Back to Journals » Risk Management and Healthcare Policy » Volume 18
Clinical Efficacy and Diagnostic Value of Metagenomic Next-Generation Sequencing (mNGS) in Hospital-Acquired Pneumonia: A Stratified Retrospective Study of Responders and Non-Responders
Authors Zhang B ![]() , Wang J, Li Q, Ge J, Zhang C, Zhou T, Guo H, Yang B, Jiang H
, Wang J, Li Q, Ge J, Zhang C, Zhou T, Guo H, Yang B, Jiang H
Received 27 June 2025
Accepted for publication 19 November 2025
Published 8 December 2025 Volume 2025:18 Pages 3803—3818
DOI https://doi.org/10.2147/RMHP.S550021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Keon-Hyung Lee
Bin Zhang, Jianjun Wang, Qing Li, Jingyi Ge, Chenxi Zhang, Ting Zhou, Haiming Guo, Bo Yang, Hongying Jiang
Department of Respiratory Rehabilitation Center, Beijing Rehabilitation Hospital of Capital Medical University, Beijing, People’s Republic of China
Correspondence: Hongying Jiang, Email [email protected]
Introduction: Hospital-acquired pneumonia (HAP) remains a major challenge in clinical practice, particularly due to polymicrobial infections and antimicrobial resistance. Traditional diagnostic methods, such as culture and PCR, are limited by low sensitivity, slow turnaround time, and inability to detect fastidious or novel pathogens. Metagenomic next-generation sequencing (mNGS) offers an unbiased approach to pathogen detection and may improve diagnostic accuracy and clinical decision-making.
Methods: We conducted a retrospective study of 300 adult HAP patients admitted to Beijing Rehabilitation Hospital, China. Bronchoalveolar lavage fluid samples were analyzed using the Illumina sequencing platform for mNGS. Detection rates, pathogen spectrum, resistance gene identification, and treatment modifications were compared with conventional culture methods.
Results: mNGS achieved a pathogen detection rate of 92%, significantly higher than the 72% achieved by culture. It identified a broader spectrum of bacteria, fungi, and viruses, including Pseudomonas, Klebsiella, and Aspergillus, which were often missed by culture. Polymicrobial infections were detected in 28% of cases, and antibiotic resistance genes were identified in 30% of samples. The median turnaround time for mNGS results was 48 hours after BAL sampling. Based on mNGS findings, treatment regimens were adjusted in 26% of patients.
Conclusion: mNGS demonstrated superior diagnostic performance compared with culture by increasing pathogen detection rates, identifying resistance genes, and guiding treatment adjustments in HAP patients. Despite its promise for precision medicine, further studies are needed to assess cost-effectiveness and generalizability, given the retrospective and single-center design of this study.
Keywords: hospital-acquired pneumonia, metagenomic next-generation sequencing, mNGS, hospital-acquired pneumonia, HAP, pathogen detection, antimicrobial resistance
Introduction
Traditional diagnostic methods for pneumonia, including culture-based techniques and polymerase chain reaction (PCR), have long been the standards for pathogen detection. Culture methods involve growing pathogens from clinical samples on selective media, which can provide information on antimicrobial susceptibility but often require several days to yield results. Moreover, these methods may fail to detect fastidious or slow-growing organisms, leading to delayed or inadequate treatment. PCR offers a faster alternative by amplifying specific genetic sequences of targeted pathogens, allowing for quicker identification. However, PCR is limited by its reliance on predefined primers, restricting its ability to detect unexpected or novel pathogens. Additionally, both culture and PCR methods struggle to identify polymicrobial infections, which are prevalent in hospital-acquired pneumonia (HAP) and can complicate treatment strategies.1,2 In contrast, metagenomic next-generation sequencing (mNGS) provides an unbiased, comprehensive approach to pathogen detection by sequencing all nucleic acids in a sample, thereby overcoming many limitations of traditional methods.
In contrast, mNGS is an unbiased approach that can detect all potential pathogens, including bacteria, viruses, fungi, and parasites, from a single sample. mNGS sequences the entire nucleic acid content of a sample, offering a more comprehensive view of the infection profile. Studies have demonstrated that mNGS not only improves pathogen detection in cases where traditional methods fail but also identifies antimicrobial resistance genes, providing crucial information for targeted therapy.2–4
The clinical application of mNGS has been largely unproblematic, with studies demonstrating its superior sensitivity and specificity compared to traditional methods, particularly in complex infections or when initial diagnostics are inconclusive.1,5 This was especially crucial for identifying rare or novel pathogens, often implicated in emerging infectious diseases and pandemics like SARS-CoV, MERS-CoV, and SARS-CoV-2. The rapid identification of causative agents is paramount in mitigating the threat of infectious disease outbreaks, and mNGS has proven instrumental in this regard, as evidenced by its pivotal role in the identification and genomic surveillance of the SARS-CoV-2 virus during the COVID-19 pandemic.3,4,6
In spite of these benefits, there are barriers to the incorporation of mNGS into routine clinical diagnostics–not the least of which is the added cost of sequencing and the requirements for bioinformatics support. Interpretation of mNGS data also requires in-depth knowledge of genomics, which is unlikely to exist in all clinical settings, as well as expertise in managing voluminous sequencing data containing a high proportion of human host DNA, which requires sophisticated computational tools to mine for pathogen-related information.3
Several studies have already evaluated the clinical utility of mNGS in different medical areas, such as the diagnosis of respiratory, central nervous system and bloodstream infections, reporting its potential to critically modify clinical management and prognosis by providing rapid and reliable diagnoses. In particular, mixed infections have been identified as one of the most significant advantages of mNGS for directing therapy and impacting patient prognosis. This supports the hypothesis that mNGS could hold a novel and important role in clinical microbiology.7,8
mNGS also yields valuable epidemiological information, such as which infections are spreading, and how pathogens are evolving or adapting to antibiotics, information that is vital for controlling outbreaks and developing prophylactic measures. That became crystal clear when the world was reeling from recent global health crises, when suspect specimens from hospitalised patients were routed through mNGS for real-time surveillance and quick responses.1,2,9
HAP and community-acquired pneumonia (CAP) represent two distinct clinical challenges in terms of diagnosis and management. CAP typically arises outside of healthcare settings, and its causative pathogens are often susceptible to commonly used antibiotics, making standard culture-based diagnostic methods relatively effective in identifying the responsible organisms. However, CAP diagnosis is not without challenges; the overlap of symptoms with other respiratory illnesses and the presence of atypical pathogens can complicate the diagnostic process.10
In contrast, hospital-acquired pneumonia (HAP), which occurs 48 hours or more after hospital admission, presents distinct challenges compared to community-acquired pneumonia (CAP). HAP is often associated with a higher incidence of multidrug-resistant (MDR) organisms, making accurate etiological diagnosis more complex. The pathogens responsible for HAP can be fastidious or have slow replication rates, leading to frequent misses by traditional culture methods. Additionally, HAP frequently involves polymicrobial infections and co-infections with fungi and viruses, particularly in immunocompromised patients, which further complicates diagnosis. This necessarily depends on advanced diagnostic techniques such as mNGS, which offers a comprehensive and unbiased identification of pathogens by sequencing all nucleic acids in a sample, thereby improving diagnostic accuracy and informing targeted treatment strategies.11
This study aims to address the critical diagnostic gap in HAP by evaluating the clinical efficacy and diagnostic value of mNGS, particularly in detecting MDR organisms and polymicrobial infections that are often missed by standard diagnostic techniques. By comparing mNGS with traditional culture methods, we aim to demonstrate the superiority of mNGS in providing rapid, accurate, and actionable data for the management of HAP, ultimately improving patient outcomes. Given the global rise of antibiotic resistance, this study holds significance in advancing pathogen detection methods, not only in hospital settings but also for cases where traditional methods fail, such as in CAP.
