Back to Journals » Cancer Management and Research » Volume 18

CAR-T Cell Therapy in Glioblastoma: Overcoming Immunological Barriers and Advancing Clinical Translation
Authors Pourmasoumi P ![]() , Pourmasoumi A, Zamani F, Amini A, Shokri S
, Pourmasoumi A, Zamani F, Amini A, Shokri S
Received 10 November 2025
Accepted for publication 13 February 2026
Published 17 February 2026 Volume 2026:18 580320
DOI https://doi.org/10.2147/CMAR.S580320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Alessandro Rizzo
Parvin Pourmasoumi,1,2 Ali Pourmasoumi,3 Farshid Zamani,4 Abdollah Amini,2 Shiva Shokri5
1Department of Biomedical Engineering, Central Tehran Branch, Islamic Azad University, Tehran, Iran; 2Department of Biology and Anatomical Sciences, Shahid Beheshti University of Medical Sciences (SBMU), Tehran, Iran; 3Department of Biology, Science and Research Branch, Islamic Azad University, Tehran, Iran; 4Department of Immunology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 5School of Medicine, Iran University of Medical Sciences, Tehran, Iran
Correspondence: Parvin Pourmasoumi; Abdollah Amini, Email [email protected]; [email protected]; [email protected]
Abstract: Chimeric antigen receptor (CAR) T-cell therapy has delivered unprecedented clinical benefit in hematological malignancies, yet its successful translation to solid tumors remains a major unmet clinical challenge. Glioblastoma (GBM), the most aggressive primary brain tumor, exemplifies the barriers confronting cellular immunotherapy, including profound intratumoral heterogeneity, antigenic escape, a highly immunosuppressive tumor microenvironment, and anatomical constraints imposed by the blood–brain barrier. Together, these features limit CAR-T cell trafficking, persistence and sustained antitumor activity in the central nervous system.In this Review, we examine recent advances in next-generation CAR-T engineering strategies aimed at overcoming these obstacles in GBM. Key developments include multi-antigen and logic-gated CAR designs to mitigate tumor immune evasion, armored CAR-T cells capable of cytokine delivery or resistance to suppressive mediators such as TGF-β, and checkpoint-resistant constructs to counteract functional exhaustion. We also highlight emerging delivery paradigms — including locoregional administration, viral vectors and nanotechnology-enabled platforms — designed to enhance blood–brain barrier penetration and intratumoral retention. Furthermore, we discuss combinatorial strategies integrating CAR-T therapy with immune checkpoint blockade, oncolytic virotherapy and other immunomodulatory interventions to remodel the hostile glioblastoma microenvironment and amplify therapeutic efficacy. Finally, we address the principal translational challenges that must be resolved for broader clinical implementation, including neurotoxicity, manufacturing scalability and the development of predictive preclinical models.Collectively, these multidisciplinary advances provide a roadmap for optimizing CAR-T cell therapy in glioblastoma and accelerating its translation toward durable clinical benefit.
Keywords: CAR-T cell therapy, glioblastoma, tumor microenvironment, blood–brain barrier, antigen heterogeneity, nanodelivery systems, immune checkpoint blockade, translational challenges
Introduction
Glioblastoma multiforme (GBM) is the most aggressive and lethal primary malignancy of the central nervous system and remains the most common form of malignant brain tumor in adults. It accounts for approximately 14.5% of all central nervous system (CNS) tumors and nearly 48.6% of malignant CNS neoplasms. Despite continuous advances in surgical and adjuvant treatment strategies, the prognosis of GBM patients remains extremely poor, with a median overall survival of approximately 15 months.1,2 Current standard management, consisting of maximal safe surgical resection followed by radiotherapy and temozolomide chemotherapy, provides only modest survival benefit and is rarely curative.3 The therapeutic failure in GBM is largely attributable to its complex biological characteristics, including diffuse infiltration into surrounding brain tissue, substantial intra- and inter-tumoral heterogeneity, and a profoundly immunosuppressive tumor microenvironment (TME).4,5 These features not only promote resistance to conventional therapies but also pose major obstacles to emerging immunotherapeutic approaches. In addition, recent studies emphasize that GBM progression is strongly shaped by evolving clinical and translational challenges, including immune escape and treatment-associated toxicity, highlighting the need for innovative therapeutic paradigms.6 In recent years, immune-based therapies have transformed the treatment landscape of several solid tumors, such as melanoma and non–small cell lung carcinoma, by enhancing endogenous immune recognition and elimination of malignant cells.7 However, GBM has shown limited responsiveness to immune checkpoint inhibitors, largely due to its immunologically “cold” phenotype, low tumor mutational burden, and anatomical constraints such as the blood–brain barrier (BBB), which restrict immune cell trafficking into the CNS. Although the BBB is partially disrupted in GBM, heterogeneous permeability and the presence of a blood–tumor barrier still limit uniform immune-cell infiltration into tumor sites. Consequently, there is a critical unmet need for alternative immune strategies capable of overcoming these barriers.8 Chimeric antigen receptor (CAR) T-cell therapy represents one of the most promising advances in cancer immunotherapy. By genetically engineering patient-derived T lymphocytes to recognize tumor-associated antigens (TAAs) in an MHC-independent manner, CAR-T therapy has achieved remarkable clinical success in hematological malignancies, including acute lymphoblastic leukemia and diffuse large B-cell lymphoma, resulting in durable remissions and long-term survival benefits.9,10 These achievements have stimulated growing interest in adapting CAR-T approaches to solid tumors, including GBM. Nevertheless, CAR-T cell therapy in glioblastoma faces distinct and multifactorial challenges. Antigenic heterogeneity within GBM promotes immune escape through antigen loss variants, while the immunosuppressive TME inhibits CAR-T activation and persistence. Moreover, limited CAR-T trafficking into brain tumors, partly mediated by BBB-associated restrictions and abnormal tumor vasculature, further reduces therapeutic efficacy.11 Early clinical and preclinical studies targeting antigens such as EGFRvIII and IL-13Rα2 have demonstrated proof-of-concept activity; however, responses remain variable and often transient, underscoring the need for improved engineering strategies.12,13
To address these limitations, recent advances have focused on multi-antigen targeting CAR designs, armored CAR-T cells capable of secreting immunostimulatory cytokines, checkpoint-resistant constructs, and novel delivery approaches that enhance CNS penetration and local persistence.14 Combination strategies that integrate CAR-T therapy with immune checkpoint blockade, oncolytic virotherapy, and nanotechnology-based delivery systems are being actively investigated to enhance antitumor immunity and improve clinical outcomes. Nanoparticle-enabled delivery platforms, in particular, offer potential to facilitate BBB penetration and promote locoregional persistence of CAR-T cells and immunomodulatory agents within the CNS tumor microenvironment.
In this review, we provide an integrative and up-to-date framework of the latest progress in CAR-T cell therapies for GBM. We discuss the major biological and physical barriers limiting therapeutic efficacy, highlight innovative engineering and combinatorial strategies developed to overcome immune suppression and antigen escape, and address key translational challenges such as toxicity management, manufacturing scalability, and clinical implementation. Ultimately, a multidisciplinary approach will be essential to advance CAR-T therapies toward effective and durable treatment options for patients with glioblastoma.
Methodology for Literature Review
A structured literature review was performed to identify recent advances in CAR-T cell therapy for glioblastoma (GBM). Electronic databases including PubMed, Scopus, and Web of Science were systematically searched for articles published between 2006 and 2026. The search strategy combined keywords and Medical Subject Headings (MeSH) such as “CAR-T cell therapy,” “glioblastoma,” “tumor microenvironment,” “blood–brain barrier,” “antigen escape,” “immune checkpoint inhibitors,” and “oncolytic virotherapy.”
The initial search yielded approximately 352 publications. After removal of duplicates, titles and abstracts were screened to exclude studies unrelated to GBM, non–CAR-based immunotherapies, conference abstracts, and articles published prior to 2010. Full-text assessment was subsequently conducted to determine eligibility.
Studies were included if they (i) investigated CAR-T engineering strategies relevant to glioblastoma, (ii) addressed immunosuppressive mechanisms within the GBM microenvironment, or (iii) evaluated combinatorial approaches such as checkpoint blockade or oncolytic virotherapy. Both preclinical and clinical studies were considered. Importantly, article selection was based on scientific relevance, originality, and translational significance, rather than journal impact factor alone, leading to the final inclusion of 74 key studies.
Tumor Immune Microenvironment of GBM
The tumor microenvironment (TME) of gliomas consists of a heterogeneous network of malignant cells interacting with various non-neoplastic components, including neural, vascular, stromal, stem, and immune cells. Within this ecosystem, the glioma tumor immune microenvironment (TIME) is a highly dynamic and complex niche that profoundly influences tumor growth, immune escape, and therapeutic resistance. Owing to its limited immune infiltration and suppressive characteristics, GBM is often classified as an “immunologically cold” or immune-hostile tumor,15 as illustrated in Figure 1.
