Back to Journals » Journal of Blood Medicine » Volume 15
Case Study: Rosai-Dorfman Disease and Its Multifaceted Aspects
Authors Werneck Rodrigues DO ![]() , Wolp Diniz R, Dentz LC, Costa MA, Lopes RH, Suassuna LF, Cintra JRD
, Wolp Diniz R, Dentz LC, Costa MA, Lopes RH, Suassuna LF, Cintra JRD ![]() , Domenge C
, Domenge C
Received 16 October 2023
Accepted for publication 15 February 2024
Published 11 March 2024 Volume 2024:15 Pages 123—128
DOI https://doi.org/10.2147/JBM.S436720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Daniela Oliveira Werneck Rodrigues,1,2 Roberta Wolp Diniz,2 Leonardo Cunha Dentz,1 Monica de Albuquerque Costa,3 Roberto Heleno Lopes,2 Lucas Fernandes Suassuna,4 Jane Rocha Duarte Cintra,2 Christian Domenge2
1Department of Medicine, Centro Universitário Presidente Antônio Carlos, Juiz de Fora, MG, Brazil; 2Department of Oncohematology, Hospital 9 de Julho/ Instituto Oncológico de Juiz de Fora, Juiz de Fora, MG, Brazil; 3Department of Dematology, Secretaria de Saúde da Prefeitura Municipal de Juiz de Fora, Juiz de Fora, MG, Brazil; 4Department of Medicine, Universidade Federal de Juiz de Fora (UFJF), Juiz de Fora, MG, Brazil
Correspondence: Daniela Oliveira Werneck Rodrigues, Instituto Oncológico de Juiz de Fora, R. Santos Dumont, 56 - Granbery, Juiz de Fora, MG, 36010-510, Brazil, Tel +553232573126, Email [email protected]
Abstract: Rosai-Dorfman Disease (RDD) is a rare non-Langerhans histiocytosis, usually self-limited and presenting with massive, painless, bilateral cervical lymphadenopathy, with or without constitutional symptoms. Extranodal disease is frequently present, and may happen in the absence of lymph node involvement, symptomatology and differential diagnosis will depend on the site affected and fatal cases may occur. The authors present two cases of Rosai-Dorfman disease (RDD), diagnosed through immunohistochemistry, with different progressions, one with complete remission and one culminating in death, highlighting the variety of presentations and the diagnostic difficulty. RDD is a rare condition with clinical presentations similar to several diseases, and should be considered in the differential diagnosis of lymphadenopathy with extranodal lesions.
Keywords: Rosai Dorfman disease, immunohistochemistry, diagnosis, differential diagnosis (MeSH-NCBI)
Introduction
Rosai-Dorfman disease (RDD) was first described by Destombes in 1965, and later by Rosai and Dorfman in 1969, as a sinus histiocytosis with massive lymphadenopathy.1–3 It is a rare non-Langerhans histiocytosis, with a prevalence of 1:200,000 and an annual incidence of 100 cases per year in the United States.1 It predominantly affects males (1.4:1), children and young adults, with a mean age of 20.6 years. RDD is classified as sporadic (classic nodal or extranodal), familial or cutaneous, and may be associated with onco-hematological neoplasms and autoimmune diseases (Figure 1).4 The disease is usually self-limited and presents with massive, painless, bilateral cervical lymphadenopathy, with or without constitutional symptoms. The prognosis is associated with the number of lymph node groups involved and the affected site, the clinical course is chronic and may alternate between episodes of exacerbation and remission.5 Its etiology and pathogenesis are unclear, and probably are not uniform across the disease spectrum, initially it was classified as a polyclonal, non-neoplastic and reactive histiocytosis, occurring from an aberrant immune response associated with infections, but studies have failed to establish a causal link, and current research may lead to future changes in disease classification.5
