Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Associations Between Albumin-Corrected Anion Gap and Mortality in Heart Failure Patients with Chronic Obstructive Pulmonary Disease: A Retrospective Cohort Study
Authors Wu G ![]() , Ke H
, Ke H ![]() , Jin Z, Shen Z
, Jin Z, Shen Z ![]() , Tong Z
, Tong Z
Received 14 October 2025
Accepted for publication 10 March 2026
Published 20 March 2026 Volume 2026:21 574358
DOI https://doi.org/10.2147/COPD.S574358
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Vanesa Bellou
Gang Wu,1– 3,* Huanya Ke,4,* Zhaoxia Jin,1– 3,* Zhengjun Shen,1– 3 Zijia Tong1– 3
1Department of Cardiology, Huanggang Central Hospital, Huanggang, People’s Republic of China; 2Department of Cardiology, Hubei Provincial Industrial Technology Research Institute of Cardio-Cerebral Co-Therapy, Huanggang, People’s Republic of China; 3Department of Cardiology, Hubei Provincial Engineering Research Center of Cardio-Cerebral Co-Therapy, Huanggang, People’s Republic of China; 4Department of Pharmacy, Huanggang Central Hospital, Huanggang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zijia Tong, Department of Cardiology, Huanggang Central Hospital, Huanggang, People’s Republic of China, Email [email protected]
Background: Heart failure (HF) and chronic obstructive pulmonary disease (COPD) are common comorbidities in intensive care unit (ICU) patients. The albumin-corrected anion gap (ACAG) has shown utility in predicting mortality across various populations; however, its impact on HF patients with COPD remains unclear. This study investigated the relationship between ACAG and mortality in this population.
Methods: We conducted a retrospective cohort study using the Medical Information Mart for Intensive Care (MIMIC)-IV database. A total of 1283 patients with heart failure and chronic obstructive pulmonary disease were included from the MIMIC-IV database. ACAG levels were assessed within 24 hours of admission. The association between ACAG and in-hospital and 30-day mortality was analyzed using Kaplan-Meier analysis, multivariate Cox regression, restricted cubic spline (RCS) analysis, subgroup analysis, and receiver operating characteristic (ROC) curve analysis.
Results: Among 1283 HF patients with COPD (54.6% male), in-hospital and 30-day mortality rates were 11.2% and 13.7%, respectively. Kaplan-Meier analysis demonstrated significantly increased mortality risk in patients with higher ACAG levels (log-rank P< 0.001). In fully adjusted Cox models, compared to the lowest ACAG group (T1), the highest group (T3) showed hazard ratios of 2.04 (95% CI: 1.18– 3.54; p=0.011) for in-hospital mortality and 1.83 (95% CI: 1.12– 2.97; p=0.015) for 30-day mortality. RCS analysis revealed a linear relationship between ACAG and mortality risk, consistent across subgroups. ROC analysis demonstrated superior discriminatory ability of ACAG for in-hospital mortality (AUC=0.693) compared to anion gap (AUC=0.571) and albumin (AUC=0.640), with similar findings for 30-day mortality.
Conclusion: ACAG is closely associated with the risk of mortality in HF patients with COPD. It appears to be a potential prognostic predictor for HF patients with COPD, aiding in risk stratification for this population. However, further prospective studies are needed to consolidate our findings.
Keywords: albumin-corrected anion gap, ACAG, heart failure, chronic obstructive pulmonary disease, mortality, MIMIC-IV database
Introduction
Heart failure (HF) remains a leading cause of global morbidity and mortality, accounting for substantial healthcare burden worldwide.1 The prognosis of HF is frequently complicated by the presence of comorbidities, among which chronic obstructive pulmonary disease (COPD) represents one of the most prevalent and clinically significant. Studies have documented that COPD affects approximately 20–30% of HF patients, with this overlap associated with worse functional status, increased hospitalization rates, and elevated mortality risk compared to HF patients without COPD.2,3 The coexistence of these conditions creates diagnostic and therapeutic challenges, as both share overlapping symptoms such as dyspnea and exercise intolerance, and their treatments may sometimes have conflicting effects on hemodynamics and respiratory function.4
Identifying reliable prognostic biomarkers in patients with both HF and COPD is essential for risk stratification and clinical decision-making. Traditional indicators of disease severity, such as left ventricular ejection fraction, natriuretic peptide levels, and spirometric parameters, provide valuable prognostic information but may not fully capture the complex metabolic derangements that occur in critically ill patients with these conditions.5,6 Acid-base disturbances and electrolyte imbalances are common in both HF and COPD exacerbations and have been associated with adverse outcomes.7,8
The anion gap (AG), calculated as the difference between measured cations and anions in serum, has traditionally been employed to evaluate acid-base balance and identify metabolic acidosis.9 AG reflects the concentration of unmeasured anions, including lactate, ketones, phosphate, and sulfate, with elevated values indicating accumulation of unmeasured acids. Recent studies have demonstrated that elevated AG is associated with increased mortality across various critical conditions, including cardiac arrest, sepsis, and acute myocardial infarction.10–12 However, AG measurements can be misleading in patients with hypoalbuminemia, a condition frequently observed in critically ill patients including those with HF and COPD.13–15 Since albumin represents a major unmeasured anion, low serum albumin levels can result in falsely low AG values, potentially masking underlying metabolic acidosis and impairing accurate prognostic assessment.16
To address this limitation, the albumin-corrected anion gap (ACAG) was developed. This correction provides a more accurate reflection of unmeasured anions and has demonstrated superior prognostic value compared to uncorrected AG in multiple clinical settings.17 Recent investigations have highlighted ACAG’s utility in predicting mortality across diverse patient populations. Hu et al reported that elevated ACAG (>21.25 mmol/L) independently predicted in-hospital mortality in septic patients admitted to intensive care units (ICU), with superior predictive accuracy compared to either AG or albumin alone.18 Similarly, Li et al demonstrated that higher ACAG levels were independently associated with increased in-hospital mortality in patients with acute pancreatitis.19 In cardiovascular diseases, studies have shown that elevated ACAG predicts both short-term and long-term mortality in acute myocardial infarction patients,20,21 and is associated with increased mortality risk in atrial fibrillation patients.22 Furthermore, Zhong et al found that elevated ACAG (>20 mmol/L) at the initiation of continuous renal replacement therapy was significantly associated with ICU mortality in acute kidney injury patients.23
Despite these findings, the prognostic significance of ACAG in patients with the specific combination of HF and COPD remains unexplored. Given the high prevalence of comorbid HF and COPD, the associated poor prognosis, and the theoretical rationale for ACAG as a marker of metabolic dysfunction in these patients, investigating the relationship between ACAG and clinical outcomes in this specific population is warranted. Therefore, this retrospective cohort study aims to evaluate the association between ACAG levels at ICU admission and mortality in patients with both HF and COPD, utilizing data from the Medical Information Mart for Intensive Care (MIMIC)-IV database.
Methods
Data Source
This population-based retrospective open-cohort study utilized data from the MIMIC-IV (version 3.1) database. MIMIC-IV is a publicly available critical care database jointly developed by the Beth Israel Deaconess Medical Center (BIDMC) and the Massachusetts Institute of Technology (MIT) Laboratory for Computational Physiology.24 The database contains detailed de-identified clinical data, including demographics, vital signs, diagnostic codes, laboratory results, clinical annotations, and mortality outcomes.
