Back to Journals » Clinical Interventions in Aging » Volume 18
Autoimmunity and Frontotemporal Lobar Degeneration: From Laboratory Study to Clinical Practice
Authors Sun Y, Zhang L, Liu P, Peng G
Received 21 October 2022
Accepted for publication 16 March 2023
Published 27 March 2023 Volume 2023:18 Pages 495—503
DOI https://doi.org/10.2147/CIA.S394286
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Zhi-Ying Wu
Yan Sun, Lumi Zhang, Ping Liu, Guoping Peng
Department of Neurology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
Correspondence: Guoping Peng, Department of Neurology, The First Affiliated Hospital, Zhejiang University School of Medicine, #79 Qingchun Road, Hangzhou, Zhejiang Province, 310003, People’s Republic of China, Tel +86 13588150613, Email [email protected]
Abstract: Frontotemporal lobar degeneration (FTLD) is a group of neurodegenerative diseases with heterogenous clinical, genetic, and pathological characteristics that show similar impairment of areas in the frontal and/or temporal lobes. Prime doctors’ lack of awareness of this complex disease makes early identification and accurate intervention difficult. Autoimmune diseases and autoantibodies are manifestations of different levels of autoimmune reactions. This review presents research findings examining the relationship between autoimmunity and FTLD in terms of autoimmune diseases and autoantibodies with a focus on identifying potential diagnosis and treatment approaches. The findings indicate that the same or similar pathophysiological mechanisms may exist from clinical, genetic, and pathological perspectives. However, the existing evidence is not sufficient to extract substantial conclusions. On the basis of the current situation, we propose future research patterns using prospective studies on large populations and combined clinical and experimental research. Autoimmune reactions or, more generally, inflammatory reactions should receive increased attention from doctors and scientists of all disciplines.
Keywords: frontotemporal lobar degeneration, autoimmunity, autoimmune disorders, autoantibodies, diagnosis, treatment
Introduction
Epidemiology of Frontotemporal Lobar Degeneration (FTLD)
FTLD is a spectrum of diseases that specifically exhibit signs of neurodegeneration in the frontal and/or temporal lobes. This disease continuum possesses different genetic backgrounds, various pathologic hallmarks, and multifarious clinical manifestations.1 FTLD ranks third among the causes of all-age-group dementia, after only Alzheimer’s disease (AD) and Lewy body dementia, and ranks second or third for early onset dementia.2 The point prevalence of FTLD ranges from 0.01 to 4.6 per 1000 persons in different studies.3 However, FTLD patients may impose greater stress and burden on their caregivers than patients with AD or other types of dementia.4 Furthermore, we have reason to believe that the actual number of patients could be greater because of the relatively poor awareness of FTLD in clinical practice worldwide.
Genetic Background of FTLD
In up to 43% of FTLD cases, a positive family history (at least one first-degree family member diagnosed with dementia) has been identified.5 The percentage of FTLD patients with evidence of autosomal dominant inheritance ranges from 10.2% to 27%.6–8 Three types of genetic mutations mainly occur, chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and granulin (GRN) mutations, which collectively account for approximately 60% of familial FTLD cases.9 Mutations in MAPT mainly lead to impaired axonal and mitochondrial function via tau protein hyperphosphorylation and aggregation and neuroinflammation;10,11 mutations in GRN are closely related to the oxidative stress response and neuroinflammation;12 and mutations in C9orf72 usually cause haploinsufficiency and metabolic abnormalities in DNA/RNA.13 Rarer causal genes include TANK-binding kinase 1 (TBK1),14 valosin containing protein (VCP),15 charged multivesicular body protein 2B (CHMP2B),16 sequestosome 1 (SQSTM1),17 ubiquilin 2 (UBQLN2),18 and coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10).19
Pathological Background of FTLD
The neuropathological classification of FTLD relies on the components of protein inclusions found in degenerative neurons, including two main types, ie, TAR DNA-binding protein 43 (TDP43) and tau, as well as three minor types, ie, FUS, EWS or TATA-binding protein-associated factor 2N (FET), ubiquitin-proteasome system (UPS), and negative for any protein inclusion. The intricate relationship among phenotypes, genotypes, and pathotypes of FTLD has been extensively described in previous reviews.1,20,21
Clinical Manifestations of FTLD
Based on clinical characteristics including patient behavior, language skill, and executive function impairment, “classical” FTLD (also known as frontotemporal dementia (FTD) as a clinical syndrome) can be divided into three main subtypes, including behavioral-variant FTD (bvFTD), progressive non-fluent aphasia (also known as non-fluent-variant primary progressive aphasia), and semantic dementia (also known as semantic-variant primary progressive aphasia).1 The commonly accepted diagnostic standards for bvFTD are the FTDC (International Behavioral Variant FTD Criteria Consortium) recommendations updated by Rascovsky et al,22 and the diagnostic standards for the other two subtypes are the criteria developed by Gorno-Tempini et al.23 Recently, an overlap between motor neuron disease, atypical parkinsonian syndromes, and FTD has been revealed. As the largest portion of motor neuron disease, approximately 10–15% of amyotrophic lateral sclerosis (ALS) patients meet the diagnostic standard for FTD, and ALS patients can also show the same pathological background of TDP43 as FTD patients do.24,25 For atypical parkinsonian syndromes, some cortical basal syndrome and progressive supranuclear palsy cases can also be clinically classified as FTD.26 However, the underlying correlation requires further investigation.