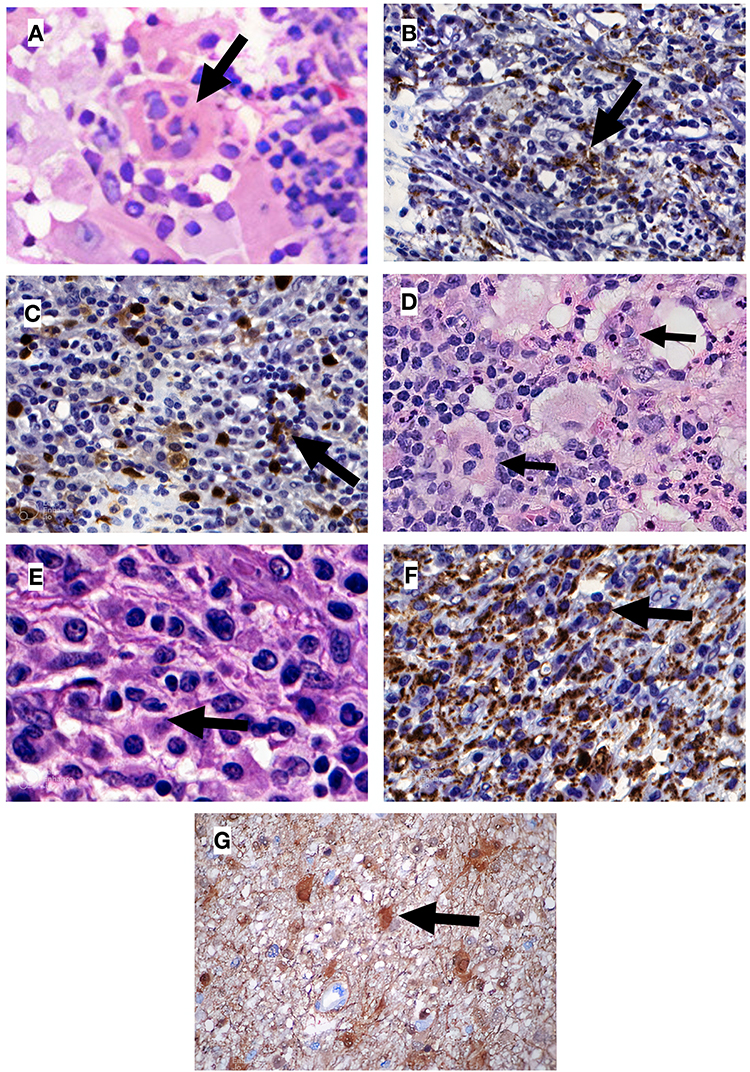 |
Figure 1 Histopathological findings for patients 1 and 2. (A) Patient 1. Non-neoplastic proliferation showing lymphocyte emperipolesis on 800x increase; (B) Patient 1. Positive immunohistochemistry for CD68 (indicating macrophages) on histiocytic infiltrate; (C) Patient 1. Immunohistochemistry with occasional positivity for S100 on histiocytic infiltrate; (D) Patient 2. Non-neoplastic proliferation showing lymphocyte emperipolesis on 400x increase; (E) Patient 2. Non-neoplastic proliferation showing lymphocyte emperipolesis on 800x increase; (F) Patient 2. Positive immunohistochemistry for CD68 (indicating macrophages) on histiocytic infiltrate; (G) Patient 2. Positive immunohistochemistry for S100; Black arrows indicate emperipolesis on (A, D and E), and indicate immunohistochemistry positivity on (B, C, F and G). |
New studies show evidence of a clonal nature in a subset of cases, generally caused by mutations in the MAPK pathway, most frequently in the MAP2K1 and KRAS genes.6 However, Durham et al performed whole exome sequencing in 17 cases of RDD and found alterations not only in the kinases of the MAPK pathway, but also in genes involved in several intracellular pathways, such as intracellular trafficking, transcriptional regulation, cell cycle regulation, DNA mismatch repair and the ubiquitin proteasome pathway.7 BRAF V600E mutations, often described in LCH, are rare but not absent in RDD, in a total of 94 cases reviewed in the literature, only 3 had alterations in the gene.1,8
The diagnosis of RDD is difficult and often delayed due to its rarity, varied presentation, and nonspecific symptoms.5,9,10 Laboratory evaluation may show normocytic and normochromic anemia, leukocytosis, thrombocytopenia, eosinophilia, hypergammaglobulinemia, and elevated ESR. Intermittent fever, night sweats, weight loss, and asthenia may be present. In the pathology, there is a marked sinusoidal expansion, which may lead to the disappearance of the nodal architecture, with a diffuse infiltrate of histiocytes with pale and ill-defined cytoplasm, and a presence of plasma cells, which may be positive for IgG4, a factor with not yet defined importance.1 The characteristic finding is emperipolesis, defined as an intact hematolymphoid cell vacuolated or floating freely in the cytoplasm of macrophages. RDD immunohistochemistry histiocytes are positive for S100, CD68, CD163 and negative for CD1a and CD207, excluding Langerhans-cell histiocytosis (LCH), the main differential diagnosis of RDD.1 Other diseases considered in the differential diagnosis of RDD are Sinus histiocytosis, a reactive process characterizes by a positivity for S-100, cyclin D1 and negative for OCT2; Erdheim-Chester disease (EDD), Juvenile xanthogranuloma, which both show abundant foamy cytoplasm with surrounding fibrosis, EDD’s histiocytes are positiver for CD68, CD163, factor XIIIa and negative for OCT2; and low grade B cell lymphoma, which is negative for OCT2. The following case reports have been approved by the Ethics Committee of the care providing institution, and all patients provided written informed consent for their personal or clinical details along with any identifying images to be published in this study.
