")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
DR10627, a Novel Dual Glucagon‑like Peptide‑1 and Gastric Inhibitory Polypeptide Receptor Agonist for the Treatment of Obesity and Type 2 Diabetes Mellitus
Authors Shao Y , Chen Y , Zhu M , Liu Y, Fang C, Wang M, Sun P, Fu W, Huang J , Sheng S, Huang Y
Received 5 January 2024
Accepted for publication 20 March 2024
Published 6 April 2024 Volume 2024:17 Pages 1563—1573
DOI https://doi.org/10.2147/DMSO.S457830
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Yujian Shao,1,2 Yonglu Chen,2 Mingyue Zhu,3 Yuanyuan Liu,2 Chen Fang,3 Minjun Wang,2 Peng Sun,3 Weiling Fu,2 Jing Huang,3 Shimei Sheng,2 Yanshan Huang2
1Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China; 2Department of Innovative Drug Discovery and Development, Zhejiang Doer Biologics Co., Ltd, Hangzhou, Zhejiang, People’s Republic of China; 3First Research Institute, Zhejiang Heze Pharmaceutical Technology Co., Ltd, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Yanshan Huang, Department of Innovative Drug Discovery and Development, Zhejiang Doer Biologics Co., Ltd, Hangzhou, Zhejiang, People’s Republic of China, Email [email protected]
Introduction: Diabetes and obesity are momentous risk factors threatening people’s lives and health. Currently available incretin analogue glucagon-like peptide 1 (GLP-1) possesses huge hypoglycemic effect with the unsatisfactory effect of weight loss. Co-agonists targeting GLP-1R plus glucagon receptor (GCGR) or gastric inhibitory polypeptide receptor (GIPR) show synergistic benefits in glycaemic control and weight loss. Here, we describe a novel dual GIP and GLP-1 receptor agonist, DR10627, and performed a preclinical assessment of it.
Methods: The agonistic ability of DR10627 was indirectly assessed by inducing cAMP accumulation in Chinese hamster ovary (CHO) cells transfected with GLP-1R or GIPR in vitro. The plasma pharmacokinetics of DR10627 were analysed in cynomolgus monkeys. The OGTTs were performed in Sprague‑Dawley (SD) rats. The glucose lowering effects were evaluated by repeated administration of DR10627 in diabetic (db/db) mice for 4 weeks. The effects of anti-obesity and improving metabolism of DR10627 were evaluated by repeated administration of DR10627 in diet-induced obesity (DIO) mice for 57 days.
Results: DR10627 had the capacity to activate both GLP-1R and GIPR in vitro. The terminal half-life of DR10627 was found to be approximately 4.19– 5.8 h in cynomolgus monkeys. DR10627 had a great improvement in oral glucose tolerance in SD rats. Moreover, DR10627 had a potent glucose-lowering effect in db/db mice, and the hypoglycemic effect of 18 nmol/kg DR10627 was better than that of 50 nmol/kg liraglutide. In addition, 10 and 30 nmol/kg DR10627 possessed the ability of potentiating the weight-loss, lipid-lowing efficacy and improving metabolism to a greater extent than 80 nmol/kg liraglutide.
Conclusion: Preclinical assessment demonstrated that administration of DR10627 resulted in glucose lowering in SD rats and db/db mice, and substantial body weight reduction and metabolism improvement in DIO mice. DR10627 is a promising agent deserving further investigation for the treatment of type 2 diabetes and obesity.
Keywords: glucagon-like peptide-1, gastric inhibitory polypeptide receptor, obesity, type 2 diabetes, inflammation
Introduction
As society has developed in recent decades, people’s exercise and dietary patterns are constantly changing, leading to an increase in overweight or obesity rates.1 Overweight [body mass index (BMI) ≥ 25 kg/m2] and obesity (BMI ≥ 30 kg/m2) are driving global morbidity and mortality, posing a substantial risk for type 2 diabetes (T2D), denoting a huge worldwide health crisis.2 Moreover, T2D increases the risk of cardiac complications. Achieving weight loss (≥5% but <10%), to a large extent, contributes to minimizing diabetes-associated complications.3
Bariatric surgery is one of the effective methods to attain long-lasting weight loss, which requires surgeons and facilities and may cause debilitating adverse effects. Pharmacotherapies based on glucagon-like peptide 1 (GLP-1) have emerged and achieved continuing clinical successes in the treatment of T2D and obesity over the past decade, profiting from the function of reducing food intake and weight via physiological satiety pathways. Among diabetes treatment options, GLP-1 analog treatment can be used safely in diabetics with heart failure and GLP-1 agonists are also safe medications in treatment of diabetics with atrial fibrillation.4 For instance, the STEP-5 trial uncovered the efficacy of semaglutide (Novo Nordisk) for weight loss, in which 2.4 mg dose of semaglutide achieved weight reduction by 13% (placebo-subtracted) over two years in adults with overweight (with at least one weight-related comorbidity) or obesity, without diabetes.5 However, this magnitude of weight loss is insufficient for those with higher grades (BMI ≥ 35 kg/m2).