Materials and Methods
Study Design and Setting
This retrospective cohort study was conducted in the Beijing Rehabilitation Hospital, affiliated to Capital Medical University, Department of Respiratory and Critical Care Medicine, China, from August 2021 to January 2024. It addressed adult patients admitted to hospital for HAP to evaluate the diagnostic value of mNGS versus culture. The study was approved by the institutional review board at the hospital. Given the retrospective design, individual consent for this study was waived by the institutional review board. All methods were performed in accordance with the relevant guidelines and regulations and the Declaration of Helsinki. However, all patients or their families had previously provided consent for the acquisition of samples for medical purposes. This study was approved by the Institutional Review Board of Beijing Rehabilitation Hospital. All data were kept anonymous, under strict confidentiality and an ethical framework.
Study Population
Patients eligible for inclusion in the study were those diagnosed with HAP based on the criteria defined by the Infectious Diseases Society of America (IDSA) and American Thoracic Society (ATS), which include being aged 18 years or older, pneumonia that develops 48 hours or more after hospital admission, new or progressive infiltrates on chest imaging, and clinical signs such as fever, leukocytosis, or purulent respiratory secretions. Radiological assessments included chest X-ray or CT (computerized tomography) scans as per standard clinical practice. An adjudication committee comprising three independent pulmonologists reviewed each case to confirm the diagnosis of HAP. All patients exhibited persistent symptoms, including fever, elevated inflammatory markers, and respiratory distress, despite initial antibiotic treatment based on sputum culture results, yet still had a poor prognosis characterised by fever and high inflammatory markers persisting two to three days after treatment. Patients were excluded if baseline or 72–96-hour inflammatory markers, respiratory parameters, or 7-day clinical outcomes were missing. Additionally, individuals with severe liver or kidney dysfunction or unstable vital signs were excluded. By further screening patients with HAP who met these criteria and had subsequently undergone mNGS pathogen diagnosis, a total of 250 responders and 50 non-responders were finally included.
Bronchoalveolar lavage fluid (BALF) samples were collected upon clinical diagnosis of HAP, following standard bronchoscopy procedures. Each sample consisted of 200 mL of saline instilled and retrieved. Positive controls (spiked with known quantities of Pseudomonas aeruginosa and Candida albicans) and negative controls (saline blanks) were included in each batch of mNGS testing to monitor for contamination and ensure sequencing accuracy.
To maintain focus on pathogen detection and drug resistance, we streamlined the clinical evaluation indicators. Key indicators included: Oxygenation Index, a measure of lung function, defined as the ratio of arterial oxygen partial pressure to fractional inspired oxygen (PaO2/FiO2); Pneumonia Severity Index (PSI), a widely used scoring system to assess the severity of pneumonia; Clinical Pulmonary Infection Score (CPIS), a scoring tool used to gauge the severity of pneumonia based on clinical, radiological, and microbiological findings. Other health indices like frailty (measured using the FRAIL scale: Fatigue, Resistance, Ambulation, Illness, and Loss of weight), comorbidity burden (Charlson Comorbidity Index), and daily living activities (Barthel Index) were recorded but simplified for clarity in the analysis of the primary focus, which was pathogen detection and treatment adjustments.
Patients who were excluded were those with serious liver or kidney dysfunction, patients discharged or died before receiving next-generation sequencing (NGS) results, those without consent for collection of bronchoalveolar lavage fluid for NGS testing, those with unstable vital signs, those with incomplete clinical data and unclear prognosis.
Data Collection
Clinical and laboratory data was obtained from the electronic medical records which had record of demographics, clinical scoring indices, as well as routine haematological and biochemical parameters. Pneumonia-related laboratory tests such as hemoglobin A1c (HbA1c), albumin (ALB), prealbumin (PA), white blood cell count (WBC), C-reactive protein (CRP), interleukin-6 (IL-6) and creatinine (Cr) were also collected. Key clinical indicators were selected based on international guidelines for HAP management, including patient age, gender, Pneumonia Severity Index (PSI), Clinical Pulmonary Infection Score (CPIS), Charlson Comorbidity Index, Barthel Index (BI), and Acute Physiology and Chronic Health Evaluation II (APACHE II). Additionally, routine biochemical parameters such as hemoglobin A1c (HbA1c), albumin (ALB), prealbumin (PA), white blood cell count (WBC), C-reactive protein (CRP), interleukin-6 (IL-6), and creatinine (Cr) were recorded.
Traditional Culture Methods
Bronchoalveolar lavage fluid (BALF) samples were cultured using both aerobic and anaerobic conditions following the Clinical and Laboratory Standards Institute (CLSI) guidelines. Aerobic cultures were incubated on Blood Agar and MacConkey Agar plates at 35°C for 48 hours, while anaerobic cultures were performed using Bacteroides Bile Esculin (BBE) Agar under anaerobic conditions for up to 7 days. Bacterial identification was conducted using the Vitek-2 automated system (bioMérieux, France), and antimicrobial susceptibility testing was performed according to CLSI standards to guide antibiotic treatment prescriptions.
