using flow cytometry and magnetic resonance imaging, respectively. The therapeutic efficacy of tPF@PCM was investigated in a renal cell carcinoma mouse model. Moreover, we explored the synergistic anti-tumor mechanism by examining the intracellular reactive oxygen species (ROS), aggregated proteins, ER stress response levels, and type of cell death.
Results: tPF@PCM had excellent tumor-targeting properties and exhibited satisfactory photothermal-enhanced tumor inhibition efficacy both in vitro and in vivo. Specifically, the phase transition temperature (45 °C) maintained using 808 nm laser irradiation significantly increased the release and catalytic activity of the peroxidase mimic Fe3O4 NPs. This strongly catalyzed the generation of hydroxyl radicals (•OH) via the Fenton reaction in the acidic tumor microenvironment. The redox imbalance subsequently resulted in an increase in the level of damaged proteins in the ER and initiated ER stress. Moreover, the pan-deubiquitinase inhibitor PR-619 blocked the “adaptive” UPR-mediated degradation of these damaged proteins, exacerbating the ER burden. Consequently, irremediable ER stress activated the “terminal” UPR, leading to apoptosis in cancer cells.
Conclusion: This ER stress-exacerbating strategy effectively suppresses tumorigenesis, offering novel directions for advances in the treatment of conventional therapy-resistant cancers.
Keywords: endoplasmic reticulum stress, unfolded protein response, nanocatalytic medicine, reactive oxygen species, deubiquitinase inhibitor, apoptosis
Introduction
The endoplasmic reticulum (ER) plays a central role in the synthesis, folding, and modification of secreted and transmembrane proteins in eukaryotic cells.1 Generally, multiple intrinsic and extrinsic disturbances imposed on cancer cells interfere with protein folding in the ER, thereby triggering ER stress.2 Under normal conditions, cancer cells can relieve this mildly elevated ER stress by activating the “adaptive” unfolded protein response (UPR), which decreases protein translation, assists protein refolding, or triggers cytoprotective autophagy and ER-associated degradation (ERAD) of unfolded or misfolded proteins.3 In contrast, continuous and irremediable ER stress often activates PERK/ATF6/IRE1α-mediated apoptosis pathways (“terminal” UPR), and directly leads to apoptosis or sequentially triggers autophagy-dependent cell death and apoptosis; thus, targeting ER stress provides a promising approach for cancer therapy.4,5 Although traditional therapies, such as cytotoxic drugs and radiation, induce ER stress to some extent, this limited disturbance is often ineffective for certain tumors, such as renal cell carcinoma (RCC), or even pro-oncogenic owing to the activation of oncogenic signaling associated with the “adaptive” UPR.6 Furthermore, these non-selective attacks may cause severe systemic toxicity. Consequently, these defects limit the large-scale application of clinical ER stress-related therapies.
Owing to their intrinsic ER stress-inducing capacity, several nanoparticles, including silver, zinc oxide, and iron oxide nanoparticles (Fe3O4 NPs), may be promising alternatives for promoting ER stress at the tumor site.7 In addition to acting as targeted nanocarriers, these nanoparticles tend to disrupt the balance of the redox state inside the ER lumen, thereby disturbing protein folding and elevating ER stress.8 Among these nanoparticles, the widely used dual enzyme-mimic nanocatalyst Fe3O4 NPs are able to conduct tumor-specific redox targeting by utilizing the different metabolic environments between the tumor and normal tissues.9,10 Specifically, Fe3O4 NPs act as a catalase to decompose H2O2 into H2O and O2 in neutral environments, eg, in blood circulation and normal tissues. In contrast, their peroxidase (POD)-like activity in the mildly acidic tumor microenvironment (TME) appears to trigger the Fenton reaction to yield toxic •OH.11,12 Moreover, owing to the photothermal properties of Fe3O4 NPs, their catalytic activity can be enhanced by laser irradiation,13,14 which offers a flexible method to amplify the production of reactive oxygen species (ROS) and contributes to more severe oxidative damage to the ER. Therefore, the hyperthermia-augmented Fe3O4 NPs constitute a reasonable choice to elevate ER stress in the acidic TME.
However, under the regulation of the initially cytoprotective “adaptive” UPR, using Fe3O4-based nano-formulations as a single-agent clinical therapy to elevate ER stress might still be challenging. Therefore, using a UPR modulator along with the ER stress-inducing Fe3O4 NPs may further augment ER stress, which might achieve a better antitumor effect. Recently, increasing evidence has suggested that the protein degradation pathway, ERAD, is uniquely important because of its negative feedback regulation of the most conserved UPR sensor.15 Among all ERAD components, deubiquitinases (DUBs) play prominent roles in maintaining protein homeostasis and have served as innovative ERAD targets.16,17 Moreover, it has been demonstrated that ROS reversibly inactivate DUBs,18 thereby allowing DUB inhibition with a lower dose of DUB inhibitor in the presence of ROS producers and mitigating the side effects. Therefore, the UPR modulator, DUB inhibitors, may have the potential to render cancer cells more sensitive to the upstream initiation of ER stress induced by photothermal-enhanced Fe3O4 NPs.
RCC, one of the most fatal urological tumors, is particularly insensitive to the traditional ER stress-related chemotherapy and radiotherapy.19 Although targeted therapy and immunotherapy have been developed for the treatment of advanced RCC, the objective response rate and overall survival of these reagents are less than satisfactory.20,21 Therefore, exploring novel therapeutic strategies for RCC is of importance.
In this study, we developed an efficient photothermal-augmented tumor therapeutic approach, by regulating redox homeostasis concurrently with the activity of DUBs, to convert the pro-survival UPR into the pro-apoptotic one (Figure 1).22 A nanocatalytic system, tLyP-1/PR-619/Fe3O4@PCM (tPF@PCM) co-loaded with Fe3O4 NPs and the pan-DUB inhibitor PR-619, was synthesized by taking advantage of a melting point-controlled thermal responsive phase-change material (PCM). Upon release from tPF@PCM at 45 °C by laser irradiation, Fe3O4 NPs were shown to be robust ROS inducers at the enhanced catalytic temperature, leading to increased damaged proteins in the ER lumen and initiating ER stress. Moreover, PR-619 cut off the essential step of ERAD, deubiquitination, and exacerbated the accumulation of damaged proteins. The increased input and simultaneously reduced output of ER stress were caught in a vicious circle, leading to prolonged activation of UPR, and ultimately causing apoptosis of RCC 786-O cells both in vitro and in vivo.
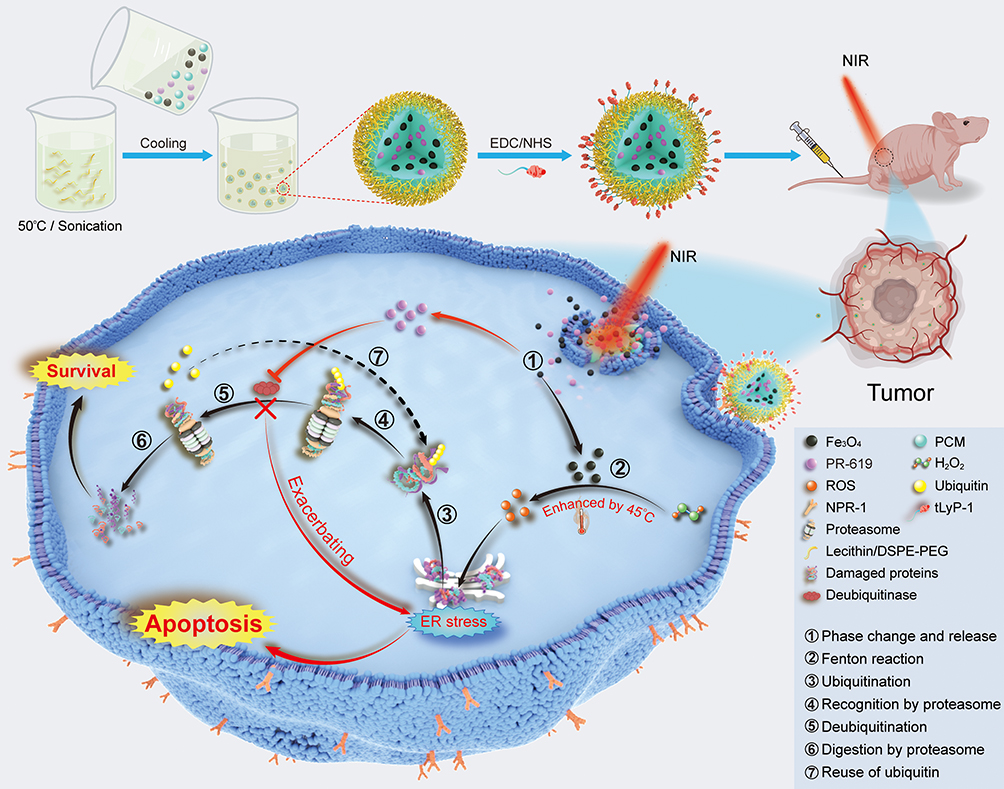 |
Figure 1 Schematic illustration for the synthesis of tPF@PCM and the synergistic mechanism for cancer therapy. Briefly, PF@PCM was first synthesized via nanoprecipitation and resolidification methods and then coupled with tLyP-1. The obtained tPF@PCM was next injected to 786-O tumor-bearing mice via the tail vein. At a local temperature of 45 °C by laser irradiation, Fe3O4 NPs were released from tPF@PCM and triggered the generation of ROS efficiently, resulting in increased unfolded or misfolded proteins in the ER lumen. The DUB inhibitor PR-619 suppressed the degradation of these damaged proteins, exacerbating ER stress, and finally leading to apoptosis of cancer cells. ① NIR-activated solid-to-liquid phase transition of tPF@PCM and release of Fe3O4 NPs and PR-619. ② Production of ROS via enhanced Fenton reaction by moderate heat of 45 °C. ③ Ubiquitination of unfolded and misfolded proteins. ④ Recognition of ubiquitinated proteins by the proteasome. ⑤ Deubiquitination of ubiquitinated proteins mediated by deubiquitinase. ⑥ Degradation of damaged proteins by the proteasome. ⑦ Recycling of ubiquitins after deubiquitination. |
Materials and Methods
Preparation of Phase-Change Materials
1-Hexadecanol (99%; Aladdin, Shanghai, China) and oleic acid (90%; Sigma-Aldrich, St. Louis, MO, USA) (3.5:1 by weight) were first dissolved in methanol at a concentration of 10 mg/mL, and the mixture was stored at –20 °C for further use.