Case Reports
Case 1
A 57-year-old white woman presented, in April 2007, with a nodular cutaneous lesion in the left breast, measuring 1.2 cm in the longest axis, without phlogistic signs or constitutional symptoms, during a routine primary care visit. Mammography and ultrasound of the breast were performed, with normal results. After nodulectomy with 2.5cm margins, the histopathological report showed histiocytic proliferation and emperipolesis. Immunohistochemistry was positive for anti-protein S-100 and anti-CD68 antibodies and negative for anti-CD1a, confirming the diagnosis of cutaneous RDD (Figure 1). Immunohistochemical panels were performed for breast cancer humoral receptors (ER, RP and HER 2) that were non-reactive. The patient was referred to the hematology service.
A chest and abdomen computed tomography for investigation of nodal disease was carried out, which did not identify other sites of disease. Considering the laboratory (Table 1), radiological and clinical data, we opted for an expectant management.
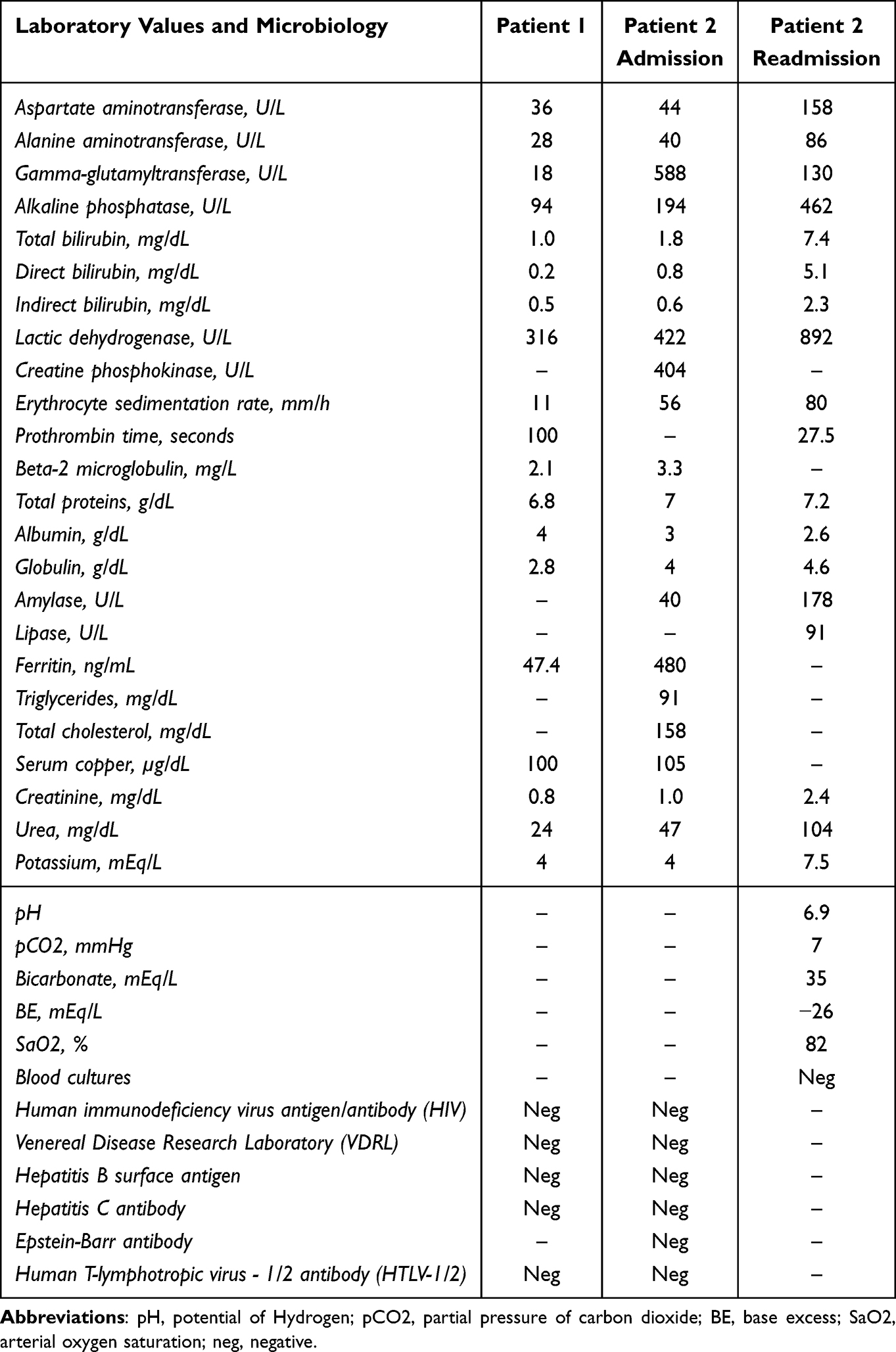 |
Table 1 Laboratory and Microbiology Results from Patients 1 and 2 |
In April 2008, a new nodule measuring 1.5 cm X 1.0 cm was observed on the excisional biopsy scar, the patient underwent a new biopsy with 2.5 cm margins, another body CT, and a bone marrow study, again showing cutaneous RDD, with S-100 positivity and negative for CD1a, with no extranodal disease.
The patient remains under outpatient onco-hematological control and has been in clinical remission for 14 years, with the last consultation in October 2022.
Case 2
A 26-year-old man of African descent was referred to the Oncological Surgery Service in January 2010, with painful right inguinal adenomegaly (3.5 X 2.5×1.5 cm), with progressive growth for 20 days, without phlogistic signs. The patient reported continuous 39°C fever, asthenia and hyporexia.