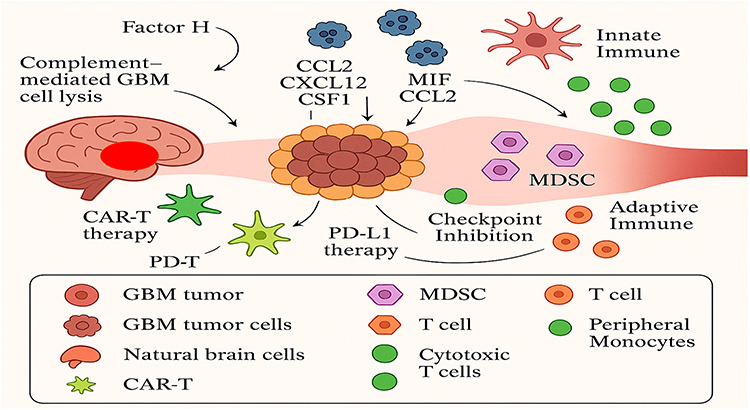 |
Figure 1 Overview of cellular and immune interactions within the glioblastoma microenvironment. Adapted from Datsi et al.15 |
The glioblastoma (GBM) tumor microenvironment contains a diverse population of immune cells, primarily tumor-associated macrophages (TAMs), microglia, myeloid-derived suppressor cells (MDSCs), dendritic cells, and various subsets of T lymphocytes. These immune components form a substantial part of the GBM microenvironment and play critical roles in modulating tumor behavior. Glioma cells release a range of metabolites and signaling molecules that reshape immune cell function, promoting the infiltration of immunosuppressive regulatory T cells (Tregs) and MDSCs into the (TIME). This remodeling process disrupts effective tumor recognition and dampens antitumor immunity. Moreover, the overexpression of metabolic enzymes further suppresses cytotoxic immune activity by limiting the recruitment of natural killer (NK) and cytolytic T cells, while enhancing inflammatory pathways that favor tumor progression.16
Characterized by invasive growth, extensive angiogenesis, chronic inflammation, hypoxic and necrotic regions, and profound immune suppression, the (TIME) establishes a permissive niche that supports glioblastoma survival and progression. A notable feature of GBM, considering its cold environment and low immunological immunity, is the strong infiltration of neoplastic cells such as immune cells, endothelial cells, and fibroblasts, whose cellular interactions with proinflammatory cytokines, chemokines, growth factors, or direct cellular interactions enhance pro -pro-tumorigenic forces at TIME.17 The accumulation and functional activation of anti-inflammatory immune populations give rise to a profoundly immunosuppressive (TIME), which facilitates glioma cell invasion while dampening effective antitumor immune responses.18 Tumor progression and immune escape in glioblastoma are largely driven by the intricate network of interactions between immune cells and signaling molecules within the (TIME). Among these regulatory components, glioblastoma-derived exosomes (GDEs) act as pivotal mediators that reprogram tumor-associated macrophages (TAMs) and other immune populations, thereby enhancing immunosuppression and promoting tumor growth and invasion.15 Tumor-associated macrophages (TAMs) display remarkable plasticity, adopting either proinflammatory, anti-tumor (M1-like) or immunosuppressive, pro-tumor (M2-like) phenotypes, with the latter predominating in glioblastoma. These cells promote tumor progression by secreting cytokines and growth factors that suppress T-cell activation, enhance angiogenesis, and support tumor proliferation. TAMs originate from two major sources—bone marrow–derived monocytes and resident microglial cells within the central nervous system.19 Glioblastoma-derived exosomes (GDEs) are detected both within the tumor microenvironment and among infiltrating immune cells. They interact with classically activated M1 macrophages, driving their phenotypic conversion toward the immunosuppressive M2 state. This reprogramming occurs through multiple signaling pathways, including the upregulation of Programmed Death-Ligand 1 (PD-L1) expression, which further dampens antitumor immune activity.20 Extracellular vesicles released by glioblastoma stem cells (GSCs) profoundly influence dendritic cell (DC) metabolism by promoting lipid accumulation and triggering ferroptotic cell death. This metabolic reprogramming is mediated through activation of the nuclear factor erythroid 2–related factor 2 (NRF2)/glutathione peroxidase 4 (GPX4) signaling pathway, ultimately leading to impaired DC function and weakened antitumor immunity.18 Altering the metabolism of dendritic cells (DCs), also known as immunometabolism, is a conducive area of research for improving DC-based immunotherapies, particularly in cancer treatment. By manipulating metabolic pathways within DCs, researchers aim to enhance their ability to activate immune responses and overcome limitations of existing therapies.18,21 This implicates targeting various metabolic processes like glycolysis, lipid metabolism, and amino acid metabolism to collimate DC function. GDEs, as mediators in the GBM tumor microenvironment, are strongly associated with immunosuppression and tumor growth enhancement due to components of the signal transducer and activator of transcription 3 (STAT3) signaling pathway. These exosomes can carry arginase-1 and Intercellular Adhesion Molecule 1 (ICAM-1), both of which are involved in immunosuppression.18 Exosomes are extracellular vesicles that act as messengers between cells, and their cargo can significantly influence the immune response. Arginase-1 is an enzyme that degrades arginine, an amino acid crucial for T cell function, thus suppressing immune activity. ICAM-1, on the other hand, is an adhesion molecule that can mediate the interaction between exosomes and immune cells, potentially facilitating immunosuppression, which macrophage migration, proliferation, and phagocytosis, eventually supporting tumorigenesis, particularly under hypoxic conditions.18,21 GBM cells secrete branched-chain ketoacids (BCKAs) into the tumor microenvironment, influencing the behavior and polarization of neighboring immune cells. Specific metabolites, such as α-ketoisocaproate (KIC) and α-keto-β-methylvalerate (KMV), have been shown to promote a pro-tumoral macrophage phenotype, whereas α-ketoisovalerate (KIV) exerts pro-inflammatory effects. This bidirectional metabolic communication highlights the intricate interplay between glioma cells and immune populations within the tumor microenvironment.22
In addition, polysialic acid (polySia)—a sialic acid homopolymer enriched in glioblastoma-derived exosomes (GDEs), which are small extracellular vesicles that mediate intercellular communication within the tumor microenvironment—interacts with sialic acid–binding immunoglobulin-like lectin 16 (Siglec-16) on tumor-associated macrophages (TAMs), triggering pro-inflammatory signaling cascades. Conversely, BCKA uptake by TAMs attenuates their phagocytic function, further contributing to immune evasion. The reprogramming of the GBM microenvironment is also tightly orchestrated by microglia, the resident immune cells of the central nervous system, which adapt their functional state in response to these tumor-derived cues.23,24
Engineering CAR-T Cells to Overcome Immunosuppression and Enhance Therapeutic Synergy in Glioblastoma
One of the major obstacles limiting the efficacy of CAR-T cell therapy in glioblastoma (GBM) is the profoundly immunosuppressive tumor microenvironment (TME). Within this hostile milieu, CAR-T cells are exposed to inhibitory cytokines such as transforming growth factor-β (TGF-β) and interleukin-10 (IL-10), metabolic stress, and suppressive immune populations including regulatory T cells (Tregs) and myeloid-derived suppressor cells. These factors collectively impair CAR-T cell activation, persistence, and cytotoxic function.25
To address these challenges, recent genetic engineering strategies have focused on enhancing CAR-T resistance to immune suppression, improving intratumoral persistence, and synergizing CAR-T activity with complementary immunotherapeutic approaches.26 An overall schematic summary of representative CAR-T targets, key immunological barriers, and emerging engineering strategies in GBM is presented in Figure 2.