Synthesis of tLyP-1/PR-619/Fe3O4@PCM
The tPF@PCM was synthesized according to a previously published protocol with some modifications.23 PF@PCM was first fabricated using nanoprecipitation and resolidification methods. Briefly, 15 mg of lecithin (98%; Aladdin, Shanghai, China) and 10 mg of DSPE‐PEG-COOH (molecular weight [MW]: 5000; J&K Scientific, Beijing, China) were dispersed in a 4% aqueous ethanol solution and heated to 50 °C. Further, Fe3O4 NPs (So-Fe Biomedical, Shanghai, China) and PR-619 (Selleck, Houston, TX, USA) were dissolved in tetrahydrofuran and DMSO, respectively. The PCM (1 mL, 10 mg/mL), Fe3O4 NPs (100 μL, 10 mg Fe/mL), and PR-619 (90 μL, 2 mg/mL) were mixed and added dropwise into the preheated aqueous ethanol solution under sonication for 10 min. Subsequently, the suspension was immediately solidified in an ice bath to form PF@PCM, which was further labeled with tumor-homing peptide tLyP-1 (CGNKRTR; Chinese Peptide Company, Hangzhou, China) via the carbodiimide coupling method. Specifically, 5 mg of PF@PCM was activated in 1 mL of PBS (pH 7.4) containing 10 mg of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) and 7 mg of N-hydroxysuccinimide (Thermo Fisher Scientific, Waltham, MA, USA) at 25 °C while stirring for 30 min. Afterward, 0.25 mg of tLyP-1 peptide was added to the suspension and stirred continuously for ~4 h on an ice bath. The resultant tPF@PCM was dialyzed at 4 °C for 24 h to remove surplus peptides and then stored in deionized water at 4 °C for further use.
Characterization of tLyP-1/PR-619/Fe3O4@PCM
The size distribution and zeta potential were analyzed using a Zetasizer Nano ZSP (Malvern Instruments, Malvern, UK). Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100 TEM (Tokyo, Japan) at 200 kV. The Fe concentration was determined using atomic absorption spectroscopy (AAS, z2000; Hitachi, Japan). The PR-619 concentration was detected using high-performance liquid chromatography (HPLC; Waters, Manchester, UK).
Photothermal Properties of tLyP-1/PR-619/Fe3O4@PCM
Different concentrations of tPF@PCM were exposed to an 808 nm laser for 10 min, and an IR thermal imaging camera (IRS-S6; IRS System, Shanghai, China) was used to monitor the temperature changes of the solutions.
In vitro Iron Ion and Drug Release Study
The in vitro release of tPF@PCM was evaluated using the dialysis bag diffusion technique. Briefly, 1 mL of tPF@PCM (2 mg/mL) in PBS with different pH values was divided into four groups: I) pH 5.5; II) pH 5.5 + laser; III) pH 7.4; and IV) pH 7.4 + laser. For the laser groups, tPF@PCM was first exposed to the laser to maintain the temperature at 45 °C for 10 min. Next, all groups were placed in separate dialysis bags (MW: 1000). Afterward, the dialysis bags were dipped into 20 mL of PBS with the same pH and shaken constantly at 37 °C for 72 h. At definite time intervals (1, 3, 6, 12, 24, 48, and 72 h), a 1-mL sample was withdrawn for analysis and replaced with the same amount of PBS. Fe was then analyzed by AAS and PR-619 by HPLC to estimate the amount of iron ions or drug released, respectively.
Peroxidase-Like Activity of tLyP-1/PR-619/Fe3O4@PCM and Bare Fe3O4 Nanoparticles
POD-like activity was measured according to a previously described method with some modifications.24 Briefly, the 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) working buffer was first prepared by mixing 496 μL of sodium citrate buffer (0.1 M, pH 4.5; Solarbio Life Sciences, Beijing, China), 2 μL of TMB (20 mg/mL; Sangon Biotech, Shanghai, China), and 2 μL of H2O2 (30%). Then, 5 μL of tPF@PCM (2 mg Fe/mL) or bare Fe3O4 NPs (2 mg Fe/mL) was added to 100 μL of TMB working buffer. After exposure to the laser to keep the temperature at 37 or 45 °C for 10 min, the absorbance spectra were measured in the range of 350–800 nm using a Synergy H1 microplate reader (BioTek, Winooski, VT, USA). The time-dependent photothermal-enhanced POD-like activity was assessed using the same method at different irradiation times (2, 4, 6, 8, and 10 min). The absorbance at 652 nm was also measured using the Synergy H1 microplate reader.
Cell Culture of 786-O, HK-2, PC-3, and Human Umbilical Vein Endothelial Cells
The authenticated short tandem repeat-profiled RCC 786-O, normal renal tubular epithelial HK-2, and prostate cancer PC-3 cell lines were purchased from the Stem Cell Bank, Chinese Academy of Sciences. Human umbilical vein endothelial cells (HUVECs) were a kind gift from the Department of Vascular Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University (SJTU) and the use of HUVECs was approved by the Ethics Committee of Renji Hospital, School of Medicine, SJTU. All cells were routinely tested for mycoplasma and were found to be negative. 786-O and PC-3 cells were cultured in RPMI-1640 (Gibco, Billings, MT, USA) with 10% FBS (Invitrogen, Waltham, MA, USA), HK-2 cells were cultured in DMEM/F12 (Gibco) with 10% FBS, and HUVECs were cultured in endothelial cell growth medium-2 (EGM-2; Lonza, Basel, Switzerland). All cells were incubated at 37 °C in 5% CO2.
Cellular Uptake of PR-619/Fe3O4@PCM and tLyP-1/PR-619/Fe3O4@PCM
Cellular uptake of PF@PCM or tPF@PCM was determined by flow cytometry and confocal laser scanning microscopy (CLSM) analyses. Briefly, 786-O cells were incubated with 50 μg Fe/mL FITC-PF@PCM or FITC-tPF@PCM at 37 °C for 1, 3, 6, 12, and 24 h. Cells were then collected, and fluorescence (FITC channel) was determined using a CytoFLEX S (Beckman Coulter, Brea, CA, USA) cytometer. For CLSM experiments, 786-O cells were incubated with 50 μg Fe/mL FITC-PF@PCM or FITC-tPF@PCM for 6 h and fixed in 4% paraformaldehyde for 20 min. Cell nuclei were stained with DAPI (C1005; Beyotime, Shanghai, China) for 5 min. Cellular uptake was observed using CLSM (Leica Microsystems, Wetzlar, Germany).
In vitro Cytotoxicity of tLyP-1/PR-619/Fe3O4@PCM
In vitro cytotoxicity was assessed using the CCK-8 assay (Dojindo Molecular Technologies, Rockville, MD, USA). Cells were seeded in 96-well plates at an initial density of 2000 cells/well in 100 μL of complete medium. After a 24-h incubation, RPMI-1640 was replaced with a fresh medium containing various formulations (control, tP@PCM, tF@PCM, or tPF@PCM) and cultured for an additional 24 h. Subsequently, for groups receiving laser irradiation, the cells were exposed to an 808 nm laser to maintain the cell temperature at 45 °C for 10 min. After another 24-h incubation, the CCK-8 assay was carried out, and the absorbance at 450 nm was determined using the Synergy H1 microplate reader. Each condition was performed in quadruplet, and each experiment was repeated three times.