Upon admission, the patient was anemic, and had increased erythrocyte sedimentation rate (ESR), creatine phosphokinase (CPK), gamma glutamyl transferase (GGT) and alkaline phosphatase (AP) (Table 1). There was a suspicion of lymphoproliferative disease, and a biopsy was performed with subsequent discharge. However, the patient’s symptomatology worsened and he was readmitted. Abdominal ultrasound showed splenomegaly (16.5 cm in the longest axis) and multiple enlarged hypoechoic lymph nodes (2.5 cm X 3 cm) in the right iliac fossa and retroperitoneum. Posteroanterior chest X-ray revealed heterogeneous opacity with a nodule in the hilar and right paracardiac regions, in addition to mediastinal right inter-cleido-hilar widening. Considering the severity of the case, prednisone, ceftazidime and gentamicin were empirically started before a definitive diagnosis. The workup for infectious diseases was negative. The patient’s general condition worsened, with generalized adenomegaly, sepsis, hepatic and renal failure, dying 15 days after the biopsy.
Histopathology of the lymph node identified a proliferative process of undetermined nature, with rounded cells and wispy cytoplasm, requiring immunohistochemistry for diagnostic definition, which confirmed the replacement of normal lymph node architecture by histiocyte proliferation, with lymphoplasmacytic infiltrate and emperipolesis, with positivity for S-100, CD68, and sparse CD45, and negative for CD1a, CD207, CD30 (K1-1), clone Ber-H2, CD21 (clone 1F8), CD20 (clone L-26), CD3 (policlonal), CD15 (clone C3D-1), CD117 (clone T595), CD34 (clone BIRMA-K3), CD23 (clone MHM6) (Figure 1). A presumptive diagnosis of RDD was established post mortem.