Access to the MIMIC-IV database was obtained after completing the required National Institutes of Health (NIH) web-based training course and passing the examination for “Protecting Human Research Participants” (certification number: 62773844). The study was approved by the Institutional Review Boards of both MIT and BIDMC.25 This study utilized the MIMIC database, in which all patient information has been de-identified in accordance with the Health Insurance Portability and Accountability Act (HIPAA) Safe Harbor provisions. According to the Measures for the Ethical Review of Life Science and Medical Research Involving Human Subjects (issued February 18, 2023, China), Article 32(1) provides that research utilizing lawfully obtained public data or data derived from unobtrusive observation of public behavior may be exempt from formal ethical review. Additionally, Article 32(2) stipulates that studies relying exclusively on anonymized information or biological specimens are similarly eligible for exemption. Given that this research was conducted entirely using publicly available, de-identified data, it was determined to be exempt from institutional ethics approval in accordance with these regulatory provisions.
Study Population
We retrospectively assessed the impact of ACAG during ICU hospitalization on mortality in patients with HF combined with COPD. HF and COPD were identified using diagnostic codes from the International Classification of Diseases, Ninth and Tenth Revisions (ICD-9 and ICD-10), Clinical Modification. Patients were excluded based on the following criteria: (1) absence of a COPD diagnosis; (2) age under 18 years; (3) missing albumin or anion gap data; (4) non-first-time ICU admission; or (5) an ICU stay of less than 24 hours. Ultimately, 1283 patients met the inclusion criteria and were categorized into three groups according to ACAG tertiles (T1: <19.55; T2: 19.55–23.5; T3: ≥23.5) (Figure 1).
 |
Figure 1 Study population selection flowchart. |
Data Collection
In this investigation, a wide range of clinical and demographic variables were obtained, including patient demographics (age, sex, body mass index [BMI], and ethnicity), vital signs (heart rate [HR], systolic blood pressure[SBP] and diastolic blood pressure [DBP], respiratory rate [RR], body temperature, and oxygen saturation [SpO2]), and commonly documented comorbidities (hypertension [HTN], coronary artery disease [CAD], chronic kidney disease [CKD], diabetes mellitus [DM], obesity, and stroke). Laboratory measures consisted of red and white blood cell counts (RBC, WBC), platelet count, red cell distribution width [RDW], hematocrit, hemoglobin, serum creatinine, blood urea nitrogen [BUN], serum albumin, bicarbonate, chloride, sodium, potassium, calcium, and blood glucose. Medication use and interventions recorded comprised beta-blockers, angiotensin-converting enzyme inhibitors [ACEI], angiotensin II receptor blockers [ARB], angiotensin receptor–neprilysin inhibitors [ARNI], diuretics, mechanical ventilation, and renal replacement therapy [RRT]. Disease severity was evaluated using two well-established scoring systems. The Sequential Organ Failure Assessment (SOFA) score quantifies organ dysfunction across six organ systems (respiratory, cardiovascular, hepatic, coagulation, renal, and neurological), with each system scored from 0 to 4 points, yielding a total score ranging from 0 to 24; higher scores indicate greater severity of organ dysfunction and are strongly associated with increased mortality in critically ill patients.26 The Simplified Acute Physiology Score II (SAPS II) is calculated from 17 variables including 12 physiological parameters, age, type of admission, and three underlying disease variables (acquired immunodeficiency syndrome, metastatic cancer, and hematologic malignancy), collected within the first 24 hours of intensive care unit admission; the score ranges from 0 to 163 points and provides an estimate of mortality risk.27 Both scores were calculated using data from the first 24 hours following ICU admission. Variables with missing rates exceeding 10% were excluded from the analysis. Among the remaining variables included in the final analysis, all had missing rates below 5%. For continuous variables with missing values, median imputation was applied. This approach was adopted based on established methodological evidence that simple imputation methods produce negligible bias when missing rates are low (<5%).28
The anion gap (AG, mmol/L) was calculated using the formula: AG = (sodium + potassium) - (chloride + bicarbonate). Subsequently, the ACAG was derived as follows: ACAG = AG + (4.4 - albumin [g/dL]) × 2.5.29 All values used in the calculations were obtained from the earliest available measurements after ICU admission.
Chronic conditions were defined according to the corresponding diagnostic codes in the ICD-9 and ICD-10. Using PostgreSQL (version 16.9), we systematically retrieved clinical information recorded during the initial 24-hour period after ICU admission from the MIMIC-IV database.
Endpoints of Interest
Patient clinical outcomes were recorded, with a primary focus on both in-hospital and 30-day all-cause mortality.
Statistical Analysis
Data are summarized as follows: normally distributed continuous variables as mean ± standard deviation, compared by independent t-test; non-normal continuous variables as median with interquartile range (IQR), compared by Mann–Whitney U-test; and categorical variables as counts (percentages), compared by chi-square test.
Survival outcomes, including in-hospital and 30-day mortality, were evaluated using Kaplan–Meier curves stratified by ACAG tertiles.
The relationship between ACAG and mortality was quantified using Cox proportional hazards models, yielding hazard ratios (HRs) with 95% confidence intervals (CIs). Covariates for the model were selected through a two-step process. First, candidate variables were identified based on their established clinical relevance to patient outcomes and prior evidence from the literature. These included demographics, vital signs, comorbidities, medications, interventions, laboratory values, and illness severity scores. Second, each candidate variable was assessed in a univariable Cox model. Variables with a significance level of p<0.05 were retained for consideration in the initial multivariable model. Final model selection involved a backward elimination procedure, maintaining variables significant at p<0.05 and those deemed clinically essential. Potential multicollinearity was examined via variance inflation factor (VIF); variables with VIF > 10 were dichotomized at their median or mean for inclusion.
Three models were specified: a crude model (Model 1); an adjusted model for demographics and anthropometry (sex, age, BMI; Model 2); and a fully adjusted model (Model 3) that additionally incorporated hemodynamic, clinical, and laboratory parameters (HR, SBP, RR, SpO2, HTN, CKD, DM, RRT, ACEI/ARB/ARNI, RBC, RDW, hematocrit, hemoglobin, WBC, creatinine, BUN, glucose, calcium, SOFA, SAPS II). ACAG was analyzed as both continuous and categorical (tertile-based) variables, with the lowest tertile as reference.
Furthermore, an RCS model was applied to examine the dose-response relationship between ACAG and mortality.
Stratified analyses assessed the consistency of ACAG’s prognostic effect across prespecified subgroups: sex, age (<65/≥65 years), key comorbidities (hypertension, chronic kidney disease, diabetes), RRT status, and ACEI/ARB/ARNI use. Effect modification was evaluated via likelihood ratio tests.
Finally, receiver operating characteristic (ROC) analysis was used to evaluate the predictive ability of ACAG for in-hospital and 30-day mortality, with the area under the curve (AUC) calculated. To compare the discriminative performance between ACAG and its individual components (anion gap and albumin), we employed DeLong’s test for comparing correlated ROC curves. The differences in AUC (ΔAUC) along with corresponding standard errors, Z statistics, and P values were reported. A two-sided P value < 0.05 was considered statistically significant.
All statistical analyses were performed with SPSS (version 26.0; IBM Corp., USA) and R software (version 4.4.1; R Foundation). A two-sided p-value < 0.05 was considered statistically significant.
Results
Baseline Characteristics
As shown in Table 1, this study included a total of 1283 patients with a median age of 72.3 years (interquartile range: 64.07–79.65), of which 45.4% were female (n=582). The median ACAG value was 21.20 (interquartile range: 18.65–24.62). The ICU in-hospital all-cause mortality rate was 11.22% (n=144), and the 30-day all-cause hospital mortality rate was 13.72% (n=176). Table 1 details the baseline characteristics of patients stratified by ACAG tertiles. In the T3 group, increased heart rate and respiratory rate were observed, along with increased white blood cell count, RDW, and serum creatinine, decreased hemoglobin, increased prevalence of chronic kidney disease and diabetes, and decreased prevalence of hypertension. With increasing ACAG tertiles, mortality rates progressively increased: in-hospital mortality (4.27% vs. 10.14% vs. 19.20%, p<0.001) and 30-day hospital mortality (5.92% vs. 13.36% vs. 21.78%, p<0.001).