Live-Dead Analysis of tLyP-1/PR-619/Fe3O4@PCM in 786-O Cells
786-O cells were seeded into 12-well plates at a density of ~50% and treated as described above. The cells were then stained with Calcein-AM and 7-aminoactinomycin D (7-AAD) staining working solution at 37 °C for 20 min in the dark. The live/dead cells were visualized using a ZOE Fluorescent Cell Imager (Bio-Rad, Hercules, CA, USA).
In vitro Measurement of Intracellular Reactive Oxygen Species in 786-O Cells
Intracellular ROS was measured using the 2ʹ,7ʹ-dichlorodihydrofluorescein diacetate (DCFH-DA) probe (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). For CLSM, 786-O cells were seeded in confocal dishes. After different treatments, cells were co-incubated with DCFH-DA (1:1000 in free RPMI-1640) at 37 °C for 20 min and then observed by CLSM. For flow cytometry analyses, different groups of 786-O cells were co-incubated with DCFH-DA and then harvested and resuspended in PBS for flow cytometry analyses.
Detection of Aggregated Proteins of 786-O Cells
Aggregated proteins were detected with the PROTEOSTAT Aggresome Detection Kit (ENZ-51035; Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturer’s instructions. Briefly, 786-O cells were seeded in 24-well culture plates. After different treatments, cells were fixed with 4% formaldehyde at 25 °C for 30 min and permeabilized with 0.5% Triton X-100 and 3 mM EDTA (pH 8.0) on ice for 30 min. Subsequently, the cells were stained with a dual detection reagent containing Hoechst 33342 (nuclear stain) and PROTEOSTAT dye reagent, and then incubated at 25 °C for 30 min. Aggregated protein imaging was performed using the ZOE Fluorescent Cell Imager.
Real-Time Polymerase Chain Reaction Assays
Total RNA was extracted from various groups of 786-O cells using a FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme, Nanjing, China). Complementary DNA (cDNA) was prepared from 500 ng of total RNA according to the reverse transcription protocol using the PrimeScript™ RT Reagent Kit (Takara, Dalian, China). Real-time Polymerase Chain Reaction (RT-PCR) analyses were performed using SYBR® Premix Ex Taq™ (Takara). Samples were analyzed using an ABI PRISM® 7900HT Real‐Time PCR System (Applied Biosystems, Waltham, MA, USA). The human mRNA primer sequences for target genes are listed in Table S1. Relative gene expression was normalized to that of the TATA box-binding protein (TBP). The relative mRNA levels were calculated using the 2−ΔΔCt method.
Western Blot Analysis
Western blot (WB) analysis was performed according to standard procedures. The antibodies used in these experiments are listed in Table S2. Briefly, cells were lysed in RIPA buffer, and 30-μg samples were loaded and separated by a 4–15% Omni-PAGE gel (EpiZyme, Shanghai, China). Proteins were then transferred to a PVDF membrane (Millipore, Billerica, MA, USA) using a fast wet-transfer apparatus (eBlot™ L1; GenScript, Nanjing, China). After blocking with 5% milk at 25 °C for 1 h, blots were incubated with primary antibodies at 4 °C overnight and then with HRP-conjugated secondary antibodies at 25 °C for 1 h. Membranes were then incubated with ECL substrate and detected by the ChemiDoc™ Imaging System (Bio-Rad).
TUNEL Staining
TUNEL staining was performed using a one-step TUNEL apoptosis assay kit (Beyotime Biotechnology) according to the manufacturer’s protocol. Briefly, cells were fixed in 4% paraformaldehyde and permeabilized with 0.3% Triton X-100 for 5 min. The sections were then subjected to a TUNEL assay at 37 °C in the dark for 1 h. Nuclei were stained with DAPI for 5 min. Randomly chosen fields were examined using the ZOE fluorescent cell imager.
Apoptosis Assays by Annexin V-PE/7-Aminoactinomycin D Double Staining
The apoptotic and necrotic cell distribution was analyzed using the Annexin V-PE/7-AAD apoptosis detection kit (Yeasen, Shanghai, China). Briefly, different groups of cells were harvested and adjusted to a concentration of 0.5–1×105 cells/100 μL using 1X binding buffer. Then, 5 μL of Annexin V-PE and 10 μL of 7-AAD were added to a 100-μL cell suspension and incubated at 25 °C for 10 min. Finally, 400 μL of 1X binding buffer was added, and the cells were analyzed by flow cytometry.
Construction of Mouse Xenograft Model
All animal studies were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University (Permit Number: SYXK-Hu-2018-0021) and performed in compliance with the Guide for Care and Use of Laboratory Animals by the Institute of Laboratory Animal Research (ILAR). Male BALB/c nude mice (4–6 weeks old) were obtained from Shanghai SLAC Laboratory Animal Co. (Shanghai, China). After acclimatization for one week, each mouse was injected with 3 million 786-O cells in a 1:1 mixture of PBS and Matrigel (#354234; BD Biosciences, Franklin Lakes, NJ, USA) in the right flank. After tumors reached ~100 mm3 (~7 weeks), 786-O tumor-bearing mice were randomly grouped for downstream experiments.
In vivo Magnetic Resonance Imaging of 786-O Tumor-Bearing Mice
In vivo magnetic resonance imaging (MRI) was performed using a 7T small animal MRI scanner. 786-O tumor-bearing mice were anesthetized with a mixture of oxygen and isoflurane, and then subjected to MRI to obtain the initial T2-weighted images. Subsequently, mice were injected with PF@PCM or tPF@PCM at 15 mg Fe/kg body weight (BW) via the tail vein (n = 3), and T2-weighted MR images were obtained at 1, 3, 6, and 24 h post-injection.
Biodistribution of tLyP-1/PR-619/Fe3O4@PCM in 786-O Tumor-Bearing Mice
786-O tumor-bearing mice were injected with tPF@PCM at 15 mg Fe/kg BW via the tail vein and were sacrificed by cervical dislocation at set time points (1, 3, 6, 24, 48, 72, and 96 h post-injection; n = 3). The main organs (heart, liver, spleen, lung, and kidney) and tumors were collected and digested with aqua regia. The Fe content of each sample was determined by AAS and calculated as the percentage of the injected dose (ID) per gram (%ID/g).
In vivo Antitumor Therapy in 786-O Tumor-Bearing Mice
786-O tumor-bearing mice were randomly divided into seven groups (n = 4): PBS; laser; tPF@PCM; tP@PCM + laser; tF@PCM + laser; tPF@PCM + laser; and sunitinib. tPF@PCM at 15 mg Fe/kg BW in 200 μL final volume was injected via the tail vein for tPF@PCM and tPF@PCM + laser groups; tF@PCM at 15 mg Fe/kg BW was for tF@PCM + laser group; and tP@PCM at 2.5 mg PR-169/kg BW was for tP@PCM + laser group. The PBS and laser groups were injected with 200 μL of PBS alone. Sunitinib was administered at 20 mg/kg BW by oral gavage in a 5-day-on/2-day-off cycle. For groups receiving laser irradiation, after 24 h, mice were exposed to an 808 nm laser to keep the temperature of tumor regions at 45 °C for 10 min. During the 15 days of treatment, the tumor volume and BW of each mouse were measured at 3-day intervals. Tumor volumes were calculated according to the following formula: Tumor volume = π/6 × (width)2 × length. Relative tumor volume was calculated by normalizing the measured tumor volume (V) to the initial value (V0).
Histochemical Evaluations of 786-O Tumor-Bearing Mice
786-O tumor-bearing mice were treated as described above. Three days after laser irradiation, mice were sacrificed. Tumors were harvested, fixed in 10% formalin for 24 h, embedded in paraffin, sliced, and stained with hematoxylin and eosin (H&E), Prussian blue, anti-ubiquitin (Ub), anti-Ki-67, anti-cleaved caspase-3, and TUNEL for further explorations according to standard procedures.
Safety Evaluation of tLyP-1/PR-619/Fe3O4@PCM in 786-O Tumor-Bearing Mice
At the end of antitumor therapy, blood was first collected from the hearts of sacrificed mice for various hematological and biochemical analyses. Subsequently, the main organs of these mice were collected for standard H&E staining.
Statistical Analyses
Statistical evaluations were conducted using Student’s t-test using Prism v8.0 (GraphPad Software, San Diego, CA, USA). All data are shown as the mean ± standard error of the mean (SEM) unless otherwise indicated. The level of significance was set at P < 0.05: *P < 0.05, **P < 0.01, and ns = P ≥ 0.05.