Analysis and Result Interpretation of mNGS
BALF samples (200 mL) were collected from each patient under sterile conditions and processed for DNA extraction using the Qiagen QIAamp DNA Mini Kit, following the manufacturer’s protocol to ensure consistent performance and prevent cross-contamination. The extracted DNA was quantified using a Qubit Fluorometer to assess concentration and purity. Library preparation was performed using the Illumina Nextera XT DNA Library Preparation Kit, which involves random fragmentation of the DNA followed by adapter ligation. Sequencing was conducted on the Illumina NextSeq 550 platform, generating paired-end reads of 150 bp in length. Rigorous quality control measures were implemented at each step, including the use of positive controls (spiked with known quantities of Pseudomonas aeruginosa and Candida albicans) and negative controls (saline blanks) to monitor for contamination and ensure sequencing accuracy. Each batch of testing included negative controls (sterile water) and positive controls (spiked with known pathogens) with strict thresholds: batches were re-evaluated if any microbe exceeded >10 reads in the negative control, and reported pathogens were required to have a reads per million (RPM) value at least 10-fold higher than that in the negative control. The positive control was required to be effectively detected (>1000 reads), otherwise the entire experiment was considered invalid. Raw sequencing data were quality-controlled using Fastp, and human sequences were filtered out using Bowtie2 (average removal rate: 92.5%). The remaining non-human sequences (averaging 20 million per sample) were subjected to microbial taxonomic classification using Kraken2 and Bracken. Pathogen identification followed composite criteria: for bacteria/fungi, a positive call required either absence in the negative control or an RPM ratio >10 with top 10 coverage within the genus/species; for viruses, ≥3 unique sequences with absence in the negative control or an RPM ratio >5; for M. tuberculosis and Cryptococcus, ≥1 specific sequence was reported. Final results were comprehensively interpreted by clinical microbiologists incorporating pathogen load, resistance genes, and clinical presentation.
Pathogen Detection and Analysis
The study evaluated the spectrum and frequency of pathogens detected by mNGS compared to traditional methods. The efficacy of each technique in identifying causative agents in patients with complex clinical presentations was assessed, with particular attention to the detection of polymicrobial infections and antibiotic resistance markers.
The positive results from mNGS were determined using both quantitative and qualitative criteria. For each sample, the microbial load was calculated based on the proportion of microbial reads out of the total sequenced reads. A pathogen was considered significant if its relative abundance exceeded 1% of the total microbial reads. This threshold has been validated using a large number of BALF samples in our laboratory and shows high concordance with conventional culture and qPCR results, indicating that this level is associated with clinically relevant infections. The clinical significance of detected pathogens was assessed through several factors: pathogen abundance, virulence (evaluated through known pathogenicity databases such as the NCBI Pathogen Database), and the presence of polymicrobial infections, where multiple pathogens exceeding the 1% threshold suggested a higher likelihood of clinical relevance. Additionally, mNGS results were cross-referenced with the detection of antibiotic resistance genes, with resistant pathogens prioritized for significance, especially in cases where these were missed by traditional culture methods. Finally, pathogen identification was corroborated with the clinical presentation of each patient, including symptom severity and inflammatory markers, ensuring that only pathogens consistent with the clinical course were considered as causative agents. This multi-factor approach minimized the risk of false positives from commensal organisms or contaminants often found in respiratory samples, ensuring a higher diagnostic accuracy.
Definition of Responders and Non-Responders
Patients were classified as responders or non-responders based on their clinical outcome following treatment adjustments informed by mNGS results. Responders were defined by the attainment of clinical stability within 7 days of initiating mNGS-guided therapy, characterized by: resolution of fever (axillary temperature <37.3°C for >48 hours), significant improvement in respiratory signs and symptoms, and a pronounced decline in systemic inflammatory markers (>50% reduction in CRP level). Non-responders failed to meet these criteria within the same timeframe, showing persistent or worsening clinical signs, ongoing fever, and inadequately controlled inflammatory response despite tailored antimicrobial treatment based on mNGS findings.
Statistical Analysis
Statistical analyses were performed using SPSS software. We used descriptive statistics to summarise patient demographics and clinical characteristics. We defined sensitivity and specificity of mNGS versus traditional culture methods to compare using Chi-square tests for categorical data and t-tests for continuous variables. Statistical significance was set at a p-value less than 0.05. Adjustments in antibiotic therapy based on mNGS findings were also analyzed to evaluate the impact on patient outcomes.
Results
Demographic and Clinical Characteristics
Analysis of demographic data (Table 1) revealed that the mean age of responders was significantly lower than that of non-responders, with responders averaging 64.7 years compared to 69.1 years for non-responders (p=0.005). Gender distribution across the groups showed a majority of males in both responders (67.2%) and non-responders (74%), however the difference was not statistically significant (p=0.4371).