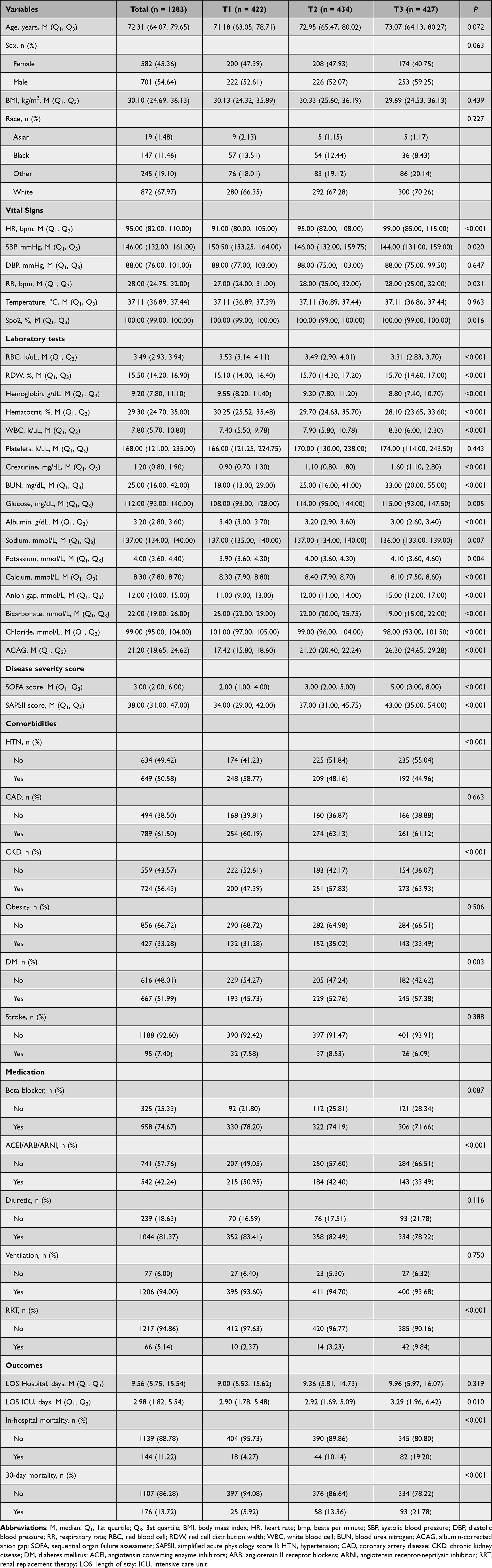 |
Table 1 Baseline Characteristics According to ACAG Index Tertiles |
Kaplan-Meier Survival Analysis
Kaplan-Meier survival analysis was performed to evaluate the effect of ACAG across different tertiles on the two endpoints. Figure 2 shows that patients in higher ACAG tertiles exhibited significantly higher mortality rates both in-hospital and at 30 days (log-rank p<0.0001).
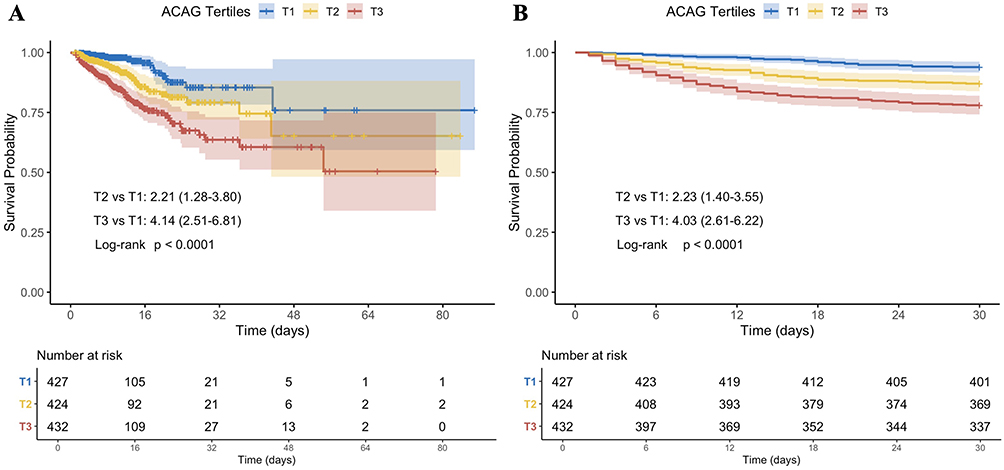 |
Figure 2 Kaplan-Meier survival curves for all-cause mortality. (A) in-hospital mortality and (B) 30-day mortality. Abbreviation: ACAG, albumin-corrected anion gap. |
Relationship Between ACAG and Clinical Outcomes
To further investigate the relationship between ACAG and mortality, we conducted Cox proportional hazards regression models (Table 2). The results showed that in the unadjusted model, each standard deviation (SD) increase in ACAG was associated with an 11% higher in-hospital mortality risk (HR 1.11, 95% CI 1.08–1.14, p<0.001), which remained significant in the partially adjusted model (HR 1.11, 95% CI 1.09–1.15, p<0.001), and was attenuated but remained significant in the fully adjusted model (HR 1.07, 95% CI 1.04–1.11, p<0.001). When ACAG was categorized as an ordinal variable, in the unadjusted model, patients in the T3 group had about four times higher mortality risk compared to the T1 group (HR 4.08, 95% CI 2.48–6.73, p<0.001), a risk that remained similarly elevated in the partially adjusted model (HR 4.08, 95% CI 2.47–6.72, p<0.001), and decreased to about two times higher in the fully adjusted model (HR 2.04, 95% CI 1.18–3.54, p<0.001), with a significant increasing trend in risk as ACAG increased (p for trend <0.05). Similar trends were observed for 30-day mortality: each SD increase in ACAG corresponded to an HR of 1.11 (95% CI 1.09–1.14, p<0.001) in the unadjusted model, 1.11 (95% CI 1.09–1.14, p<0.001) in the partially adjusted model, and 1.07 (95% CI 1.03–1.10, p<0.001) in the fully adjusted model. Compared to the T1 group, patients in the T3 group had significantly higher 30-day mortality risk, with an HR of 4.00 (95% CI 2.59–6.18) indicating approximately four times higher risk in the unadjusted model, an HR of 3.89 (about 3.9 times higher risk; 95% CI 2.52–6.02, p<0.001) in the partially adjusted model, and an HR of 1.83 (about 1.8 times higher risk; 95% CI 1.12–2.97, p<0.001) in the fully adjusted model, all showing a significant upward trend (p for trend <0.05).
