Back to Journals » Journal of Inflammation Research » Volume 15
Clinicopathological and Gene Mutation Analysis of 27 Cases with Extranodal Rosai–Dorfman Disease
Authors Tang M, Gu XZ, Wu PC, Yang XT
Received 4 March 2022
Accepted for publication 14 April 2022
Published 29 April 2022 Volume 2022:15 Pages 2775—2787
DOI https://doi.org/10.2147/JIR.S365098
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ming Tang,1,* Xue-Zhong Gu,2,* Peng-Chun Wu,3 Xuan-Tao Yang1
1Department of Pathology, the First People’s Hospital of Yunnan Province, the Affiliated Hospital of Kunming University of Science and Technology, Kunming, 650032, People’s Republic of China; 2Department of Hematology, the First People’s Hospital of Yunnan Province, Yunnan Blood Disease Clinical Medical Research Center, the Affiliated Hospital of Kunming University of Science and Technology, Kunming, 650032, People’s Republic of China; 3Department of Pathology, Kunming Kingmed Institute for Clinical Laboratory, Kunming, 650506, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xuan-Tao Yang, Department of Pathology, the First People’s Hospital of Yunnan Province, the Affiliated Hospital of Kunming University of Science and Technology, No. 157 of Jinbi Road, Xishan District, Kunming, 650032, People’s Republic of China, Tel +86 871 63638441, Email [email protected] Peng-Chun Wu, Department of Pathology, Kunming Kingmed Institute for Clinical Laboratory, K13-K14, Daliyuan Road, Chenggong District, Kunming, 650506, People’s Republic of China, Tel +86 871 68172222, Email [email protected]
Objective: To investigate the clinicopathological features, and mutations of NRAS, KRAS, BRAF and MAP2K1 genes in extranodal Rosai–Dorfman disease (RDD).
Methods: The clinic opathological features of 27 patients with extranodal RDD were retrospectively analyzed, and the NRAS, KRAS, BRAF and MAP2K1 genes mutation were detected by Sanger sequencing.
Results: The male to female ratio was 1.7:1. The average age was 46.9 years. There were skin lesions in 12 cases (44.4%) and head and neck lesions in 8 cases (29.6%). Microscopically, those patients with skin RDD had lesions characterized by clear and dark intervals and obvious emperipolesis, while in other parts, the background was more complex. About 21.1% (4/19) had mutations, including 3 mutations in NRAS 2 exon and 1 mutation in KRAS 2 exon. Two of the three NRAS mutations were located in the skin, accounting for 20% (2/10) of skin RDD.
Conclusion: Extranodal RDD was more common in males than in females, and might occur in all ages, with a greater incidence in skin, head, and neck. Besides the obvious microscopic characteristics in those with skin RDD, the background of other parts was complex and easily missed or misdiagnosed. Some RDD with gene mutations, mainly in NRAS 2 exon, especially in skin RDD, support partial RDD is a clonal disease.
Keywords: Rosai–Dorfman disease, clinical pathology, gene mutation, NRAS, KRAS, BRAF, MAP2K1, diagnosis, identification
Corrigendum for this paper has been published
Introduction
Rosai–Dorfman disease (RDD) is also known as sinus histiocytosis with massive lymphadenopathy. It is a rare non-Langerhans cell histiocytosis with unknown etiology and pathogenesis. It was previously believed to be a polyclonal proliferative disease. In recent years, some studies have found that RDD has different proportions of multiple gene mutations involved in the MAPK/ERK pathway, so at least part of RDD is considered to be tumor.1 RDD has a variety of clinical manifestations, including fever, leukocytosis, accelerated erythrocyte sedimentation rate, and polyclonal gamma globulinemia. It can be divided into the lymph node type, extranodal type, and mixed type, of which the extranodal type is the rarest.2–4 Under the microscope, the forms of extranodal RDD are diverse with complex backgrounds. Inadequate knowledge of the disease may lead to misdiagnosis and mistreatment. In the present study, the clinicopathological features of 27 cases with extranodal RDD were summarized, and literature analysis and discussion were combined to improve the understanding of extranodal RDD to avoid missed diagnosis and misdiagnosis. Meanwhile, the NRAS/KRAS/BRAF/MAP2K1 genes of 22 patients with paraffin blocks were sequenced by fluorescence PCR-capillary electrophoresis sequencing method, and the molecular pathological characteristics of RDD were preliminarily explored to provide theoretical basis for relevant targeted therapy.