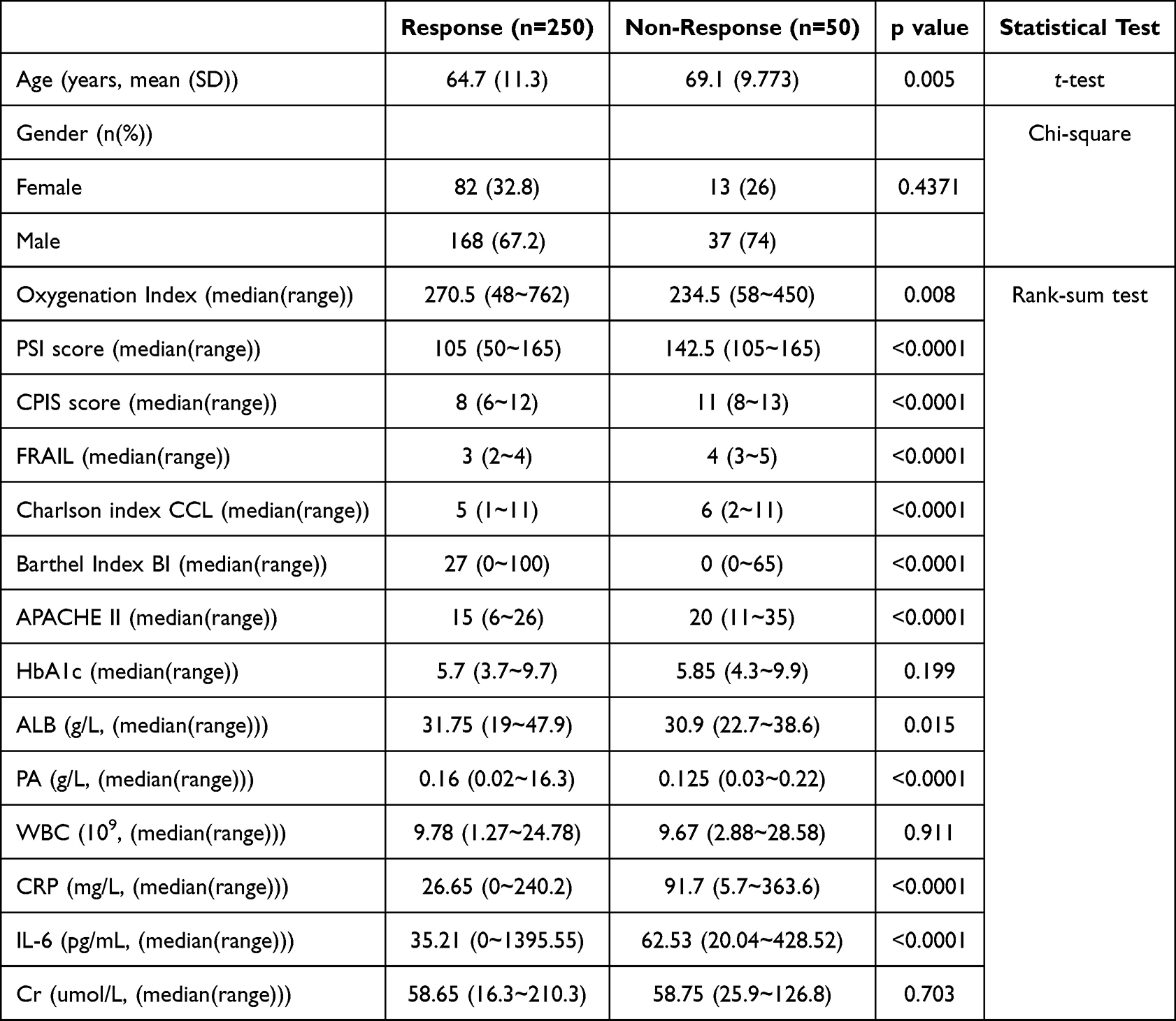 |
Table 1 Demographic and Clinical Characteristics of Study Participants |
A range of clinical scores and health indices were assessed (Table 1), revealing notable differences between responders and non-responders. The median oxygenation index was significantly higher in responders. Critical clinical scores such as the PSI and CPIS were considerably lower in responders, suggesting less severe clinical presentations compared to non-responders. The PSI scores ranged from 50 to 165 for responders and 105 to 165 for non-responders, showing a significant difference (p<0.0001). Similarly, CPIS scores also differed significantly, with responders averaging lower scores indicative of better clinical status.
Health indices further demonstrated disparities between the two groups. The FRAIL scale, used to assess frailty, showed that responders were less frail compared to non-responders, with scores significantly differing (p<0.0001). The Charlson Comorbidity Index and the BI, which measures daily living activities, also reflected better health status in responders. Likewise, the APACHE II scores, which estimate ICU mortality risk, were lower in responders, suggesting a better prognosis (p<0.0001).
Laboratory findings complemented these clinical assessments, with several key biomarkers showing significant differences between the groups. Notably, ALB and PA levels were lower in non-responders, reflecting potentially poorer nutritional status or more severe disease states. Inflammatory markers such as CRP and IL-6 were significantly elevated in non-responders, indicating higher levels of systemic inflammation and potentially more severe or uncontrolled infectious processes (p<0.0001 for both). However, no significant differences were observed in HbA1c and Cr levels between the groups, suggesting similar baseline metabolic and renal functions (Table 1).
Pathogen Detection
The analysis revealed that mNGS identified a higher diversity of pathogens compared to traditional culture methods, achieving a pathogen detection rate of 92%, significantly higher than the 72% detection rate with traditional culture. Traditional culture methods detected pathogens in 216 out of 300 samples, with the most frequently identified bacteria being Pseudomonas, Klebsiella, Acinetobacter, Streptococcus, and Staphylococcus. In contrast, mNGS detected a broader range of bacteria, fungi, and viruses, including Candida, Aspergillus, Pneumocystis, Rhizopus, HSV1, human cytomegalovirus (CMV), Epstein-Barr virus (EBV), varicella zoster virus, Primate erythroparvovirus 1, human herpesvirus 7, and human adenovirus B (Figures 1 and S1).
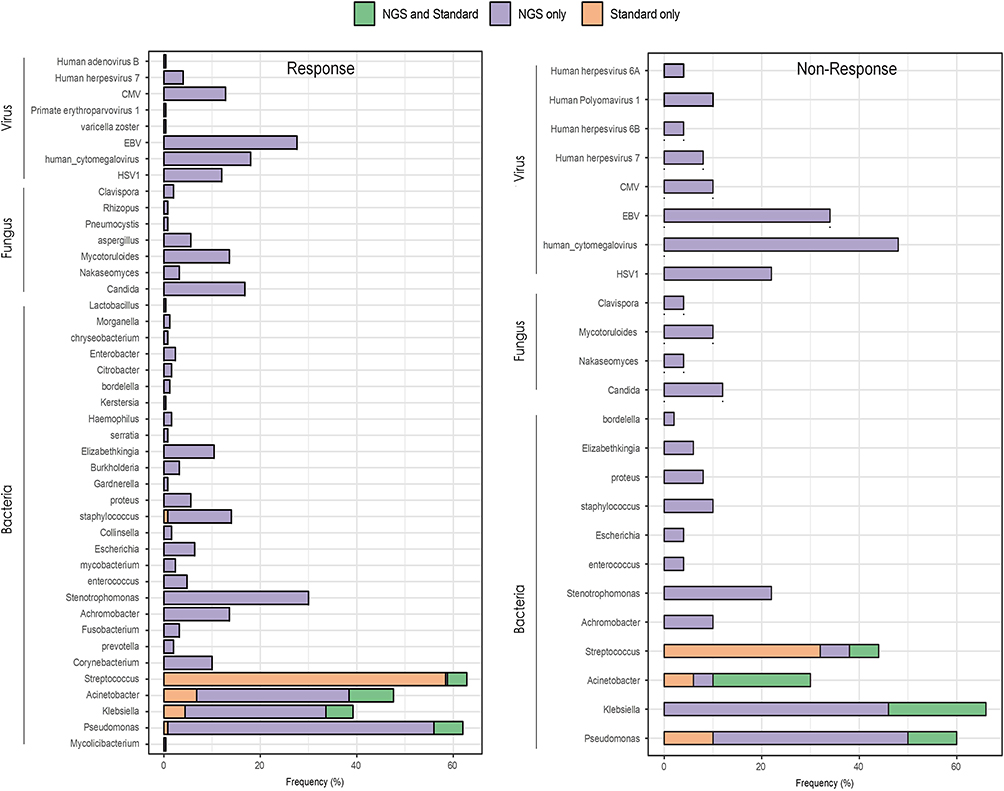 |
Figure 1 Pathogen Profiles Detected in Responders and Non-Responders. The distribution of detected pathogens between responders and non-responders is shown, with mNGS consistently identifying a broader spectrum of bacterial, fungal, and viral pathogens in both groups. However, in non-responders, mNGS was particularly valuable, identifying critical pathogens that traditional methods missed, including drug-resistant strains. This higher detection rate in non-responders underscores mNGS’s role in guiding more precise treatment adjustments for patients who fail to respond to initial therapies. |
mNGS detected a wide range of pathogens in both responder and non-responder groups. The frequency of bacterial detection was higher in the responder group compared to non-responders (Figure 1). Notably, Pseudomonas and Klebsiella were the predominant bacteria found in both groups. Candida was the most frequently detected fungus in both groups. Aspergillus and Pneumocystis were more prevalent in the non-responder group. HSV1, CMV, and EBV were among the most common viruses detected. The presence of these viruses was higher in non-responders compared to responders (Figure 1).