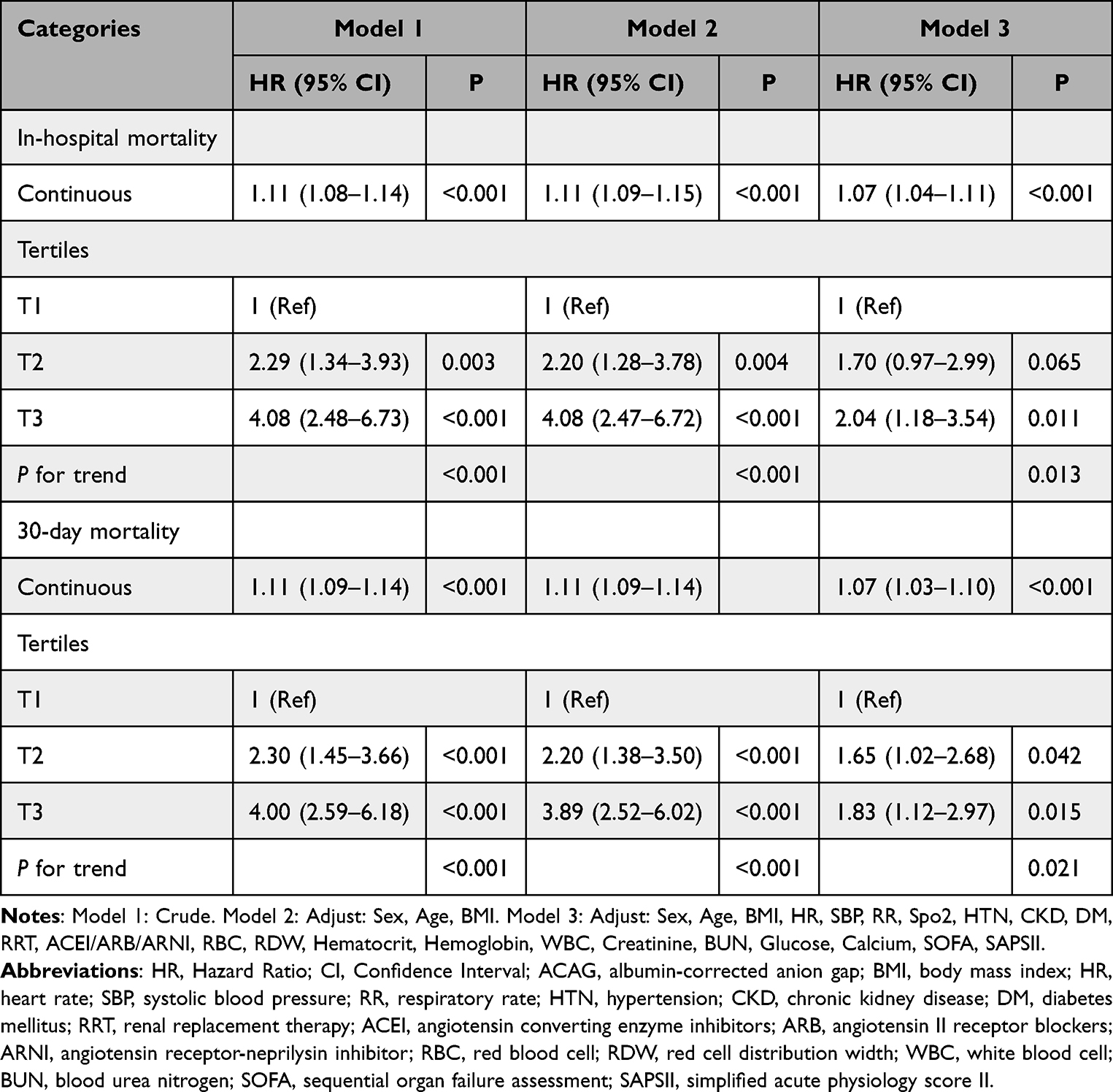 |
Table 2 Cox Proportional Hazard Models for In-Hospital Mortality and 30-Day Mortality |
The Detection of Nonlinear Relationships
Additionally, we employed the RCS model to evaluate the potential nonlinear relationship between ACAG and all-cause mortality (Figure 3). The RCS analysis revealed a positive linear association between ACAG and both in-hospital mortality risk and 30-day mortality risk, with no significant nonlinearity detected (p for nonlinear > 0.05).
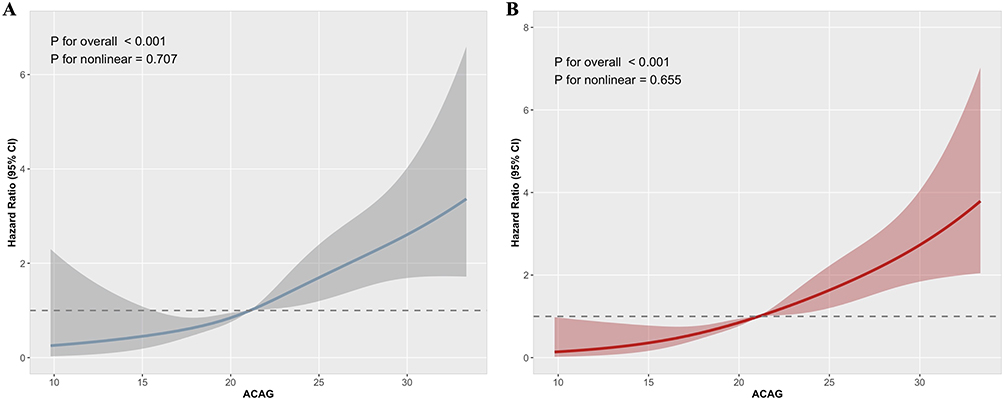 |
Figure 3 RCS analysis of the association between ACAG and all-cause mortality. (A) in-hospital mortality and (B) 30-day mortality. Abbreviation: ACAG, albumin-corrected anion gap. |
Subgroup Analysis
We further evaluated the prognostic value of ACAG for in-hospital and 30-day all-cause mortality across various subgroups (Figure 4), including sex, age, hypertension, CKD, diabetes, RRT, and ACEI/ARB/ARNI use. The results indicated no significant interaction between ACAG and any of the subgroups for either in-hospital or 30-day outcomes (interaction p-values: 0.137–0.937). This suggests that the association between ACAG and clinical outcomes remains consistent in patients with heart failure complicated by chronic obstructive pulmonary disease.
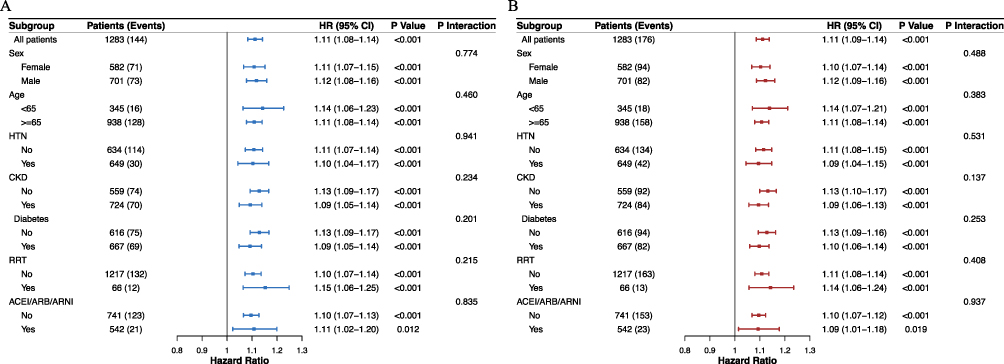 |
Figure 4 Subgroup analysis of the association between ACAG and all-cause mortality. (A) in-hospital mortality and (B) 30-day mortality. Abbreviations: HR, Hazard Ratio; CI, Confidence Interval; HTN, hypertension; CKD, chronic kidney disease; RRT, renal replacement therapy; ACEI, angiotensin converting enzyme inhibitors; ARB, angiotensin II receptor blockers; ARNI, angiotensin receptor-neprilysin inhibitor. |
The Predictive Significance of ACAG for All-Cause Mortality
To assess the prognostic value of ACAG and its individual components, we conducted ROC curve analysis comparing ACAG with albumin and anion gap (Figure 5). For in-hospital mortality, ACAG demonstrated an AUC of 0.693, which was numerically higher than that of anion gap (AUC = 0.571) and albumin (AUC = 0.640). Formal comparison using DeLong’s test for correlated ROC curves revealed that ACAG’s discriminative performance was significantly superior to that of anion gap (ΔAUC = 0.122, p< 0.001), but not statistically superior to albumin alone (ΔAUC = 0.053, p = 0.083). Similarly, for 30-day mortality, ACAG (AUC = 0.680) exhibited significantly greater AUC values compared to both anion gap (ΔAUC = 0.106, p < 0.001) and albumin (ΔAUC = 0.059, p = 0.037) (Tables 3 and S1). These quantitative comparisons confirm the superior discriminative ability of the composite ACAG metric over its individual components, particularly for 30-day mortality.