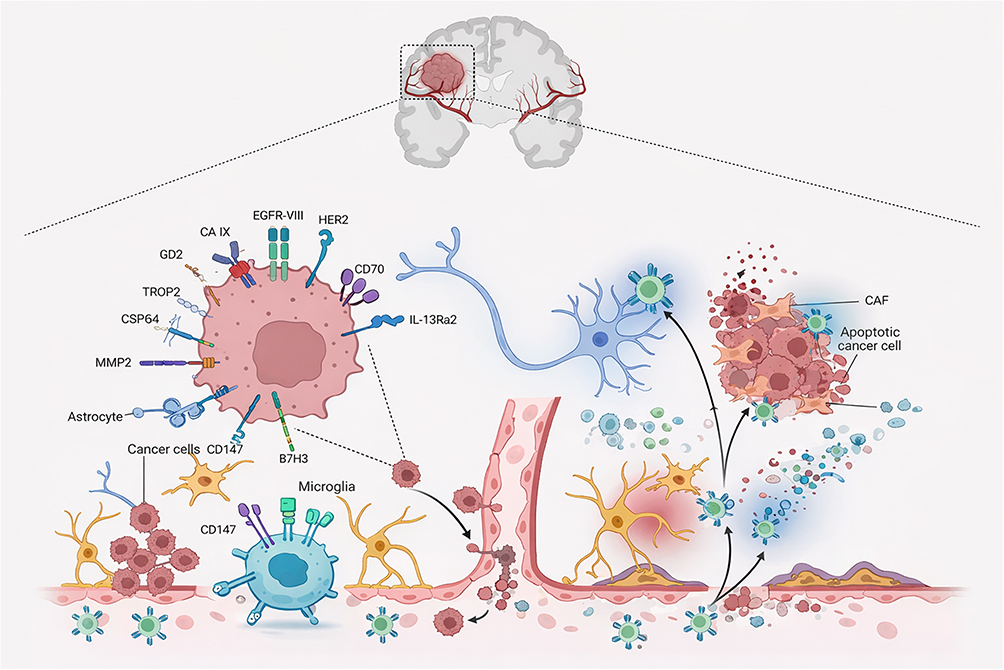 |
Figure 2 Schematic illustration of chimeric antigen receptor (CAR) T-cell therapy in glioblastoma (GBM), highlighting key therapeutic mechanisms, representative target antigens, and major translational barriers. Tumor-associated antigens, including epidermal growth factor receptor variant III (EGFRvIII), human epidermal growth factor receptor 2 (HER2), disialoganglioside GD2, interleukin-13 receptor alpha 2 (IL-13Rα2), and matrix metalloproteinase-2 (MMP2), are recognized by CAR-T cells to induce tumor cell killing (center). The schematic also illustrates critical challenges (right), such as limited CAR-T cell infiltration, tumor microenvironment (TME)–mediated immunosuppression, neuronal collateral damage, tumor lysis syndrome (TLS), and blood–brain barrier (BBB) disruption. Created with BioRender.com. |
Engineering CAR-T Cells to Overcome Immunosuppression and Enhance Therapeutic Synergy in Glioblastoma
Given the profound immunosuppressive nature of the glioblastoma microenvironment, CAR-T cells rapidly become dysfunctional after tumor entry, exhibiting impaired expansion, early exhaustion, and limited persistence. To overcome these limitations, next-generation CAR-T engineering approaches have focused on enhancing intrinsic T-cell fitness, resisting inhibitory cytokine signaling, and promoting sustained effector function within the CNS tumor niche.27
Genetic Modifications to Improve CAR-T Persistence and Function in GBM
CAR-T cells are engineered T lymphocytes designed to recognize tumor-associated antigens in an MHC-independent manner. Despite their transformative success in hematologic malignancies, CAR-T therapies in glioblastoma remain severely limited by impaired expansion, early exhaustion, and restricted persistence within the profoundly immunosuppressive CNS tumor microenvironment. Consequently, next-generation strategies have increasingly focused on genetic modifications that enhance CAR-T effector function, long-term fitness, and resistance to inhibitory signaling pathways.27,28 One of the most established approaches to improve CAR-T efficacy in GBM involves optimizing intracellular co-stimulatory signaling domains, including 4–1BB and OX40, which play central roles in reinforcing T-cell activation, survival, and durability under suppressive tumor conditions. Preclinical studies have demonstrated that incorporation of these domains significantly enhances CAR-T persistence and cytotoxic activity within hostile TME settings, thereby reducing functional exhaustion and improving therapeutic durability.29 In addition to tumor-intrinsic immune suppression, CAR-T cell performance in glioblastoma is further constrained by CNS-specific anatomical barriers that limit effective trafficking and infiltration into tumor sites. Tight junction proteins such as claudin-5, together with immunomodulatory signaling from astrocytes and microglia, contribute to an inhibitory cytokine milieu dominated by mediators such as TGF-β and IL-10, which collectively impair CAR-T activation and persistence after tumor entry.30
To overcome these limitations, advanced engineering strategies have focused on enhancing CAR-T migratory capacity through chemokine receptor reprogramming. For example, modification of CAR-T cells with receptors such as CXCR2 or CCR2 has shown improved homing toward chemokine-enriched tumor regions, resulting in significantly enhanced intratumoral accumulation and antitumor efficacy in preclinical solid tumor models.31 Together, these genetic engineering approaches represent critical steps toward improving CAR-T durability and functionality in the uniquely immunosuppressive landscape of glioblastoma.
Function of Co-Stimulatory Molecules (4-1BB, OX40)
Several co-stimulatory molecules enhance the anti-tumor activity of CAR-T cells by promoting T cell activation, proliferation, and persistence, while also reducing exhaustion. CD28 and 4–1BB are the most common and are included in FDA-approved CAR-T cell therapies. Other promising candidates include OX40, CD27, GITR, and ICOS.32 These molecules, when incorporated into CAR constructs, can improve the efficacy and longevity of CAR-T cell responses, particularly against solid tumors. In CAR-T cell therapy for GBM, 4–1BB and OX40 are co-stimulatory molecules that enhance the anti-tumor activity of CAR-T cells.18 They achieve this by boosting T cell activation, survival, and proliferation, while also potentially inhibiting regulatory T cell (Treg) function, which can suppress the immune response. The expansion and survival of T cells, which are generated through 4–1BB costimulatory receptor signaling, leads to a reduction in antigen-positive relapse. The 4–1BB signaling can also enhance memory T cell formation and reduce CAR T cell depletion, while OX40, a member of the TNFR family, OX40 signaling can boost T cell activation and survival, outstandingly improving CAR-T cell persistence and anti-tumor activity, and potentially counteract Treg-mediated immunosuppression. To equip CAR-T cells with resistance to immunosuppressive agents like TGF-β, researchers are exploring several strategies, including modifying CAR-T cells to express dominant-negative TGF-β receptors, using cytokines to counteract TGF-β’s effects, or engineering CAR- T cells that can redirect TGF-β signaling to promote anti-tumor activity.32 Smith et al showed that by throwing into disarray or bypassing TGF-β signaling, they saw increased stability of CAR-T cells and enhanced antitumor function in preclinical models.32 They successfully engineered CAR-T cells to evade TGF-β signaling pathways, which resulted in significantly improved antitumor responses in preclinical GBM models. Researchers are actively investigating the use of these and other co-stimulatory domains to optimize CAR-T cell design and improve treatment effectiveness.32 These advancements are particularly relevant in challenging tumor microenvironments like those found in glioblastoma multiforme (GBM) and triple-negative breast cancer (TNBC). Research is ongoing to identify and incorporate novel co-stimulatory molecules into CAR constructs.
Development in Genetic Alteration for Cytokine-Secreting CAR-T Cells (IL-12, GM-CSF)
Chimeric antigen receptor (CAR) T cells are genetically engineered lymphocytes designed to enhance cancer immunotherapy. These modified T cells express synthetic receptors that enable them to specifically recognize and bind antigens on tumor cells, facilitating targeted cytotoxic activity. In addition to direct tumor recognition, CAR-T cells can be further engineered to secrete immunostimulatory cytokines such as interleukin-12 (IL-12) and granulocyte-macrophage colony-stimulating factor (GM-CSF), thereby amplifying local immune activation and recruiting other effector immune cells to the tumor microenvironment. This strategy, as reported by Zhang et al, has shown encouraging outcomes in preclinical models, highlighting its potential to overcome the immunosuppressive landscape of solid tumors,33 This approach helps mitigate the tumor microenvironment’s immunosuppressive effects (TME), thereby increasing the overall effectiveness of CAR-T cell therapy. Interleukin-12 (IL-12), as a proinflammatory cytokine, in particular, acts as a key role by suppressing the expression of FoxP3 and TGF-β, both of which are related to the extension of regulatory T cells (Tregs) that facilitate immune escape. It acts as a key regulator of both innate and adaptive immunity, particularly in the context of cell-mediated immunity.32 Additionally, IL-12 reduces the proliferation of myeloid-derived suppressor cells (MDSCs) and promotes the infiltration of cytotoxic T lymphocytes (CTLs) into the tumor site, further strengthening antitumor immune responses. It plays a crucial role in enhancing cellular immunity by promoting Th1 cell differentiation and the production of IFN-γ, which is vital for clearing pathogens like Mycobacterium tuberculosis (Mtb). IL-12 also activates signaling pathways that contribute to the development and regulation of inflammatory diseases within the central nervous system (CNS).34
CAR-T cells engineered for constitutive interleukin-12 (IL-12) secretion have been shown to exhibit enhanced persistence, decreased exhaustion, and greater infiltration into tumor tissues. When combined with TGF-β signaling inhibitors, these IL-12-producing CAR-T cells display synergistic antitumor effects by counteracting immunosuppressive mechanisms within the tumor microenvironment (TME), thereby amplifying therapeutic potency.35
Chimeric Antigen Receptor Therapies with Immune Checkpoint Inhibitors: A Combining Approach