Results and Discussion
Synthesis and Characteristics of tLyP-1/PR-619/Fe3O4@PCM
We synthesized tPF@PCM in three steps.23 PCM with a melting point of 45 °C was first prepared by mixing 1-Hexadecanol with oleic acid (3.5:1 by weight) in methanol (as described in the Methods and Materials section). Next, PR-619 and Fe3O4 NPs were loaded into the PCM to obtain PF@PCM via nanoprecipitation and resolidification methods.25 Lastly, the cyclic peptide tLyP-1, a robust and selective tumor-homing peptide, was coupled to PF@PCM to yield tPF@PCM using carbodiimide chemistry for better tumor-targeting delivery.26 The final nano-formulation tPF@PCM exhibited a near-spherical morphology with a uniform size distribution of ~130 nm (Figure 2A). The hydrodynamic size determined by dynamic light scattering (DLS) increased from ~170 to 177 nm after the peptide modification of PF@PCM (Figure 2B), and the corresponding zeta potential changed from –25 to –13 mV (Figure S1). Moreover, the loading efficiency was 8.58 wt.% for Fe, as determined by AAS, and ~1.43 wt.% for PR-619, as determined by HPLC. Further, the encapsulation efficiency of PR-619 was ~88.3%. The tPF@PCM remained stable in various solutions at 37 °C for 72 h (Figure 2C), allowing for subsequent in vitro and in vivo applications. We also synthesized PR-619-loaded (tP@PCM) or Fe3O4 NPs-loaded (tF@PCM) particles using similar approaches as controls.
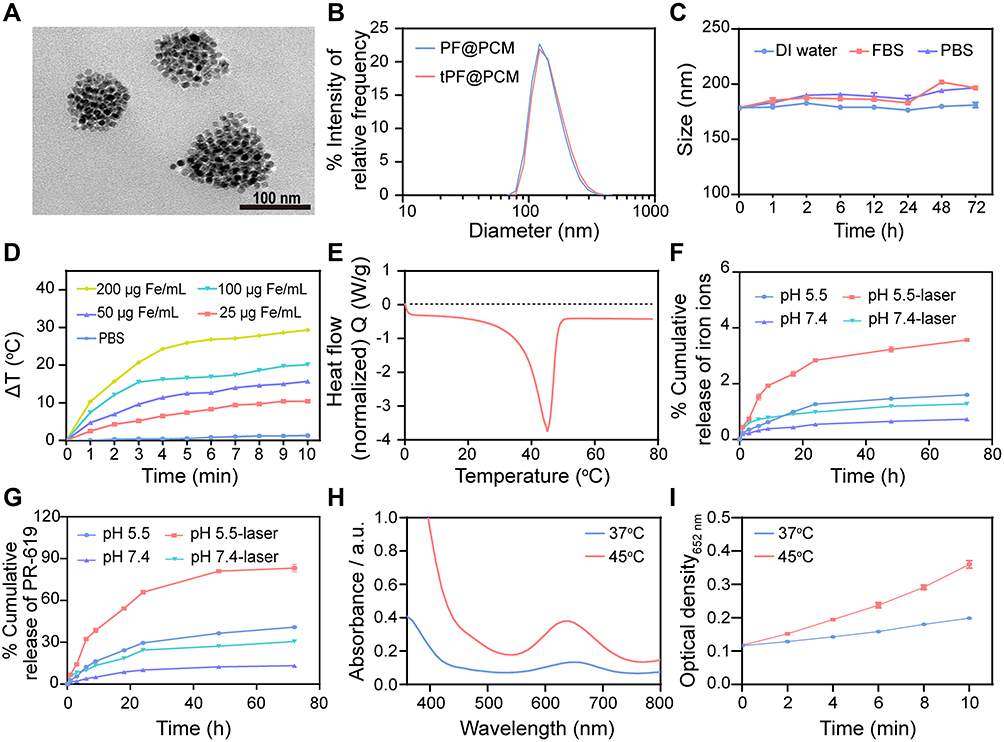 |
Figure 2 Characterizations of tPF@PCM. (A) TEM image of tPF@PCM. Scale bar represents 100 nm. (B) Size distribution of PF@PCM and tPF@PCM in distilled (DI) water. (C) Stability of tPF@PCM in DI water, fetal bovine serum (FBS), and PBS at 37 °C for 72 h, respectively. (D) Photothermal heating curve of tPF@PCM at different concentrations. (E) Characterization of phase transformation of PCM measured by DSC. (F, G) Cumulative release of iron ions (F) and PR-619 (G) from tPF@PCM at pH 5.5 and 7.4 with or without NIR laser irradiation, respectively. (H) POD-like activity of tPF@PCM at 37 °C and 45 °C maintained by laser irradiation for 10 min. (I) Absorbance variation of tPF@PCM at 652 nm at different temperatures. Data are presented as mean ± SEM. |
Efficient drug release and catalytic capability are two important features of a potent antitumor nanocatalytic platform. According to accumulating evidence, PCMs exhibit solid-liquid phase transition at a certain temperature; moreover, appropriate hyperthermia can enhance the POD-like activity of Fe3O4 NPs.13,27 Accordingly, we reasoned that the release of PR-619 and Fe3O4 NPs might be facilitated by photothermal stimulation, with simultaneous enhancement of the enzyme activity of Fe3O4 NPs. Therefore, we first assessed the photothermal effect of tPF@PCM under 808 nm laser irradiation. As in most Fe3O4-based nano-formulations previously reported,28 the increase in temperature was dependent on both concentration and irradiation time (Figures 2D and S2). Subsequently, differential scanning calorimetry (DSC) experiments confirmed that the phase change temperature of tPF@PCM was 45 °C (Figure 2E).
Based on the considerable photothermal performance and thermal phase transition property of tPF@PCM, we next evaluated the release pattern of iron and PR-619 at pH 5.5 and pH 7.4, respectively, with or without initial laser irradiation (45 °C for 10 min). The results showed that the acidic environment favored the release of both iron and PR-619, which was subsequently significantly enhanced by laser intervention (Figure 2F and G).
We next assessed the influence of the photothermal effect on POD-like activity of tPF@PCM. The tPF@PCM was exposed at 37 or 45 °C by laser irradiation for 10 min in the presence of H2O2, while TMB was used as the substrate, which had an absorption peak at 652 nm after oxidation by •OH.12 Consistent with other Fe3O4-based nano-formulations,24,29 tPF@PCM showed enhanced POD-like activity at 45 °C (Figure 2H). Moreover, the production of •OH increased with irradiation time (Figure 2I). To exclude the possible influence of the release behavior of Fe3O4 NPs on the catalytic activity, bare Fe3O4 NPs were used to assess POD-like activity under the same conditions. As expected, the catalytic activity of the bare Fe3O4 NPs was likewise enhanced by hyperthermia (Figure S3).
In conclusion, the heat simulation (45 °C for 10 min) was sufficient for both encapsulated content release and •OH production, which might be beneficial for the treatment of cancers. Therefore, irradiation conditions were fixed to maintain the temperature at 45 °C for 10 min in the following cell and animal experiments. Based on these results, we successfully synthesized tPF@PCM with good photothermal performance and controllable phase transition properties, which could be applied for photothermal-enhanced cancer therapy.
Cellular Uptake and Cytotoxicity of tLyP-1/PR-619/Fe3O4@PCM
Neuropilin-1 and −2 (NRP1/2) are overexpressed on the membrane of numerous tumor cells and/or tumor lymphatics, including brain cancer, prostate cancer, and RCC.30–32 Therefore, to improve the tumor-targeting property, the tLyP-1 peptide, as a ligand of both NPR1 and NPR2, was coupled to the catalytic nanoparticles. The targeting property of tPF@PCM was evaluated using the NPR1 positive RCC cell line 786-O.33 The uptake of FITC-labeled nanoparticles was examined by flow cytometry. The fluorescence intensity of FITC-tPF@PCM in 786-O cells increased significantly with incubation time and was stronger than that of non-targeting FITC-PF@PCM at each time point from 1 to 24 h (Figures 3A and B and S4). CLSM further corroborated the results that FITC-tPF@PCM was considerably more internalized by 786-O cells than FITC-PF@PCM (Figure 3C). Overall, the excellent tumor-targeting property of tPF@PCM for 786-O cells establishes a foundation for further targeting therapy.