According to the safety profile of the STEP program trials, gastrointestinal disorders were the most common adverse events with semaglutide, typically transient, of mild-to-moderate severity, occurring during dose escalation, and such events led to permanent treatment discontinuation.5 With enhanced efficacy and enlarged therapeutic benefits, co-agonists appealed to researchers and drug companies, which possess activation activity of GLP-1 receptor plus an additional one such as gastric inhibitory polypeptide receptor (GIPR) or glucagon receptor (GCGR).6 GIP is an insulin stimulating hormone that is secreted from K cells in the upper small intestines and its postprandial level reached four times that of GLP-1 under normal physiological conditions,7,8 stimulating insulin under hyperglycaemic conditions in a glycaemic-dependent and dose-dependent manner.9–13 Additionally, GIP may promote storage of dietary lipids by boosting the healthy expansion of white adipose tissue (WAT) and reducing food intake and body weight through its actions in the central nervous system (CNS).14,15 GIP agonists are beneficial in treating both obesity and T2D, and clinical studies have shown the potential of GIP receptor agonists to be used in combination with other glucose-lowering peptides in the treatment of obese type 2 diabetes.16 Current research data confirmed the synergistic benefits of GIP on improving lipid and glucose metabolism when paired with the anorexigenic mechanism of GLP-1.17,18 The SURPASS trials revealed that tirzepatide (Lilly), a GLP1 and GIP receptor co-agonist, is effective in improving glycaemia and reducing weight in T2D, superior to semaglutide at the 1 mg weekly dose that is licensed for T2D.19 Besides, the SURMOUNT-4 trial demonstrated that the overall mean weight reduction from week 0 to 88 was 25.3% for tirzepatide and 9.9% for placebo.20
Here, we characterize a novel dual GIP and GLP-1 receptor agonist DR10627, also known as HZ010. Preclinical assessment demonstrated that administration of DR10627 resulted in glucose lowering in Sprague‑Dawley (SD) rats and diabetic (db/db) mice, and substantial body weight reduction and metabolism improvement in diet-induced obesity (DIO) mice. Thus, DR10627 is promising in the treatment of diabetes and obesity and is currently undergoing Phase I clinical trials.
Materials and Methods
Human GLP-1 Receptor and GIP Receptor Agonistic Activity Assay in vitro
To test the ability of DR10627 to activate GLP-1R and GIPR, the cAMP accumulation has been measured using a cell-based luciferase reporter gene assay based on CHO-K1 cell cotransfected with cDNA for each individual receptor and a luciferase reporter gene construct fused with a cAMP response element. CHO-K1 was obtained from ATCC (Manassas, VA, USA). Briefly, the density of the cotransfected cells after digestion and resuspension using slab culture medium (DMEM/F-12 + 0.5%FBS) was adjusted to a slab density of 2.5 × 105 cells/mL, and then added the cells to the all-white 96-well luminescent plate and incubate at 37°C and 5% CO2 for 18–20 hours overnight. Subsequently, serial dilutions of DR10627 (DMEM/F-12 + 0.5%FBS + 2%BSA) were mixed with the cells in the 96-well plates. Following incubation at 37°C and 5% CO2 for further 6 hours, the luciferase activity of the CHO-K1 cells was measured using a Bright-Glo™ Luciferase Assay System purchased from Promega Biotech Co., Ltd (Beijing, China), according to the manufacturer’s instructions.
Pharmacokinetics Assay Studies in Cynomolgus Monkeys
Cynomolgus monkeys, half male and half female, were assigned into 3 groups (n=6/group). All animals were injected s.c. with a single dose of DR10627 (1 nmol/kg, 5 nmol/kg or 26 nmol/kg), and blood samples were collected at 0 (pre-dose), 0.5, 1, 2, 4, 8, 12, 16, 20, 24, 30, 38 and 48 h after administration. The serum concentrations of DR10627 were measured by LC-MS/MS. In brief, chromatographic separation was done with a C18 column (4.6 × 50 mm). Mobile phase consisted of solvent A (0.5% formic acid) and solvent B (acetonitrile consisted of 0.5% formic acid), with a gradient elution (0–0.1 min, 35%; 0.1–1.5 min, 35–90%; 1.5–3.2 min, 95% of solvent B) and the flow rate was 0.8 mL/min. The column temperature was maintained at 60°C. Mass spectral analysis was performed on a QTRAP 6500 mass spectrometer (AB/Sciex) equipped with an electrospray ionization (ESI) source and operated in positive ion mode. A calibration solution was used to calibrate the instrument. The curtain gas (CUR), nebulizer gas (GS1) and turbo gas (GS2) were set at 45 psi, 70 psi and 70 psi, respectively. The electrospray voltage was 6 kV, and the turbo ion spray source temperature was 400°C. Nitrogen was employed as the collision gas. The mass spectrometer was operated in the MRM full scan mode with a 1115.3 → 396 m/z range for DR10627. Based on the biological analysis data, the pharmacokinetic parameters of DR10627 were calculated with Phoenix WinNonlin 8.1 using the method of non-atrioventricular model analysis.
Effect of a Single DR10627 Administration on Oral Glucose Tolerance in SD Rats
Male SD rats were randomly assigned into 5 groups (body weight, 247~325 g; n=12/group) and each group of the rats fasted overnight were dosed orally with D-glucose solution at a dose of 2 g glucose/kg of animal body weight following s.c. injection of the respective doses of vehicle, liraglutide (50 nmol/kg) or DR10627 (1 nmol/kg, 4 nmol/kg, 12 nmol/kg). The blood samples were collected from the tail vein at point 0 (prior to glucose administration), 15, 30, 60, 90, 120 min post-glucose challenge using ONETOUCH®UltraEasyTM blood glucometer (Johnson & Johnson China Ltd, China).
Glucose-Lowering Effects of Repeated DR10627 Administrations in db/db Mice
Male db/db mice (7 weeks old) were randomly assigned into 5 groups (body weight, 29.7~36.5 g; n=10/group), including a model control group (group 2), a liraglutide group (50 nmol/kg, group 3) and 3 doses groups of DR10627 (2 nmol/kg, 6 nmol/kg, 18 nmol/kg, group 4–6). Another 10 male wild-type mice were used as a normal control group (group 1).
Mice in group 2–6 were treated once daily with dosing at a volume of 5 mL/kg subcutaneously for 4 weeks. Blood glucose was measured before administration to 24 h after administration on D1 and D28. Random blood glucose was measured once at about 9:30~10:30 a.m. on D6, D13 and D20. Six-hour fasting blood glucose was measured after the 6-h fasting at about 16:30~17:30 p.m. on D7, D14, D20 and D25, the fasting started about 1 h after the administration.