Autoimmunity in Nervous System Diseases
Autoimmunity is the process by which the immune system identifies the body’s own tissues as antigens and responds to them, leading to tissue damage and function impairment. Autoantibodies and/or sensitized lymphocytes are typically the causes of autoimmunity onset. With advances in testing technology, there is increasing evidence that autoimmune responses are involved in or directly contribute to a variety of central and peripheral nervous system disorders. Among diseases with acute to subacute onset, autoimmune encephalitis could be associated with N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptor autoantibodies27 and leucine-rich glioma-inactivated protein 1 (LGI1)28 and myelin oligodendrocyte glycoprotein antibodies;29 neuromyelitis optica spectrum disorders could be related to aquaporin 4 (AQP4) autoantibodies.30 As a neurodegenerative disease with a chronic course, antibodies targeting melanin,31 α-synuclein,32 and GM1 ganglioside33 have been found to be responsible for Parkinson’s disease, while in AD, autoantibodies against glial fibrillary acidic protein and S100b could affect astrocytes.34
Recently, an overlap between FTLD and autoimmune disease has been discovered, generating a new line of inquiry into the causes and potential treatments for FTLD. This review mainly presents evidence of a link between FTLD and autoimmune diseases or autoantibodies, describes active attempts to identify therapies targeting autoimmunity, and offers suggestions for future research.
FTLD and Autoimmune Disorders
Autoimmune disorders either specifically influence one organ or system (eg, Hashimoto thyroiditis, autoimmune encephalitis) or systematically influence the whole human body (eg, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA)). The commonly accepted view is that immune responses triggered by genetic predisposition and environmental factors can cause autoimmune diseases.35 Two classic mechanisms of environmental factors are molecular mimicry36 and epitope spreading,37 both of which result in the loss of immune tolerance. Because FTLD is influenced by genetic background and could be affected by the environment, the same immune-induced pathways might lead to both disease continuums.
Clinical Comorbidity of FTLD and Autoimmune Disease
In FTLD-TDP43 patients, the prevalence of overall autoimmune disease varies from 18% to 23%.38,39 A case report and a small-scale study40,41 reported an increased prevalence of primary progressive aphasia in men with a history of vasectomy. These results suggest that autoimmune mechanisms are involved in the pathogenesis of FTLD because constant resorption of sperm provides a sufficient supply of the antigens responsible for many immune events.42 Over the past 10 years, studies examining the clinical correlation between FTLD and autoimmune diseases based on a broader population have been performed (Table 1). The influence of autoimmune thyroid diseases (including Graves’ disease and Hashimoto disease), as the most frequently diagnosed autoimmune diseases in the entire population,43 on the incidence of FTLD has not been clarified.38,44 In contrast, some relatively uncommon autoimmune diseases have shown a strong relationship with FTD, including inflammatory arthritis, cutaneous conditions, and gastrointestinal disorders.38,39 Therefore, non-thyroid disease “clusters” might serve as a more accurate indicator for FTLD.
 |
Table 1 Available Data of Cohort Studies on FTLD and Autoimmunity Diseases |
Clinicians should enhance the awareness of comorbidities between FTLD and autoimmune diseases, not only waiting for the occurrence passively, but also monitoring relative markers actively. For example, neurologists could pay more attention to FTLD patients’ complaint on joint pain, abdominal pain or hematochezia, and test blood markers as antinuclear antibody more frequently. Also, rheumatologists could also concern more about their patients’ cognitive function by means of neuropsychological scales like MMSE and MoCA. With more information gathered on this area, the goal of early recognition and intervention might be achieved earlier.
Genetic Factors Related to FTLD and Autoimmune Disease
FTLD and autoimmune diseases might share some genetic risk factors. A genome-wide association study helped reveal two novel loci associated with the human leukocyte antigen and the lysosomal and autophagy pathways, separately, at 6p21.3 and 11q14, providing strong evidence for the involvement of autoimmune responses at the genetic level.45 Another genome-wide association study including 192,886 cases and controls showed evident genetic enrichment between FTLD and autoimmune diseases (including RA, ulcerative colitis, type 1 diabetes, and celiac disease), ranging from 160-fold to 270-fold. Furthermore, the human leukocyte antigen region on chromosome 6, which is crucial for modulation of microglial function, was found to exhibit genetic enrichment in FTD and the autoimmune diseases mentioned above.46 Phospholipase C gamma 2 (PLCG2) plays a potential role in inflammation-related pathways.47 However, a gain-of-function or deletion mutation in the PLCG2 gene was associated with autoimmune disease occurrence.48–50 Furthermore, rs72824905-G, a rare PLCG2 mutation, was protective against FTD.51 Although the precise mechanism remains unknown, PLCG2 could be a connection between autoimmune disease and FTD.