The integration of chimeric antigen receptor (CAR) T-cell therapy with immune checkpoint inhibitors (ICIs) has emerged as a promising approach to overcome the individual limitations of each treatment, particularly in the context of solid tumors. CAR-T cells are genetically modified to recognize tumor-associated antigens with high specificity, whereas ICIs—such as anti-PD-1 and anti-PD-L1 antibodies—function by blocking inhibitory pathways that restrain T-cell activity. The combination of these modalities is designed to enhance CAR-T cell persistence and cytotoxicity, mitigate the effects of the immunosuppressive tumor microenvironment (TME), and ultimately improve overall therapeutic efficacy.36 Combining CAR T-cell therapy with immune checkpoint inhibitors in glioblastoma aims to dominate the tumor’s immunosuppressive environment and enhance CAR T-cell effectiveness. This strategy involves using checkpoint inhibitors to counteract the inhibitory signals within the GBM microenvironment that suppress CAR T-cell activity, ultimately boosting their anti-tumor response.36 Recent advancements in immune checkpoint inhibition have expanded beyond PD-1 and CTLA-4 (Cytotoxic T-lymphocyte Associated Protein 4) inhibitors, with promising candidates targeting additional immune checkpoints to overcome the immunosuppressive microenvironment in GBM. These emerging strategies aim to rejuvenate exhausted T cells and enhance anti-tumor immunity.37
CAR-T Cells and Immune Checkpoint Inhibitors (ICIs)
Immune checkpoints are inhibitory receptors found on the surface of immune cells—most prominently on T lymphocytes—that serve as essential modulators of immune activity. They play a pivotal role in preserving immune tolerance and preventing excessive immune activation that could lead to autoimmunity.35 Under normal physiological conditions, immune checkpoint pathways are indispensable for maintaining self-tolerance and safeguarding healthy tissues from immune-mediated injury. Therapeutically, blockade of these pathways through immune checkpoint inhibitors (ICIs)—particularly those targeting programmed cell death protein 1 (PD-1)—has been shown to restore the function of exhausted CAR-T cells and enhance their antitumor efficacy. This synergistic effect was demonstrated in preclinical studies conducted by Johnson et.31 Their findings show that merging PD-1 blockade with CAR-T cell therapy cause greater tumor reduction and improved endurance in GBM animal models. These preclinical insights are consistent with current clinical trials assessing the treatment potential of CAR-T and immune checkpoint inhibitor co-administration in GBM patients. In comparison to normal brain tissues, glioblastoma is overexpressed with certain type of cytokine receptors known as IL13Rα2 (Interleukin-13 Receptor Subunit Alpha-2) that exhibit high affinity for IL-13. By blocking the PD-1 and PD-L1 signaling axis, the antitumor activity of CAR-T cells particularly those targeting IL13Rα2is significantly improved, resulting in more favorable therapeutic responses, as reported by Lee et al.38 To achieve a sustained antitumor effect in glioblastoma, it is critical to ensure that therapeutic agents remain localized within tumor tissues. This challenge can be effectively addressed through the use of nanocarriers, particularly those that are surface-functionalized to enhance tumor penetration and retention. Over the past few decades, nanotechnology-based drug delivery systems have garnered significant attention for the treatment of various malignancies, including glioblastoma multiforme (GBM).39 Among potential molecular targets, the interleukin-13 receptor alpha 2 (IL13Rα2) has emerged as one of the most extensively investigated due to its high expression in GBM cells and negligible presence in normal brain tissue. The restricted distribution of IL13Rα2 in healthy tissues renders it an attractive and selective target for glioblastoma-directed nanotherapeutic and immunotherapeutic strategies.38
Virotherapy and CAR-T Cells in Cancer Treatment
Oncolytic viruses (OVs) are viruses engineered to selectively target and destroy cancer cells, while leaving healthy cells unharmed. Genetic engineering plays a crucial role in enhancing the efficacy and safety of OVs by modifying their ability to replicate in tumors, express therapeutic genes, and stimulate the immune system.40 This approach, known as oncolytic virotherapy, holds significant promise for cancer treatment. Oncolytic virotherapy, which utilizes genetically modified viruses to preferentially infect and destroy tumor cells, has been examined as a complementary approach to CAR-T cell therapy. Furthermore, to their direct cancer treatment effects, these viruses enable the secretion of tumor-associated antigens (TAAs) into the tumor microenvironment (TME), thereby boosting immune activation and ameliorating CAR-T cell efficacy. This synergistic approach has been established in animal studies by Wang et al.40 Studies have demonstrated that oncolytic viruses can reshape the tumor microenvironment (TME), rendering it more supportive of immune activity and thereby increasing the proliferation and persistence of CAR-T cells. Moreover, specific viral platforms such as adenoviruses possess the capability to traverse the blood–brain barrier (BBB), allowing direct infection of GBM cells within the CNS. This dual action contributes to both localized immune stimulation and tumor cell lysis. These observations are consistent with the results reported by Davis et al.41 They confirmed that the integration of oncolytic virotherapy with CAR-T cell treatment significantly amplified antitumor responses in GBM preclinical models. Oncolytic virotherapy combined with CAR-T cell therapy has shown promise in amplifying antitumor responses in glioblastoma (GBM). This combination approach leverages the strengths of both therapies to overcome challenges associated with treating solid tumors like GBM.
Progresses in CAR-T Layout for Brain Tumor Immunotherapy
Receptors of chimeric antigen are synthetic fusion proteins engineered to potentiate T cell responses against malignant cells. Progress in CAR-T cell therapy for brain tumors involves advancements in CAR-T structure, identifying suitable tumor targets, and combining CAR-T with other immunotherapies.41 A standard CAR structure comprises three fundamental elements: an extracellular domain responsible for identifying tumor-associated antigens (TAAs), a transmembrane and spacer region for structural support and flexibility, and an intracellular signaling module that initiates T cell activation. Second-generation CARs are enhanced with co-stimulatory domains such as 4–1BB or CD28 to boost activation and longevity, while third-generation constructs incorporate multiple co-stimulatory signals for superior functionality. CAR-T cells can be precisely engineered to distinguish and attack specific TAAs, including IL13Rα2, HER2, and EGFRvIII, that are commonly upregulated in GBM and other brain cancers. Johnson et al31 and Brown et al42 have highlighted the demonstration of the therapeutic potential of these targeted designs, which have proved preclinical success in targeting these antigens, particularly IL13Rα2, showing promise in overcoming tumor-associated challenges like antigen heterogeneity and immune evasion. To further counteract the effects of the immunosuppressive tumor microenvironment (TME), CAR-T cells have been genetically altered to yield therapeutic agents such as cytokines like interleukin-12 (IL-12) that amplify their antitumor activity. In parallel, advances in CAR architecture have aimed to address structural and physiological barriers, most remarkably the BBB, which presents a main problem in treating central nervous system tumors such as GBM. Improved trafficking across the blood-brain barrier (BBB) can significantly advance the treatment of central nervous system (CNS) disorders. By enhancing drug delivery to the brain, researchers can develop more effective therapies for conditions like brain tumors, Alzheimer’s disease, and other neurological diseases. They have shown that incorporating modifications for improved trafficking across the BBB can advance the efficiency of CAR-T cells in brain cancer therapy.43 Each component of the Structure of CAR contributes to receptor stability, structural adaptability, and the precise identification of tumor-associated antigens such as IL13Rα2, HER2, and EGFRvIII, which are frequently expressed in brain tumors. The intracellular domain integrates CD3ζ signaling, often supplemented with co-stimulatory molecules such as CD28, 4–1BB, OX40, CD27, or ICOS, which collectively enhance T-cell activation, persistence, and cytotoxic efficiency. These elements are crucial for sustaining long-term CAR-T cell responses and overcoming the immunosuppressive microenvironment in brain tumor therapy.42