Effects of Repeated DR10627 Administrations in DIO Mice
Male C57BL/6 mice (7 weeks old) were fed a high-fat diet (60% kcal% fat, Research Diets, D12492i) for 11 weeks. Fifty mice (body weight, 42.2~48.7 g) were screened and assigned randomly into five groups (n = 10/group): a model control group (group 2), a liraglutide group (80 nmol/kg, group 3), 3 doses groups of DR10627 (3 nmol/kg, 10 nmol/kg, 30 nmol/kg, group 4–6). Another 10 male C57BL/6 mice fed an SPF-grade diet were used as a normal control group (group 1). Mice in group 2–6 were treated once daily with a dosing volume of 5 mL/kg subcutaneously for 57 days.
Body weight and food take of all groups throughout the study were measured and recorded after administration. Body weight changes and cumulative food intake were calculated. Abdominal fats including subcutaneous fat, perirenal fat, epididymal fat, mesenteric fat and subcutaneous brown fat of scapula were harvested after the final blood sample collection, blotted with filter paper, and weighed. Blood samples were collected on D0 (pre-dose), D28, D41 and D48 from the tail veins of mice fasted overnight for further biochemical analyses including total cholesterol (TC), triglyceride (TG) and low-density lipoprotein (LDL).
Statistical Analysis
Statistical analysis was performed using one-way or two-way analysis of variance (ANOVA) followed by LSD, Kruskal–Wallis or Mann–Whitney test. Results are presented as the mean ± SD. Differences with p values less than 0.05 were considered statistically significant and are identified with an asterisk.
Ethics Approval
Animal experiments in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of JOINN Laboratories (China) Co., Ltd. or InnoStar Bio-tech Nantong (China) Co., Ltd. All experiments were performed according to Guide for the Care and Use of Laboratory Animals (8th ed., 2011) and Regulations on the management of experimental animals (Revised in 2017).
Results
Chemical Structure of DR10627
The peptide DR10627 is a chemically synthesized linear chain consisting of 39 amino acids. N-terminal amino acid sequence of DR10627 (1–29) is based on GLP-1, with mutation sites related to GIP activity introduced, and the 10 amino acids in the C-terminal are borrowed from exenatide, known as tryptophan cage, to enhance metabolic stability, and the C-terminus is amidated. A palmitoyl group is conjugated to DR10627 via a -γ-glutamyl-linker connected to the Lys10 position. Palmitoylation endows albumin binding, prolonging half-life (Figure 1). The molecular weight of DR10627 is 4457.96 Da.
 |
Figure 1 Amino acid sequence of GLP-1R/GIPR coagonist, DR10627 and the related peptides GLP-1, GIP, exenatide and liraglutide. Differences in amino acids from native GLP-1 and GIP are denoted in red. K* indicates a palmitoyl group conjugated to the ε-N of K10 via a -γ-glutamyl-linker (γE-C16) (A). Structure schematic of DR10627 (B). |
DR10627 is a Dual GLP-1 and GIP Receptor Agonist in vitro
DR10627 was designed to be fully recombinantly expressed as a unimolecular polypeptide with dual GLP-1/GIP receptor agonistic activities. In receptor activation assay using cell lines with recombinantly expressed GIPR or GLP-1R, DR10627 effectively stimulates cAMP accumulation by either receptor in a concentration-dependent manner. The potency (EC50) value of DR10627 was 17.65 pM for GLP-1R, which was approximately 41.93% of the potency (EC50 = 7.40 pM) of native GLP-1. The potency value of DR10627 for GIPR was 0.26 nM, which was approximately 714.83% of the potency (EC40 = 1.88 nM) of native GIP (Figure 2). These results showed DR10627 could activate both GLP-1R and GIPR in vitro.
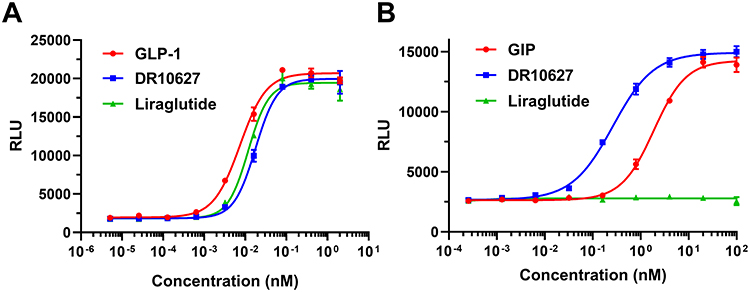 |
Figure 2 Representative concentration–response curves of DR10627, liraglutide, GLP-1 or GIP in cAMP accumulation assays of CHO cell lines expressing human GLP-1 receptors (A) or human GIP receptors (B). Values are presented as the mean (± SD) from duplicate analyses fitted with a 4-parameter logistic fit to determine the EC50 value. |
DR10627 Has an Extended Half‑life in vivo
To explore the concentration–time dynamics of DR10627 in vivo, the pharmacokinetics of DR10627 were assessed in cynomolgus monkeys. After s.c. administration of single doses of 1 nmol/kg, 5 nmol/kg and 26 nmol/kg, the increase of Cmax (the maximal observed plasma concentration) and AUCINF_obs (the area under the plasma concentration curve from zero to infinity) in cynomolgus monkeys was proportional to the dose. The pharmacokinetic parameters for DR10627 in plasma are presented in Table 1. The maximum DR10627 concentration observed occurred within 6–8 h after dosing and the terminal half-life of DR10627 was found to be approximately 4.19–5.8 h (Figure 3), thus supporting a once-daily dosing regimen.