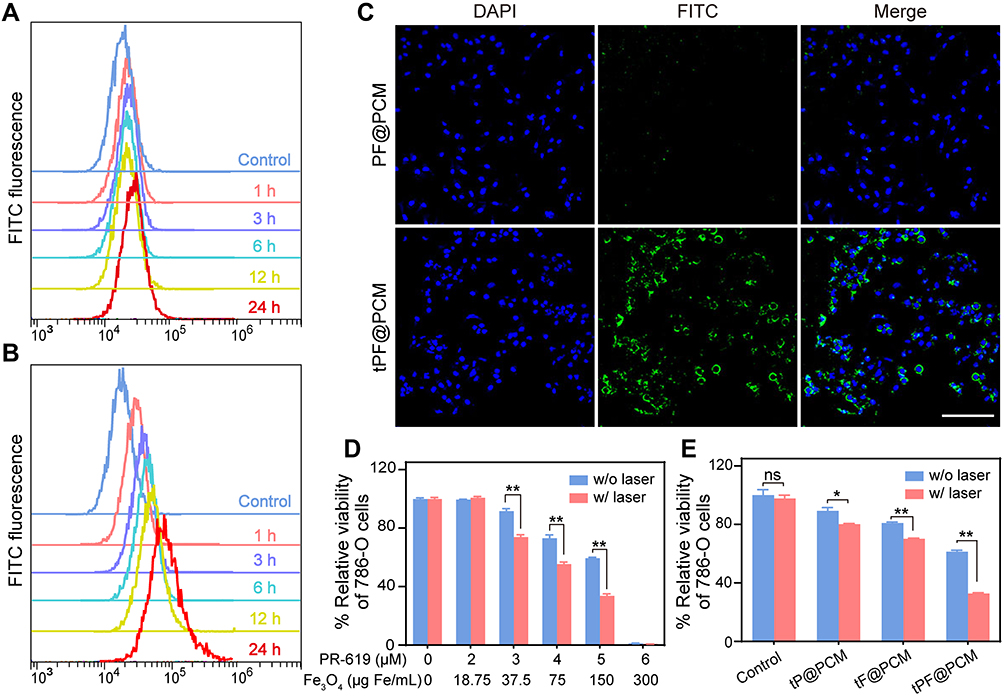 |
Figure 3 Cellular uptake and cytotoxicity of tPF@PCM. (A, B) Cellular uptake of FITC-labeled PF@PCM (A) and tPF@PCM (B) determined by flow cytometry. (C) CLSM images of 786-O cells co-incubated with FITC-labeled PF@PCM (top row) and tPF@PCM (bottom row) at 50 μg Fe/mL for 6 h. Cell nuclei were stained with DAPI (blue). Scale bar represents 200 μm. (D) Cytotoxicity evaluation of tPF@PCM with different concentrations of PR-619 and Fe3O4 NPs against 786-O cells with or without laser irradiation. **P < 0.01. (E) Cytotoxicity evaluation of tP@PCM, tF@PCM, and tPF@PCM (5 μM for PR-619; 150 μg Fe/mL for Fe3O4 NPs) against 786-O cells with or without laser irradiation. *P < 0.05; **P < 0.01; ns = P ≥ 0.05. |
Subsequently, we evaluated the therapeutic effect of tPF@PCM in vitro using CCK-8 assays. To first investigate the biosafety of the PCM vehicle (without drug loading), different cell lines (HUVECs, HK-2, PC-3, and 786-O) were co-incubated with different concentrations of PCM. All cells showed negligible cytotoxicity, even when co-incubated with the PCM vehicle at a high concentration (4 mg/mL) for 48 h, indicating excellent biocompatibility with the PCM (Figure S5). Subsequently, to evaluate the cytotoxicity of tPF@PCM with or without laser irradiation, 786-O cells were first incubated with tPF@PCM containing various concentrations of PR-619 (0–6 μM) and Fe3O4 NPs (0–300 μg Fe/mL) for 24 h. An 808 nm laser was later applied to maintain the cells at 45 °C for 10 min for the laser groups. All cells were then cultured for an additional 24 h. The cell viability decreased to ~60% in the presence of 5 μM PR-619 with 150 μg Fe/mL Fe3O4 NPs. In particular, at concentrations of 6 μM for PR-619 and 300 μg Fe/mL for Fe3O4 NPs, virtually no cells survived. Notably, laser irradiation significantly enhanced cytotoxicity in groups containing 3, 4, or 5 μM PR-619 (Figure 3D). Therefore, in the subsequent in vitro experiments, we chose concentrations of 5 μM for PR-619 and 150 μg Fe/mL for Fe3O4 NPs for further exploration.
Next, we determined the synergistic therapeutic effect of tPF@PCM and laser-induced hyperthermia. 786-O cells were divided into eight groups: I) control; II) tP@PCM; III) tF@PCM; IV) tPF@PCM; V) laser; VI) tP@PCM + laser; VII) tF@PCM + laser; and VIII) tPF@PCM + laser. The tF@PCM + laser group exhibited moderate cytotoxicity (>70% survival). However, PR-619 increased the cytotoxicity in the tPF@PCM + laser group, with only ~30% of living cells being observed, indicating that PR-619 could serve as a reinforcer to increase cell death induced by photothermal-enhanced Fe3O4 NPs (Figure 3E). A visual representation of this phenomenon was later exhibited by staining 786-O cells with the live/dead cell indicator Calcein-AM and 7-AAD following different treatments. In this case, fewer living cells (green fluorescence) were left in the tPF@PCM + laser group than in other groups (Figure S6). Moreover, this therapeutic strategy had much milder cytotoxicity against HK-2 cells (Figure S7), verifying the safety of tPF@PCM + laser for normal cells. Together, these results demonstrate a satisfactory synergistic effect of photothermal-enhanced Fe3O4 NPs and PR-619 on 786-O RCC cells.
Exacerbated Endoplasmic Reticulum Stress Induces Apoptosis in 786-O Cells
We further explored the synergistic antitumor mechanism of our therapeutic strategy. The upregulation of ROS induced by Fe3O4 NPs and other nano-formulations is capable of disrupting protein folding, leading to an increase in ER stress.34–36 Under normal conditions, the unfolded or misfolded proteins could be handled by the detoxification process, ERAD, which is mediated by the “adaptive” UPR and involves ubiquitination, transportation to proteasomes, deubiquitination, and degradation.37 However, PR-619 can block ERAD at the step of deubiquitination and lead to an accumulation of polyubiquitinated damaged proteins in the proteasome. In this way, the damaged proteins that could not be degraded in the ER lumen cause severe ER stress, thereby activating the “terminal” UPR and eventually inducing the apoptosis of cancer cells (Figure 4A).
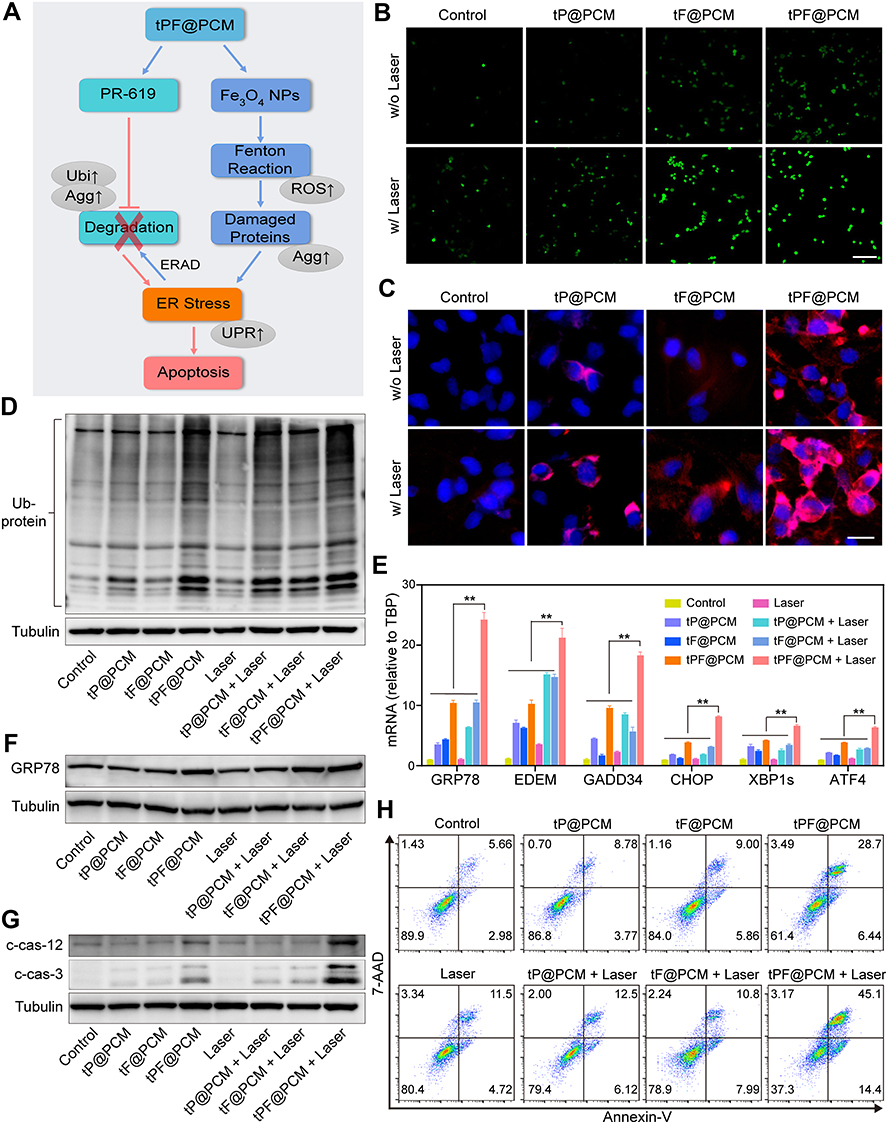 |
Figure 4 Exacerbating ER stress by tPF@PCM with laser irradiation induces apoptosis in 786-O cells. (A) Schematic overview of the mechanism of synergistic apoptosis. Fe3O4 NPs-triggered Fenton reaction led to increased damaged proteins in the ER lumen and caused initial ER stress. ERAD pathway was then activated to degrade these proteins (blue arrows). However, PR-619 inhibited the degradation of the stacked damaged proteins, which further exacerbated the existing ER stress. Ultimately, unsalvageable ER stress induced apoptosis in 786-O RCC cells (red arrows). The grey ellipses represent the corresponding intracellular biological changes. (B) Confocal laser scanning microscopy images of intracellular ROS detection in 786-O cells from different groups. Scale bar represents 200 μm. (C) Fluorescence images of protein aggregates in 786-O cells after various treatments. Cell nuclei were stained with Hoechst 33342 (blue) and protein aggregates were stained with PROTEOSTAT dye (red). Scale bar represents 25 μm. (D) Western blot analysis of total ubiquitinated proteins in 786-O cells after different treatments. Tubulin was used as a loading control. (E) mRNA expression of UPR target genes in 786-O cells after different treatments. TBP mRNA levels were used as internal controls. **P < 0.01. (F–G) Western blot analysis of GRP78 (F), cleaved caspase-12 (c-cas-12), and cleaved caspase-3 (c-cas-3) (G) expression in 786-O cells after different treatments. Tubulin was used as a loading control. (H) Apoptosis detection of 786-O cells after various treatments using annexin V-PE/7-AAD kit by flow cytometry. Early apoptotic cells were analyzed as annexin V+/7-AAD−, whereas late apoptotic/necrotic cells were determined as annexin V+/7-AAD+. Abbreviations: ROS, reactive oxygen species; Agg, aggresome; UPR, unfolded protein response; Ubi, ubiquitination. |
To validate this assumption, we first examined intracellular ROS levels with different treatments using DCFH-DA staining. CLSM showed moderate fluorescence intensity in cells treated with tF@PCM, which was further enhanced by laser irradiation owing to the photothermal-enhanced POD-like activity of Fe3O4 NPs. Moreover, the addition of PR-619 expanded this trend, and tPF@PCM + laser-treated cells displayed the strongest intensity. In contrast, cells in groups without Fe3O4 NPs showed either a slight or insignificant increase in fluorescence (Figure 4B). These observations were further validated by flow cytometry, where the tPF@PCM + laser presented the highest efficiency in intracellular ROS production (Figure S8). These findings provide evidence that a large amount of intracellular ROS was synergistically produced in response to photothermal-enhanced Fe3O4 NPs and the ERAD inhibitor, PR-619, which possibly delivered a constant supply of unfolded or misfolded proteins.