As the most common genetic background associated with FTLD, C9orf72 also contributes to the incidence of autoimmune diseases. However, whether C9orf72 mutation is a protective factor or a risk factor for autoimmune disease remains unclear. Some results support that FTLD patients without C9orf72 mutation have a greater likelihood of suffering from autoimmune diseases than FTLD patients with C9orf72 mutation,52,53 while another study suggests the opposite.39 In animal experiments, the C9orf72−/− genotype could induce myeloid expansion, T cell activation, and plasma cell proliferation in mice, and also an elevated autoantibody level.54 Although hundreds of thousands of repeated expansion units are common in C9orf72-related FTLD, new evidence has illustrated that intermediate expansion, defined as hexanucleotide units ranging from 9 to 30, is closely related to autoimmune diseases, such as SLE, RA, and kidney-involved diseases.55,56
Environmental Factors Related to FTLD and Autoimmune Disease
Studies of familial FTLD have shown that the GGGCCC hexanucleotide repeat amplification sequence of C9orf72 has incomplete penetrance,57,58 which indicates that other factors (such as environmental factors) can change an individual’s disease risk. Therefore, for people who have a genetic susceptibility, early intervention against hazardous factors may help prevent the occurrence of FTLD.
Infection, as a common and well-studied factor that can induce autoimmune diseases, also acts as an environmental factor that could modify the risk of FTLD. Burberry et al showed that reducing the abundance of bacteria that can produce immune stimulation in the environment can reduce the risk of premature death in mice with C9orf72 gene mutations.59 This study also provided evidence supporting intestinal microbe regulation therapy.
Environmental pollutants such as planar polychlorinated biphenyls and polycyclic aromatic hydrocarbons, which have been shown to be risk factors for autoimmune disease, could cause the pathologic accumulation of TDP43 in mice by activating the aryl hydrocarbon receptor.60 Occupational exposure to aluminum, pesticides, dyes, paints, or thinners was found to be related to an increased incidence of FTD in a case-control study in Northern Italy.61 Exposure to these substances also leads to an increased risk of autoimmune diseases such as SLE,62 Graves’ disease,63 and multiple sclerosis.64 In contrast to common views, smoking and alcohol consumption, which act as risk factors for most autoimmune diseases,65,66 seem to be protective factors against FTLD.67
FTLD and Autoantibodies
Autoantibodies refer to antibodies against an organism’s own tissues, organs, cells, and/or cellular components. Autoantibodies exist at low levels in healthy individuals without causing any disease. However, when the titer of some autoantibodies exceeds a given level, they may cause direct damage to the body. In other cases, elevation of the levels of some autoantibodies is a secondary result of an autoimmune reaction.68 FTLD patients without clinical diagnostic autoimmune diseases can also present an abnormal autoantibody titer (Table 2).
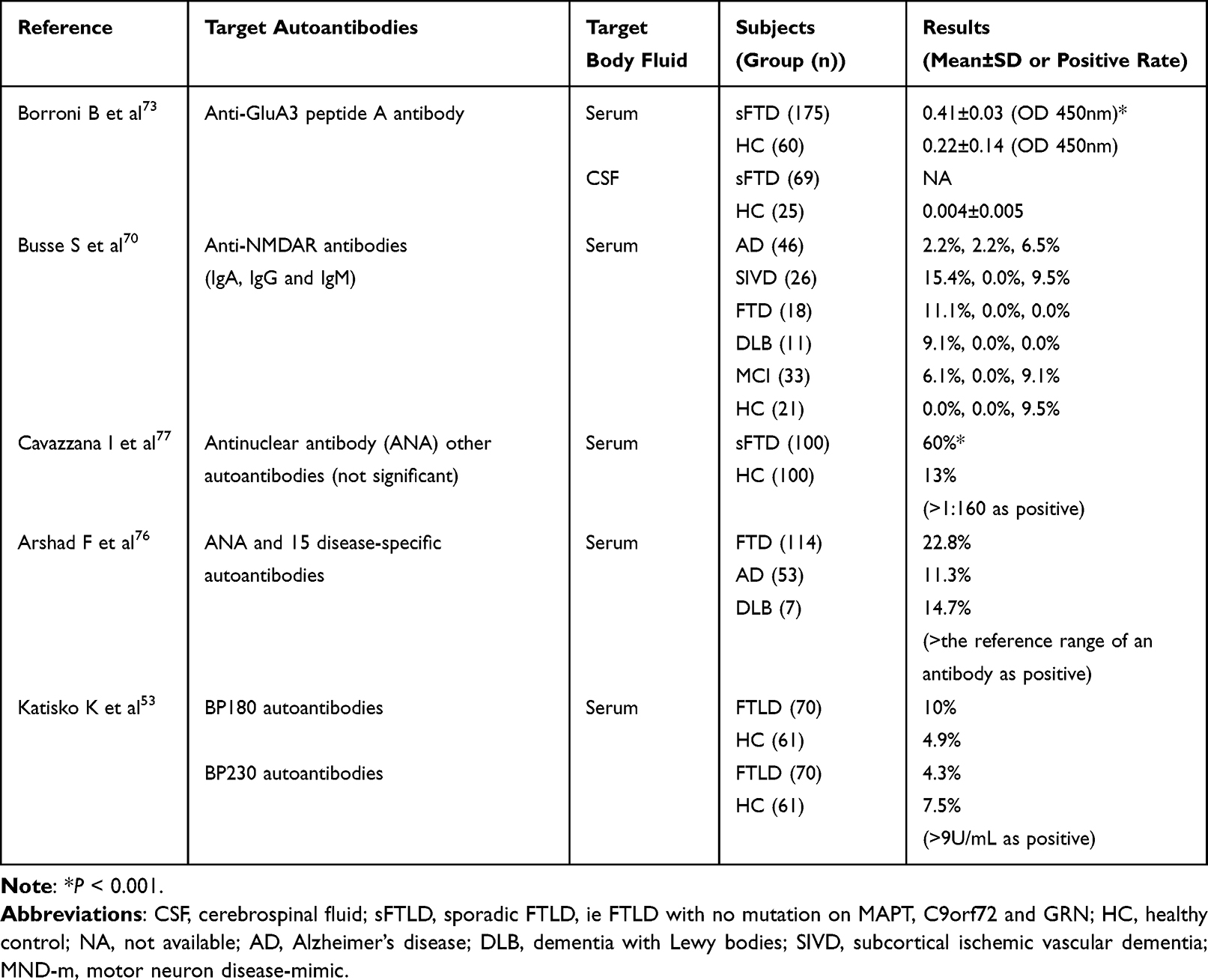 |
Table 2 Available Data of Cohort Studies on FTLD and Autoantibodies |
Anti-Glutamatergic Receptor Autoantibodies Related to FTLD