Venn diagrams were used to illustrate the overlap and unique detections between mNGS and standard methods. They highlighted the added value of mNGS in identifying additional pathogens, which played a crucial role in guiding targeted antimicrobial therapy (Figure 2).
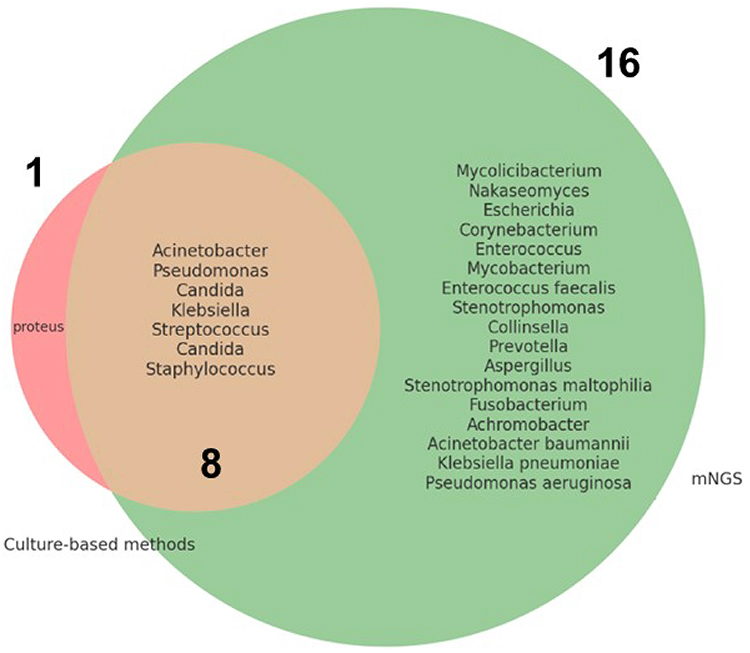 |
Figure 2 Overlap and Unique Detections Between mNGS and Culture-based Methods. Among the pathogens detected, 1 was uniquely identified by culture-based methods, 17 were uniquely detected by mNGS-based methods, and 7 were detected by both methods. |
Treatment Adjustments
Based on the results of mNGS, treatment regimens were adjusted for both responders and non-responders. Treatment regimens were modified in 64 (25.6%) of the 250 responders and 14 (28%) of the 50 non-responders, showing no significant difference between the groups (p=0.86). The addition of antimicrobial agents was necessary in a majority of cases, with 186 (74.4%) of responders and 36 (72%) of non-responders receiving additional agents (Table 2).
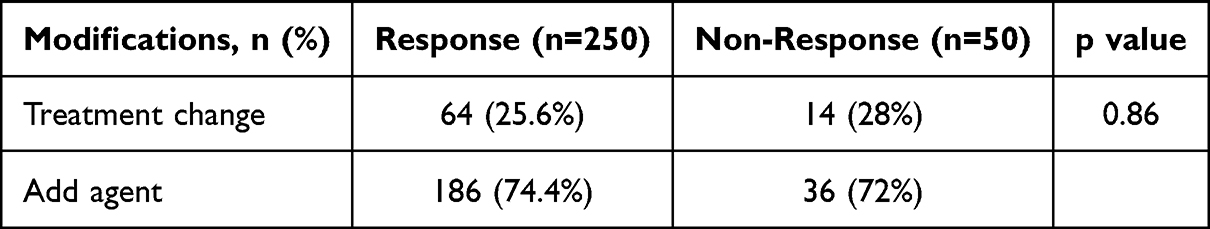 |
Table 2 Treatment Adjustments Based on mNGS Results |
Clinical Outcomes
Clinical outcomes were assessed and compared between responders and non-responders. The median duration of ICU stay was 18 days for responders (range: 0–55 days) compared to 15.5 days for non-responders (range:2–31 days), although this difference was not statistically significant (p=0.786). Responders required a significantly shorter duration of mechanical ventilation (median: 3.5 days, range: 0–30 days) compared to non-responders (median: 12 days, range: 0–26 days) (p=0.014). Additionally, a higher proportion of responders underwent tracheostomy (64.8%) compared to non-responders (46%) (p=0.019) (Table 3). These findings suggest that mNGS-guided treatment adjustments may contribute to more efficient respiratory support and procedural interventions, potentially improving patient outcomes.