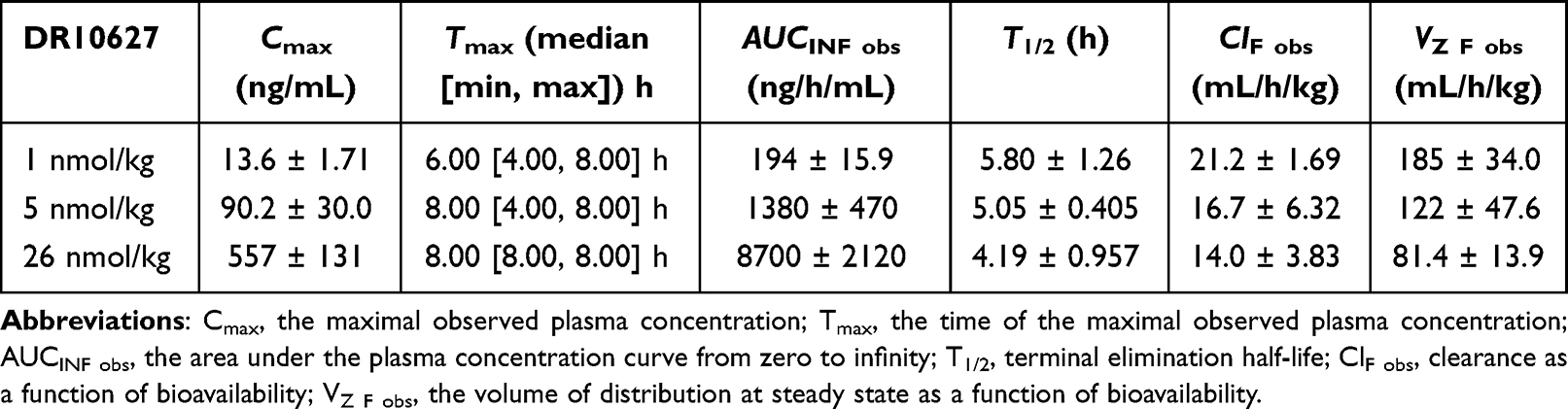 |
Table 1 Pharmacokinetic Parameters in Cynomolgus Monkeys (Mean ± Standard Deviation) |
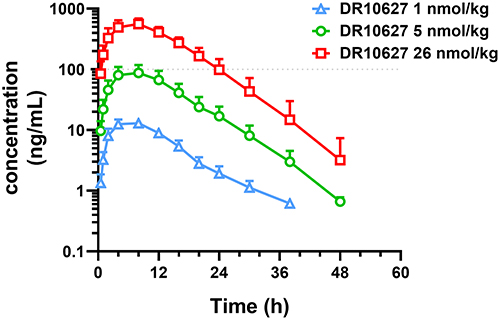 |
Figure 3 Pharmacokinetics of DR10627 in cynomolgus monkeys. Blood samples were collected at the indicated time points after a single s.c. injection of DR10627 (1 nmol/kg, 5 nmol/kg, 26 nmol/kg) in cynomolgus monkeys (n = 6). The serum concentrations of DR10627 were measured by LC-MS/MS. Pharmacokinetic parameters of DR10627 were calculated with Phoenix WinNonlin 8.1 using the method of non-atrioventricular model analysis. Values are presented as mean (± SD). |
DR10627 Improves Oral Glucose Tolerance in SD Rat
To evaluate the effect of DR10627 on glucose tolerance, we performed an OGTT in SD rats. As shown in Figure 4, 4 nmol/kg DR10627 significantly lowered blood glucose level at 30, 60, and 90 min (p < 0.001) and 12 nmol/kg DR10627 significantly lowered blood glucose level at 30, 60, 90 and 120 min (p < 0.001) compared with that achieved with vehicle. Moreover, significant reductions in glucose level were induced by 12 nmol/kg DR10627 compared to 50 nmol/kg liraglutide at 30, 60 min (p < 0.01–0.001) (Figure 4A). There was no difference in glucose-lowering effect between 1 nmol/kg DR10627 and vehicle. Meanwhile, a significant decrease in glucose response, measured by OGTT AUC (0–2 h), was observed in 4 nmol/kg and 12 nmol/kg DR10627 compared with vehicle, and 12 nmol/kg DR10627 AUC (0–2 h) was significantly lower than 50 nmol/kg liraglutide (Figure 4B). Therefore, these results indicated that DR10627 had a great improvement on oral glucose tolerance in SD rat, and 12 nmol/kg DR10627 was more potent than 50 nmol/kg liraglutide.
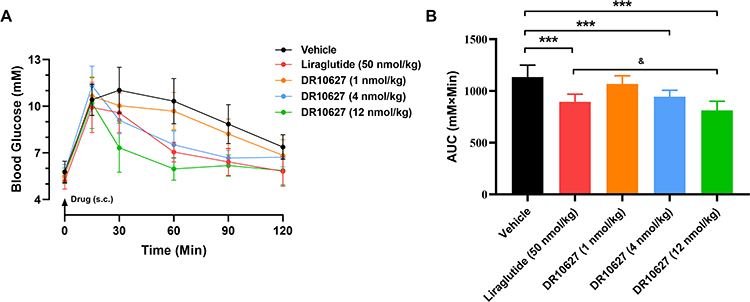 |
Figure 4 Effects of single doses of DR10627 and liraglutide on glucose tolerance in OGTT in SD rats. Glucose levels of rats at 0, 15, 30, 60, 90 and 120 min post-glucose challenge (A) and overall area under the curve (AUC) values (B). Male SD rat fasted overnight were dosed orally with D-glucose solution at a dose of 2 g glucose/kg of animal body weight following subcutaneous injection of the respective doses of vehicle, liraglutide (50 nmol/kg) or DR10627 (1 nmol/kg, 4 nmol/kg, 12 nmol/kg). Blood samples were collected at the indicated time points. Time 0 was immediately prior to glucose challenge. The values represent the mean ± SD. n = 12 mice/group. ***p < 0.001 compared to vehicle; &p < 0.05 compared to liraglutide (50 nmol/kg). |
DR10627 Has a Potent Glucose-Lowering Effect in db/db Mice
To confirm the glucose-lowering effect of DR10627, we explored the effect of DR10627 on glycemic control in type 2 diabetic db/db mice that were treated with either a daily s.c. administration of the respective doses of DR10627, liraglutide or vehicle for 4 weeks. As shown in Figure 5A, blood glucose levels were similar among all groups before administration on D1, and decreased significantly after treatment with DR10627 or liraglutide. Figure 5B shows that before administration on D28, the blood glucose levels treated with 2, 6 and 18 nmol/kg DR10627 or 50 nmol/kg liraglutide were significantly lower than that of vehicle (24.3 ± 2.9, 22.1 ± 4.5, 18.4 ± 5.4, 27.0 ± 3.9 mM vs 33.6 ± 6.4 mM, p < 0.05–0.001). The trend of blood glucose change treated with 2 nmol/kg DR10627 was similar to that of 50 nmol/kg liraglutide. The blood glucose decreased to the lowest level at 2 h after administration of 12 nmol/kg DR10627, significantly lower than that of vehicle (24.1 ± 2.5 vs 34.5 ± 8.2 mM, p < 0.001), and the hypoglycemic effect lasted for 24 h. From 0.5 h to 24 h after treatment, the blood glucose treated with 6 and 18 nmol/kg DR10627 was significantly lower than that of vehicle (p < 0.05–0.001), and the hypoglycemic effect was dose-dependent. Moreover, the blood glucose treated with 18 nmol/kg DR10627 was significantly lower than that of 50 nmol/kg liraglutide (p < 0.05–0.001) at 0.5h, 1h, 12h and 24h after treatment.