The glutamate neurotransmitter system plays an important part in the pathogenesis of FTLD, which has been thoroughly discussed in previous reviews.69 Briefly, there are two forms of glutamatergic receptors at the synaptic level, ionotropic and metabotropic glutamate receptors. The former consists of three subtypes, NMDA receptors (NMDARs), AMPA receptors (AMPARs) and kainic acid receptors, while the latter is a receptor family that is coupled with G proteins. Peripheral autoantibodies against NMDARs have been detected in serum samples from patients with FTD as well as in patients with other neurodegenerative diseases and healthy older adults.70 Among these antibodies, IgA and IgG anti-NMDAR antibodies are responsible for decreasing the NMDAR density. However, no measurement of the anti-NMDAR level in the cerebral spinal fluid (CSF) of FTLD patients is available. Therefore, the existence of anti-NMDAR antibodies in serum can only be treated as a warning of possible future development of FTLD.
AMPARs consist of four types of subunits referred to as GluA1–A4, among which GluA3 has been the focus in the study of FTLD.71 The first case was reported in 2017; a man developed clinically diagnosed FTLD after a vasectomy and had detectable serum levels of anti-AMPAR antibodies.72 Later, Borroni et al73 estimated the positive anti-GluA3 dosage to be 0.64 (optical density 450 nm, unit unmentioned) in serum and 0.019 (optical density 450 nm, unit unmentioned) in CSF, which could distinguish 29% and 21.7% of patients among healthy controls. The serum and CSF levels of anti-GluA3 antibodies are closely related. An increased anti-GluA3 level indicates a younger age of onset in FTLD cases. In vitro, rat primary hippocampal neurons processed with anti-GluA3 antibodies showed a significant decrease in GluA3 subunit levels at postsynaptic sites. Similar results were found in human-induced pluripotent stem cells,73,74 which were later supported by findings from post-mortem specimens.52 In vivo, a decreased level of intracortical facilitation in humans via transcranial magnetic stimulation was reported.75 To compensate for dysregulation of the glutamatergic system, D-Ser, L-Ser, and L-Glu levels in the CSF could increase, which could also serve as an indicator of this disequilibrium.75
Other Autoantibodies Related to FTLD
Because a wide spectrum of autoantibodies has been found in FTLD patients, the connection of autoimmunity with FTLD seems to be more substantial than that for other neurodegenerative diseases causing dementia.76 The presence of antinuclear antibody might be the initial disruptive event in the immune system. Then, changes in other autoantibodies with more directivity (eg, anti-dsDNA, anti-Sm) indicating a particular autoimmune disease could occur.77
GRN acts as a key modulator for immune regulation and development of inflammatory reactions, probably by partly blocking binding of tumor necrosis factor to its receptor.78,79 Despite the absence of direct evidence linking FTLD with GRN mutation and autoimmunity, an increased anti-progranulin level was detected in patients with autoimmune diseases including SLE, RA, and vasculitis compared with that in healthy controls.80
Additional sporadic studies without received adequate validation have been conducted. Anti-TDP43 antibodies were significantly higher in an ALS cohort than in FTLD, AD, and healthy control cohorts,81 but a decreased level of high-affinity anti-TDP43 NAbs and IgM antibodies was reported in another ALS cohort.82 More FTLD patients than healthy controls tested positive for the autoantibodies BP180 and BP230, which are responsible for bullous pemphigoid, especially in the C9orf72-carrier subgroup.53 An elevated level of antibodies against neuronal voltage-gated calcium channels, such as P/Q- and N-type calcium channel antibodies, which are usually found in paraneoplastic autoimmune encephalitis, could result in subacute onset of bvFTD-like clinical symptoms and neuroimaging results.83 Likewise, autoantibody-related encephalitis, which mimics the manifestations and imaging alterations of FTD, could also be induced by voltage-gated potassium channel antibodies.84,85 The connections among these factors are still unknown.
