Back to Journals » ImmunoTargets and Therapy » Volume 9
Targeting Prostate Cancer Using Intratumoral Cytotopically Modified Interleukin-15 Immunotherapy in a Syngeneic Murine Model
Authors Papaevangelou E ![]() , Smolarek D
, Smolarek D ![]() , Smith RA, Dasgupta P, Galustian C
, Smith RA, Dasgupta P, Galustian C ![]()
Received 15 April 2020
Accepted for publication 26 May 2020
Published 27 July 2020 Volume 2020:9 Pages 115—130
DOI https://doi.org/10.2147/ITT.S257443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Efthymia Papaevangelou,1 Dorota Smolarek,1 Richard A Smith,1 Prokar Dasgupta,1,2 Christine Galustian1
1Peter Gorer Department of Immunobiology, School of Immunology and Microbial Sciences, King’s College London, Guy’s Hospital, London, UK; 2Urology Centre, Guy’s Hospital, London, UK
Correspondence: Efthymia Papaevangelou
Peter Gorer Department of Immunobiology, School of Immunology and Microbial Sciences, Guy’s Hospital, King’s College London, Great Maze Pond, London SE1 9RT, UK
Tel +44 20 7188 8568
Email [email protected]
Background: The prostate cancer microenvironment is highly immunosuppressive; immune cells stimulated in the periphery by systemic immunotherapies will be rendered inactive once entering this environment. Immunotherapies for prostate cancer need to break this immune tolerance. We have previously identified interleukin-15 (IL-15) as the only cytokine tested that activates and expands immune cells in the presence of prostate cancer cells. In the current study, we aimed to identify a method of boosting the efficacy of IL-15 in prostate cancer.
Methods: We engineered, by conjugation to a myristoylated peptide, a membrane-localising form of IL-15 (cyto-IL-15) and the checkpoint inhibitor antibodies cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death ligand 1 (PD-L1) (cyto-abs) to enable them to bind to cell surfaces by non-specific anchoring to the phospholipid bilayer. The efficacy of these agents was investigated by intratumoral administration either alone (cyto-IL-15 or cyto-abs) or in combination (cyto-combo) in subcutaneous TRAMP-C2 prostate tumors in C57BL/6J mice and compared with their non-modified equivalents in vivo. Following the survival endpoint, histological analyses and RNA sequencing were performed on the tumors.
Results: Intratumoral injection of cyto-IL-15 or cyto-combo delayed tumor growth by 50% and increased median survival to 28 and 25 days, respectively, compared with vehicle (17 days), whereas non-modified IL-15 or antibodies alone had no significant effects on tumor growth or survival. Histological analysis showed that cyto-IL-15 and cyto-combo increased necrosis and infiltration of natural killer (NK) cells and CD8 T cells in the tumors compared with vehicle and non-modified agents. Overall, the efficacy of cyto-combo was not superior to that of cyto-IL-15 alone.
Conclusion: We have demonstrated that intratumoral injection of cyto-IL-15 leads to prostate cancer growth delay, induces tumor necrosis and increases survival. Hence, cytotopic modification in combination with intratumoral injection appears to be a promising novel approach for prostate cancer immunotherapy.
Keywords: IL-15, checkpoint blockade, prostate cancer, cytotopic modification, NK cells
Key Points
Membrane-localising IL-15, injected directly into prostate tumors in mice induces tumor death and improves mouse survival while circumventing toxicities seen with cytokine immunotherapies administered systemically. This modified IL-15 could have a great impact in treating prostate cancer patients.
Introduction
Prostate cancer is the second leading cause of cancer-related death behind lung cancer, and the most common malignancy diagnosed in men.1 Treatments for prostate cancer include surgery, radiation, chemotherapy and androgen deprivation therapy. However, despite the high rates of progression-free survival, almost half of the patients progress to castration-resistant prostate cancer (CRPC).2
It has previously been suggested that prostate cancer can be immunogenic. This is supported by the presence of numerous tumor-associated antigens in the prostate, including prostate-specific antigen (PSA) and prostatic acid phosphatase (PAP), as well as tumor infiltrating lymphocytes (TILs), such as CD4 and CD8 T cells, natural killer (NK) cells, dendritic cells (DCs) and macrophages within tumors.3–5 However, the prostate cancer immunogenicity is hampered by the highly immunosuppressive microenvironment, which is probably due to improper functionality of TILs (anergy, exhaustion or senescence) or the presence of regulatory T cells (Tregs).6 High-grade prostate cancer with poorer prognosis has low infiltration of T cells and DCs and high occurrence of Tregs and tumor-associated macrophages;7–9 whereas, elevated infiltration of NK cells within tumors is associated with low risk of prostate cancer progression.10 Immunotherapy, which aims to overcome the immunosuppressive microenvironment and generate or enhance immune responses against tumor cells, has emerged as a promising alternative therapeutic approach for the treatment of prostate cancer.