Materials and Methods
Clinical Data
Clinical data of 27 patients with extranodal RDD diagnosed from January 2014 to January 2022 were collected, including gender, age, lesion location, treatment, and follow-up duration. Among them, 6 cases were from the First People’s Hospital of Yunnan Province, 9 cases were from the Kunming Jinyu Medical Laboratory, 2 cases were from the Yunnan Provincial Hospital of Traditional Chinese Medicine, and 10 cases were from the Changhai Hospital affiliated to the Second Military Medical University. All cases were surgically resected. No informed consent was required after review by the Ethics Committee of the First People's Hospital of Yunnan Province.
Immunohistochemical Staining
All specimens were fixed with 10% of neutral formalin, routinely dehydrated, and paraffin-embedded. Sections with a thickness of 4 μm were prepared for routine hematoxylin and eosin staining. The diagnosis was reviewed by two experienced pathologist (X.-T. Yang et al) The EnVision method was used for immunohistochemical staining. The primary antibodies used includes S-100, CD68, CDla, CD30, CD20, CD3, IgG and IgG4, all of which are mouse monoclonal antibodies. They were purchased from Fuzhou Maixin Biotechnology Development Co.LTD. The second antibody was purchased from Dako, Denmark. Manual manipulation was referred to instructions. Positive and negative controls were routinely set up.
Sanger Sequencing
NRAS exons 2/3/4, KRAS exons 2/3/4, MAP2K1 exons 2/3/6 and BRAF exon 15 were detected by fluorescence PCR and capillary electrophoresis sequencing. All mutation detection kits are manufactured by Beijing Jingzhun Medical Technology Co., LTD. PCR reaction system was 25μL: PCR reaction solution was 12.5μL, primer mixture was 10.5μL, DNA template was 2.0μL. The amplification conditions were as follows: anti-contamination reaction of UDG enzyme at 37°C for 10min, pre-denaturation at 95°C for 10min, denaturation at 95°C for 15s, annealing at 62°C for 20s, extension at 72°C for 30s, a total of 40 cycles, and then melting curve analysis. According to whether there is a melting peak at the target temperature to determine whether there is a target product. Enzymolysis purification: 3μL PCR amplification product and 2 µL enzymolysis mixture were mixed and reacted at 37°C for 60min, then incubated at 80°C for 15min for enzyme denaturation. Sequencing system configuration: enzymolysis amplification product 2.0μL, sequencing primer 3.0μL, sequencing reagent 1.0μL, a total of 6.0μL sequencing reaction system. The sequencing reaction conditions were as follows: pre-denaturation at 96°C for 1min, denaturation at 96°C for 10s, annealing at 50°C for 5s, and extension at 60°C for 2min, with a total of 30 cycles. The sequencing products were purified by sodium acetate - ethanol precipitation method and sequenced by ABI 3500Dx gene analyzer. The sequencing results were compared with the genome sequence, and the NRAS reference serial number was NM_002524.5. KRAS reference serial number is NM_004985; MAP2K1 reference serial number is NM_002755; The BRAF reference serial number is NM_004333.6.
Results
Clinical Characteristics
There were 27 patients with extranodal RDD, including 17 males and 10 females, with a male to female ratio of 1.7:1. The ages ranged from 10 to 68 years, with an average age of 46.9 years and a median age of 50 years. There were 12 cases of lesions in the skin (44.4%), 8 cases in the head and neck (accounting for 29.6%, including 4 cases in the nasopharynx sinus, 2 cases in the central nervous system, 1 case in the orbit, and 1 case in the salivary gland), 2 cases in the bone, 2 cases in the breast, 1 case in the trachea, 1 case in the colon, and 1 case in the scrotum. Imaging data were available in all cases except those located in the skin. Two cases in the bone showed local osteolytic destruction of the centrum and soft tissue swelling (Figure 1). Other radiographs showed a local solid mass. Superficial mass was considered the main symptom to be treated in all 12 patients with skin lesions. Other clinical manifestations and signs were connected to the lesion sites. The specific clinical conditions characteristics are shown in Table 1.