Outlook and Challenges In Brain Tumors CAR-T Cell Therapy
The application of chimeric antigen receptor (CAR) T-cell therapy in brain tumors is hindered by several unique challenges, including inefficient trafficking of effector cells to the tumor site and the intricate nature of the central nervous system (CNS) tumor microenvironment. Additional obstacles, such as limited T-cell infiltration, off-target toxicity, cytokine release syndrome (CRS), and tumor lysis syndrome (TLS), further complicate therapeutic success. Although CAR-T cell therapy has revolutionized the treatment of hematologic malignancies, translating this success to solid tumors—particularly glioblastoma (GBM)—remains a formidable task. Two major barriers define this difficulty: the blood–brain barrier (BBB), which restricts the migration of CAR-T cells into the CNS, and the immunosuppressive tumor microenvironment (TME), which severely impairs their persistence and cytotoxic function (Figure 3).44,45
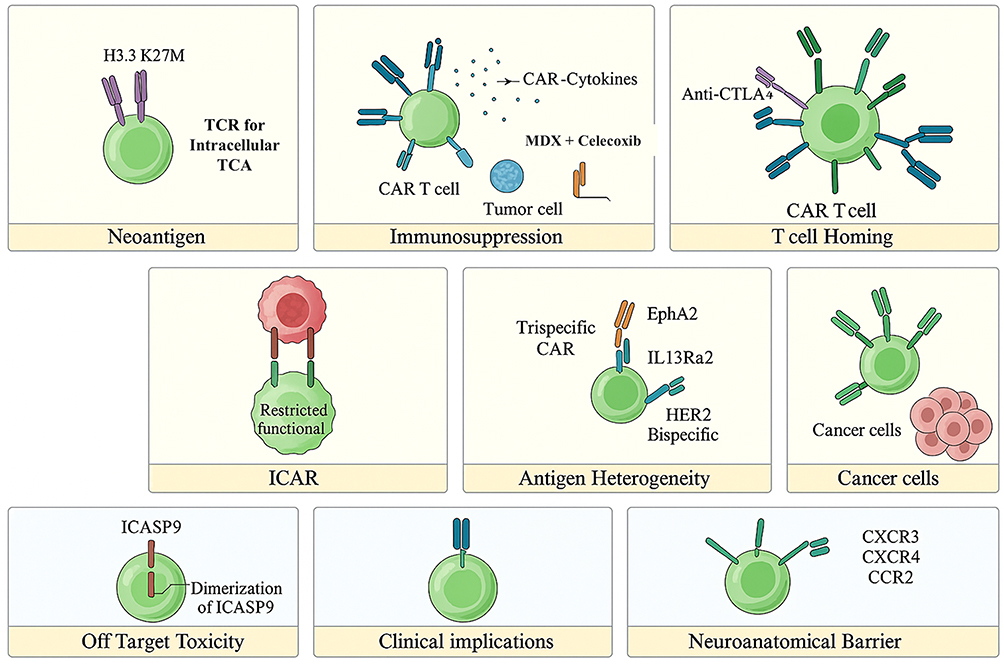 |
Figure 3 Schematic overview of key challenges and emerging strategies in CAR-T cell therapy for glioblastoma and other brain tumors. Major barriers include limited CAR-T infiltration across the blood–brain barrier (BBB), antigenic heterogeneity, and immunosuppressive signaling within the tumor microenvironment (TME). Novel engineering approaches such as bispecific and trispecific CAR designs, chemokine receptor–modified CAR-T cells, immune checkpoint blockade, and safety switch mechanisms aim to enhance tumor targeting, persistence, and therapeutic control. Adapted from.46 |
These challenges include limited T-cell infiltration due to the blood-brain barrier (BBB), immune evasion mechanisms within the tumor microenvironment (TME), and antigenic heterogeneity. Novel approaches such as bispecific and trispecific CARs, chemokine receptor-expressing CARs, and immune checkpoint blockade enhance tumor targeting, persistence, and adaptability within the immunosuppressive TME. Bispecifics are engineered to overcome the limitations of traditional CAR T-cell therapies. The primary function of tandem CARs is to extend the therapeutic reach of CAR T-cells by targeting malignant cells that evade single-target therapies.46 This approach focuses on cell-to-cell interactions between cancer cells and CAR T-cells, allowing for the targeting of multiple antigens. This strategy is particularly useful in treating cancers that may escape initial therapies. Additionally, safety mechanisms like inhibitory CARs (iCARs) and inducible apoptosis switches (iCASP9) mitigate off-target toxicity, ensuring controlled CAR-T activation. These improvements collectively enhance the therapeutic ability of CAR-T cells in fighting brain tumors while addressing their inherent complexities.46
Immune Preferences in the CNS Parenchyma
The brain’s immune system is not entirely isolated; instead, it actively interacts with the body’s immune system through specialized “niches” and pathways. These interactions are crucial for brain health, repair, and even behavior. While the brain has protective barriers, it’s now understood that it’s not immune-privileged in the traditional sense. The brain parenchyma’s immune system primarily consists of myeloid cells, specifically CNS-associated macrophages. Perivascular macrophages and microglia patrol the brain parenchyma, while the meningeal spaces have a diverse immune repertoire.22 Research is ongoing to understand how immune cells move from the parenchyma to the subarachnoid space and then into meningeal lymphatic vessels. This complex interplay highlights the intricate immune surveillance within the central nervous system. The CNS parenchyma exhibits immune privilege, meaning it has a reduced immune response compared to other body tissues, due to several factors, including the blood-brain barrier (BBB) and specific immune cell behavior. This presentation helps protect the delicate neural tissue from damaging inflammation, but also presents challenges for treating infections and tumors within the CNS.46 Beyond the physical and immunological barriers, several intrinsic tumor characteristics—such as hypoxia, T-cell exhaustion, and pronounced intra- and inter-tumoral heterogeneity—further impede the therapeutic response in gliomas. Together, these factors underlie the modest clinical efficacy observed with current immunotherapeutic strategies for brain tumors.Importantly, the diverse population of innate and adaptive immune cells residing within the meningeal and parenchymal compartments of the central nervous system (CNS) should not be regarded solely as a potential source of neuroinflammation. Rather, they function as tightly regulated partners that sustain neuroimmune communication and contribute to neural homeostasis. Advances in neuroimmunology have recently elucidated the pivotal roles of both microglia and meningeal immune networks in maintaining normal brain physiology and orchestrating immune responses under pathological conditions (Figure 4).22
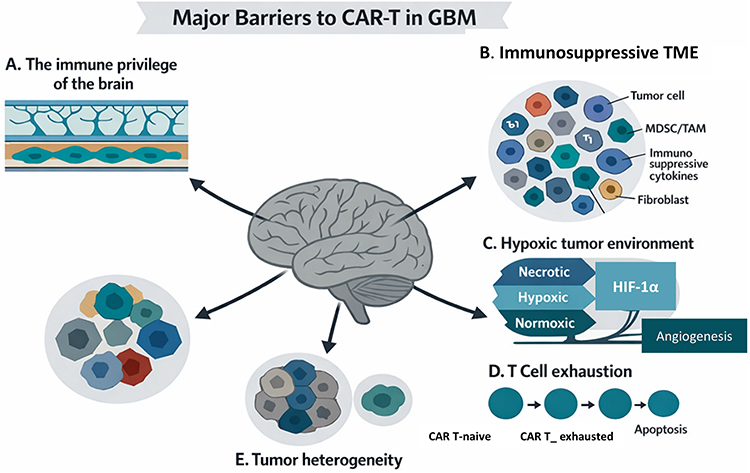 |
Figure 4 Key challenges of CAR-T cell therapy in glioma. CAR-T cell efficacy in glioma is limited by multiple structural and immunological barriers. The blood–brain barrier (BBB) restricts T-cell migration into the tumor site, while antigen-presenting cells in perivascular niches play a minimal role in reactivating infiltrating lymphocytes. Within the tumor microenvironment (TME), abundant myeloid populations, immunosuppressive cytokines, and checkpoint ligands collectively inhibit CAR-T activation and persistence. Additional constraints include hypoxia, which deprives T cells of oxygen and nutrients, chronic antigen exposure leading to exhaustion, and pronounced inter- and intra-tumoral heterogeneity, complicating target selection and CAR design.29 |
Penetrating the Blood-Brain Barriers
Overcoming the blood-brain barrier (BBB) is a significant challenge in CAR-T cell therapy for glioblastoma (GBM), as the BBB restricts the delivery of therapeutic CAR-T cells to the tumor site. Several strategies are being explored to enhance CAR-T cell delivery to GBM, including invasive and non-invasive methods, as well as targeting specific antigens and managing the BBB itself.29 Importantly, the BBB in glioblastoma is not uniformly intact. GBM-associated angiogenesis and tumor-driven remodeling lead to partial disruption of the BBB and formation of a heterogeneous blood–brain tumor barrier (BBTB). While this altered vasculature may allow limited immune-cell entry in certain tumor regions, it remains highly variable and insufficient for consistent CAR-T trafficking and intratumoral distribution. Therefore, strategies aimed at enhancing delivery across both the intact BBB and the heterogeneous BBTB remain essential for effective CAR-T therapy in GBM.47
For CAR-T cell therapy to achieve clinical effectiveness in GBM, modified T cells must successfully overpass the blood–brain barrier (BBB) to admission tumor sites. To boost this process, several strategies are under investigation, including the improvement of CAR-T cell migratory capacity and the use of nanoparticle-assisted delivery platforms aimed at improving central nervous system targeting and overall treatment effectiveness.29 Recent studies have highlighted that functionalized nanocarriers can substantially improve drug transport and tumor targeting in glioblastoma. For example, folic acid–conjugated exosomes co-loaded with temozolomide (TMZ) and quercetin have demonstrated enhanced BBB penetration and selective accumulation in GBM cells, resulting in increased apoptosis and tumor growth inhibition in preclinical models.48 These findings provide valuable insight into how nanocarrier-based platforms could be integrated with CAR-T therapy to enhance CNS infiltration, improve T-cell persistence, and potentiate immune responses within the tumor microenvironment. Even with the progression of the tumor, the structure and function of the BBB are changed after the rupture of the tumor membrane and the deterioration of the tumor. Eventually, BBB is replaced by the blood-brain tumor barrier (BBTB), which hinders the delivery of most anti-tumor drugs. Research focuses on innovative strategies to overcome this barrier and improve CAR T-cell therapy success in GBM. This involves exploring methods to enhance CAR T-cell penetration into the brain tissue to target the tumor effectively. Nanotechnologies that enable drug delivery across the BBB hold great promise for the treatment of GBM. An overview of BBB and drug delivery strategies to the CNS, examples of nanocarriers for drug delivery, such as polymeric nanoparticles, liposomes, dendrimers, Nano capsules, and so on. CAR-T constructs capable of simultaneously targeting multiple tumor-associated antigens, thereby reducing the risk of immune evasion and relapse.29,49