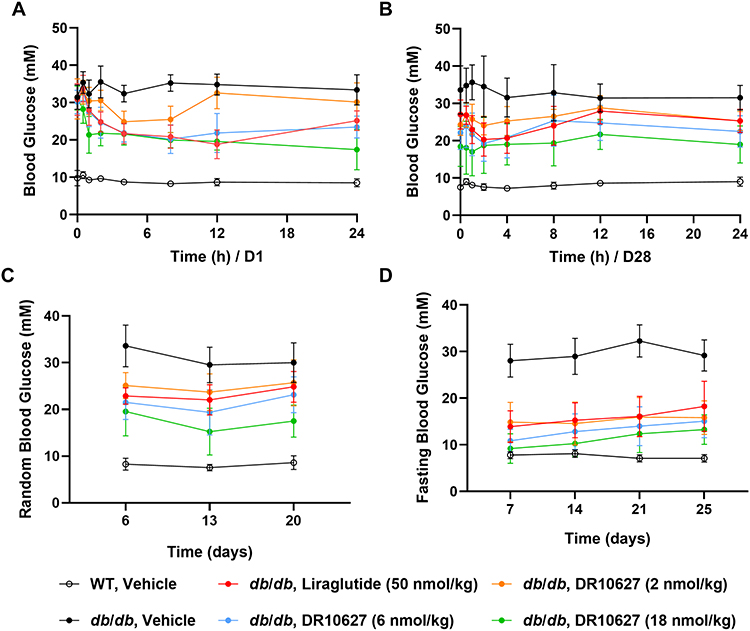 |
Figure 5 Effects of repeated-dose treatment with DR10627 and liraglutide on glucose tolerance in db/db mice. Male db/db mice were treated with either a daily s.c. administration the respective doses of vehicle, liraglutide (50 nmol/kg) or DR10627 (2 nmol/kg, 6 nmol/kg, 18 nmol/kg) for 4 weeks. Blood glucose was measured before administration to 24 h after administration on D1 (A) and D28 (B), random blood glucose was measured on D6, D13 and D20 (C). Fasting blood glucose was measured on D7, D14, D20 and D25 (D). The values represent the mean ± SD. n = 10 mice/group. |
Besides, the random blood glucose (RBG) and fasting blood glucose (FBG) levels were measured during the study. The RBG level of the model control group was significantly higher than that of the normal control group on D6, D13 and D20, while DR10627 significantly lowered RBG level in a dose-dependent way. In addition, 18 nmol/kg DR10627 was more potent than 50 nmol/kg liraglutide in decreasing the RBG level (Figure 5C). Similar to the RGB level, DR10627 significantly lowered the FBG level in a dose-dependent way and 18 nmol/kg DR10627 exhibited enhanced effect than 50 nmol/kg liraglutide on decreasing the FBG level (Figure 5D). These results demonstrated that DR10627 had a potent glucose-lowering effect in db/db mice and the hypoglycemic effect of 18 nmol/kg DR10627 was better than that of 50 nmol/kg liraglutide.
DR10627 Reduces Body Weight and Improves Metabolism in DIO Mice
To characterize the metabolic effects of DR10627, we conducted daily doses of DR10627 or liraglutide to DIO mice for 57 days. DR10627 and liraglutide in DIO mice significantly reduced the body weight from D5 to D57 compared to the vehicle. The mean body weight of the vehicle-treated animals increased by 9 ± 3.81% on D57, whereas administrations of 3, 10 and 30 nmol/kg DR10627 led to more reductions in the mean body weight than that of 80 nmol/kg liraglutide (15 ± 6.35%, 20 ± 7.75%, 24 ± 4.24% vs 8 ± 4.87%, p < 0.05–0.001) (Figure 6A). In addition to body weight, all dose of DR10627 and liraglutide also lead to a reduction in food intake. The cumulative total food intake of DIO mice received repeated treatment with 3, 10 and 30 nmol/kg DR10627 was significantly lower than that of the vehicle treated mice (32.96 ± 3.13, 29.73 ± 2.44, 30.63 ± 3.71 vs 42.12 ± 1.97, p < 0.001). Additionally, 10 and 30 nmol/kg DR10627 exhibited enhanced effect on reduction of cumulative total food intake compared with 80 nmol/kg liraglutide (29.73 ± 2.44, 30.63 ± 3.71 vs 34.05 ± 1.97, p < 0.05–0.001) over the course of study (Figure 6B).