Discussion
The cases presented demonstrate the variable manifestations and evolution of RDD. In the first case, there was cutaneous involvement in the left breast, without lymph node disease or other site of injury, with recrudescence after surgical excision, and complete remission after a second excision. This behavior is typical of the cutaneous involvement of RDD, an article that followed 202 patients showed that after surgical excision, about 40% of patients do not achieve complete remission. However, the disease remained localized and did not present malignant evolution.11
Extranodal disease is present in up to 43% of all RDD cases, and may occur in the absence of lymph node involvement. The sites most commonly affected are skin (10%), nasal cavity and paranasal sinuses (11%), bone (5–10%), orbital tissue (11%) and central nervous system (5%).1,5 Symptomatology and differential diagnosis will depend on the site affected (Table 2). Cutaneous RDD is considered a distinct clinical entity, being more common in women (2:1) around 43.5 years, with a higher incidence in Caucasians and Asians, it is generally not associated with systemic or extracutaneous disease, and tends to remain localized, even after long periods of evolution.11
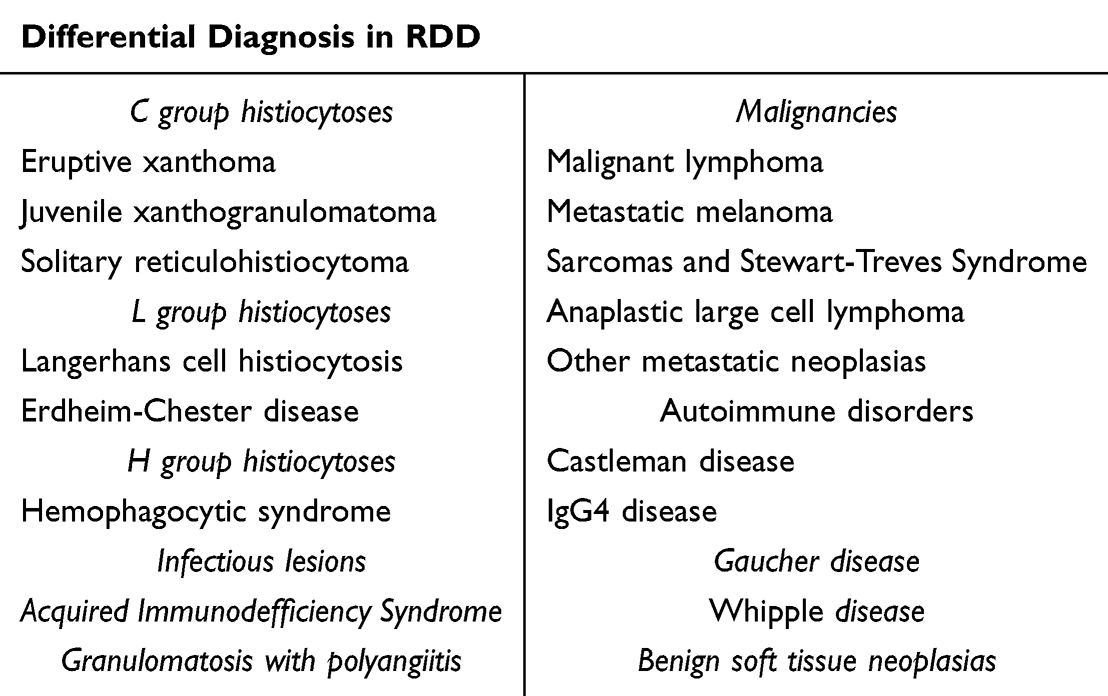 |
Table 2 Differential Diagnosis for Rosai-Dorfman Disease |
In the second case, there was an aggressive presentation, with multifocal lymphadenopathy, intense constitutional symptoms and laboratory markers indicating severe liver and pancreatic damage, with initial suspicion of non-Hodgkin’s lymphoma, and rapid progression to death. The aggressiveness of a normally benign disease, the absence of risk factors or associated diseases show a case of exception. Extranodal disease, particularly the intracranial phenotype, is associated with a poor prognosis, deaths in RDD occur due to direct complications of the disease, infections and amyloidosis. There was not enough time to carry out an adequate investigation of an extranodal condition, and the real cause of splenomegaly and multi-organ failure is not definitively known. Several conditions were considered in the differential diagnosis, such as Langerhans cell histiocytosis, Erdheim-Chester disease and Hemophagocytic Lymphohistiocytosis (HLH), however, the immunohistochemistry result suggested RDD. The serology for Epstein-Barr, an infection commonly associated with HLH, was negative. Thus, based on the patient’s initial clinical manifestations and immunohistochemistry results, the possibility of liver and kidney involvement is considered in an attempt to justify the unfavorable evolution.