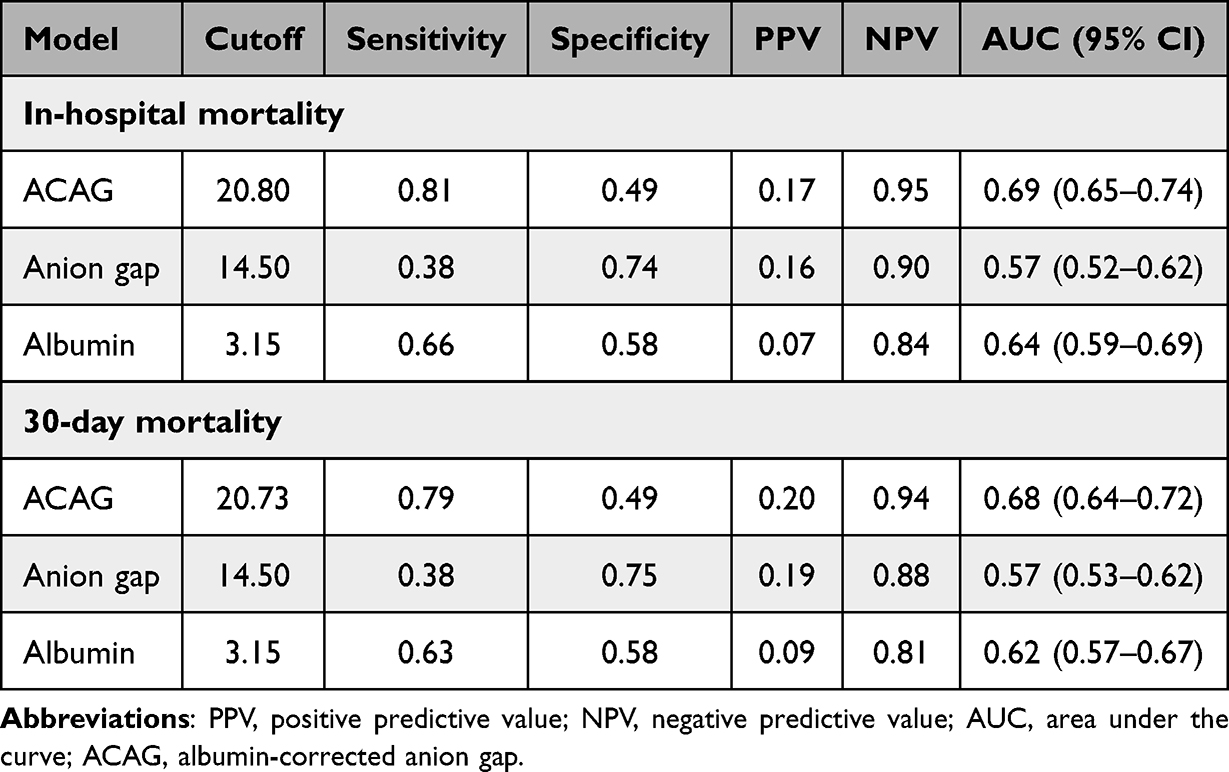 |
Table 3 Prognostic Accuracy of the ACAG, Anion Gap and Albumin |
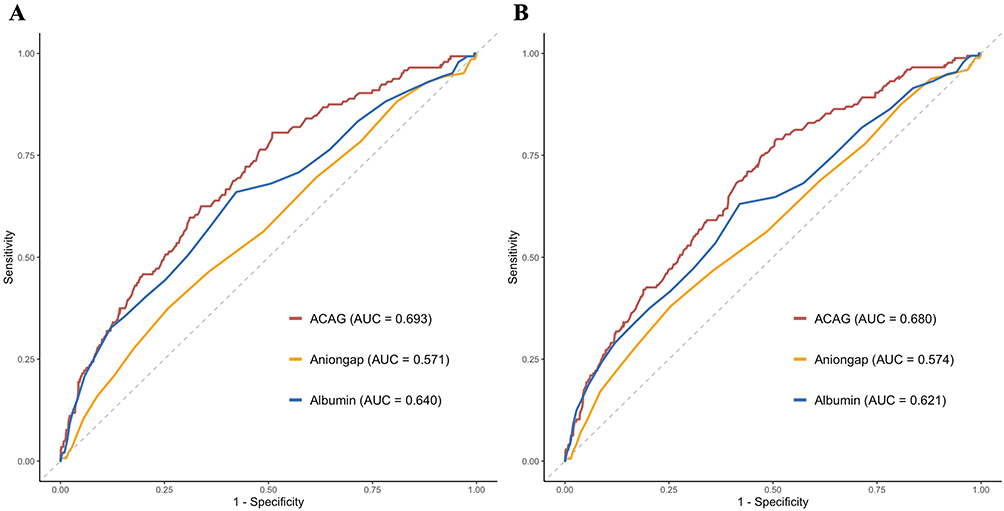 |
Figure 5 ROC curves for predicting all-cause mortality. (A) in-hospital mortality and (B) 30-day mortality. Abbreviations: AUC, area under the curve; ACAG, albumin-corrected anion gap. |
Discussion
This retrospective cohort study utilized data from 1283 patients in the MIMIC-IV database, representing the first study to demonstrate a significant independent association between ACAG and mortality in patients with HF combined with COPD. Our primary findings revealed that after comprehensive adjustment for demographic, clinical, and laboratory confounding factors, each standard deviation increase in ACAG corresponded to a 7% increase in in-hospital mortality risk. Furthermore, patients in the highest ACAG tertile (≥23.5 mmol/L) had more than double the mortality risk compared to those in the lowest tertile, with in-hospital mortality rates of 19.20% and 4.27%, respectively (p<0.001). These associations remained robust across multiple sensitivity analyses and different patient subgroups, underscoring the potential utility of ACAG as a readily available prognostic tool in this high-risk population.
The superior predictive performance of ACAG compared to its individual components warrants particular emphasis. Our ROC curve analysis demonstrated that ACAG achieved an AUC of 0.693 for in-hospital mortality and 0.680 for 30-day mortality, significantly outperforming both uncorrected anion gap (AUC 0.571 and 0.574, respectively) and albumin alone (AUC 0.640 and 0.621, respectively). Formal comparison using DeLong’s test confirmed that ACAG demonstrated significantly greater discriminative ability than anion gap for both outcome measures, underscoring the value of integrating albumin to correct the anion gap. Relative to albumin alone, ACAG yielded consistently higher AUC values, with the difference attaining statistical significance for 30‑day mortality but not for in‑hospital mortality. This lack of significance for in‑hospital mortality may reflect the lower number of in‑hospital mortality events and the consequently reduced statistical power, or it could be due to the greater clinical heterogeneity and influence of competing factors in hospital‑based outcomes, which might attenuate the prognostic signal of any single biomarker. Nevertheless, the consistent trend and the significant improvement for 30-day mortality reinforce the concept that ACAG is a more robust composite biomarker than either component alone. This enhanced discriminative ability reflects ACAG’s capacity to simultaneously capture two critical pathophysiological domains: the accumulation of unmeasured anions (reflected in the anion gap) and the nutritional/inflammatory status (reflected in albumin levels). In hypoalbuminemic patients, which comprised a substantial proportion of our cohort (median albumin 3.20 g/dL), conventional anion gap systematically underestimates the true acid-base derangement by approximately 2.5 mmol/L for every 1 g/dL decrease in albumin.30,31 By correcting for this artifact, ACAG provides a more accurate assessment of metabolic dysfunction and unmeasured anion burden, thereby improving prognostic stratification.