Preclinical and Clinical Studies
Ongoing clinical trials targeting EGFRvIII, IL13Rα2, and HER2 in GBM have further explored the potential of CAR-T cell therapy (Table 1).50,51 Some trials have demonstrated clinical benefits, with tumor regression observed in certain patients. However, a review by Guzman G et al showed that the duration of response is often limited, and tumor recurrence remains a significant challenge.44 For example, trials using EGFRvIII-targeting CAR-T cells showed tumor regression in a subset of patients, but the efficacy was inconsistent, and some patients experienced severe neurotoxicity.42,50–53 Other trials targeting alternative antigens such as B7-H3, CD133, and HER2 have faced similar hurdles with antigen variability and difficulties in overcoming the BBB.43,54,55 Yuan et al discussed how these trials emphasize the critical need for improving CAR-T cell delivery methods, including direct intracerebral injection, to achieve better penetration of the BBB. Additionally, combining CAR-T cells with ICIs has emerged as a promising strategy to overcome the immunosuppressive TME and enhance therapeutic outcomes in brain tumor treatments.56
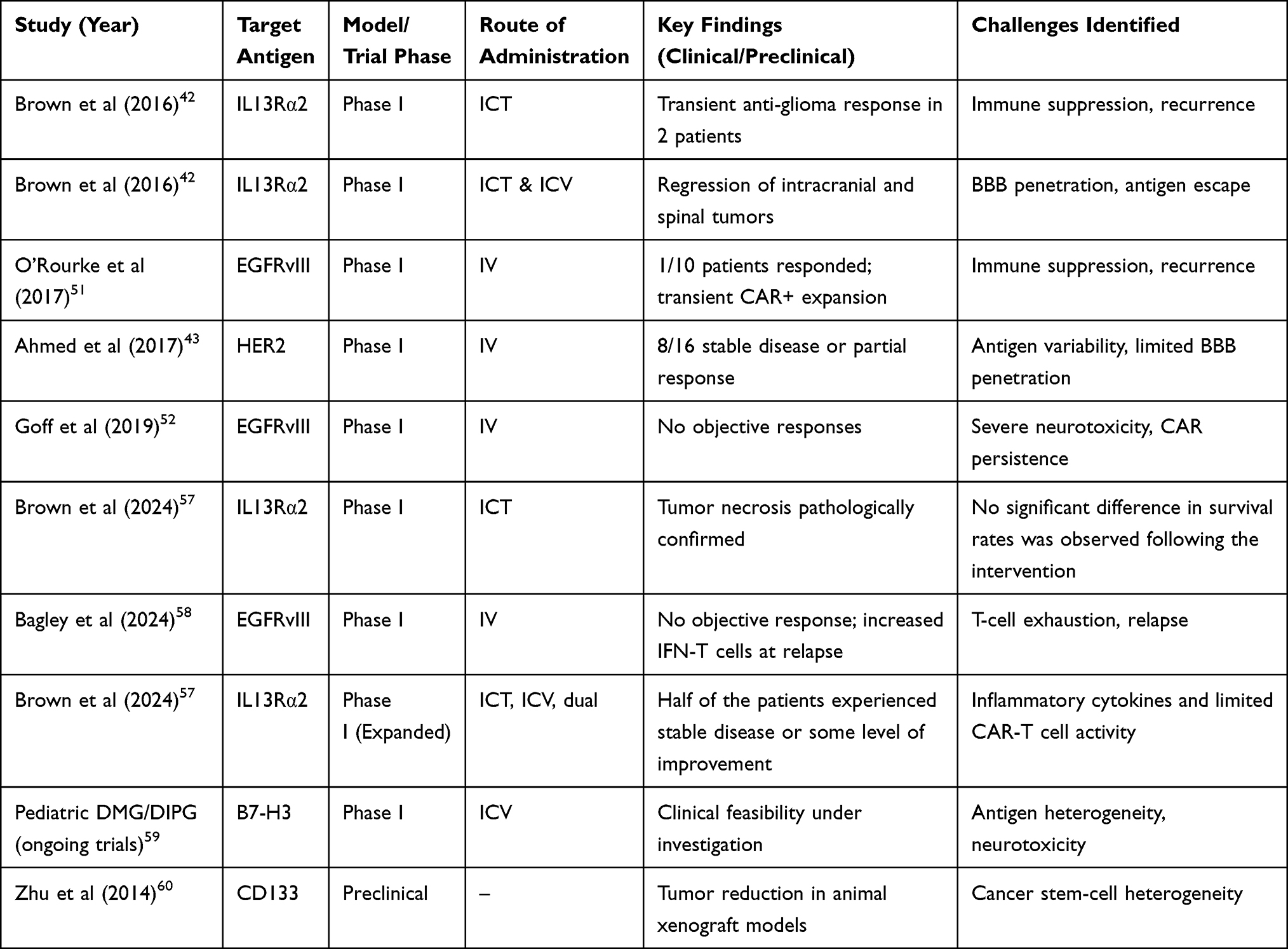 |
Table 1 Remarkable Findings from Clinical Trials and Preclinical Studies on CAR-T Therapy for Brain Tumors |
The clinical and preclinical experiences summarized in Table 1 illustrate both the promise and the persistent barriers limiting the success of CAR-T cell therapy in glioblastoma. While targeting antigens such as EGFRvIII, IL13Rα2, and HER2 has demonstrated feasibility and occasional tumor regression, therapeutic benefit has generally been modest, heterogeneous, and rarely durable. A central limitation is antigen escape, driven by marked intratumoral heterogeneity and immune-selective loss of target expression following CAR-T pressure. Moreover, restricted trafficking across the blood–brain barrier, inadequate intratumoral persistence, and profound suppression within the glioblastoma microenvironment collectively impair sustained effector activity. Importantly, neurotoxicity
Advancement for Next-Generation CAR-T Cells with Different Approaches
Advancements in next-generation CAR-T cell therapies are focused on improving efficacy and reducing relapse. Bispecific approaches are being explored to address antigen loss, a common cause of relapse. Research in CAR-NK cells is another promising area of study. Additionally, developments include T cell-targeted CAR generation in situ. The FDA has approved several CAR-T cell therapies for hematologic malignancies, demonstrating the clinical progress in this field. These advancements aim to enhance treatment outcomes for patients. Advances in CAR-T cell engineering focus on improving antigen specificity, reducing off-target effects, and enhancing persistence within the tumor microenvironment (TME). One encouraging approach is the development of dual- and multi-target CAR-T cells, which can recognize multiple tumor antigens, thereby alleviating the problem of antigen escape.61 Furthermore, novel CAR designs such as TRUCKs (T cell Redirected for Universal Cytokine Killing), switchable CARs, and inhibitory CARs (iCARs) present enhanced flexibility, ameliorated safety, and better adaptation to the complex GBM TME. Engineering CAR-T cells with cytokine support, such as IL-12 or IL-15-secreting CAR-T cells, has shown potential in restoring their persistence and anti-tumor efficacy.62
Discussion
This review highlights the potential of genetic modifications to enhance CAR-T cell therapy for glioblastoma multiforme (GBM), focusing on key strategies such as co-stimulatory signaling, TGF-β resistance, and cytokine secretion to overcome the immunosuppressive tumor microenvironment (TME). Our findings align with growing evidence suggesting that reprogramming CAR-T cells to counteract inhibitory signals within the TME is critical for improving therapeutic efficacy. Consistent with the work of Debinski et al63 and Smith et al,32 which demonstrated that incorporating co-stimulatory molecules and modifying signaling pathways can significantly enhance CAR-T cell survival and function, we emphasize that molecules like 4–1BB and OX40 play a key role in enhancing CAR-T cell persistence and tumor-killing ability in the hostile GBM microenvironment.64 One of the major obstacles to CAR-T therapy in solid tumors, particularly GBM, is the immunosuppressive nature of the TME. Our analysis further supports the idea that TGF-β signaling is a key mechanism of immune resistance. Strategies to bypass or neutralize TGF-β have shown promise in enhancing CAR-T cell persistence and effector function. As shown by Smith et al,32 genetic modifications conferring TGF-β resistance have significantly improved antitumor activity in preclinical models. These results indicate that next-generation CAR-T cells engineered to resist key immunosuppressive pathways could offer more durable responses in GBM.65
We also examined the synergistic potential of combining CAR-T therapy with immune checkpoint inhibitors (ICIs), particularly PD-1 inhibitors, to reverse T-cell exhaustion and enhance antitumor responses. The findings of Johnson et al19 and Lee et al19 and Lee et al38 support the hypothesis that blocking the PD-1/PD-L1 axis enhances CAR-T cell activity by preventing functional exhaustion, thereby leading to prolonged tumor regression. Our review highlights those ongoing clinical trials are actively evaluating this combination approach in GBM, and emerging data suggest that integrating ICIs with CAR-T therapy may offer a viable strategy to overcome the suppressive TME.
Additionally, this study explored the emerging role of oncolytic virotherapy as an adjunct to CAR-T therapy. Preclinical investigations by Wang et al40 and Davis et al41 indicate that oncolytic viruses not only directly lyse tumor cells but also release tumor-associated antigens (TAAs), thereby amplifying the immune response and promoting CAR-T cell expansion within the TME. Importantly, some oncolytic viruses, such as engineered adenoviruses, can cross the blood-brain barrier (BBB) and selectively infect GBM cells, providing a dual mechanism of direct oncolysis and immune stimulation. This combination strategy holds great promise for enhancing CAR-T cell efficacy and improving treatment outcomes.
Despite these promising advancements, several critical challenges remain. Tumor heterogeneity continues to pose a significant hurdle for CAR-T therapy in GBM, as antigen loss or clonal evolution can enable tumor cells to evade immune recognition. Future research should focus on developing multi-antigen targeting CAR-T cells, as well as biomarker-driven approaches to personalize antigen selection and improve therapeutic precision.66 Additionally, while oncolytic virotherapy has demonstrated preclinical success, concerns regarding viral resistance, off-target effects, and long-term safety must be addressed through rigorous clinical evaluation. More studies are needed to determine optimal virus-CAR-T combinations and mechanisms to enhance viral persistence within the GBM microenvironment.67 Another major challenge is the scalability and manufacturing of CAR-T therapies for GBM.68 Unlike hematologic malignancies, where CAR-T cell production is relatively standardized, solid tumor applications require more complex engineering, enhanced persistence, and optimized delivery mechanisms.69 Establishing standardized protocols for CAR-T manufacturing, expansion, and genetic modification will be essential for clinical translation and widespread adoption of this therapy.70
Future Directions and High-Priority Research Gaps
Despite rapid advances, several priorities remain central for achieving durable clinical benefit in GBM. First, biomarker-guided antigen selection and spatially informed multi-target designs are needed to mitigate antigen heterogeneity and adaptive escape. Second, strategies that sustain intratumoral persistence—through resistance to myeloid suppression, metabolic stress, and exhaustion—should be integrated early in construct design. Third, delivery optimization (locoregional approaches and BBB-permissive platforms) must be paired with real-time monitoring to balance exposure and neurotoxicity risk. Finally, scalable manufacturing workflows and clinically predictive preclinical models will be essential to standardize product quality and de-risk translation across patient subsets.