Immunotherapies fall into four categories: adoptive T cell therapy, cancer vaccines, checkpoint inhibitors and cytokines.11 The only FDA-approved vaccine for prostate cancer is Sipuleucel-T (Provenge®), a DC-based vaccine that activates an antitumor response against PAP.12 However, Sipuleucel-T only extends patient survival by few months.13 One of the most studied immune checkpoint targets is cytotoxic T lymphocyte antigen 4 (CTLA-4), which downregulates T cell activation. The anti-CTLA-4 blocking antibody ipilimumab is FDA-approved for the treatment of advanced melanoma and is undergoing clinical trials for prostate cancer. So far, ipilimumab showed no benefit in overall survival, but it slightly improved progression-free survival in metastatic CRPC.14 Programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-L1) are also checkpoint targets as their interaction (PD-1/PD-L1) mediates immunosuppression, and thus is a crucial mechanism by which tumors escape immune response.15 To date, the FDA has approved at least six antibodies targeting PD-1/PD-L1, for the treatment of patients with melanoma and solid tumors, such as non-small cell lung cancer and urothelial cancer.16 The anti-PD-1 antibody pembrolizumab is FDA-approved for metastatic melanoma and solid tumors with mismatch repair deficiency or microsatellite instability, including metastatic CRPC, for which it has shown a favourable side effect profile.17 However, immunotherapy in prostate cancer has shown less efficacy than in other tumors, probably due to the low somatic mutation burden and suppressive tumor microenvironment.18–20 Therefore, immunotherapies that lead to activation and proliferation of NK and T cells within the tumor microenvironment, used alone or combined with other types of immunotherapies, may prove to be more efficacious than those aimed at a single target.
One of the most promising cytokines for cancer immunotherapy is interleukin-15 (IL-15), which has several biological functions equivalent to those of interleukin-2 (IL-2) – another known cytokine used for cancer immunotherapy – but without any significant sequence homology. IL-15 mediates its functions through the IL-2/IL-15Rβ-chain and common gamma (γc)-chain that it shares with the IL-2 receptor, and its own unique IL-15Rα-chain.21,22 Similar functions of IL-15 and IL-2 are: stimulation of T cell proliferation, generation of cytotoxic T lymphocytes (CTLs), stimulation of immunoglobulin synthesis by B cells, and proliferation and activation of NK cells.23,24 IL-2 and IL-15 have also distinct functions crucial for maintaining homeostasis of adaptive immune responses. IL-2 inhibits T cell responses by eliminating self-reactive T cells through activation-induced cell death (AICD) of CD8 effector T cells, and is involved in the maintenance and retention of Tregs.25,26 In contrast, IL-15 has a role in T and NK cell activation, in the survival of CD8 memory T cells, in inhibiting AICD, while it has no effect on Tregs. Those distinct functions of IL-15 make it a more suitable cytokine for cancer immunotherapy.21,27,28
We have recently shown that IL-15 is the only cytokine among a panel of agents that can expand and activate immune cells and that the presence of prostate cancer cells augments its capability.29 Moreover, a study in a TRAMP-C2 prostate tumor model showed that IL-15 combined with anti-CTLA-4 and anti-PD-L1 antibodies decreased tumor growth and prolong survival of tumor-bearing animals.30 Other preclinical studies have shown that IL-15 treatment leads to less toxicities with limited vascular capillary leak compared to IL-2,31 while the T cell-mediated antitumor potency of IL-15 is greater than that of IL-2.32
In the present study, the immunotherapeutic potential of IL-15 was explored as monotherapy or in combination with two checkpoint inhibitor antibodies, CTLA-4 and PD-L1, administered intratumorally in prostate cancer. To enhance and prolong the therapeutic capabilities of IL-15, anti-CTLA-4 and anti-PD-L1 antibodies, we created novel cytotopic (membrane-localising) forms of these agents that can adhere to cell surfaces by non-specific anchoring to the phospholipid bilayer, while maintaining the ability to recognise and bind to their receptors. Initially, the binding efficiency and the activity of these proteins were investigated in vitro. The efficacy of the agents was then explored in a syngeneic subcutaneous prostate cancer mouse model and tumor response was characterized using histopathology, cytokine analysis and RNA sequencing on tumor extracts.