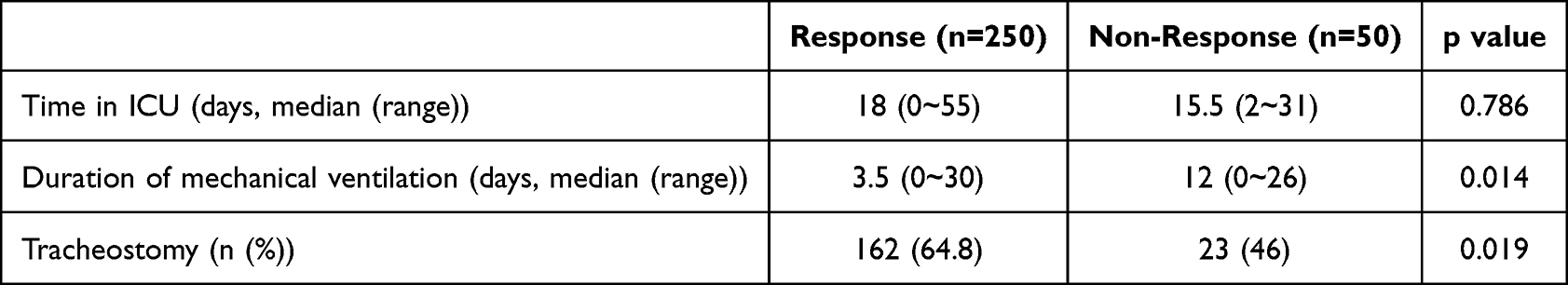 |
Table 3 Clinical Outcomes in Responders and Non-Responders |
Resistance Genes
Our analysis identified a variety of resistance genes associated with the pathogens detected in both responders and non-responders (Table 4). The analysis revealed the presence of multiple resistance genes, highlighting the complexity of treating infections in these patients.
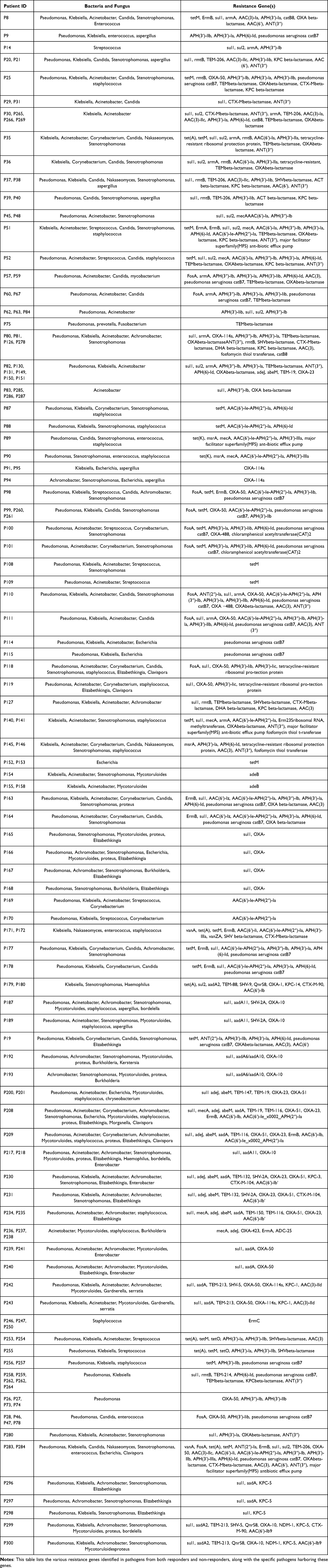 |
Table 4 Distribution of Resistance Genes Among Detected Pathogens |
Several resistance genes were frequently identified, including tetM, ErmB, sul1, and various beta-lactamases such as OXA and TEM. These genes were associated with resistance to tetracyclines, macrolides, sulfonamides, and beta-lactam antibiotics, respectively. The detailed distribution of resistance genes among the patients showed that multiple pathogens harbored these genes, contributing to multidrug-resistant infections. For example, Pseudomonas, Klebsiella, and Acinetobacter were commonly found with resistance genes such as tetM, sul1, and OXA beta-lactamase. Staphylococcus species frequently carried mecA, encoding methicillin resistance. Candida and other fungal pathogens also exhibited resistance, complicating antifungal therapy (Table 4).
Some patients had complex resistance profiles involving multiple genes. For instance, Patient P8 harbored tetM, ErmB, sul1, armA, AAC(3)-Ia, APH(3′)-Ia, catB8, OXA beta-lactamase, and others; Patient P9 had APH(3′)-llb, APH(3′)-la, APH(6)-Id, and Pseudomonas aeruginosa catB7. Many patients exhibited mixed infections involving multiple resistant pathogens, further complicating treatment strategies (Table 4).
Discussion
As our study shows, mNGS has the potential to change the diagnostic approach to HAP, generating an unbiased detection of pathogens that would not be covered by traditional culture methods. This is in line with the increasing literature consensus that mNGS will radically change the landscape of infectious disease diagnosis by providing an unbiased picture of pathogens.7,8,12,13
In the context of our study, it became clear that i) mNGS had a much greater sensitivity and can detect a much larger array of pathogens, including fastidious (difficult-to-culture) bacteria, fungi and viruses, compared with traditional culture methods.9 ii) Conventional culture-based diagnostics miss many fastidious organisms which pose challenges to culture and often necessitate special growth conditions. Wilson et al found that routine microbiologic testing is often insufficient to detect all potential neuroinvasive pathogens in CSF. Herein, they utilised mNGS of CSF collected from patients with meningitis or encephalitis to diagnose more neurologic infections and provided actionable information for a subset of patients.14 This aligns with your study’s findings on the superiority of mNGS over traditional culture methods in detecting a wider array of pathogens, including those in culture-negative samples. This advantage of mNGS offering enhanced sensitivity is crucial for accurate and timely diagnosis in critically ill patients.
The clinical implications of mNGS are profound, as evidenced by our study’s findings. In responders, 25.6% of cases had their treatment regimens adjusted based on mNGS results, and similarly, 28% of non-responders experienced treatment changes. This underscores mNGS’s role in facilitating targeted antimicrobial therapy, aligning with our study’s objective to evaluate its diagnostic value in HAP. Furthermore, the identification of a broader spectrum of pathogens, including polymicrobial infections and antibiotic-resistant strains, directly addresses the diagnostic challenges inherent in HAP management. These findings are consistent with those of Gu et al, who demonstrated the value of blood mNGS in infectious patients with mild and non-specific symptoms, where therapeutic regimens were altered in 70.3% of cases based on mNGS results.15 By enabling the detection of pathogens that traditional culture methods miss, mNGS provides critical information that can lead to more effective and timely antimicrobial interventions.