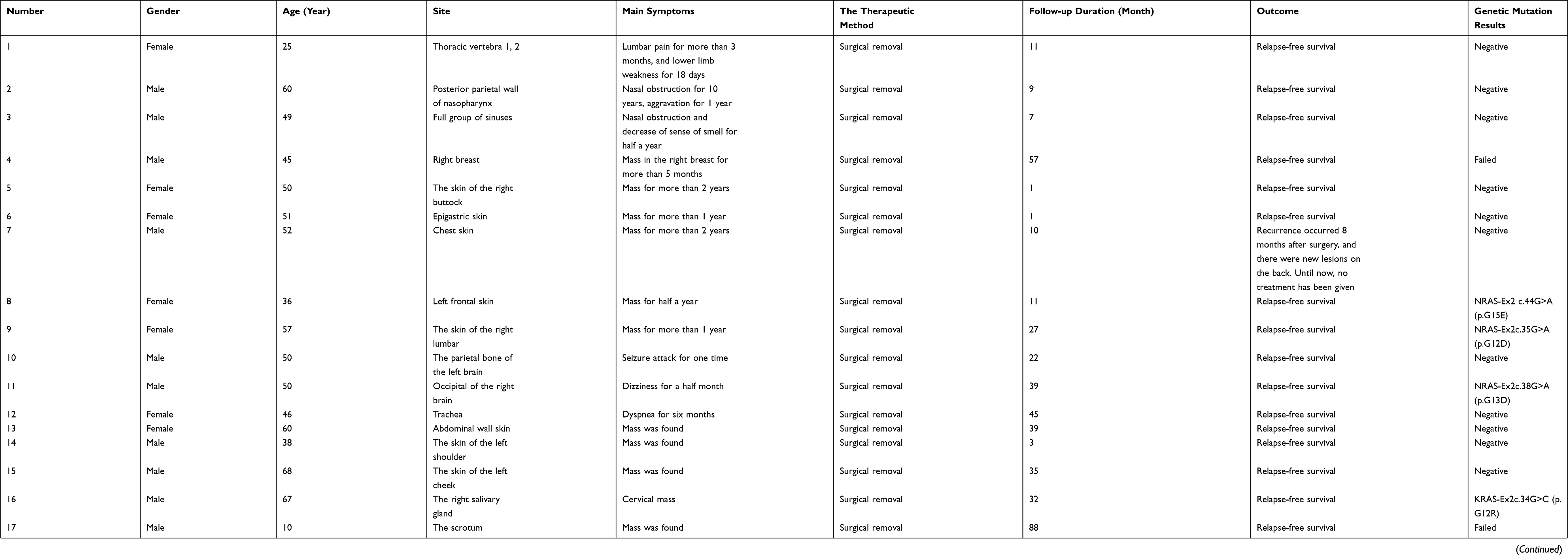 |
Table 1 Clinical Data of 27 Patients with Rosai–Dorfman Disease |
 |
Figure 1 MRI indicated abnormal signal of thoracic vertebra 1, 2 and soft tissue around the appendix of the vertebra. |
Pathological Examinations
In 12 patients with a skin lesion, a distinctive dermal or subcutaneous light and dark pattern were observed under the microscope, with the light and dark areas simulating lymph node structures (Figure 2). The bright area was widened, with large tissue cells appearing mainly flake-like or in a syncytial pattern. The cytoplasm was rich, large in volume, and lightly stained. The nuclei were round or oval, with fine chromatin and small nucleoli. The phenomena of phagocytic lymphocytes, plasma cells, and neutrophils were visible in the cytoplasm. These are also known as extension phenomenon (Figure 3). The dark area was dominated by lymphoplasma cells and some follicles. Different degrees of fibrous tissue hyperplasia were visible in all cases with skin RDD, and some cases had significant local fibrous tissue hyperplasia and showed a striated structure (Figure 4). In two cases located in bone, the bright and dark areas were not featured, and a variety of inflammatory cells were mixed with many neutrophil infiltrations, resembling the chronic suppurative inflammation (Figure 5). This could easily be misdiagnosed. However, careful observation showed a huge volume of tissue cells was still visible, and some of the cytoplasm was foamy and showed extension. Two cases located in the central nervous system did not obviously feature bright and dark areas but showed diffused infiltration of many lymphocytes and plasma cells accompanied by small vascular proliferation (Figure 6). The four cases located in the nasopharynx of the nasal sinus had the characteristic structure of bright and dark areas, but the number of RDD tissue cells decreased in some areas, with an obvious inflammatory background accompanied by fibroblasts/myofibroblasts hyperplasia. This was easily missed by those with less experience. In the remaining cases, different degrees of dark and bright structures, extension, and fibrous hyperplasia appeared. No obliterating phlebitis was found in any case.