Autoimmune-Mediated Therapies for FTLD
There is still no precise evidence to support any efficient therapy against the impairment of social function and cognitive capacity in FTLD patients. Although the evidence mentioned above suggests a relationship between FTLD and NMDARs, the NMDAR antagonist memantine has not shown efficacy against FTLD.86 Currently, antipsychotics are still the most frequently prescribed medications used to treat abnormalities in mood or behavior.
Plasma exchange, glucocorticoids, and intravenous immunoglobulin are classic treatments widely used for acute or subacute neurological diseases related to immune dysregulation, such as autoimmune encephalitis and Guillain–Barre syndrome. Although the deterioration of FTLD patients is chronic in most cases, one patient had sudden onset of an FTD-like disorder that was cured using the treatments mentioned above.83 Rituximab worked well as a second-line treatment in a case of severe delayed NMDAR encephalitis.87 If long-term autoimmune encephalitis and the chronic course of FTLD are somewhat comparable, monoclonal antibody therapy might offer a new direction for exploration.
Conclusions
On the basis of the available findings, the association between FTLD and autoimmunity has not been thoroughly clarified. The existence of autoimmune diseases or elevated autoantibody levels can only provide information but cannot act as diagnostic criteria. In addition, unlike the C9orf72 and GRN genotypes (usually showing TDP43 pathology), FTLD with mutation of MAPT (usually showing tau pathology) and other genes has been rarely discussed.
Because most studies were designed to review participants’ histories of autoimmune disease or detect their autoantibody level rather than track their immune function during a prospective course, there are still many questions that need to be addressed. It is unknown whether autoimmune disease or FTLD occurs first or whether they are both expressions of a genetic mutation or pathologic change. Because of the variety of the FTLD disease spectrum, determination of a superficial relationship between clinically diagnosed FTLD and the existence of autoimmune disease or autoantibodies may not be satisfying. More promising pathological or genetic targets for diagnosis or treatment could be hidden. Moreover, some results could be presented incorrectly as a trend or discrepancy without precise diagnosis of the FTLD subtype.
With the increasing burden of FTLD worldwide, deficits in its early identification and medical treatment urgently need to be amended. We believe that autoimmunity is a promising and worthy target for improved understanding and management of FTLD after more thorough clinical and laboratory investigations have been performed to obtain more substantial results. Clinical studies based on large-scale and long-term tracked cohorts will be necessary in the future. Multi-center databases such as the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) and the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) databases will also offer convenience for further investigation. Experiments in vivo and in vitro, along with corresponding pathological evidence from brain autopsy or biopsy, might provide a more comprehensive protocol for research.
Acknowledgments
This work was funded by National Natural Science Foundation (82071182) and Zhejiang Provincial Natural Science Foundation (LY20H090014).
We thank Lisa Kreiner, PhD, from Liwen Bianji, (Edanz) (www.liwenbianji.cn), for editing the English text of a draft of this manuscript.
Disclosure
The authors declare no competing interests in this work.
References
1. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672–1682. doi:10.1016/S0140-6736(15)00461-4
2. Vieira RT, Caixeta L, Machado S, et al. Epidemiology of early-onset dementia: a review of the literature. Clin Pract Epidemiol Ment Health. 2013;9:88–95. doi:10.2174/1745017901309010088
3. Hogan DB, Jetté N, Fiest KM, et al. The prevalence and incidence of frontotemporal dementia: a systematic review. Can J Neurol Sci. 2016;43(Suppl 1):S96–S109. doi:10.1017/cjn.2016.25
4. Kawano Y, Terada S, Takenoshita S, et al. Patient affect and caregiver burden in dementia. Psychogeriatrics. 2020;20(2):189–195. doi:10.1111/psyg.12487
5. Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(Pt 9):2016–2022. doi:10.1093/brain/awg204
6. Rohrer J, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):18. doi:10.1212/WNL.0b013e3181bf997a
7. Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65(11):1817–1819. doi:10.1212/01.wnl.0000187068.92184.63
8. Seelaar H, Kamphorst W, Rosso SM, et al. Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71(16):1220–1226. doi:10.1212/01.wnl.0000319702.37497.72
9. Le Ber I. Genetics of frontotemporal lobar degeneration: an up-date and diagnosis algorithm. Rev Neurol (Paris). 2013;169(10):811–819. doi:10.1016/j.neurol.2013.07.014
10. Zhang CC, Xing A, Tan MS, Tan L, Yu JT. The role of MAPT in neurodegenerative diseases: genetics, mechanisms and therapy. Mol Neurobiol. 2016;53(7):4893–4904. doi:10.1007/s12035-015-9415-8
11. Butzlaff M, Hannan SB, Karsten P, et al. Impaired retrograde transport by the dynein/dynactin complex contributes to tau-induced toxicity. Hum Mol Genet. 2015;24(13):3623–3637. doi:10.1093/hmg/ddv107
12. Galimberti D, Bonsi R, Fenoglio C, et al. Inflammatory molecules in frontotemporal dementia: cerebrospinal fluid signature of progranulin mutation carriers. Brain Behav Immun. 2015;49:182–187. doi:10.1016/j.bbi.2015.05.006
13. Sansan T, Jun L, Lan T, Jintai Y. Genetics of frontotemporal lobar degeneration: from the bench to the clinic. J Alzheimers Dis. 2016;52(4). doi:10.3233/JAD-160236