The detection of polymicrobial infections and antibiotic resistance genes through metagenomic mNGS represents a significant advancement in the diagnosis and management of HAP. Traditional culture-based diagnostic methods often fail to identify polymicrobial infections due to their inability to simultaneously grow multiple pathogens under standard laboratory conditions. In our study, mNGS identified polymicrobial infections in 28% of cases, a detection rate substantially higher than that of culture-based methods. Polymicrobial infections are clinically significant in the hospital setting as they are frequently associated with more severe disease progression and higher rates of treatment failure. This underscores the utility of mNGS as a diagnostic tool capable of providing a comprehensive pathogen profile, thereby enabling more targeted and effective treatment strategies. Additionally, the detection of antibiotic resistance genes in 30% of samples highlights mNGS’s ability to inform antimicrobial stewardship by identifying resistance patterns that are crucial for selecting appropriate therapies. The simultaneous identification of pathogens and their resistance profiles is a distinct advantage of mNGS over traditional methods, which typically require separate assays for antimicrobial susceptibility testing. This real-time detection facilitates timely adjustments in antimicrobial therapy, thereby reducing the risk of prolonged ineffective treatment and limiting the spread of resistant organisms. These findings not only align with our study’s objectives but also contribute meaningfully to the broader context of improving diagnostic accuracy and treatment efficacy in hospital-acquired pneumonia management.
Identification of antibiotic resistance genes was the key finding of our study. Resistance genes tetM, ErmB and a number of beta-lactamases were picked up, enabling comprehensive data for antibiotic stewardship, in line with another mNGS study conducted by Gan et al who reported the ability of mNGS to identify resistance genes targeting different antibiotics in the treatment of severe pneumonia in paediatric patients.16
Our work confirms that mNGS has an important impact on patient clinical outcome by identifying the pathologies, where conventional methods are frequently negative, leading to an increase in the detection of severe infection-causing, and multi-drug resistant (hard-to-treat) bacteria, such as Pseudomonas, Klebsiella and Acinetobacter, as well as fungal pathogens such as Candida and Aspergillus. These results are confirmed by the major studies in the field, highlighting the key role of broad pathogen detection to decide targeted antibiotic and antifungal therapies.17,18
The identification of viral pathogens, including HSV1, CMV, and EBV, further emphasizes the diagnostic utility of mNGS, particularly in immunocompromised patients. While these viruses are commonly found in the population, their detection in a hospital setting is clinically significant as they can cause opportunistic infections in immunosuppressed individuals. In this study, the presence of these viral pathogens was confirmed in patients with underlying conditions that could predispose them to viral reactivation. Their detection provided important information for clinicians to tailor antiviral therapies, particularly in cases where bacterial or fungal pathogens alone could not explain the severity of the illness. Prompt and accurate identification of these viruses may lead to better patient outcomes, reduced hospital stays, and lower healthcare costs.6,19
The cost-effectiveness of mNGS remains a subject of ongoing debate. While the initial capital investment for sequencing equipment and the operational costs of high-throughput sequencing are substantial, these may be offset by reductions in hospital stays, more targeted antimicrobial therapies, and improved patient outcomes. Jing et al forecast that costs will continue to decrease as sequencing technologies advance and become more widely adopted.20 Additionally, the implementation of portable sequencing devices and point-of-care mNGS platforms has the potential to revolutionize infectious disease diagnostics by enabling rapid pathogen detection directly at the patient’s bedside.21 However, several challenges must be addressed to facilitate the widespread integration of mNGS into routine clinical practice. These include the need for specialized bioinformatics infrastructure and expertise to analyze and interpret vast amounts of sequencing data, ensuring data privacy and security, and developing standardized protocols for sample preparation and data reporting. Furthermore, the clinical utility of mNGS must be consistently demonstrated through larger, multicenter studies to validate its benefits across diverse healthcare settings. Overcoming these challenges is essential for mNGS to transition from a research tool to a standard diagnostic modality in clinical microbiology.
Although currently limited to specialized centers with the required infrastructure and bioinformatics expertise, the adoption of mNGS is expected to grow over the next 5 to 10 years as sequencing technology becomes more cost-effective and accessible. In addition, ongoing improvements in automated data analysis tools will reduce the need for highly specialized bioinformatics staff, further accelerating the integration of mNGS into routine clinical microbiology. Public health efforts to combat antimicrobial resistance are likely to drive this shift, as mNGS provides rapid, comprehensive data on pathogen detection and resistance profiles. Thus, mNGS is anticipated to become a staple in diagnostic laboratories, particularly in hospitals dealing with high-risk infections, as early as the next decade.
While these results are promising, several key limitations of our study need to be recognized. The retrospective design and our single-center setting are likely to generate selection bias and, therefore, restrict the generalizability of our outcomes. Secondly, as this is a retrospective study, very few patients had their BALF samples used for antimicrobial susceptibility testing (AST), which is an important assay for evaluating the clinical relevance of genotype-phenotype resistance prediction. Another major limitation of this study is the relatively small sample size, particularly in the non-response group, which may limit statistical power. In the future, larger-scale, multicenter, prospective cohort studies are needed to confirm our findings and to evaluate the cost-effectiveness of mNGS in clinical practice.
Conclusion
In conclusion, our study demonstrated the superior diagnostic performance of mNGS over traditional culture methods in HAP. mNGS not only provided comprehensive pathogen detection, identifying a wider array of bacteria, fungi, and viruses, but also facilitated the profiling of antibiotic resistance genes, thereby enhancing targeted antimicrobial therapy. These capabilities make mNGS a valuable tool in modern infectious disease control and management.
Abbreviations
mNGS, Metagenomic Next-Generation Sequencing; HAP, Hospital-Acquired Pneumonia; CAP, Community-Acquired Pneumonia; CSF, Cerebrospinal Fluid; BALF, Bronchoalveolar Lavage Fluid; PSI, Pneumonia Severity Index; CPIS, Clinical Pulmonary Infection Score; APACHE II, Acute Physiology and Chronic Health Evaluation II; HbA1c, Hemoglobin A1c; ALB, Albumin; PA, Prealbumin; WBC, White Blood Cell Count; CRP, C-Reactive Protein; IL-6, Interleukin-6; Cr, Creatinine; MDR, Multidrug-Resistant; HSV1, Herpes Simplex Virus 1; CMV, Cytomegalovirus; EBV, Epstein-Barr Virus; PCR, polymerase chain reaction; IDSA, Infectious Diseases Society of America; ATS, American Thoracic Society; antimicrobial susceptibility testing, AST; CT, computerized tomography; FRAIL, Fatigue, Resistance, Ambulation, Illness, and Loss of weight; NGS, Next-Generation Sequencing; BI, Barthel Index; CLSI, Clinical and Laboratory Standards Institute; BBE, Bacteroides Bile Esculin; RPM, reads per million.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Informed Consent Statement
Given the retrospective design, individual consent for this study was waived by the Institutional Review Board of Beijing Rehabilitation Hospital.