 |
Figure 2 Lesions in the bright and dark areas of the dermis stimulate the lymph node structure (HE). |
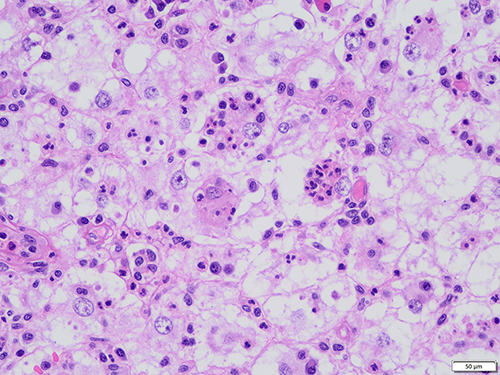 |
Figure 3 The phenomenon of phagocytic lymphocytes, plasma cells, and neutrophils in the cytoplasm (HE). |
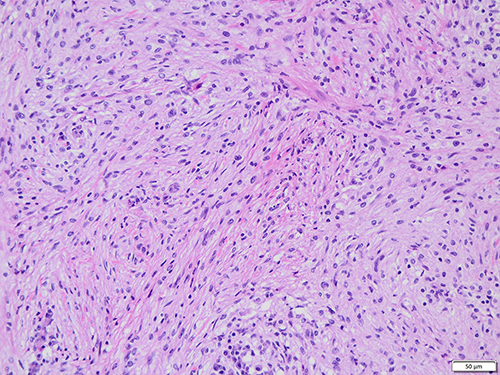 |
Figure 4 Obvious fibrous tissue hyperplasia with storiform structure in some areas (HE). |
 |
Figure 5 The features of bright and dark areas were not obvious with extensive neutrophil infiltration (HE). |
 |
Figure 6 Diffused infiltration of a large number of lymphocytes and plasma cells accompanied by small vascular proliferation. |
Immunohistochemical Results
The large tissue cells were positive for S–100 (Figure 7) and CD68. Conversely, CD1a and CD30 were negative. The background lymphocytes were positive for CD20 and CD3. A small number of IgG4-positive cells (about 10–30 per high-powered field [HPF]) were detected in 1 case with RDD in the central nervous system, 8 case with RDD in the skin, 1 case with RDD in the salivary gland, and 1 case with RDD in the trachea, with the IgG4/IgG ratio <40%. All the remaining cases were negative for IgG4.
 |
Figure 7 Positive for S100 in the histiocytes. The En Vision two-step method. |
Sanger Sequencing Results
Among 27 patients, 5 cases were sent to our hospital for consultation and could not obtain paraffin blocks, so a total of 22 cases were sequenced. However, there were 3 cases with poor DNA quality and sequencing failure due to old paraffin blocks or improper preservation, so there were actually 19 cases with sequencing results. Single gene mutation was detected in 4 cases, accounting for 21.1% (4/19), of which 3 cases (15.8%, 3/19) were mutations in NRAS 2 exon, which were c.44G > A (p.G15E), c.35G > A (p.G12D) and c.38G >A (p.G13D) (Figures 8–10). One case (5.3%, 1/19) was KRAS 2 exon mutation, c.34G> C (p.G12R) (Figure 11). Two of the three NRAS mutations (Cases 8 and 9) were located in the skin, accounting for 20% (2/10) of skin RDD, and one case (case 11) was located in the occipital part of the right brain. KRAS mutation was located in the right submandibular gland (case 16). BRAF and MAP2K1 gene mutations were not detected in all cases. See Table 1 for details.