Sporadic RDD spontaneously remisses in up to 50% of all cases, mortality is around 10%, occurring due to direct complications of the disease, infections and amyloidosis. Extranodal disease presenting as intracranial or mediastinal RDD seems to have a poorer prognosis, being responsible for 50% of the deaths in a literature review, nosocomial infections, kidney RDD and generalized amyloidosis were also mentioned as the cause of death in case reports.1,12–14
Expectant management is recommended for oligosymptomatic patients and those with unifocal cutaneous RDD. Surgical excision is usually restricted for patients with unifocal extranodal disease. Patients with unresectable multifocal extra nodal RDD and disease affecting airways, intracranial or intraspinal space require systemic therapy, which remains without a standardized regimen, and can include corticosteroids, sirolimus, radiotherapy, chemotherapy (cladribine, methotrexate, 6-mercaptopurine, vinca alkaloids), and immunomodulators (thalidomide, lenalidomide, rituximab, imatinib mesylate).1,5
Conclusion
RDD is rare and presents in a multisystemic form, requiring its inclusion in the differential diagnosis of suspected lymphoproliferative and histiocytic diseases, as well as in skin lesions with difficult characterization and fever of unknown origin.
Acknowledgments
The authors thank the Anticorpos laboratory for their availability in analyzing the samples.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73(11):697–705. PMID: 32591351. doi:10.1136/jclinpath-2020-206733
2. Destombes P. Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases). Bull Soc Pathol Exot Filiales. 1965;58(6):1169–1175.
3. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87(1):63–70.
4. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. doi:10.1182/blood-2016-01-690636
5. Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131(26):2877–2890. doi:10.1182/blood-2018-03-839753
6. Garces S, Medeiros LJ, Patel KP, et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai–Dorfman disease. Modern Pathol. 2017;30(10):1367–1377. doi:10.1038/modpathol.2017.55
7. Durham BH, Lopez Rodrigo E, Picarsic J. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nature Med. 2019;25(12):1839–1842. doi:10.1038/s41591-019-0653-6
8. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6(2):154–165. doi:10.1158/2159-8290.CD-15-0913
9. Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105(2):348–357. doi:10.3324/haematol.2019.219626
10. Albano D, Bosio G, Bertagna F. 18F-FDG PET/CT Follow-up of Rosai-Dorfman disease. Clin Nucl Med. 2015;40(8):e420–e422. doi:10.1097/RLU.0000000000000853
11. Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166–173. doi:10.1016/j.anndiagpath.2019.02.004
12. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1984;7:19–73.
13. Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am J Hematol. 2002;69(1):67–71. PMID: 11835335. doi:10.1002/ajh.10008
14. Lauwers GY, Perez-Atayde A, Dorfman RF, et al. The digestive system manifestations of Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy): review of 11 cases. Hum Pathol. 2000;31(3):380–385. doi:10.1016/s0046-8177(00)80254-3
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinicopathological and Gene Mutation Analysis of 27 Cases with Extranodal Rosai–Dorfman Disease
Tang M, Gu XZ, Wu PC, Yang XT
Journal of Inflammation Research 2022, 15:2775-2787
Published Date: 29 April 2022
CD133, but Not CD44, May Serve as a Novel Biomarker for Differential Diagnosis Between Basal Cell Carcinoma and Trichoblastomas
Bi Y, Shi X, Chen D, Zhao Y
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1517-1526
Published Date: 2 August 2022
Dermoscopy of Primary Localized Cutaneous Nodular Amyloidosis
Pulgarin LM, De Pellegrin A, Stinco G, Errichetti E
Clinical, Cosmetic and Investigational Dermatology 2024, 17:395-398
Published Date: 8 February 2024
Analysis and Perception of Chlamydia psittaci Pneumonia: Novel Insights of the Rare Disease in Infectiology
Huang H, Liu Y, Liu Y, Lin J, Guo H, Xu Q, Zhang H, Li Z, Zhu H, Ding M
Infection and Drug Resistance 2024, 17:5261-5275
Published Date: 28 November 2024
Dermoscopic Features of Erosive Pustular Dermatosis of the Scalp: A Comparative Multicentric Retrospective Study in Bald and Hairy Patients
Plozner N, Zelin E, Zalaudek I, Lallas A, Piraccini BM, Starace M, Errichetti E
Clinical, Cosmetic and Investigational Dermatology 2025, 18:669-676
Published Date: 20 March 2025