14. Gijselinck I, Van Mossevelde S, van der Zee J, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85(24):2116–2125. doi:10.1212/WNL.0000000000002220
15. WattsGDJ, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381. doi:10.1038/ng1332
16. Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. doi:10.1038/ng1609
17. Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations–bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014;325(1):27–37. doi:10.1016/j.yexcr.2014.01.020
18. Dillen L, Van Langenhove T, Engelborghs S, et al. Explorative genetic study of UBQLN2 and PFN1 in an extended Flanders-Belgian cohort of frontotemporal lobar degeneration patients. Neurobiol Aging. 2013;34(6):
19. Bannwarth S, Ait-El-Mkadem S, Chaussenot A, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137(Pt 8):2329–2345. doi:10.1093/brain/awu138
20. Van Mossevelde S, Engelborghs S, van der Zee J, Van Broeckhoven C. Genotype-phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol. 2018;14(6):363–378. doi:10.1038/s41582-018-0009-8
21. Boeve BF, Boxer AL, Kumfor F, Pijnenburg Y, Rohrer JD. Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022;21(3):258–272. doi:10.1016/S1474-4422(21)00341-0
22. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. doi:10.1093/brain/awr179
23. Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi:10.1212/WNL.0b013e31821103e6
24. Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918–1929. doi:10.1111/ene.14393
25. Burrell JR, Halliday GM, Kril JJ, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388(10047):919–931. doi:10.1016/S0140-6736(16)00737-6
26. Deutschländer AB, Ross OA, Dickson DW, Wszolek ZK. Atypical parkinsonian syndromes: a general neurologist’s perspective. Eur J Neurol. 2018;25(1):41–58. doi:10.1111/ene.13412
27. Nissen MS, Ryding M, Meyer M, Blaabjerg M. Autoimmune encephalitis: current knowledge on subtypes, disease mechanisms and treatment. CNS Neurol Disord Drug Targets. 2020;19(8):584–598. doi:10.2174/1871527319666200708133103
28. Dürr M, Nissen G, Sühs KW, et al. CSF findings in acute NMDAR and LGI1 antibody-associated autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm. 2021;8(6):e1086. doi:10.1212/NXI.0000000000001086
29. Salama S, Khan M, Pardo S, Izbudak I, Levy M. MOG antibody-associated encephalomyelitis/encephalitis. Mult Scler. 2019;25(11):1427–1433. doi:10.1177/1352458519837705
30. Hinson SR, Lennon VA, Pittock SJ. Autoimmune AQP4 channelopathies and neuromyelitis optica spectrum disorders. Handb Clin Neurol. 2016;133:377–403. doi:10.1016/B978-0-444-63432-0.00021-9
31. Double KL, Rowe DB, Carew-Jones FM, et al. Anti-melanin antibodies are increased in sera in Parkinson’s disease. Exp Neurol. 2009;217(2):297–301. doi:10.1016/j.expneurol.2009.03.002
32. Papachroni KK, Ninkina N, Papapanagiotou A, et al. Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J Neurochem. 2007;101(3):749–756. doi:10.1111/j.1471-4159.2006.04365.x
33. Zappia M, Crescibene L, Bosco D, et al. Anti-GM1 ganglioside antibodies in Parkinson’s disease. Acta Neurol Scand. 2002;106(1):54–57. doi:10.1034/j.1600-0404.2002.01240.x
34. Gruden MA, Davidova TB, Malisauskas M, et al. Differential neuroimmune markers to the onset of Alzheimer’s disease neurodegeneration and dementia: autoantibodies to Abeta((25–35)) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007;186(1–2):181–192. doi:10.1016/j.jneuroim.2007.03.023
35. Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. 2015;278(4):369–395. doi:10.1111/joim.12395
36. Blank M, Barzilai O, Shoenfeld Y. Molecular mimicry and auto-immunity. Clin Rev Allergy Immunol. 2007;32(1):111–118. doi:10.1007/BF02686087
37. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2(2):85–95. doi:10.1038/nri724
38. Miller ZA, Rankin KP, Graff-Radford NR, et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry. 2013;84(9):956–962. doi:10.1136/jnnp-2012-304644
39. Miller ZA, Sturm VE, Camsari GB, et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: completing the picture. Neurol Neuroimmunol Neuroinflamm. 2016;3(6):e301. doi:10.1212/NXI.0000000000000301
40. Weintraub S, Fahey C, Johnson N, et al. Vasectomy in men with primary progressive aphasia. Cogn Behav Neurol. 2006;19(4):190–193. doi:10.1097/01.wnn.0000213923.48632.ab
41. Decker DA, Heilman KM. Steroid treatment of primary progressive aphasia. Arch Neurol. 2008;65(11):1533–1535. doi:10.1001/archneur.65.11.1533
42. Sotolongo JR. Immunological effects of vasectomy. J Urol. 1982;127(6):1063–1066. doi:10.1016/s0022-5347(17
43. Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84(3):223–243. doi:10.1006/clin.1997.4412
44. Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC. Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosurg Psychiatry. 2003;74(11):1574–1576. doi:10.1136/jnnp.74.11.1574
45. Ferrari R, Hernandez DG, Nalls MA, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13(7):686–699. doi:10.1016/S1474-4422(14
46. Broce I, Karch CM, Wen N, et al. Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. PLoS Med. 2018;15(1):e1002487. doi:10.1371/journal.pmed.1002487
47. Tsai AP, Dong C, Lin PBC, et al. PLCG2 is associated with the inflammatory response and is induced by amyloid plaques in alzheimer’s disease. Genome Med. 2022;14(1):17. doi:10.1186/s13073-022-01022-0
48. Ombrello MJ, Remmers EF, Sun G, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. 2012;366(4):330–338. doi:10.1056/NEJMoa1102140
49. Yu P, Constien R, Dear N, et al. Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity. 2005;22(4):451–465. doi:10.1016/j.immuni.2005.01.018
50. Borders C, Sajjadi A. Association of inflammatory disorders with degenerative neuropathologies. Neurology. 2021;96(15SUPPL 1):1.