 |
Figure 8 NRAS-Ex2 c.44G>A (p.G15E). |
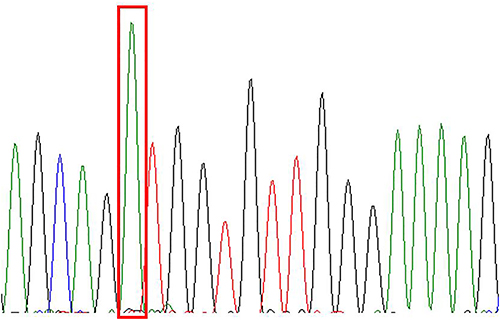 |
Figure 9 NRAS-Ex2c.35G>A (p.G12D). |
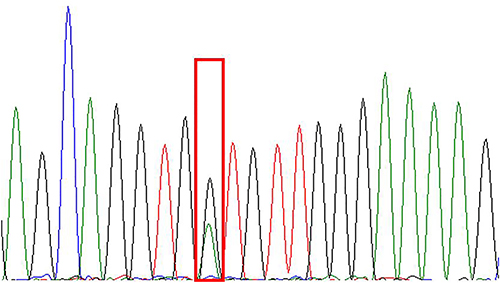 |
Figure 10 NRAS-Ex2c.38G>A (p.G13D). |
 |
Figure 11 KRAS-Ex2 c.34G>C (p.G12R). |
Follow-Up
The minimum follow-up duration was one month, the maximum was 88 months, and the median was 31 months. Six cases were lost during the follow-up period. One patient with skin lesions had local recurrence and new lesions 8 months after surgery, and until now, no treatment has been given. There was no recurrence in other cases.
Discussion
Destombes first reported RDD in 1965, and it was named by Rosai and Dorfman in 1969.5 The incidence is approximately 0.005%, and it affects all age groups. In 2016, the International Society for Tissue-cell Tumor Research classified the tissue cells into R groups according to their origin and divided them into familial, tumor-related, immune-related, and IgG4-related RDD.3 According to the different sites involved, the lesions can be divided into the lymph node type, extranodal type, and mixed type. The lymph node type and mixed type are the most common, while the extranodal type is the least common (approximately 10%–20%).3,4 The male to female ratio of 27 patients with extranodal RDD in the present group was 1.7:1. The age range was 10–68 years, with an average age of 46.9 years and a median age of 50 years. The skin was the most commonly affected location (44.4%), followed by the head and neck (29.6%).
The etiology of RDD is unknown. It may involve viral infections, malignant tumors, host immune disorders, IgG4-related diseases, and various autoimmune diseases, but no conclusion as to its cause has been reached. Olmedo–Reneaum et al6 reported a case of a person with HIV, RDD, and simultaneous infection with Salmonella and Mycobacterium avium who eventually died. Gorodetskiy et al7 reported a case of a person with Sjögren’s syndrome accompanied by RDD and lymph node marginal region lymphoma in which the Epstein–Barr virus was detected in both the RDD and lymphoma tissues. It is suggested that viral infection may create an immune environment that ultimately leads to activation of the histiocyte–macrophage system or tumor.
In recent years, some researchers have paid attention to the relationship between IgG4-related diseases and RDD. Some researchers believe there is a morphological overlap between the two with the number of plasma cells in RDD increased and an IgG4/IgG ratio of >40%, so the two diseases are considered being correlated.8 However, most researchers believe the two diseases are distinct, with different clinical manifestations and therapeutic methods. Infrequently, an increase of IgG4-positive cells may occur in RDD, and more often, the two diseases only overlap in morphology and need to be distinguished.9,10 A small number of IgG4-positive cells (approximately 10–30 per HPF) were observed in 11 of the 27 patients in the present group, and the IgG4/IgG ratio was <40%. The other cases were negative for IgG4, and no occlusive phlebitis or storiform structures were found in the morphology. Therefore, it was not enough to prove an association between RDD and IgG4-related diseases.