Our finding that ACAG demonstrates a linear dose-response relationship with mortality, as confirmed by restricted cubic spline analysis, has important clinical implications. This linear association suggests that ACAG functions as a continuous risk marker rather than simply a threshold indicator, supporting its use for granular risk stratification rather than binary classification. Notably, the relationship between ACAG and mortality persisted across all examined subgroups, including patients stratified by age, sex, hypertension, chronic kidney disease, diabetes, renal replacement therapy status, and ACEI/ARB/ARNI use, with no significant interactions detected. This consistency suggests that ACAG’s prognostic value is independent of these common comorbidities and treatments, enhancing its generalizability across the heterogeneous HF combined with COPD population.
The mechanistic underpinnings linking elevated ACAG to mortality in patients with HF combined with COPD are multifaceted and reflect the convergence of several pathophysiological processes. First, tissue hypoperfusion resulting from reduced cardiac output in heart failure leads to inadequate oxygen delivery, triggering anaerobic metabolism and lactate accumulation.32,33 Second, pulmonary dysfunction in COPD contributes to ventilation-perfusion mismatch, hypoxemia, and hypercapnia, further exacerbating cellular hypoxia and metabolic acidosis.34–36 Third, renal dysfunction, present in 56.43% of our cohort, impairs the excretion of organic acids, sulfate, and phosphate, leading to their systemic accumulation.37,38 The prevalence of chronic kidney disease increased progressively across ACAG tertiles (47.39% in T1 versus 63.93% in T3), suggesting that renal impairment plays a pivotal role in ACAG elevation. Notably, emerging evidence further supports the clinical relevance of ACAG in renal and cardiorenal syndromes. Recent studies have demonstrated that elevated ACAG is independently associated with acute kidney injury and adverse outcomes in patients with heart failure, underscoring a potential bidirectional interaction between acid–base disturbances, renal dysfunction, and cardiovascular disease progression.39,40 Fourth, systemic inflammation, a hallmark of both COPD and decompensated heart failure, promotes the generation of inflammatory mediators and reactive oxygen species that interfere with cellular metabolism and contribute to organ dysfunction.41,42 Finally, the use of loop diuretics, which were administered to 81.37% of our patients, can induce contraction alkalosis and electrolyte disturbances that paradoxically coexist with metabolic acidosis from other sources, creating complex acid-base disorders that ACAG helps to unmask.43
Beyond reflecting disease severity, elevated ACAG may directly contribute to adverse outcomes through several mechanisms. Metabolic acidosis impairs myocardial contractility by reducing calcium sensitivity of contractile proteins and inhibiting beta-adrenergic receptor responsiveness, potentially precipitating hemodynamic decompensation.44,45 Acidosis also promotes arrhythmogenesis by altering ion channel function and prolonging action potential duration, increasing susceptibility to ventricular tachyarrhythmias.46 Furthermore, acidemia induces pulmonary vasoconstriction, thereby increasing right ventricular afterload, a particularly detrimental effect in patients with pre-existing COPD-related pulmonary hypertension.47,48 At the cellular level, metabolic acidosis activates inflammatory cascades, enhances oxidative stress, promotes apoptosis, and impairs mitochondrial function, collectively contributing to multi-organ failure.49,50 These direct pathophysiological effects underscore that ACAG is not merely a marker of illness severity but potentially a mediator of adverse outcomes.
The baseline characteristics of our cohort provide additional insights into the pathobiology of ACAG elevation in patients with HF combined with COPD. Patients with higher ACAG levels exhibited several features indicative of more severe illness: elevated heart rate and respiratory rate, higher white blood cell counts and red cell distribution width, increased serum creatinine and blood urea nitrogen, decreased hemoglobin and albumin levels, and higher SOFA and SAPS II scores. Notably, the prevalence of diabetes increased across ACAG tertiles (45.73% in T1 versus 57.38% in T3), while hypertension prevalence paradoxically decreased (58.77% versus 44.96%). This inverse relationship with hypertension may reflect the competing effects of chronic antihypertensive medication use versus acute hemodynamic decompensation, or potentially survivor bias whereby patients with more severe multimorbidity achieve less stringent blood pressure control. The progressive increase in renal replacement therapy utilization across tertiles (2.37% in T1 versus 9.84% in T3) further emphasizes the critical role of kidney dysfunction in ACAG pathogenesis.
The clinical implications of our findings are multifold. Our focus on the specific population of patients with HF combined with COPD addresses an important gap in the literature, as previous ACAG studies predominantly examined single-disease cohorts. ACAG represents an easily obtainable, cost-effective prognostic marker that requires no additional testing beyond routine electrolyte and albumin measurements universally performed in ICU patients. Its superior predictive performance compared to conventional anion gap or albumin alone supports its preferential adoption in clinical practice. Elevated ACAG levels, particularly values exceeding 23.5 mmol/L, may warrant increased clinical attention and closer monitoring. However, given the modest discriminatory ability of ACAG alone (AUC<0.7), it should be interpreted in conjunction with other clinical parameters and biomarkers rather than used as a standalone predictor for guiding therapeutic decisions. Such interventions might include optimization of cardiac output through inotropic support or mechanical circulatory assistance, correction of hypoxemia through supplemental oxygen or ventilatory support, judicious fluid management to balance volume status without exacerbating pulmonary congestion, and consideration of renal replacement therapy in patients with severe metabolic acidosis and renal failure. Furthermore, ACAG assessment, when integrated with other established prognostic indicators, may contribute to comprehensive risk evaluation and facilitate discussions regarding patient prognosis. Nevertheless, its limited predictive accuracy as a single marker suggests that clinical decisions should not rely solely on ACAG values.
Nevertheless, several limitations merit acknowledgment. First, the retrospective observational design precludes definitive causal inference, as unmeasured confounding and selection bias cannot be entirely eliminated. Although we adjusted for numerous covariates, residual confounding from variables not captured in the database, such as duration and severity of chronic diseases, medication adherence, socioeconomic factors, and quality of care, may influence results. Second, the discriminative ability of ACAG as a standalone predictor was modest, with an area under the receiver operating characteristic curve (AUC) below 0.7 for the primary outcomes. This underscores that its clinical utility lies in being integrated into a comprehensive assessment rather than being used in isolation. Third, ACAG values were derived from laboratory measurements obtained within the first 24 hours of ICU admission, representing a single time point. We did not examine the prognostic value of serial ACAG measurements or temporal trends, which might provide additional insight into disease trajectory and treatment response. Dynamic changes in ACAG following interventions could potentially offer greater prognostic discrimination than baseline values alone. Fourth, the database lacks detailed characterization of heart failure phenotype (heart failure with reduced versus preserved ejection fraction) and COPD severity (GOLD classification), limiting our ability to assess whether ACAG’s prognostic value varies according to these disease-specific characteristics. Fifth, the specific causes of death were not systematically analyzed, precluding determination of whether ACAG preferentially predicts cardiovascular versus non-cardiovascular mortality. Finally, the study population comprised exclusively patients from the United States, predominantly of white ethnicity (67.97%), potentially limiting generalizability to other geographic and ethnic populations where disease epidemiology and healthcare delivery may differ.