 |
Figure 6 Effects of 11 weeks of repeated-dose treatment with DR10627 on body weight change (A), cumulative food intake (B), abdominal fat to body weight ratio (C), cholesterol (D), triglyceride (E) and LDLC (F) in male DIO mice. Male DIO mice were treated with either a daily s.c. administration the respective doses of vehicle, liraglutide (80 nmol/kg) or DR10627 (3 nmol/kg, 10 nmol/kg, 30 nmol/kg) for 11 weeks. Abdominal brown fat was harvested after the final blood sample collection. Blood samples were collected on D0 (pre-dose), D28, D41 and D48 from the tail veins of mice fasted overnight. The values represent the mean ± SD. n = 10 mice/group. *p < 0.05, **p < 0.01, ***p < 0.001 compared to vehicle; &p < 0.05; &&p < 0.01 compared to liraglutide (80 nmol/kg). |
Moreover, we measured body fat accumulation biochemical parameters including cholesterol, triglyceride and LDLC. As shown in Figure 6C, 3, 10 and 30 nmol/kg DR10627 significantly reduced abdominal fat-to-body ratio in a dose-dependent manner compared to vehicle (3.863 ± 1.344, 3.473 ± 0.302, 2.343 ± 0.490 vs 6.838 ± 1.169, p < 0.001). Moreover, 10 and 30 nmol/kg DR10627 exhibited enhanced effect on reduction of abdominal fat-to-body ratio compared with 80 nmol/kg liraglutide (3.473 ± 0.302, 2.343 ± 0.490 vs 4.507 ± 1.032, p < 0.05–0.001). Meantime, all dose of DR10627 showed the effect of reducing cholesterol, triglyceride and LDLC, which was similar to or slightly stronger than 80 nmol/kg liraglutide (Figure 6D–F). Since the liver is the central pivot of fat metabolism, we evaluated the liver lesions by liver weighing and physiological observation. Results showed that DR10627 significantly reduced the liver weight (Supplementary Figure 1) and improved the liver steatosis (Supplementary Table 1 and Supplementary Figure 2) of the DIO mice in a dose-dependent way more than liraglutide. Collectively, these results demonstrated that 10 and 30 nmol/kg DR10627 possessed the ability to potentiate the weight-loss, lipid-lowering efficacy and improve metabolism to a greater extent than 80 nmol/kg liraglutide.
Discussion
Benefiting from the positive effects of GLP-1 on insulin secretion and suppression of glucagon secretion as well as β-cell survival, GLP-1 possesses a potent hypoglycemic effect on the treatment of T2D.21–23 Besides, GLP-1R is expressed in the brain, where GLP-1 takes the effect of slowing gastric emptying and intestinal transit as well as promoting satiety by activation of GLP-1Rs at central and peripheral enteric neurons. Hence, pharmacotherapies based on GLP-1 on treatment of obesity come into being.24,25 In 2014, liraglutide (marketed as Saxenda for obesity) as the first GLP-1RA was approved by the FDA for managing obesity, without concurrent diabetes. The SCALE trials demonstrated that 2-year liraglutide-receiving contributed to 5–10% on weight loss in non-diabetic participants.26 Nevertheless, any positive effects on weight loss can be eliminated by drug discontinuation due to gastrointestinal side-effects.27–29
The combination of GLP-1 and GIP seems to be a promising solution towards enhancing weight loss efficacy while attenuating GLP-1RA-induced adverse events theoretically. Not only is GIP able to strengthen glucose-stimulated insulin secretion working with GLP-1 but it can also act in the CNS to lower food intake and reduce body weight as well as attenuate GLP-1RA-induced adverse events such as nausea and emesis when combined with GLP-1.30,31
To apply the theory to practice, we designed a GLP-1R and GIPR co-agonist, DR10627, the sequence of which is based on GLP-1 with several mutation sites related to GIP activity substituted (Figure 1), possessing activation potency for both GLP-1R and GIPR with a bias towards GLP-1R activation in vitro (Figure 2). Of note, there are no unnatural amino acids in the sequence of DR10627, considering that introducing unnatural amino acids may increase the risk of immunogenicity increasing, for instance, taspoglutide, which contains Aib (a-amino isobutyric acid) in the sequence, caused 49% rate of antitaspoglutide antibodies detection in its Phase 3 trial.32
The half-life of GLP-1 and GIP in the circulation is approximately a few minutes on account of fast renal clearance and rapid enzymatic inactivation such as dipeptidyl peptidase-4 (DPP-4).33 Many strategies to prolong half-life have emerged, such as nonnatural amino acids substitution, PEG modification and FC fusion.34 For DR10627, Tyr at position 10 of which is substituted by Lys compared to GLP-1 or GIP in order to crosslink a palmitoyl group via a -γ-glutamyl-linker (Figure 1), possesses enhanced activity and prolonged half-life (data not shown). Table 1 shows that the terminal half-life of DR10627 in cynomolgus monkeys was 4.19–5.8 h (Figure 3), which is suitable for once-daily dosing. Of note, tirzepatide, which is also a GLP-1 and GIP receptor co-agonist, has shown great success in the treatment of T2D and has huge potential in the treatment of obesity. Different from DR10627, tirzepatide is a 39 amino acid linear peptide, conjugated to a C20 fatty diacid moiety via a linker connected to the lysine,20 containing two non-coded amino acid residues (Aib) at positions 2 and 13, providing a once-weekly dosing regimen in humans (Supplementary Figure 3).35 The success of tirzepatide demonstrates the therapeutic value of the GLP-1 and GIP co-agonist, and we look forward to the exciting results of DR10627 in future clinical trials.
The OGTT in SD rat showed DR10627 had a great improvement on oral glucose tolerance in a dose-dependent way, especially 12 nmol/kg DR10627 was more potent than 50 nmol/kg liraglutide, suggesting that GLP-1 and GIP synergistically exert hypoglycemic effects (Figure 4). This synergy effect was further verified by administration of respective doses of DR10627, liraglutide or vehicle in type 2 diabetic db/db mice. Blood glucose levels were improved by 4-week administration of DR10627 and liraglutide, including RBG and FBG, and 18 nmol/kg DR10627 was also superior to 50 nmol/kg liraglutide (Figure 5). These results showed that the GLP-1R and GIPR co-agonist DR10627 had greater potential than GLP-1RA in treatment of T2D.