Because of the extremely low incidence of RDD, there are relatively few studies on its pathogenesis. Most of the studies are first-generation or second-generation sequencing studies of individual cases, and no consensus has been reached. In recent years, a relatively large number of case studies have found mutually exclusive gene mutations of Kirsten rat sarcoma viral oncogene homolog (KRAS) and mitogen-activated protein kinase (MAPK) kinase (MEK) 1 (MAP2K1) in RDD. Garces et al1 investigated 134 genes commonly associated with tumors of the lymphohematopoietic system in 21 patients with RDD, and mutations were detected in 33% (7/21) of patients. Four of the patients had the KRAS mutation and three had the MAP2K1 mutation. In all cases, B-Raf proto-oncogene serine/threonine kinase (BRAF) gene mutations were not detected. Statistically, patients with the mutated gene were younger and experienced RDD in the head and neck more commonly than those without the mutation. Three cases with the MAP2K1 mutation were accompanied by phosphor–extracellular signal-regulated kinase (p–ERK) expression. Therefore, the authors believed that, at least in some patients, the development of RDD might be related to the MAPK/ERK pathway, and it might be a clonal disease. Mastropolo et al11 detected the BRAF–V600E mutation in a child with both RDD and Langerhans’ histiocytosis. After 3 months of treatment with the BRAF inhibitor, the clinical and imaging changes were improved. Deletion of exon 12 of the BRAF gene has been detected in RDD involving the central nervous system.12 One patient with the KRAS 117N mutation showed atypical histological changes and an invasive clinical course.13 Therefore, the pathogenesis of RDD needs to be studied in a larger population of cases.
In this study, gene sequencing results were obtained from 19 cases, which is a relatively large number of sequencing studies. About 21.1% (4/19) were found to have mutations, including 3 mutations in exon NRAS 2 and 1 mutation in exon KRAS 2. NRAS 2 exon mutation was found in 2 (20%) of the 10 skin RDD with sequencing results. Nras-ex2 C.38g >A (P.G13d) mutation was found in one case of RDD located in the occipital part of the right brain. One case of mandibular gland RDD had KRAS 2 exon C.34G>C (P.G12r) mutation. Our results further support the view that some RDD is monoclonal, disease and NRAS-EX2 mutations are predominant, especially in skin RDD. Wu et al14 found that about 57.1% (4/7) of skin RDD had NRAS mutations. In our study, the proportion of NRAS mutations was relatively low, which may be due to our large number.
Clinical manifestations vary according to the site of RDD involvement. Patients with lymph node involvement may mainly manifest symptoms like painless enlargement of lymph nodes with or without fever, night sweats, weight loss, etc. The common extranodal sites include the skin, head and neck, and bone, as well as the urogenital system, the central nervous system, and other soft tissues, which are manifested by symptoms and signs at corresponding sites.4,5,12,13 Patients with skin RDD involvement mainly manifested symptoms as a local mass and skin lesions on the body surface. Patients with the nose involved mainly manifested symptoms as nasal obstruction, epistaxis, nasal back malformation, a nasal mass, reduced hearing, and loss of the sense of smell. Those with RDD involvement in the central nervous system might manifest symptoms of facial paralysis, diplopia, headache, and somatosensory and functional disorders. Patients with bone RDD involvement might mainly present with local pain, accompanied by limb dysfunction. Radiographically, RDD might manifest as osteolytic and accompanied by a soft tissue mass, tumor-like, or an infectious disease. In the present group, infectious lesions were diagnosed by imaging in cases 1 and 3, and a malignant tumor was considered in case 2.
The diagnosis of RDD was confirmed mainly by pathological examination. Due to the rarity and the complex morphology of some cases, the literature shows that the number of biopsies required for diagnosis of RDD is 1–6, with a median of 2.2 Under low magnification, the lymph node follicles and sinus structures were visible, showing interspersed bright areas and dark areas, which is an important background suggestive of RDD. Under high magnification, the tissue cells in the bright area are grouped or flaky, with large cell volumes. The cytoplasm is abundant, eosinophilic or transparent, and foamy with ill-defined cell boundaries. The nuclei are round or oval, with light nuclear chromatin. Sometimes, the nucleoli are prominent and the karyokinesis aspect is rare. The dark area is mainly composed of lymphocytes, plasma cells, and a few neutrophils, with occasional germinal centers of lymphatic follicles. Another important histological feature of RDD is lymphocyte extension, which is the phagocytosis of lymphocytes, plasma cells, or neutrophils in the tissue cytoplasm.
The results in the present study showed that in patients with extranodal RDD in the skin, the microscopic light-dark interphase feature was obvious, and lymphocytes were easily visible. In patients with extranodal RDD in other locations, the background was more complex, especially in lesions located in the bones, sinus nasopharynx, and the central nervous system. In the later cases, more neutrophils, lymphocytes, plasma cell infiltrations with fibrous tissue, and small blood vessel hyperplasia were observed, which interfered with the diagnosis. Sometimes, the inflammatory cells in the pathological background were prominent, especially when many neutrophil infiltrations existed, which might easily be misdiagnosed as chronic suppuritis by those with insufficient experience. Some patients with RDD have a long history that may include significant fibrosis. The reduction of typical RDD cells, coupled with increased inflammatory cell infiltration, is also easily missed in diagnosis. Immunohistochemistry showed the tissue cells were positive S–100 and CD68, while negative for CD1a, langerin, and CD30.