Therefore, we further analyzed the unfolded or misfolded proteins in 786-O cells after different treatments using the PROTEOSTAT Aggresome detection kit. The PROTEOSTAT dye specifically intercalates into structures typically found in misfolded or polyubiquitinated proteins, resulting in red fluorescence with distinct distribution patterns.38 Specifically, the accumulation of misfolded proteins in the ER results in diffuse cytoplasmic staining, whereas an increase in polyubiquitinated proteins in the proteasome leads to discrete punctate staining of perinuclear structures.39 As expected, the tPF@PCM + laser group exhibited the brightest fluorescence with both staining patterns of aggregated proteins. In contrast, cells treated with tF@PCM and tF@PCM + laser exhibited only diffuse cytoplasmic staining, whereas the tP@PCM and tP@PCM + laser groups exhibited punctate red fluorescence (Figure 4C). Consistently, the elevated polyubiquitinated protein levels in the proteasome were confirmed by WB, which showed that the ubiquitination level was substantially increased when the cells were treated with tPF@PCM + laser (Figure 4D). In summary, our results demonstrate that the combination strategy resulted in a considerable accumulation of polyubiquitinated proteins in the proteasome and unfolded or misfolded proteins in the ER lumen, which potentially contributed to a drastic increase in ER stress.
To verify the induction of ER stress by our therapeutic strategy, the activation of the ER stress response (specifically UPR) was further evaluated.40 Multiple UPR target genes were first detected by RT-PCR, and the tPF@PCM + laser group displayed the highest efficiency in activating this specific stress response pathway (Figure 4E). Among these target genes, chaperone GRP78 plays an initiating role in the UPR.3 Accordingly, the expression of GRP78 in different 786-O cell groups was subsequently measured by WB. The results revealed that the tPF@PCM + laser group yielded the most significant increase of this master regulator for ER stress (Figure 4F). In summary, these results indicate that the combinatorial use of photothermal-enhanced Fe3O4 NPs and PR-619 potently activated UPR signaling induced by severe ER stress.
Generally, the UPR is triggered to maintain cell survival or induce cell death, depending on the level of ER stress. If the UPR fails to lessen the ER burden, ER-related apoptosis occurs.41 Because there was a significant population of dead cells in the tPF@PCM + laser group (Figures 3E and S6), we inferred that the overloaded ER stress induced by the tPF@PCM + laser eventually led to the apoptosis of 786-O cells. To confirm this hypothesis, the total proteins of 786-O cells in different groups were first collected and WB was performed with anti-cleaved caspase-3, as the hallmark of apoptosis, and anti-cleaved caspase-12, an indicator of ER-specific apoptosis.42,43 Both cleaved caspase-3 and −12 levels were markedly increased in the combination group (Figure 4G), implying that the tPF@PCM + laser potentially caused apoptosis in 786-O cells. Furthermore, flow cytometry analyses with annexin V-PE/7-AAD staining were consistent with this result. Cells treated with the tPF@PCM + laser showed the highest level of apoptosis (59.5% annexin V-positive cells; Figure 4H). Finally, a similar trend was observed in the TUNEL assay, and the combination treatment exhibited a considerable increase in the number of TUNEL-positive cells (Figure S9). Overall, the combination of photothermal-enhanced oxidative damage and ERAD inhibition induced ER stress-related apoptosis in 786-O cells.
Taken together, these results confirmed that DUB inhibition by PR-619 disabled the self-protection mechanism of 786-O cells suffering from redox dysregulation induced by the photothermal-augmented Fe3O4 NPs, and the resulting accumulation of ER stress directed the cells toward apoptosis. Following the impaired ERAD, autophagy is an alternative to restore ER homeostasis; thus, it might further improve the antitumor efficacy by co-modulating ER stress and autophagy.5 Interestingly, some natural compounds, such as kaempferol, could regulate autophagy and ER stress simultaneously, offering simpler options for more effective antitumor therapy.44 Nevertheless, our understanding of the interaction between autophagy and ER stress is far from comprehensive, and additional studies will be needed in the future.
In vivo Magnetic Resonance Imaging and Antitumor Efficacy of tLyP-1/PR-619/Fe3O4@PCM in 786-O Tumor-Bearing Mice
Owing to its good T2-weighted MRI ability (r2 = 63.59 mM−1·s−1; Figure S10), we next explored the in vivo tumor-targeting performance of tPF@PCM using a 7T small animal MRI scanner. 786-O tumor-bearing mice were first injected with PF@PCM or tPF@PCM at 15 mg Fe/kg BW via the tail vein and then subjected to MRI 0–24 h post-injection. tPF@PCM significantly accumulated at the tumor site within 1 h post-injection and increased gradually over the observed time of 24 h. However, after injection with the non-targeting PF@PCM, only a small decrease in the T2 signal was detected at the tumor site at the corresponding time points, and the signal was barely detectable after 24 h (Figure 5A). These results indicate the excellent tumor-targeting capacity of tPF@PCM, which has potential future use as in vivo cancer therapy.
 |
Figure 5 In vivo magnetic resonance imaging (MRI) and synergetic anticancer therapy. (A) T2-weighted MR images of 786-O tumor-bearing mice (n = 3) at 0, 1, 3, 6, and 24 h post-injection of 15 mg Fe/kg body weight PF@PCM (top row) or tPF@PCM (bottom row). The white dotted circles represent tumors. (B) Schematic of the treatment regimen for 786-O tumor-bearing mice. (C) Tumor volume changes (n = 4) after different treatments. **P < 0.01. (D) Photos of the tumors extracted from mice in different groups at the end of treatments (day 15). |
Subsequently, to understand the distribution of tPF@PCM in vivo, 786-O tumor-bearing mice were injected with the same dose of tPF@PCM. At 1–96 h post-injection, the main organs (heart, liver, spleen, lung, and kidney) and tumors were harvested to measure the concentration of Fe by AAS. tPF@PCM mainly accumulated in the liver and spleen, which is in line with other reports of nano-formulations.45,46 Furthermore, there was a moderate aggregation of tPF@PCM at the tumor site, with the highest level observed at 24 h post-injection (Figure S11), which reveals the ideal timing for the initiation of reliable tumor-targeting photothermal therapy.