Conclusion
Recent advancements in Chimeric Antigen Receptor (CAR) T-cell therapy, particularly when combined with immune checkpoint inhibitors (ICIs) and oncolytic virotherapy, have shown promising potential in the treatment of glioblastoma multiforme (GBM). These innovative therapeutic strategies aim to overcome critical barriers such as immune evasion, limited CAR-T persistence, and the profoundly immunosuppressive tumor microenvironment (TME), which remain central challenges in GBM therapy. Innovations in co-stimulatory signaling, cytokine modulation, and genetic strategies to counteract suppressive pathways such as TGF-β are increasingly enhancing the functionality of CAR-T cells within this uniquely hostile immunological landscape.
Despite substantial progress, major obstacles continue to limit durable clinical success, including the restrictive influence of the blood–brain barrier (BBB), marked intratumoral antigen heterogeneity, and the scalability of CAR-T manufacturing. However, recent preclinical developments, including dual- and multi-antigen targeting constructs (such as EGFRvIII and IL13Rα2), together with emerging clinical trials of CAR-T platforms such as MB-101, provide encouraging evidence that these barriers may be progressively surmountable In addition, next-generation approaches incorporating gene therapy, armored CAR-T designs, and checkpoint-resistant constructs further reinforce the therapeutic potential of engineered cellular immunotherapy for GBM.
Importantly, future progress will depend on the integration of multi-target CAR strategies to mitigate antigen escape and better address the adaptive evolution of glioblastoma under immune pressure. Advances in armored CAR-T cells capable of sustained cytokine delivery or resistance to inhibitory mediators represent critical steps toward maintaining effector activity within the immunosuppressive CNS tumor niche. Moreover, improving CAR-T delivery across the BBB remains a key translational priority. Emerging nanotechnology-enabled delivery systems and locoregional administration approaches may offer effective solutions to enhance CAR-T trafficking, intratumoral retention, and therapeutic control within the brain.
At the same time, careful clinical optimization will be required to balance efficacy with safety, particularly given the risk of neurotoxicity and inflammatory complications unique to CNS-directed immunotherapies. Furthermore, broader clinical implementation will require advances in scalable manufacturing workflows, standardized engineering protocols, and the development of clinically predictive preclinical models.
Collectively, these multidisciplinary innovations provide a roadmap for transforming CAR-T therapy from an experimental proof-of-concept into a clinically viable and durable treatment modality, offering renewed hope for achieving meaningful long-term benefit for patients with glioblastoma.
Abbreviations
GBM, Glioblastoma multiforme; CNS, central nervous system; OS, overall survival; TME, tumor microenvironment; TIME, tumor immune microenvironment; BBB, blood–brain barrier; BBTB, blood–brain tumor barrier; CAR, chimeric antigen receptor; CAR-T, CAR T-cell therapy; TAAs, tumor-associated antigens; EGFRvIII, epidermal growth factor receptor variant III; HER2, human epidermal growth factor receptor 2; GD2, disialoganglioside GD2; IL13Rα2, interleukin-13 receptor alpha 2; MMP2, matrix metalloproteinase-2; TLS, tumor lysis syndrome; TGF-β, Tregs, transforming growth factor-β; regulatory T cells; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells; NK, natural killer; GDEs, cells glioblastoma-derived exosomes; ICIs, immune checkpoint inhibitors; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; OVs, oncolytic viruses; TMZ, temozolomide; DCs, dendritic cells; GSCs, glioblastoma stem cells; NRF2, nuclear factor erythroid 2–related factor 2; GPX4, glutathione peroxidase 4; STAT3, signal transducer and activator of transcription 3; ICAM-1, intercellular adhesion molecule 1; (BCKAs), branched-chain ketoacids; KIC, α-ketoisocaproate; KMV, α-keto-β-methylvalerate; KIV, α-ketoisovalerate; polySia, polysialic acid; Siglec-16, sialic acid–binding immunoglobulin-like lectin 16; CXCR4, C-X-C chemokine receptor type 4; CCR7, C-C chemokine receptor type 7; IL-12, interleukin-12; GM-CSF, granulocyte-macrophage colony-stimulating factor; CTLs, cytotoxic T lymphocytes; IFN-γ, interferon-γ; CRS, cytokine release syndrome; ICT, intracerebral/intratumoral; ICV, intraventricular; IV, intravenous; iCASP9, inducible caspase 9; iCAR, inhibitory CAR.
Data Sharing Statement
The data analyzed in this study are available upon reasonable request from both corresponding authors.
Acknowledgments
The authors have reviewed and approved all content included in this manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors declare that they have no competing interests.
References
1. Grochans S, Cybulska AM, Simińska D, et al. Epidemiology of glioblastoma multiforme–literature review. Cancers. 2022;14(10):2412. doi:10.3390/cancers14102412
2. Hanif F, Muzaffar K, Perveen K, et al. Glioblastoma multiforme: a review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac J Cancer Prev. 2017;18:3. doi:10.22034/APJCP.2017.18.1.3
3. Kurdi M, Alkhotani A, Alsinani T, et al. Effects of radiotherapy alone versus concomitant radiotherapy with temozolomide chemotherapy on the outcome of idh-wildtype glioblastoma patients. Clin Oncol. 2025;38:103741. doi:10.1016/j.clon.2024.103741
4. Onciul R, Brehar F-M, Toader C, et al. Deciphering glioblastoma: fundamental and novel insights into the biology and therapeutic strategies of gliomas. Curr. Issues Mol. Biol. 2024;46(3):2402–17. doi:10.3390/cimb46030153
5. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer cell.. 2017;31(3):326–341
6. Satragno C, Schiavetti I, Cella E, et al. Systemic inflammatory markers and volume of enhancing tissue on post-contrast T1w MRI images in differentiating true tumor progression from pseudoprogression in high-grade glioma. Clin Transl Radiat Oncol. 2024;49:100849. doi:10.1016/j.ctro.2024.100849
7. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi:10.1126/science.aar4060
8. Sampson JH, Gunn MD, Fecci PE, et al. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20(1):12–25. doi:10.1038/s41568-019-0224-7
9. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
10. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
11. Zarychta J, Kowalczyk A, Marszołek A, et al. Strategies to overcome tumor microenvironment immunosuppressive effect on the functioning of CAR-T cells in high-grade glioma. Therapeut Adv Med Oncol. 2024;16:17588359241266140. doi:10.1177/17588359241266140
12. Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi:10.1158/1078-0432.CCR-15-0428
13. Bagley SJ, Logun M, Fraietta JA, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: Phase 1 trial interim results. Nature Med. 2024;30(5):1320–1329. doi:10.1038/s41591-024-02893-z
14. Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nature Biotechnol. 2019;37(9):1049–1058. doi:10.1038/s41587-019-0192-1
15. Datsi A, Sorg RV, Garg AD. The conundrum of CD8+ T cell trajectories in low antigenic tumors: how to overcome a hypofunctional state distinct from antigen-driven exhaustion? Genes Immun. 2024;25(5):353–355. doi:10.1038/s41435-024-00299-y
16. Dana N, Dabiri A, Najafi MB, et al. Advances in bioengineered CAR T/NK cell therapy for glioblastoma: overcoming immunosuppression and nanotechnology-based strategies for enhanced CAR T/NK cell therapy. Bioeng. Transl. Med. 2025;10(2):e10716. doi:10.1002/btm2.10716
17. Nicholson JG, Fine HA. Diffuse glioma heterogeneity and its therapeutic implications. Cancer Discovery. 2021;11(3):575–590. doi:10.1158/2159-8290.CD-20-1474
18. Vallieri N, Datsi A. Immune Cell Interplay in the Fight Against GBM. Cancers. 2025;17(5):817. doi:10.3390/cancers17050817
19. Yang J, Zhang M, Zhang X, et al. Glioblastoma-derived exosomes promote lipid accumulation and induce ferroptosis in dendritic cells via the NRF2/GPX4 pathway. Front Immunol. 2024;15:1439191. doi:10.3389/fimmu.2024.1439191
20. Gabrusiewicz K, Li X, Wei J, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7(4):e1412909. doi:10.1080/2162402X.2017.1412909
21. Mastantuono S, Manini I, Di Loreto C, et al. Glioma-derived exosomes and their application as drug nanoparticles. Int J Mol Sci. 2024;25(23):12524. doi:10.3390/ijms252312524
22. Cai Z, Li W, Brenner M, et al. Branched-chain ketoacids derived from cancer cells modulate macrophage polarization and metabolic reprogramming. Front Immunol. 2022;13:966158. doi:10.3389/fimmu.2022.966158
23. Silva LS, Poschet G, Nonnenmacher Y, et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT 1 modulate macrophage phenotype. EMBO Rep. 2017;18(12):2172–2185. doi:10.15252/embr.201744154