Extranodal RDD should be differentiated from the following diseases: (1) Chronic inflammation. The lesions were characterized by infiltration of lymphocytes and plasma cells and interstitial fibrous tissue hyperplasia. The hyperplasia of tissue cells was not obvious, and there was no lymphocyte extension; (2) Langerhans’ cell histiocytosis. There is a characteristic eosinophil background with relatively small tissue cells and a coffee-like nucleus with furrows and inconspicuous nucleoli. Under low magnification, there was no alternating light and dark area and no lymphocyte extension; (3) Erdheim–Chester disease. It is a rare, systemic disease of non-Langerhans’ cell histiocytosis that mainly affects the skeleton. Histology showed a characteristic infiltration of lipid-rich histiocytic cells in the bone marrow lumen, negative for S–100 and CD1a; (4) Rhinoscleroma, which should be distinguished from RDD occurring in the nose and sinuses. The vacuolated tissue cells, neutrophils, and plasma cells were visible microscopically in Rhinoscleroma, but the vacuolated tissue cells were S–100 negative, and bacillus rhinoscleromatis might be found by Giemsa staining; (5) Lymphoma. All the above differential diagnoses were based mainly on immunohistochemistry. The tissue cells in RDD lesions expressed CD68 and S–100 but did not express CD1a, CD30, anaplastic lymphoma kinase, etc. The background lymphocytes and plasma cells showed polyclonal expression of T cells; B cells; and plasma cell markers, such as CD2, CD3, CD20, CD19, CD38, CD138, κ, and λ.
The prognosis of RDD has not been determined. A few cases are self-limited, while most are not self-healing and are prone to recurrence with involvement of the surrounding tissues. Some cases of refractory or death have been reported.11–13,15 In 2016, the International Society for Histiocytic Oncology (including pathologists, pediatricians, oncologists, hematologists, internal medicine, and other clinicians) first proposed multidisciplinary consensus recommendations for the diagnosis and clinical management of RDD.3
The recommendations state that uncomplicated lymphadenopathy or asymptomatic skin RDD should be monitored first, since approximately 20%–50% of lymph node/skin RDD will spontaneously resolve. Surgery is usually limited to biopsy or excision of a single lesion that can be cured.
For patients with respiratory tract obstructions with lesions in the nasopharynx and sinus or other important areas, such as intracranial or in the spinal cord, surgical treatment should be the first choice. Glucocorticoid therapy can be used in patients requiring systemic therapy, but the reliability and duration of efficacy are uncertain.3,16 Radiotherapy can be recommended as adjuvant therapy in some patients, but the dose and cycle of radiotherapy have not been determined.3 Pagel et al17 reported that a patient with CD20-positive RDD received rituximab treatment leading to remission.
In recent years, with the discovery of gene mutations, there have been reports of the benefits of BRAF or MEK inhibitors.11,18 All 27 patients in the present group were resected by operation. One patient relapsed 8 months after surgery, and until now, no treatment has been given. Twenty cases survived without recurrence, and six cases did not survive the follow-up interval.
Conclusion
RDD is a rare histiocytoproliferative disease with no characteristic changes in clinical manifestations and imaging, and the diagnosis is mainly based on pathological examination. Especially in patients with extranodal RDD, the background was complex and often lacked characteristic histological findings. Immunohistochemistry S–100 and CD68 might be used for differential diagnosis. Our study found that about 21.1% of RDD had gene mutations, mainly in NRAS 2 exon, which accounting for 20% in skin RDD. A few mutations were located in KRAS 2 exon. It indicates that NRAS and KRAS mutation in MAPK/ERK pathway may involve the pathogenesis of RDD.Therefore, we believe that some RDD is a clonal disease. At the same time, this study provides a theoretical basis for taking relevant targeted therapy.
Data Sharing Statement
All data generated or analysed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Ethics Approval and Consent to Participate
The current study met the requirements of the Declaration of Helsinki of the World Medical Association. Since this study did not require any intervention or experiment related to patients, no informed consent was required after review by the Ethics Committee of the First People's Hospital of Yunnan Province.