51. van der Lee SJ, Conway OJ, Jansen I, et al. A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 2019;138(2):237–250. doi:10.1007/s00401-019-02026-8
52. Katisko K, Solje E, Koivisto AM, et al. Prevalence of immunological diseases in a Finnish frontotemporal lobar degeneration cohort with the C9orf72 repeat expansion carriers and non-carriers. J Neuroimmunol. 2018;321:29–35. doi:10.1016/j.jneuroim.2018.05.011
53. Katisko K, Kokkonen N, Krüger J, et al. The association between frontotemporal lobar degeneration and bullous pemphigoid. J Alzheimers Dis. 2018;66(2):743–750. doi:10.3233/JAD-180624
54. Atanasio A, Decman V, White D, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. doi:10.1038/srep23204
55. Fredi M, Cavazzana I, Biasiotto G, et al. C9orf72 intermediate alleles in patients with amyotrophic lateral sclerosis, systemic Lupus erythematosus, and Rheumatoid arthritis. Neuromolecular Med. 2019;21(2):150–159. doi:10.1007/s12017-019-08528-8
56. Fredi M, Biasiotto G, Cavazzana I, et al. PS1:3 analysis of c9orf72 expansions in patients with systemic lupus erythematosus and rheumatoid arthritis: preliminary data. Lupus Sci Med. 2018;5(Suppl):1. doi:10.1136/lupus-2018-abstract.52
57. Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135(Pt 3):765–783. doi:10.1093/brain/aws004
58. Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14(9):544–558. doi:10.1038/s41582-018-0047-2
59. Burberry A, Wells MF, Limone F, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 2020;582(7810):89–94. doi:10.1038/s41586-020-2288-7
60. Ash PEA, Stanford EA, Al Abdulatif A, et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol Neurodegener. 2017;12(1):35. doi:10.1186/s13024-017-0177-9
61. Adani G, Filippini T, Garuti C, et al. Environmental risk factors for early-onset alzheimer’s dementia and frontotemporal dementia: a case-control study in Northern Italy. Int J Environ Res Public Health. 2020;17(21):7941. doi:10.3390/ijerph17217941
62. Dooley MA, Hogan SL. Environmental epidemiology and risk factors for autoimmune disease. Curr Opin Rheumatol. 2003;15(2):99–103. doi:10.1097/00002281-200303000-00002
63. Antonelli A, Ferrari SM, Ragusa F, et al. Graves’ disease: epidemiology, genetic and environmental risk factors and viruses. Best Pract Res Clin Endocrinol Metab. 2020;34(1):101387. doi:10.1016/j.beem.2020.101387
64. Alfredsson L, Olsson T. Lifestyle and environmental factors in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(4):a028944. doi:10.1101/cshperspect.a028944
65. Dong YH, Fu DG. Autoimmune thyroid disease: mechanism, genetics and current knowledge. Eur Rev Med Pharmacol Sci. 2014;18(23):3611–3618.
66. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2(1):16039. doi:10.1038/nrdp.2016.39
67. Tremolizzo L, Bianchi E, Susani E, et al. Voluptuary habits and risk of frontotemporal dementia: a case control retrospective study. J Alzheimers Dis. 2017;60(2):335–340. doi:10.3233/JAD-170260
68. Williams RC, Malone CC, Silvestris F. Autoantibodies as chameleons. Scand J Rheumatol. 1997;26(2):73–78. doi:10.3109/03009749709115822
69. Benussi A, Alberici A, Buratti E, et al. Toward a glutamate hypothesis of frontotemporal dementia. Front Neurosci. 2019;13:304. doi:10.3389/fnins.2019.00304
70. Busse S, Brix B, Kunschmann R, Bogerts B, Stoecker W, Busse M. N-methyl-d-aspartate glutamate receptor (NMDA-R) antibodies in mild cognitive impairment and dementias. Neurosci Res. 2014;85:58–64. doi:10.1016/j.neures.2014.06.002
71. Italia M, Ferrari E, Di Luca M, Gardoni F. GluA3-containing AMPA receptors: from physiology to synaptic dysfunction in brain disorders. Neurobiol Dis. 2021;161:105539. doi:10.1016/j.nbd.2021.105539
72. Borroni B, Manes MA, Alberici A, et al. Autoimmune frontotemporal dementia: a new nosological entity? Alzheimer Dis Assoc Disord. 2017;31(3):259–262. doi:10.1097/WAD.0000000000000180
73. Borroni B, Stanic J, Verpelli C, et al. Anti-AMPA GluA3 antibodies in frontotemporal dementia: a new molecular target. Sci Rep. 2017;7(1):6723. doi:10.1038/s41598-017-06117-y
74. Scheggia D, Stanic J, Italia M, et al. GluA3 autoantibodies induce alterations in dendritic spine and behavior in mice. Brain Behav Immun. 2021;97:89–101. doi:10.1016/j.bbi.2021.07.001
75. Palese F, Bonomi E, Nuzzo T, et al. Anti-GluA3 antibodies in frontotemporal dementia: effects on glutamatergic neurotransmission and synaptic failure. Neurobiol Aging. 2020;86:143–155. doi:10.1016/j.neurobiolaging.2019.10.015
76. Arshad F, Varghese F, Paplikar A, et al. Role of autoantibodies in neurodegenerative dementia: an emerging association. Dement Geriatr Cogn Disord. 2021;50(2):153–160. doi:10.1159/000517238
77. Cavazzana I, Alberici A, Bonomi E, et al. Antinuclear antibodies in frontotemporal dementia: the tip’s of autoimmunity iceberg? J Neuroimmunol. 2018;325:61–63. doi:10.1016/j.jneuroim.2018.10.006
78. Bosch X. Progranulin: a growth factor, a novel TNFR ligand and a drug target. Pharmacol Ther. 2012;133(1):124–132. doi:10.1016/j.pharmthera.2011.10.003
79. Hu Y, Xiao H, Shi T, Oppenheim JJ, Chen X. Progranulin promotes tumour necrosis factor-induced proliferation of suppressive mouse CD4+ Foxp3+ regulatory T cells. Immunology. 2014;142(2):193–201. doi:10.1111/imm.12241
80. Thurner L, Preuss KD, Fadle N, et al. Progranulin antibodies in autoimmune diseases. J Autoimmun. 2013;42:29–38. doi:10.1016/j.jaut.2012.10.003
81. Conti E, Sala G, Diamanti S, et al. Serum naturally occurring anti-TDP-43 auto-antibodies are increased in amyotrophic lateral sclerosis. Sci Rep. 2021;11(1):1978. doi:10.1038/s41598-021-81599-5
82. Nielsen AK, Folke J, Owczarek S, et al. TDP-43-specific autoantibody decline in patients with amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):e937. doi:10.1212/NXI.0000000000000937
83. Younes K, Lepow LA, Estrada C, Schulz PE. Auto-antibodies against P/Q- and N-type voltage-dependent calcium channels mimicking frontotemporal dementia. SAGE Open Med Case Rep. 2018;6:2050313X17750928. doi:10.1177/2050313X17750928
84. Kotagal V, Lorincz MT, Bohnen NI. A frontotemporal dementia-like syndrome mimicking postpartum depression detected by 18F fluorodeoxyglucose positron emission tomography. Clin Nucl Med. 2012;37(9):e223–e224. doi:10.1097/RLU.0b013e31824440a1
85. Freund B, Maddali M, Lloyd TE. A case of Morvan syndrome mimicking amyotrophic lateral sclerosis with frontotemporal dementia. J Clin Neuromuscul Dis. 2016;17(4):207–211. doi:10.1097/CND.0000000000000118
86. Young JJ, Lavakumar M, Tampi D, Balachandran S, Tampi RR. Frontotemporal dementia: latest evidence and clinical implications. Ther Adv Psychopharmacol. 2018;8(1):33–48. doi:10.1177/2045125317739818
87. Leypoldt F, Gelderblom M, Schöttle D, Hoffmann S, Wandinger KP. Recovery from severe frontotemporal dysfunction at 3 years after N-methyl-d-aspartic acid (NMDA) receptor antibody encephalitis. J Clin Neurosci. 2013;20(4):611–613. doi:10.1016/j.jocn.2012.03.036
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Nanotechnology: A Promising Approach for Cancer Diagnosis, Therapeutics and Theragnosis
Dessale M, Mengistu G, Mengist HM
International Journal of Nanomedicine 2022, 17:3735-3749
Published Date: 26 August 2022
Updated Perspectives on the Diagnosis and Management of Onychomycosis
Falotico JM, Lipner SR
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1933-1957
Published Date: 15 September 2022
Nontuberculous Mycobacteria Lung Disease (NTM-LD): Current Recommendations on Diagnosis, Treatment, and Patient Management
Pathak K, Hart S, Lande L
International Journal of General Medicine 2022, 15:7619-7629
Published Date: 1 October 2022
Challenges in the Early Diagnosis and Treatment of Chronic Inflammatory Demyelinating Polyradiculoneuropathy in Adults: Current Perspectives
van Doorn IN, Eftimov F, Wieske L, van Schaik IN, Verhamme C
Therapeutics and Clinical Risk Management 2024, 20:111-126
Published Date: 14 February 2024
Managing Opioid Withdrawal Symptoms During the Fentanyl Crisis: A Review
Weber AN, Trebach J, Brenner MA, Thomas MM, Bormann NL
Substance Abuse and Rehabilitation 2024, 15:59-71
Published Date: 10 April 2024