The study in DIO mice showed administration of 10 nmol/kg and 30 nmol/kg DR10627 for 57 days exhibited enhanced effect on weight loss and reduction of food intake better than 80 nmol/kg liraglutide (Figure 6A and B). The underlying mechanism is that GIP action in the CNS may enhance the GLP-1-mediated metabolic effect. In addition, 10 nmol/kg and 30 nmol/kg DR10627 also induced greater lipid lowering, including reduction in abdominal fat mass compared to 80 nmol/kg liraglutide. Furthermore, DR10627 treatment decreased TC, TG and LDL during the course of the study (Figure 6). The curative effect of DR10627 is probably in association with GIP’s capacity to target WAT to improve lipid uptake and storage as well as energy metabolism. Moreover, non-alcoholic fatty liver disease (NAFLD) is closely related to T2D, and the liver is an important regulatory organ of glucose and lipid metabolism, playing a key role in maintaining the homeostasis of blood sugar and blood lipids. Our results demonstrated that DR10627 significantly reduced the liver weight (Supplementary Figure 1) and improved the liver steatosis (Supplementary Table 1 and Supplementary Figure 2) of the DIO mice in a dose-dependent way more than liraglutide, probably due to the DR10627-reduced improvement of lipid metabolism. These results suggest that GLP-1R and GIPR co-agonists can reduce the risk of developing serious liver disease in patients with diabetes.
Chronic tissue inflammation has emerged as a key feature of obesity and T2D and is observed in insulin target tissues, such as adipose tissue, liver, muscle, and pancreatic islets, which results in long-term complications of diabetes, including non-alcoholic fatty liver disease (NAFLD), retinopathy, cardiovascular disease, and nephropathy.36,37 Clinical studies confirmed the beneficial effects of anti-inflammatory treatment in diabetes and associated complications.38 Preclinical studies showed that GLP-1RAs have inhibitory effects on many inflammatory pathways. GLP-1RAs exert their positive effects beyond the reduction of glucose control or weight loss and can be used as first-line drugs in diabetic patients with high cardiovascular risk, probably profiting from their anti-inflammatory properties.39 On the other hand, GIP is associated with the control of inflammation and energy homeostasis.40,41 These research findings make the dual GLP-1 GIP receptor co-agonists amazing candidate for therapy in obesity or T2D and associated complications.
Above all, DR10627, as a GLP-1R and GIPR co-agonist, has excellent potency for glycaemic improvement in db/db mice as well as weight-loss and lipid-lowering in DIO mice, which outbalances liraglutide. Besides, DR10627 is well tolerated in cynomolgus monkeys (data not shown). Based on these findings and safety data from preclinical studies, DR10627 was approved for phase I clinical trials by the National Medical Products Administration (NMPA) in January 2022, and the efficacy of DR10627 in combination with other drugs is also worth exploring.
Conclusion
In conclusion, our study describes a novel dual glucagon‑like peptide‑1 and gastric inhibitory polypeptide receptor agonist, DR10627. Preclinical assessment demonstrated that DR10627 possessed the ability of glucose lowering, substantial body weight reduction and metabolism improvement in rodents, deserving further investigation for the treatment of type 2 diabetes and obesity.
Acknowledgments
We thank Zhejiang Doer Biologics Corporation, Zhejiang Heze Pharmaceutical Technology Co., Ltd and Zhejiang University for their support. We gratefully acknowledge the subjects who were involved in conducting the project.
Disclosure
Mrs Yonglu Chen and Dr Yanshan Huang report patents (CN202010269596.0 and JP2022-561670) issued to Zhejiang Doer; patents (US17916169 and EP217849991) pending to Zhejiang Doer. The authors report no other conflicts of interest in this work.
References
1. World Obesity Federation. World obesity atlas; 2023. Available from: https://data.worldobesity.org/publications/?cat=19.
2. Caballero B. Humans against obesity: who will win? Adv Nutr. 2019;10(suppl_1):S4–S9. doi:10.1093/advances/nmy055
3. Aktas G, Atak Tel BM, Tel R, Balci B. Treatment of type 2 diabetes patients with heart conditions. Expert Rev Endocrinol Metab. 2023;18(3):255–265. doi:10.1080/17446651.2023.2204941
4. Apovian CM, Okemah J, O’Neil PM. Body weight considerations in the management of type 2 diabetes. Adv Ther. 2019;36(1):44–58. doi:10.1007/s12325-018-0824-8
5. Garvey WT, Batterham RL, Bhatta M, et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat Med. 2022;28(10):2083–2091. doi:10.1038/s41591-022-02026-4
6. Clemmensen C, Finan B, Müller TD, DiMarchi RD, Tschöp MH, Hofmann SM. Emerging hormonal-based combination pharmacotherapies for the treatment of metabolic diseases. Nat Rev Endocrinol. 2019;15(2):90–104. doi:10.1038/s41574-018-0118-x
7. Nauck MA, Meier JJ. Incretin hormones: their role in health and disease. Diabetes Obes Metab. 2018;20(Suppl 1):5–21. doi:10.1111/dom.13129
8. Vollmer K, Holst JJ, Baller B, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes. 2008;57(3):678–687. doi:10.2337/db07-1124
9. Meier JJ, Gallwitz B, Siepmann N, et al. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia. 2003;46(6):798–801. doi:10.1007/s00125-003-1103-y
10. Christensen M, Calanna S, Sparre-Ulrich AH, et al. Glucose-dependent insulinotropic polypeptide augments glucagon responses to hypoglycemia in type 1 diabetes. Diabetes. 2015;64(1):72–78. doi:10.2337/db14-0440