Encouraged by the excellent tumor-targeting ability of tPF@PCM, the in vivo therapeutic efficacy was then evaluated. Twenty-eight 786-O tumor-bearing mice were randomized into seven groups and treated respectively with PBS, laser, tPF@PCM, tP@PCM + laser, tF@PCM + laser, tPF@PCM + laser, or sunitinib (first-line agent recommended for the treatment of advanced RCC) (Figures 5B and S12). All agents were administered through the tail vein, except for sunitinib, which was administered by oral gavage on a 5-day-on/2-day-off cycle. Post-injection (24 h), the tumor regions were irradiated with the laser to keep the temperature at 45 °C for 10 min for the groups receiving laser intervention. Consistent with the in vitro experiments, the tPF@PCM + laser had the most significant effect on tumor inhibition, with over half of the tumors nearly disappeared by day 15 (Figure 5C and D). Moreover, this combination therapy displayed better antitumor efficacy than the conventional dose of sunitinib in 786-O tumor-bearing mice, suggesting a promising approach for the treatment of refractory RCC. Over the course of treatment, none of the mice showed significant changes in BW (Figure S13), indicating a favorable safety profile of this treatment strategy.
To further assess the mechanism involved in the antitumor effect of synergistic therapy, various histological analyses of tumor tissues harvested at day 3 post-laser irradiation were performed (Figure 6). H&E staining first showed the histological morphology of each section, and the most significant tissue damage was observed in the tPF@PCM + laser group. Subsequently, Prussian blue staining and anti-Ub immunohistochemistry were performed to characterize the presence of Fe3O4 NPs or PR-619. Correspondingly, Prussian blue-positive staining was detected in Fe3O4 NPs-containing groups, whereas Ub-moderate/strong-positive staining was found in PR-619-containing groups. Consistent with the in vitro WB experiments, the tPF@PCM + laser group exhibited the strongest Ub-positive staining owing to the massive generation of ubiquitinated damaged proteins due to a dysfunctional clearance mechanism. These results imply that the combination of photothermal-enhanced Fe3O4 NPs and PR-619 exhibited an excellent synergistic antitumor effect. The lowest levels of Ki67, a cellular marker for proliferation, were present in the tPF@PCM + laser group. Finally, cleaved caspase-3 and TUNEL staining demonstrated that tumors treated with tPF@PCM + laser had considerably more apoptotic areas than those treated with either tF@PCM + laser, tP@PCM + laser, or tPF@PCM, whereas other groups presented with minor, if any, evidence of apoptosis. In summary, our photothermal-augmented nanocatalytic and UPR-modulated ER stress strategy showed promising results as an apoptosis inducer in RCC treatment.
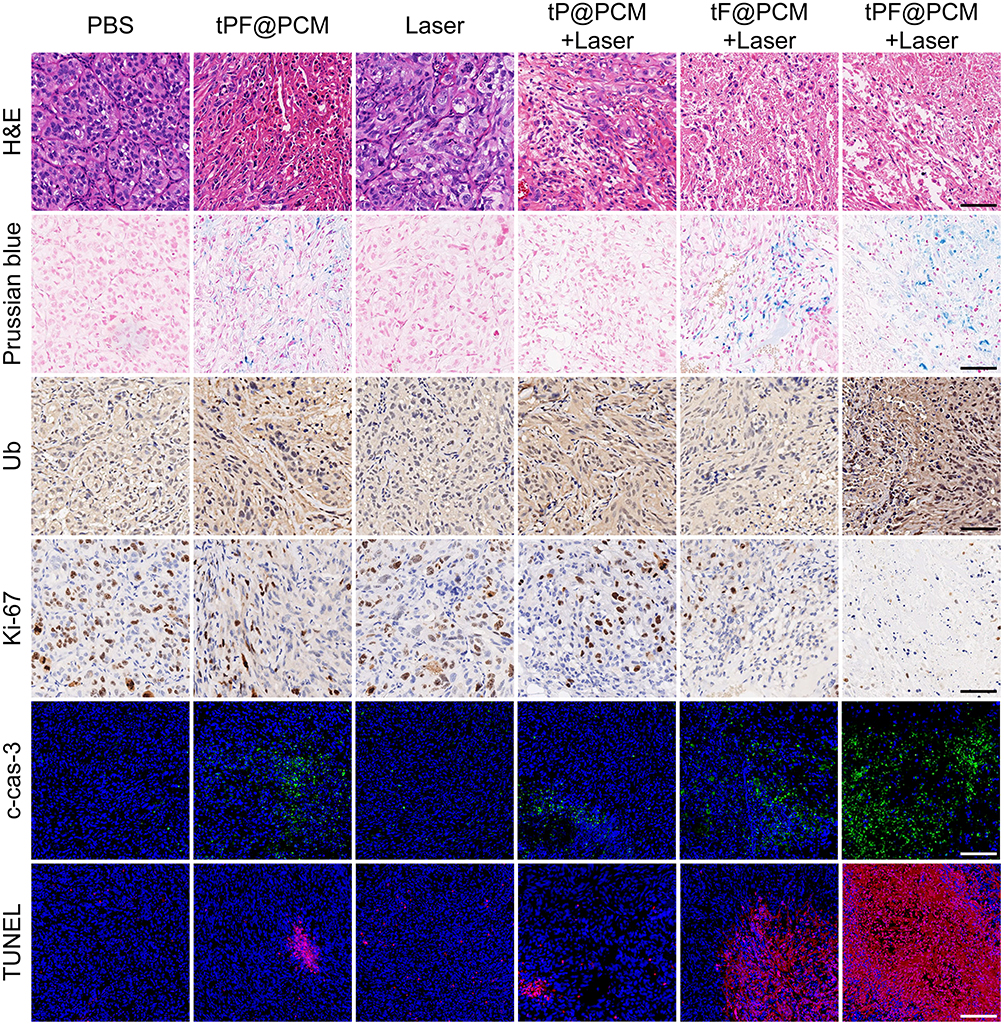 |
Figure 6 Histochemical images of xenografted 786-O tumor tissue sections at day 3 after laser irradiation. For cleaved caspase-3 (c-cas-3) and TUNEL immunofluorescence staining, nuclei were stained blue (DAPI staining) and apoptotic cells were stained green (c-cas-3 staining) or red (TUNEL staining). Scale bars represent 100 μm. |
Biosafety Evaluation of tLyP-1/PR-619/Fe3O4@PCM in 786-O Tumor-Bearing Mice
At the end of the respective treatments, the routine blood tests, renal function, and liver function of mice in the PBS, tPF@PCM, and tPF@PCM + laser groups were analyzed. All laboratory measurements of vital functions yielded normal results (Figure 7A). Furthermore, major organs dissected from these mice were subjected to H&E staining to evaluate organ toxicity. Although tPF@PCM + laser group showed the strongest tumoricidal effect, no notable changes were observed in the morphology of the heart, liver, spleen, lung, and kidney under this treatment regimen (Figure 7B). Altogether, these results verify the excellent in vivo biocompatibility of tPF@PCM at the dose used in our investigations.
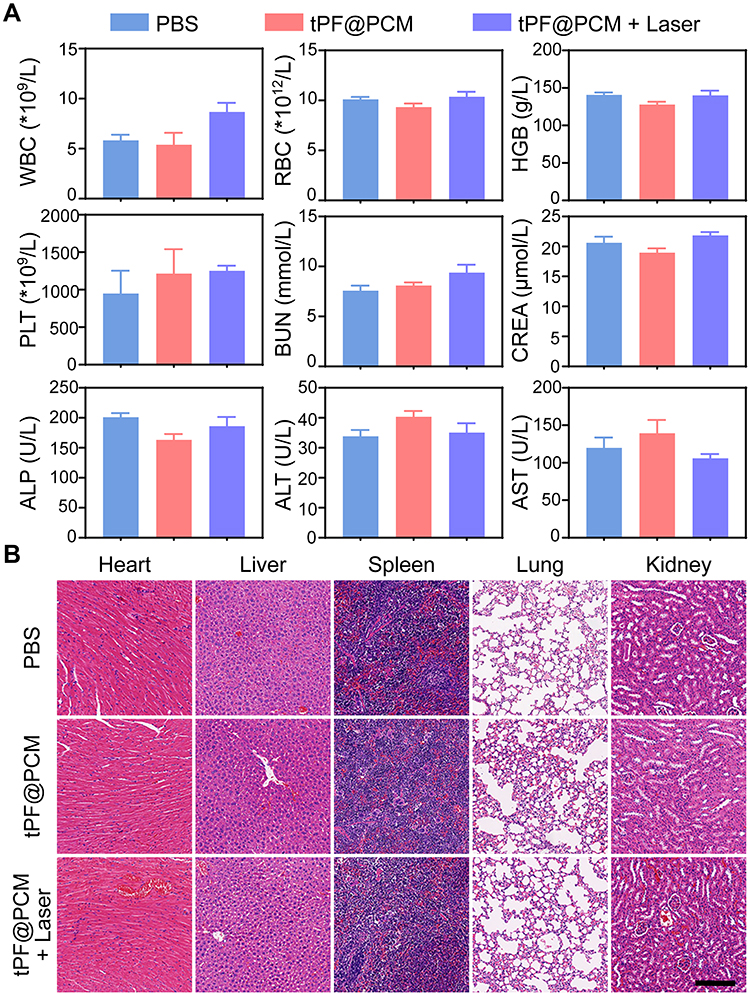 |
Figure 7 In vivo toxicity evaluations of tPF@PCM. (A) Hematological and biochemistry analyses of peripheral blood from different groups of mice at day 15 (n = 3). The results show the mean and standard error of the mean (SEM) of white blood cells (WBC), red blood cells (RBC), hemoglobin (HGB), platelets (PLT), blood urea nitrogen (BUN), creatinine (CREA), alkaline phosphatase (ALP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST). All values were within normal limits. (B) H&E staining of major organs dissected from treated mice at day 15 (n = 3). Scale bar represents 200 μm. |
Conclusion
Cancer cells handle different types of stress by activating self-protection mechanisms, such as UPR, to maintain cellular homeostasis. This is currently considered a major challenge in the treatment of refractory cancers. In this study, we developed a combination therapy using a recently developed nanocatalytic approach with a UPR modulator to significantly exacerbate ER stress and induce apoptosis in cancer cells. Multiple experiments demonstrated that our theranostic approach combining tPF@PCM and laser irradiation was successfully achieved, and both in vitro and in vivo results validated the favorable synergistic antitumor efficacy without obvious side effects. Further studies into ER stress-related cell death modalities (apoptosis, autophagy, and necrosis) will improve our understanding of its underlying antitumor mechanism. Finally, we expect that the modulation of UPR-associated factors could have a considerable impact on the treatment of cancers in the foreseeable future.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81901747, 81571729), National Key Research and Development Program of China (2017YFE0124400, 2020YFA0909000), Shanghai Science and Technology Commission Research Project (18ZR1423200), Innovation Research Plan supported by Shanghai Municipal Education Commission (2019-01-07-00-02-E00064), Med-Engineering Crossing Foundation from Shanghai Jiao Tong University (YG2017ZD05), Basic Oncology Research Program from Bethune Charitable Foundation (BCF-NH-ZL-20201119-024), and the Incubating Program for Clinical Research and Innovation of Renji Hospital (PYXJS16-008, PYIII20-07). The authors thank Shuping Wang (Department of Pharmacy, Renji Hospital) for the assistance with HPLC analyses. We would also like to thank Editage for English language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21(2):71–88.
2. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335.
3. Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22(16):622–626.
4. Siwecka N, Rozpędek W, Pytel D, et al. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int J Mol Sci. 2019;20(18):4354.
5. Ashrafizadeh M, Mohammadinejad R, Tavakol S, Ahmadi Z, Roomiani S, Katebi M. Autophagy, anoikis, ferroptosis, necroptosis, and endoplasmic reticulum stress: potential applications in melanoma therapy. J Cell Physiol. 2019;234(11):19471–19479.
6. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: from oncogenesis to chemoresistance. Biol Cell. 2019;111(1):1–17.
7. Khan AA, Allemailem KS, Almatroudi A, et al. Endoplasmic reticulum stress provocation by different nanoparticles: an innovative approach to manage the cancer and other common diseases. Molecules. 2020;25(22):5336.
8. Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–748.
9. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med. 2013;45(10):e45–e45.
10. Yang B, Shi J. Ascorbate tumor chemotherapy by an iron-engineered nanomedicine-catalyzed tumor-specific pro-oxidation. J Am Chem Soc. 2020;142(52):21775–21785.
11. Cai Y, Cao C, He X, et al. Enhanced magnetic resonance imaging and staining of cancer cells using ferrimagnetic H-ferritin nanoparticles with increasing core size. Int J Nanomed. 2015;10:2619–2634.
12. Chen Z, Yin JJ, Zhou YT, et al. Dual enzyme-like activities of iron oxide nanoparticles and their implication for diminishing cytotoxicity. ACS Nano. 2012;6(5):4001–4012.
13. Qian X, Zhang J, Gu Z, Chen Y. Nanocatalysts-augmented Fenton chemical reaction for nanocatalytic tumor therapy. Biomaterials. 2019;211:1–13.
14. Yang Z, Luo Y, Hu Y, et al. Photothermo-promoted nanocatalysis combined with H2S-mediated respiration inhibition for efficient cancer therapy. Adv Funct Mater. 2021;31(8):2007991.
15. Hwang J, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci. 2018;43(8):593–605.
16. Harris IS, Endress JE, Coloff JL, et al. Deubiquitinases maintain protein homeostasis and survival of cancer cells upon glutathione depletion. Cell Metab. 2019;29(5):1166–1181.
17. D’Arcy P, Linder S. Molecular pathways: translational potential of deubiquitinases as drug targets. Clin Cancer Res. 2014;20(15):3908–3914.
18. Lee JG, Baek K, Soetandyo N, Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun. 2013;4:1568.
19. Barata PC, Rini BI. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin. 2017;67(6):507–524.
20. Posadas EM, Limvorasak S, Figlin RA. Targeted therapies for renal cell carcinoma. Nat Rev Nephrol. 2017;13(8):496–511.
21. Xu W, Atkins MB, McDermott DF. Checkpoint inhibitor immunotherapy in kidney cancer. Nat Rev Urol. 2020;17(3):137–150.
22. Orlowski RZ. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999;6(4):303–313.
23. Zhang S, Li Q, Yang N, et al. Phase‐change materials based nanoparticles for controlled hypoxia modulation and enhanced phototherapy. Adv Funct Mater. 2019;29(49):1906805.
24. Li S, Shang L, Xu B, et al. A nanozyme with photo-enhanced dual enzyme-like activities for deep pancreatic cancer therapy. Angew Chem Int Ed Engl. 2019;58(36):12624–12631.
25. Li K, Liu B. Polymer-encapsulated organic nanoparticles for fluorescence and photoacoustic imaging. Chem Soc Rev. 2014;43(18):6570–6597.
26. Morita Y, Sakurai R, Wakimoto T, et al. tLyP-1-conjugated core-shell nanoparticles, Fe3O4NPs@mSiO2, for tumor-targeted drug delivery. Appl Surf Sci. 2019;474:17–24.
27. Qiu J, Huo D, Xia Y. Phase-change materials for controlled release and related applications. Adv Mater. 2020;32(25):e2000660.
28. Chu M, Shao Y, Peng J, et al. Near-infrared laser light mediated cancer therapy by photothermal effect of Fe3O4 magnetic nanoparticles. Biomaterials. 2013;34(16):4078–4088.
29. Xu C, Bing W, Wang F, Ren J, Qu X. Versatile dual photoresponsive system for precise control of chemical reactions. ACS Nano. 2017;11(8):7770–7780.
30. Caunt M, Mak J, Liang W-C, et al. Blocking Neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. 2008;13(4):331–342.
31. Tse BWC, Volpert M, Ratther E, et al. Neuropilin-1 is upregulated in the adaptive response of prostate tumors to androgen-targeted therapies and is prognostic of metastatic progression and patient mortality. Oncogene. 2017;36(24):3417–3427.
32. Fakhari M, Pullirsch D, Abraham D, et al. Selective upregulation of vascular endothelial growth factor receptors neuropilin-1 and −2 in human neuroblastoma. Cancer. 2002;94(1):258–263.
33. Cao Y, Wang L, Nandy D, et al. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008;68(21):8667–8672.
34. Chen R, Huo L, Shi X, et al. Endoplasmic reticulum stress induced by zinc oxide nanoparticles is an earlier biomarker for nanotoxicological evaluation. ACS Nano. 2014;8(3):2562–2574.
35. Yang Q, Wang Y, Yang Q, et al. Cuprous oxide nanoparticles trigger ER stress-induced apoptosis by regulating copper trafficking and overcoming resistance to sunitinib therapy in renal cancer. Biomaterials. 2017;146:72–85.
36. Li W, Yang J, Luo L, et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat Commun. 2019;10(1):3349.
37. Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7(8):766–772.
38. Navarro S, Ventura S. Fluorescent dye ProteoStat to detect and discriminate intracellular amyloid-like aggregates in Escherichia coli. Biotechnol J. 2014;9(10):1259–1266.
39. Kitakaze K, Taniuchi S, Kawano E, et al. Cell-based HTS identifies a chemical chaperone for preventing ER protein aggregation and proteotoxicity. eLife. 2019;8:e43302.
40. Kennedy D, Samali A, Jäger R. Methods for studying ER stress and UPR markers in human cells. Methods Mol Biol. 2015;1292:3–18.
41. Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–3077.
42. Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403(6765):98–103.
43. Lazovic J, Guo L, Nakashima J, et al. Nitroxoline induces apoptosis and slows glioma growth in vivo. Neuro Oncol. 2015;17(1):53–62.
44. Ashrafizadeh M, Tavakol S, Ahmadi Z, Roomiani S, Mohammadinejad R, Samarghandian S. Therapeutic effects of kaempferol affecting autophagy and endoplasmic reticulum stress. Phytother Res. 2020;34(5):911–923.
45. Xie S, Sun W, Zhang C, et al. Metabolic control by heat stress determining cell fate to ferroptosis for effective cancer therapy. ACS Nano. 2021;15(4):7179–7194.
46. Tsai MF, Lo YL, Huang YC, et al. Multi-stimuli-responsive dox released from magnetosome for tumor synergistic theranostics. Int J Nanomed. 2020;15:8623–8639.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.