Acknowledgments
A previous version of this manuscript was published as a preprint on Research Square. The preprint’s DOI is: https://www.researchsquare.com/article/rs-5235477/v1.22
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work has received specialized funding for scientific research from Beijing Rehabilitation Hospital of Capital Medical University (2022-007).
Disclosure
The authors declare that there is no conflicts of interest regarding the publication of this article.
References
1. He S, Xiong Y, Tu T, et al. Diagnostic performance of metagenomic next-generation sequencing for the detection of pathogens in cerebrospinal fluid in pediatric patients with central nervous system infection: a systematic review and meta-analysis. BMC Infect Dis. 2024;24(1):103. doi:10.1186/s12879-024-09010-y
2. Chen H, Huang Q, Wu W, et al. Assessment and clinical utility of metagenomic next-generation sequencing for suspected lower respiratory tract infections. Eur J Med Res. 2024;29(1):213. doi:10.1186/s40001-024-01806-7
3. Miao Q, Ma Y, Wang Q, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clinl Infect Dis. 2018;67(suppl_2):S231–S40. doi:10.1093/cid/ciy693
4. Wang C, Yan D, Huang J, et al. The clinical application of metagenomic next-generation sequencing in infectious diseases at a tertiary hospital in China. Front Cell Infect Microbiol. 2022;12:957073. doi:10.3389/fcimb.2022.957073
5. Edward P, Handel AS. Metagenomic next-generation sequencing for infectious disease diagnosis: a review of the literature with a focus on pediatrics. J Pediatric Infect Dis Soc. 2021;10(Supplement_4):S71–S7. doi:10.1093/jpids/piab104
6. Gu W, Deng X, Lee M, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27(1):115–124. doi:10.1038/s41591-020-1105-z
7. Yu C, Guo W, Zhang Z, et al. The impact of mNGS technology in the etiological diagnosis of severe pneumonia in children during the epidemic of covid-19. Infect Drug Resist. 2023;16:2395–2402. doi:10.2147/IDR.S403851
8. Han D, Yu F, Zhang D, et al. Applicability of bronchoalveolar lavage fluid and plasma metagenomic next-generation sequencing assays in the diagnosis of pneumonia. Open Forum Infect Dis. 2024;11(1):ofad631. doi:10.1093/ofid/ofad631
9. Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol. 2019;4(4):663–674. doi:10.1038/s41564-018-0349-6
10. Micek ST, Kollef KE, Reichley RM, Roubinian N, Kollef MH. Health care-associated pneumonia and community-acquired pneumonia: a single-center experience. Antimicrob Agents Chemother. 2007;51(10):3568–3573. doi:10.1128/aac.00851-07
11. Pneumonia NMSH-A. Health care-associated pneumonia, ventilator-associated pneumonia, and ventilator-associated tracheobronchitis: definitions and challenges in trial design. Clinl Infect Dis. 2010;51(Supplement_1):S12–S7. doi:10.1086/653035
12. Heitz M, Levrat A, Lazarevic V, et al. Metagenomics for the microbiological diagnosis of hospital-acquired pneumonia and ventilator-associated pneumonia (Hap/Vap) in Intensive Care Unit (Icu): a proof-of-concept study. Respir Res. 2023;24(1):285. doi:10.1186/s12931-023-02597-x
13. Sun T, Wu X, Cai Y, et al. Metagenomic next-generation sequencing for pathogenic diagnosis and antibiotic management of severe community-acquired pneumonia in immunocompromised adults. Front Cell Infect Microbiol. 2021;11:661589. doi:10.3389/fcimb.2021.661589
14. Wilson MR, Sample HA, Zorn KC, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N Engl J Med. 2019;380(24):2327–2340. doi:10.1056/NEJMoa1803396
15. Zhang H, Liang R, Zhu Y, et al. Metagenomic next-generation sequencing of plasma cell-free DNA improves the early diagnosis of suspected infections. BMC Infect Dis. 2024;24(1):187. doi:10.1186/s12879-024-09043-3
16. Gan M, Zhang Y, Yan G, et al. Antimicrobial resistance prediction by clinical metagenomics in pediatric severe pneumonia patients. Ann Clinic Microbiol Antimicrob. 2024;23(1):33. doi:10.1186/s12941-024-00690-7
17. Chen S, Hou C, Kang Y, Li D, Rong J, Li Z. Application of metagenomic next-generation sequencing in the diagnosis and resistome analysis of community-acquired pneumonia pathogens from bronchoalveolar lavage samples. J Appl Microbiol. 2023;134(6):lxad102. doi:10.1093/jambio/lxad102
18. Pang F, Xu W, Zhao H, et al. Comprehensive evaluation of plasma microbial cell-free DNA sequencing for predicting bloodstream and local infections in clinical practice: a multicenter retrospective study. Front Cell Infect Microbiol. 2024;13:1256099. doi:10.3389/fcimb.2023.1256099
19. Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14:319–338. doi:10.1146/annurev-pathmechdis-012418-012751
20. Jing C, Chen H, Liang Y, et al. Clinical evaluation of an improved metagenomic next-generation sequencing test for the diagnosis of bloodstream infections. Clin Chem. 2021;67(8):1133–1143. doi:10.1093/clinchem/hvab061
21. Bloemen B, Gand M, Vanneste K, Marchal K, Roosens NHC, De Keersmaecker SCJ. Development of a portable on-site applicable metagenomic data generation workflow for enhanced pathogen and antimicrobial resistance surveillance. Sci Rep. 2023;13(1):19656. doi:10.1038/s41598-023-46771-z
22. Zhang B, Wang J, Li Q, et al. Clinical efficacy and diagnostic value of metagenomic next-generation sequencing (mNGS) in hospital-acquired pneumonia: a stratified retrospective study of responders and non-responders. Preprint at Research Square. 2024. doi:10.21203/rs.3.rs-5235477/v1.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Bacterial Epidemiology and Antimicrobial Resistance Profiles of Respiratory Specimens of Children with Pneumonia in Hainan, China
Mai W, Liu Y, Meng Q, Xu J, Wu J
Infection and Drug Resistance 2023, 16:249-261
Published Date: 12 January 2023