11. Christensen M, Vedtofte L, Holst JJ, Vilsbøll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes. 2011;60(12):3103–3109. doi:10.2337/db11-0979
12. Christensen MB, Calanna S, Holst JJ, Vilsbøll T, Knop FK. Glucose-dependent insulinotropic polypeptide: blood glucose stabilizing effects in patients with type 2 diabetes. J Clin Endocrinol Metab. 2014;99(3):E418–26. doi:10.1210/jc.2013-3644
13. Pederson RA, Brown JC. Interaction of gastric inhibitory polypeptide, glucose, and arginine on insulin and glucagon secretion from the perfused rat pancreas. Endocrinology. 1978;103(2):610–615. doi:10.1210/endo-103-2-610
14. Rudovich N, Kaiser S, Engeli S, et al. GIP receptor mRNA expression in different fat tissue depots in postmenopausal non-diabetic women. Regul Pept. 2007;142(3):138–145. doi:10.1016/j.regpep.2007.02.006
15. DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010;53(7):1270–1287. doi:10.1007/s00125-010-1684-1
16. Bailey CJ. GIP analogues and the treatment of obesity-diabetes. Peptides. 2020;125:170202. doi:10.1016/j.peptides.2019.170202
17. Finan B, Ma T, Ottaway N, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5(209):209ra151. doi:10.1126/scitranslmed.3007218
18. NamKoong C, Kim MS, Jang BT, Lee YH, Cho YM, Choi HJ. Central administration of GLP-1 and GIP decreases feeding in mice. Biochem Biophys Res Commun. 2017;490(2):247–252. doi:10.1016/j.bbrc.2017.06.031
19. Frías JP, Davies MJ, Rosenstock J, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6):503–515. doi:10.1056/nejmoa2107519
20. Aronne LJ, Sattar N, Horn DB, Bays HE, Wharton S, Lin WY. Continued treatment with tirzepatide for maintenance of weight reduction in adults with obesity: the SURMOUNT-4 randomized clinical trial. JAMA. 2024;331(1):38–48. doi:10.1001/jama.2023.24945
21. Madsbad S. Liraglutide Effect and Action in Diabetes (LEAD) trial. Expert Rev Endocrinol Metab. 2009;4(2):119–129. doi:10.1586/17446651.4.2.119
22. Parks M, Rosebraugh C. Weighing risks and benefits of liraglutide--The FDA’s review of a new antidiabetic therapy. N Engl J Med. 2010;362(9):774–777. doi:10.1056/nejmp1001578
23. Tuchscherer RM, Thompson AM, Trujillo JM. Semaglutide: the newest once-weekly GLP-1 RA for type 2 diabetes. Ann Pharmacother. 2018;52(12):1224–1232. doi:10.1177/1060028018784583
24. Chao AM, Tronieri JS, Amaro A, Wadden TA. Semaglutide for the treatment of obesity. Trends Cardiovasc Med. 2023;33(3):159–166. doi:10.1016/j.tcm.2021.12.008
25. Iepsen EW, Torekov SS, Holst JJ. Liraglutide for type 2 diabetes and obesity: a 2015 update. Expert Rev Cardiovasc Ther. 2015;13(7):753–767. doi:10.1586/14779072.2015.1054810
26. Davies MJ, Bergenstal R, Bode B, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE diabetes randomized clinical trial. JAMA. 2015;314(7):687–699. doi:10.1001/jama.2015.9676
27. Smits MM, Van Raalte DH. Safety of semaglutide. Front Endocrinol. 2021;12:645563. doi:10.3389/fendo.2021.645563
28. Xie ZY, Yang SS, Deng WS, Li JJ, Chen JS. Efficacy and safety of liraglutide and semaglutide on weight loss in people with obesity or overweight: a systematic review. Clin Epidemiol. 2022;2022(14):1463–1476. doi:10.2147/clep.s391819
29. Consoli A, Formoso G. Potential side effects to GLP-1 agonists: understanding their safety and tolerability. Expert Opin Drug Saf. 2015;14(2):207–218. doi:10.1517/14740338.2015.987122
30. Samms RJ, Coghlan MP, Sloop KW. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol Metab. 2020;31(6):410–421. doi:10.1016/j.tem.2020.02.006
31. Lafferty RA, Flatt PR, Irwin N. GLP-1/GIP analogs: potential impact in the landscape of obesity pharmacotherapy. Expert Opin Pharmacother. 2023;24(5):587–597. doi:10.1080/14656566.2023.2192865
32. Rosenstock J, Balas B, Charbonnel B, et al. The fate of taspoglutide, a weekly GLP-1 receptor agonist, versus twice-daily exenatide for type 2 diabetes: the T-emerge 2 trial. Diabetes Care. 2013;36(3):498–504. doi:10.2337/dc12-0709
33. Scheen AJ. GLP-1 receptor agonists or DPP-4 inhibitors: how to guide the clinician? Ann Endocrinol. 2013;74(5–6):515–522. doi:10.1016/j.ando.2012.06.002
34. Wang W, Wen X, Duan W, et al. DR10601, a novel recombinant long-acting dual glucagon-like peptide-1 and glucagon receptor agonist for the treatment of obesity and type 2 diabetes mellitus. J Endocrinol Invest. 2020;43(5):653–662. doi:10.1007/s40618-019-01153-z
35. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3–14. doi:10.1016/j.molmet.2018.09.009
36. Aktas G. Association between the prognostic nutritional index and chronic microvascular complications in patients with type 2 diabetes mellitus. J Clin Med. 2023;12(18):5952. doi:10.3390/jcm12185952
37. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55(1):31–55. doi:10.1016/j.immuni.2021.12.013
38. Kurtkulagi O. Neutrophil to Lymphocyte ratio is significantly reduced after Sodium glucose cotransporter-2 inhibitor treatment in patients with type 2 diabetes mellitus. Int J Endocrinol. 2022;18(2):86–89. doi:10.22141/2224-0721.18.2.2022.1151
39. Bendotti G, Montefusco L, Lunati ME, et al. The anti-inflammatory and immunological properties of GLP-1 Receptor Agonists. Pharmacol Res. 2022;182:106320. doi:10.1016/j.phrs.2022.106320
40. Mantelmacher FD, Zvibel I, Cohen K, et al. GIP regulates inflammation and body weight by restraining myeloid-cell-derived S100A8/A9. Nat Metab. 2019;1(1):58–69. doi:10.1038/s42255-018-0001-z
41. Heimbürger SM, Bergmann NC, Augustin R, Gasbjerg LS, Christensen MB, Knop FK. Glucose-dependent insulinotropic polypeptide (GIP) and cardiovascular disease. Peptides. 2020;125:170174. doi:10.1016/j.peptides.2019.170174
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.