Future research should address several key questions. First, prospective multicenter validation studies encompassing diverse geographic and demographic populations are necessary to confirm our findings and establish generalizable ACAG thresholds for risk stratification. Second, investigations examining serial ACAG measurements and their relationship to clinical trajectories could elucidate whether declining ACAG values indicate therapeutic response and improved prognosis. Third, interventional trials evaluating whether targeting ACAG correction through specific therapeutic strategies (eg, aggressive treatment of the underlying cause, bicarbonate administration, or renal replacement therapy) improves outcomes would determine whether ACAG represents a modifiable risk factor versus merely a risk marker. Fourth, metabolomic and proteomic studies identifying the specific unmeasured anions contributing to ACAG elevation in patients with HF combined with COPD could reveal novel pathobiological insights and therapeutic targets. Fifth, integration of ACAG with other established prognostic markers—including natriuretic peptides, troponin, lactate, and clinical severity scores—into comprehensive risk prediction models could enhance prognostic accuracy beyond what any single marker achieves. Finally, health economic analyses examining whether ACAG-guided risk stratification improves resource allocation and cost-effectiveness would inform its incorporation into clinical practice guidelines.
Conclusion
This study established a linear relationship between ACAG and both in-hospital and 30-day mortality in patients diagnosed with HF and COPD. It is the first to highlight the prognostic significance of ACAG in critically ill patients with HF and COPD, suggesting its potential role in risk stratification at admission. However, to support the validity of these findings, larger-scale prospective studies with extended follow-up periods are necessary.
Data Sharing Statement
The data utilized in this study originate from the publicly accessible MIMIC-IV database (version 3.1), which can be accessed at https://mimic.mit.edu/.
Ethics Statement
The research involving human participants underwent review and approval by the Institutional Review Board of the Massachusetts Institute of Technology and Beth Israel Deaconess Medical Center. Written informed consent for participation was not deemed necessary for this study in compliance with national legislation and institutional requirements.
Acknowledgments
The authors express their sincere gratitude to the Free Statistics team for providing essential technical support and analytical tools that facilitated data processing and visualization.
Funding
No funds were received in support of this work.
Disclosure
The authors declare no competing interests associated with this work.
References
1. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. 2023;118(17):3272–16. doi:10.1093/cvr/cvac013
2. Hawkins NM, Petrie MC, Jhund PS, Chalmers GW, Dunn FG, McMurray JJ. Heart failure and chronic obstructive pulmonary disease: diagnostic pitfalls and epidemiology. Eur J Heart Fail. 2009;11(2):130–139. doi:10.1093/eurjhf/hfn013
3. Franssen FM, Soriano JB, Roche N, et al. Lung function abnormalities in smokers with ischemic heart disease. Am J Respir Crit Care Med. 2016;194(5):568–576. doi:10.1164/rccm.201512-2480OC
4. Mentz RJ, Wojdyla D, Fiuzat M, Chiswell K, Fonarow GC, O’Connor CM. Association of beta-blocker use and selectivity with outcomes in patients with heart failure and chronic obstructive pulmonary disease (from OPTIMIZE-HF). Am J Cardiol. 2013;111(4):582–587. doi:10.1016/j.amjcard.2012.10.041
5. McDonagh TA, Metra M, Adamo M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42(36):3599–3726. doi:10.1093/eurheartj/ehab368
6. Agustí A, Celli BR, Criner GJ, et al. Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Eur Respir J. 2023;61(4):2300239. doi:10.1183/13993003.00239-2023
7. Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2(1):162–174. doi:10.2215/CJN.03020906
8. Schricker S, Schanz M, Alscher MD, Kimmel M. [Metabolic acidosis: diagnosis and treatment]. Med Klin Intensivmed Notfmed. 2020;115(4):275–280. doi:10.1007/s00063-019-0538-y Danish
9. Emmett M, Narins RG. Clinical use of the anion gap. Medicine. 1977;56(1):38–54. doi:10.1097/00005792-197756010-00002
10. Chen J, Dai C, Yang Y, et al. The association between anion gap and in-hospital mortality of post-cardiac arrest patients: a retrospective study. Sci Rep. 2022;12(1):7405. doi:10.1038/s41598-022-11081-3
11. Jing L, Shi X, Xu L, Zhao X, Li F, Qin L. Association between serum anion gap trajectory and mortality in hospitalized patients with sepsis: an analysis of the MIMIC-IV database. Front Endocrinol. 2025;16:1578078. doi:10.3389/fendo.2025.1578078
12. Lu J, Zhong L, Yuan M, Min J, Xu Y. Association between serum anion gap and all-cause mortality in patients with acute myocardial infarction: a retrospective study based on MIMIC-IV database. Heliyon. 2023;9(7):e17397. doi:10.1016/j.heliyon.2023.e17397
13. Nicholson JP, Wolmarans MR, Park GR. The role of albumin in critical illness. Br J Anaesth. 2000;85(4):599–610. doi:10.1093/bja/85.4.599
14. Ghazal M, Khalife WI. Hypoalbuminemia in heart failure: pathophysiology, clinical implications, and management strategies. Heart Fail Rev. 2025;30(6):1407–1414. doi:10.1007/s10741-025-10558-3
15. Ling M, Huiyin L, Shanglin C, et al. Relationship between human serum albumin and in-hospital mortality in critical care patients with chronic obstructive pulmonary disease. Front Med. 2023;10:1109910. doi:10.3389/fmed.2023.1109910
16. Nanji AA, Campbell DJ, Pudek MR. Decreased anion gap associated with hypoalbuminemia and polyclonal gammopathy. JAMA. 1981;246(8):859–860. doi:10.1001/jama.1981.03320080045027
17. Kraut JA, Nagami GT. The serum anion gap in the evaluation of acid-base disorders: what are its limitations and can its effectiveness be improved? Clin J Am Soc Nephrol. 2013;8(11):2018–2024. doi:10.2215/CJN.04040413
18. Hu T, Zhang Z, Jiang Y. Albumin corrected anion gap for predicting in-hospital mortality among intensive care patients with sepsis: a retrospective propensity score matching analysis. Clin Chim Acta. 2021;521:272–277. doi:10.1016/j.cca.2021.07.021
19. Li P, Shi L, Yan X, et al. Albumin corrected anion gap and the risk of in-hospital mortality in patients with acute pancreatitis: a retrospective cohort study. J Inflamm Res. 2023;16:2415–2422. doi:10.2147/JIR.S412860
20. Jian L, Zhang Z, Zhou Q, Duan X, Xu H, Ge L. Association between albumin corrected anion gap and 30-day all-cause mortality of critically ill patients with acute myocardial infarction: a retrospective analysis based on the MIMIC-IV database. BMC Cardiovasc Disord. 2023;23(1):211. doi:10.1186/s12872-023-03200-3
21. Sheng H, Lu J, Zhong L, Hu B, Sun X, Dong H. The correlation between albumin-corrected anion gap and prognosis in patients with acute myocardial infarction. ESC Heart Fail. 2024;11(2):826–836. doi:10.1002/ehf2.14639