Acknowledgments
We are particularly grateful to all the people who have given us help on our article. Especially thanks to Miao-xia He for providing case data, who is the chief pathologist in Department of Pathology of Changhai Hospital affiliated to the Second Military Medical University. And sincerely thanks to Tao Liu for providing case data and rechecking diagnosis, who is the chief pathologist in Yunnan Hospital of Traditional Chinese Medicine.
Funding
The study was supported by the Yunnan Health Training Project of High Level Talents (NO. D-2018018), Yunnan Applied Basic Reserch Projects-Joint fund for Applied Basic Research of Kunming Medical University, Yunnan Provincial Department of Science and Technology (NO.2018FE001[-113])and Open Project of Yunnan Clinical Medical Center (NO.2020LCZXKF-XY02).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Garces S, Medeiros LJ, Patel KP, et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai–Dorfman disease. Mod Pathol. 2017;30(10):1367–1377. doi:10.1038/modpathol.2017.55
2. Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105(2):348–357.
3. Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131(26):2877–2890.
4. Al-Khateeb TH. Cutaneous Rosai-Dorfman Disease of the Face: a Comprehensive Literature Review and Case Report. J Oral Maxillofac Surg. 2016;74(3):528–540.
5. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87(1):63–70.
6. Olmedo-Reneaum A, Molina-Jaimes A, Conde-Vazquez E, Montero-Vazquez S. Rosai-Dorfman disease and superinfection due to Salmonella enterica and Mycobacterium avium complex in a patient living with HIV. IDCases. 2020;19:e00698.
7. Gorodetskiy VR, Klapper W, Probatova NA, Vasilyev VI, Rozhnova EV. Simultaneous Occurrence of Rosai-Dorfman Disease and Nodal Marginal Zone Lymphoma in a Patient with Sjögren’s Syndrome. Case Rep Hematol. 2018;2018:7930823.
8. Tracht J, Reid MD, Xue Y, et al. Rosai-Dorfman Disease of the Pancreas Shows Significant Histologic Overlap With IgG4-related Disease. Am J Surg Pathol. 2019;43(11):1536–1546.
9. Chen LYC, Slack GW, Carruthers MN. IgG4-related disease and Rosai-Dorfman-Destombes disease. Lancet. 2021;398(10307):1213–1214.
10. Liu L, Perry AM, Cao W, et al. Relationship between Rosai-Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol. 2013;140(3):395–402.
11. Mastropolo R, Close A, Allen SW, McClain KL, Maurer S, Picarsic J. BRAF-V600E-mutated Rosai-Dorfman-Destombes disease and Langerhans cell histiocytosis with response to BRAF inhibitor. Blood Adv. 2019;3(12):1848–1853.
12. Richardson TE, Wachsmann M, Oliver D, et al. BRAF mutation leading to central nervous system Rosai-Dorfman disease. Ann Neurol. 2018;84(1):147–152.
13. Jafri ZA, Reddy SP, Cassarino DS. KRAS 117N positive Rosai-Dorfman disease with atypical features. J Cutan Pathol. 2021;48(1):147–150.
14. Wu KJ, Li SH, Liao JB, et al. Be Involved in the Pathogenesis of Cutaneous Rosai Dorfman Disease: a Pilot Study. Biology. 2021;10(5):396.
15. Dalia S, Sagatys E, Sokol L, Kubal T. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21(4):322–327.
16. Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am J Hematol. 2002;69(1):67–71.
17. Pagel JM, Lionberger J, Gopal AK, Sabath DE, Loeb K. Therapeutic use of Rituximab for sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Am J Hematol. 2007;82(12):1121–1122.
18. Jacobsen E, Shanmugam V, Jagannathan J. Rosai-Dorfman Disease with Activating KRAS Mutation - Response to Cobimetinib. N Engl J Med. 2017;377(24):2398–2399.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Case Study: Rosai-Dorfman Disease and Its Multifaceted Aspects
Werneck Rodrigues DO, Wolp Diniz R, Dentz LC, Costa MA, Lopes RH, Suassuna LF, Cintra JRD, Domenge C
Journal of Blood Medicine 2024, 15:123-128
Published Date: 11 March 2024