24. Thiesler H, Gretenkort L, Hoffmeister L, et al. Proinflammatory macrophage activation by the polysialic acid-Siglec-16 axis is linked to increased survival of patients with glioblastoma. Clin Cancer Res. 2023;29(12):2266–2279. doi:10.1158/1078-0432.CCR-22-1488
25. Abdalsalam NMF, Ibrahim A, Saliu MA, et al. MDSC: a new potential breakthrough in CAR-T therapy for solid tumors. Cell Commun Signaling. 2024;22(1):612. doi:10.1186/s12964-024-01995-y
26. Tu Z, Xiao C, Ma M, et al. Barriers and solutions for CAR-T therapy in solid tumors. Cancer Genet Ther. 2025;32:1–12. doi:10.1038/s41417-024-00834-z
27. Moreno-Sanchez PM, Blanpain C, et al. Immunosuppressive mechanisms and therapeutic interventions shaping glioblastoma immunity. Nat Cancer. 2026;7:1–14. doi:10.1038/s43018-025-01096-w
28. Wang S, Li W, Li J, et al. Antigen escape in CAR -T therapy: a tumor microenvironment perspective from hematological to solid tumor. Int. J. Cancer. 2026;158(4):835–846. doi:10.1002/ijc.70117
29. Montoya M, Gallus M, Phyu S, et al. A roadmap of CAR-T-cell therapy in glioblastoma: challenges and future perspectives. Cells. 2024;13(9):726. doi:10.3390/cells13090726
30. Hanif S, Muhammad P, Chesworth R, et al. Nanomedicine-based immunotherapy for central nervous system disorders. Acta Pharmacol. Sin. 2020;41(7):936–953. doi:10.1038/s41401-020-0429-z
31. Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci, trans med. 2015;7(275):275ra22–275ra22. doi:10.1126/scitranslmed.aaa4963
32. Smith TT, Moffett HF, Stephan SB, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. 2017;127(6):2176–2191. doi:10.1172/JCI87624
33. Zhang L, Morgan RA, Beane JD, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21(10):2278–2288. doi:10.1158/1078-0432.CCR-14-2085
34. Liu Z, Zhou Z, Dang Q, et al. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. 2022;12(14):6273. doi:10.7150/thno.76854
35. Zong Y, Deng K, Chong WP. Regulation of Treg cells by cytokine signaling and co-stimulatory molecules. Front Immunol. 2024;15:1387975. doi:10.3389/fimmu.2024.1387975
36. Liang T, Song Y, Gu L, et al. Insight into the progress in CAR-T cell therapy and combination with other therapies for Glioblastoma. Int J Gene Med. 2023;Volume 16:4121–4141. doi:10.2147/IJGM.S418837
37. Fu M, Xue B, Miao X, et al. Overcoming immunotherapy resistance in glioblastoma: challenges and emerging strategies. Front Pharmacol. 2025;16:1584688. doi:10.3389/fphar.2025.1584688
38. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi:10.1016/S0140-6736(14)61403-3
39. Pourmasoumi P, Banihashemian SA, Zamani F, et al. Nanoparticle-based approaches in the diagnosis and treatment of brain tumors. J Clin Med. 2024;13(23):7449. doi:10.3390/jcm13237449
40. Wang G, Kang X, Chen KS, et al. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun. 2020;11(1):1395. doi:10.1038/s41467-020-15229-5
41. Davis JJ, Wang L, Dong F, et al. Oncolysis and suppression of tumor growth by a GFP-expressing oncolytic adenovirus controlled by an hTERT and CMV hybrid promoter. Cancer Gene Ther. 2006;13(7):720–723. doi:10.1038/sj.cgt.7700944
42. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497
43. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor-modified virus-specific t cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. doi:10.1001/jamaoncol.2017.0184
44. Guzman G, Pellot K, Reed MR, et al. CAR T-cells to treat brain tumors. Brain Res Bull. 2023;196:76–98. doi:10.1016/j.brainresbull.2023.02.014
45. Lin YJ, Mashouf LA, Lim M. CAR T Cell therapy in primary brain tumors: current investigations and the future. Front Immunol. 2022;13:817296. doi:10.3389/fimmu.2022.817296
46. Hegde M, Mukherjee M, Grada Z, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–3052. doi:10.1172/JCI83416
47. Cai Q, Li X, Xiong H, et al. Optical blood-brain-tumor barrier modulation expands therapeutic options for glioblastoma treatment. Nat Commun. 2023;14(1):4934. doi:10.1038/s41467-023-40579-1
48. Pourmasoumi P, Abdouss M, Farhadi M, et al. Co-delivery of temozolomide and quercetin with folic acid-conjugated exosomes in glioblastoma treatment. Nanomedicine. 2024;19(27):2271–2287. doi:10.1080/17435889.2024.2395234
49. Hou AJ, Shih RM, Uy BR, et al. IL-13Rα2/TGF- β bispecific CAR-T cells counter TGF- β -mediated immune suppression and potentiate anti-tumor responses in glioblastoma. Neuro Oncol. 2024;26(10):1850–1866. doi:10.1093/neuonc/noae126
50. Mount CW, Majzner RG, Sundaresh S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med. 2018;24(5):572–579. doi:10.1038/s41591-018-0006-x
51. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi:10.1126/scitranslmed.aaa0984.
52. Goff SL, Morgan RA, Yang JC, et al. Pilot Trial of adoptive transfer of chimeric antigen receptor-transduced t cells targeting egfrviii in patients with glioblastoma. J Immunother. 2019;42(4):126–135. doi:10.1097/CJI.0000000000000260
53. Khan SH, Choi Y, Veena M, et al. Advances in CAR T cell therapy: antigen selection, modifications, and current trials for solid tumors. Front Immunol. 2024;15:1489827. doi:10.3389/fimmu.2024.1489827
54. Flem-Karlsen K, Fodstad Ø, Tan M, et al. B7-H3 in cancer - beyond immune regulation. Trends Cancer. 2018;4(6):401–404. doi:10.1016/j.trecan.2018.03.010
55. Wang Y, Chen M, Wu Z, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7(7):e1440169. doi:10.1080/2162402X.2018.1440169
56. Yuan Y, Ding T, Wang S, et al. Current and emerging therapies for primary central nervous system lymphoma. Biomark Res. 2021;9(1):32. doi:10.1186/s40364-021-00282-z
57. Brown CE, Hibbard JC, Alizadeh D, et al. Locoregional delivery of IL-13Ralpha2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial (June, 10.1038/s41591-024-02875-1, 2023). Nature Med. 2024;30(4):1501. doi:10.1038/s41591-024-02928-5
58. Bagley SJ, Binder ZA, Lamrani L, et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. 2024;5(3):517–531. doi:10.1038/s43018-023-00709-6
59. Geurts M, Preusser M. Locoregional Delivery of Chimeric Antigen Receptor-T Cells: Breaking the Spell in Glioblastoma? Oxford University Press US; 2024:1177–1180.
60. Zhu X, Prasad S, Gaedicke S, et al. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2014;6(1):171. doi:10.18632/oncotarget.2767
61. Genßler S, Burger MC, Zhang C, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. 2016;5(4):e1119354. doi:10.1080/2162402X.2015.1119354
62. Soler DC, Kerstetter-Fogle A, McCormick TS, et al. Using chimeric antigen receptor T-cell therapy to fight glioblastoma multiforme: past, present and future developments. J Neuro-oncol. 2021;1–16.
63. Debinski W, Gibo DM, Hulet SW, et al. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999;5(5):985–990.
64. Tang OY, Binder ZA, O’Rourke DM, et al. Optimizing CAR-T therapy for glioblastoma. Mol Diagn Ther. 2023;27(6):643–660. doi:10.1007/s40291-023-00671-0
65. Alvanou M, Lysandrou M, Christophi P, et al. Empowering the potential of CAR-T cell immunotherapies by epigenetic reprogramming. Cancers. 2023;15(7):1935. doi:10.3390/cancers15071935
66. Huang Z, Dewanjee S, Chakraborty P, et al. CAR T cells: engineered immune cells to treat brain cancers and beyond. Mol Cancer. 2023;22(1):22. doi:10.1186/s12943-022-01712-8
67. Chen C, Park AK, Monroy I, et al. Using oncolytic virus to retask cd19-chimeric antigen receptor-t cells for treatment of pancreatic cancer: toward a universal chimeric antigen receptor-t cell strategy for solid tumors. J Am Coll Surg. 2023;
68. Olivet MM, Brown MC, Reitman ZJ, et al. Clinical applications of immunotherapy for recurrent glioblastoma in adults. Cancers. 2023;15(15):3901. doi:10.3390/cancers15153901
69. Capolla S, Rasool M, Toffoli G, et al. CAR‐T cell manufacturing for hematological and solid tumors: from the preclinical to clinical point of view. Cancer Med. 2025;14(5):e70726. doi:10.1002/cam4.70726
70. Colina AS, Shah V, Shah RK, et al. Current advances in experimental and computational approaches to enhance CAR T cell manufacturing protocols and improve clinical efficacy. Front. mol. med. 2024;4:1310002. doi:10.3389/fmmed.2024.1310002
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Glycolysis-Driven Immune Evasion in Microsatellite Instability-High Colorectal Cancer: An Integrated Single-Cell and Spatial Transcriptomics Study
Li C, Cai P, Zeng H, Li J, Hu H, Zhang J, Wu Z, Qin G, Deng Y
OncoTargets and Therapy 2025, 18:1027-1042
Published Date: 18 September 2025
Advances in PLGA-Based Drug Delivery Systems for Glioblastoma Treatment
Roy S, Alday D, Cai Q
International Journal of Nanomedicine 2025, 20:16125-16147
Published Date: 31 December 2025
Immunological Biomarkers in Glioblastoma: Targeting T and NK Cells for Enhanced Diagnosis and Prognosis
Akbari AM, Bourbour S, Seyedmehdi MS, Shiari S, Rustamzadeh A, Seif F, Farivar S
Biologics: Targets and Therapy 2026, 20:603545
Published Date: 29 May 2026