22. Xu J, Wang Z, Wang Y, Chen X, Ma L, Wang X. Association of elevated albumin-corrected anion gap with all-cause mortality risk in atrial fibrillation: a retrospective study. BMC Cardiovasc Disord. 2025;25(1):55. doi:10.1186/s12872-025-04518-w
23. Zhong L, Xie B, Ji XW, Yang XH. The association between albumin corrected anion gap and ICU mortality in acute kidney injury patients requiring continuous renal replacement therapy. Intern Emerg Med. 2022;17(8):2315–2322. doi:10.1007/s11739-022-03093-8
24. Johnson AEW, Bulgarelli L, Shen L, et al. MIMIC-IV, a freely accessible electronic health record dataset. Sci Data. 2023;10(1):1. doi:10.1038/s41597-022-01899-x
25. Goldberger AL, Amaral LA, Glass L, et al. PhysioBank, PhysioToolkit, and PhysioNet: components of a new research resource for complex physiologic signals. Circulation. 2000;101(23):E215–220. doi:10.1161/01.CIR.101.23.e215
26. Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707–710. doi:10.1007/BF01709751
27. Le Gall JR, Lemeshow S, Saulnier F. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA. 1993;270(24):2957–2963. doi:10.1001/jama.1993.03510240069035
28. Schafer JL. Multiple imputation: a primer. Stat Methods Med Res. 1999;8(1):3–15. doi:10.1177/096228029900800102
29. Huang S, Zhang Q, Liu L, Kutryk MJB, Zhang J. Relationship between albumin-corrected anion gap and short- and medium-term all-cause mortality in heart failure patients with a single ICU admission. Front Cardiovasc Med. 2025;12:1608383. doi:10.3389/fcvm.2025.1608383
30. Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807–1810. doi:10.1097/00003246-199811000-00019
31. Figge J, Bellomo R, Egi M. Quantitative relationships among plasma lactate, inorganic phosphorus, albumin, unmeasured anions and the anion gap in lactic acidosis. J Crit Care. 2018;44:101–110. doi:10.1016/j.jcrc.2017.10.007
32. Gunnerson KJ, Saul M, He S, Kellum JA. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Crit Care. 2006;10(1):R22. doi:10.1186/cc3987
33. Vincent JL, Quintairos ESA, Couto L Jr, Taccone FS. The value of blood lactate kinetics in critically ill patients: a systematic review. Crit Care. 2016;20(1):257. doi:10.1186/s13054-016-1403-5
34. Csoma B, Vulpi MR, Dragonieri S, et al. Hypercapnia in COPD: causes, Consequences, and Therapy. J Clin Med. 2022;11(11):3180. doi:10.3390/jcm11113180
35. Gernhold L, Neurohr C, Tsitouras K, Lutz N, Briese S, Ghiani A. Hypercapnia and lung function parameters in chronic obstructive pulmonary disease. BMC Pulm Med. 2024;24(1):345. doi:10.1186/s12890-024-03151-1
36. Martin R, Nora M, Anna L, et al. Altered hypoxia-induced cellular responses and inflammatory profile in lung fibroblasts from COPD patients compared to control subjects. Respir Res. 2024;25(1):282. doi:10.1186/s12931-024-02907-x
37. Fenves AZ, Emmett M. Approach to patients with high anion gap metabolic acidosis: core curriculum 2021. Am J Kidney Dis. 2021;78(4):590–600. doi:10.1053/j.ajkd.2021.02.341
38. Zheng CM, Liu WC, Zheng JQ, et al. Metabolic acidosis and strong ion gap in critically ill patients with acute kidney injury. Biomed Res Int. 2014;2014:819528. doi:10.1155/2014/819528
39. Ruan AF, Zheng JW, Sun SQ, Liu XZ, Chen TL. The association of albumin-corrected anion gap and acute kidney injury in heart failure patients: a competing risk model analysis. BMC Cardiovasc Disord. 2025;25(1):277. doi:10.1186/s12872-025-04723-7
40. Huang S, Zhang Q, Wei F, Kutryk MJB, Zhang J. Association of albumin-corrected anion gap with mortality in ICU patients with heart failure and acute kidney injury: analysis of the MIMIC-IV database. Eur J Med Res. 2025;30(1):745. doi:10.1186/s40001-025-03035-y
41. de Miguel Díez J, Chancafe Morgan J, Jiménez García R. The association between COPD and heart failure risk: a review. Int J Chron Obstruct Pulmon Dis. 2013;8:305–312. doi:10.2147/COPD.S31236
42. Rutten FH, Cramer MJ, Grobbee DE, et al. Unrecognized heart failure in elderly patients with stable chronic obstructive pulmonary disease. Eur Heart J. 2005;26(18):1887–1894. doi:10.1093/eurheartj/ehi291
43. Palmer BF. Metabolic complications associated with use of diuretics. Semin Nephrol. 2011;31(6):542–552. doi:10.1016/j.semnephrol.2011.09.009
44. Orchard CH, Kentish JC. Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol. 1990;258(6 Pt 1):C967–981. doi:10.1152/ajpcell.1990.258.6.C967
45. Katz AM. Influence of altered inotropy and lusitropy on ventricular pressure-volume loops. J Am Coll Cardiol. 1988;11(2):438–445. doi:10.1016/0735-1097(88)90113-1
46. Kohlhardt M, Haap K, Figulla HR. Influence of low extracellular pH upon the Ca inward current and isometric contractile force in mammalian ventricular myocardium. Pflugers Arch. 1976;366(1):31–38. doi:10.1007/BF02486557
47. Marshall BE, Marshall C. A model for hypoxic constriction of the pulmonary circulation. J Appl Physiol. 1988;64(1):68–77. doi:10.1152/jappl.1988.64.1.68
48. Naeije R, Brimioulle S. Physiology in medicine: importance of hypoxic pulmonary vasoconstriction in maintaining arterial oxygenation during acute respiratory failure. Crit Care. 2001;5(2):67–71. doi:10.1186/cc989
49. Kraut JA, Madias NE. Metabolic acidosis: pathophysiology, diagnosis and management. Nat Rev Nephrol. 2010;6(5):274–285. doi:10.1038/nrneph.2010.33
50. Kellum JA, Song M, Li J. Science review: extracellular acidosis and the immune response: clinical and physiologic implications. Crit Care. 2004;8(5):331–336. doi:10.1186/cc2900
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Short-Term Oxygen Therapy Outcomes in COPD
Soumagne T, Maltais F, Corbeil F, Paradis B, Baltzan M, Simão P, Abad Fernández A, Lecours R, Bernard S, Lacasse Y
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:1685-1693
Published Date: 28 July 2022
The Association of Renin-Angiotensin System Blockades and Mortality in Patients with Acute Exacerbation of Chronic Obstructive Pulmonary Disease and Acute Respiratory Failure: A Retrospective Cohort Study
Ruan Z, Li D, Hu Y, Qiu Z, Chen X
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2001-2011
Published Date: 1 September 2022
Optimal Management of Heart Failure and Chronic Obstructive Pulmonary Disease: Clinical Challenges
Cuthbert JJ, Pellicori P, Clark AL
International Journal of General Medicine 2022, 15:7961-7975
Published Date: 25 October 2022
Comorbid Heart Disease in Patients with COPD is Associated with Increased Hospitalization and Mortality – A 15-Year Follow-Up
Giezeman M, Sundh J, Athlin Å, Lisspers K, Ställberg B, Janson C, Montgomery S, Kisiel MA, Nager A, Sandelowsky H, Hasselgren M
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:11-21
Published Date: 9 January 2023
Relationship Between Systemic Immune-Inflammation Index and Risk of Respiratory Failure and Death in COPD: A Retrospective Cohort Study Based on the MIMIC-IV Database
Zhang Y, Tan X, Hu S, Cui Z, Chen W
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:459-473
Published Date: 19 February 2024