Methods
Cytotopic Modifications of Proteins
The doubly myristoylated peptide (2.36 kDa) (named PTL3146) consisted of two fatty acid myristoyl chains attached to the N-terminus of the L-lysine residue, a positively charged peptide and an activated thiol group;33 sequence provided in Figure S1 (Supplementary File). The myristoyl chains insert spontaneously into the phospholipid bilayer upon interaction with cell surfaces, the peptide interacts with the negatively charged membrane, and the thiol group forms a disulphide bridge with the free cysteine within the protein of interest.
IL-15: Recombinant IL-15 containing the full human IL-15 sequence and a C-terminus linker with a free cysteine (total molecular mass of 17.68 kDa) was expressed in E.coli and purified by GenScript Corporation (New Jersey, USA); sequence provided in Figure S1 (Supplementary File). For the cytotopic modification, IL-15 was partially reduced at the C-terminus with tris(2-carboxyethyl)phosphine (TCEP) (ThermoFisher) at a final concentration of 300µM for 1 h. The excess of reducing agent was removed using a G-25 spin column (GE Healthcare, Little Chalfont, UK). The reduced IL-15 was then incubated with the PTL3146 peptide (10mM in DMSO) at a 1:3 protein to peptide molar ratio at room temperature (RT) for 60 to 90 min and subsequently at 4°C overnight. The unbound PTL3146 was removed with dialysis using a Spectra-Por Micro float-A-Lyzer device (Sigma-Aldrich, Dorset, UK) against phosphate-buffered saline (PBS) (Severn Biotech Ltd., Kidderminster, UK). The cytotopically-modified IL-15 was termed cyto-IL-15 in the present study and currently a patent has been filed (application number GB 1,913,804.9) with the name Haptoleukin-15.
Anti-PD-L1 and anti-CTLA-4 antibodies: Rat monoclonal anti-mouse PD-L1 (B7-H1) antibody (10F.9G2, BioXcell, West Lebanon, USA) and Armenian hamster monoclonal anti-mouse CTLA-4 (CD152) antibody (UC10-4F10-11, BioXcell) were cytotopically modified by applying a method using 2-iminothiolane.34 Briefly, the antibody (either anti-PD-L1 or anti-CTLA-4) was incubated with a 100 mM freshly prepared solution of 2-iminothiolane hydrochloride (2-IT.HCl) (Sigma-Aldrich) for 30 min at RT (all solutions used were degassed). Then, the PTL3146 peptide (10mM in DMSO) was added (1:3 protein to peptide molar ratio) and the reaction was monitored by measuring the absorbance of pyridine-2-thione (reaction by-product) at 343 nm. The reaction was continued until one to two molecules of peptide were attached to each antibody molecule. The modified antibody was purified using a G-25 spin column to remove residual 2-IT.HCl and peptide. The cytotopically modified antibodies (abs) were termed cyto-PD-L1 and cyto-CTLA-4 (cyto-abs).
Concentration of the cytotopically-modified proteins was determined using a Pierce BCA protein assay Kit (ThermoFisher) according to manufacturer’s instructions. Cyto-IL-15 concentration was also measured using a human IL-15 ELISA MAX (Biolegend, London, UK) according to manufacturer’s instructions.
Cell Culture
Transgenic adenocarcinoma of the mouse prostate (TRAMP)-C2 cells, obtained from American Type Culture Collection (ATCC, Teddington, UK), were maintained in Dulbecco’s Modified Eagle’s culture medium (DMEM) supplemented with 2 mM
Cell Proliferation Assay
The activity of IL-15 was investigated using a CTLL-2 cell proliferation assay. CTLL-2 cells were incubated for 4 hours in phenol-free RPMI complete medium without T-STIM. Subsequently, 3.5x104 cells/well were seeded in 96-well plates and treated with IL-15 (Genscript) or cytotopically-modified IL-15 at a range of 0–1.6 ng/mL concentration. Commercial IL-15, which is recombinant human IL-15 (without a C-terminus linker) was purchased from PeproTech EC Ltd. (London, UK) and used as control. After 3 days, cells were incubated with CellTiter 96 AQueous One Solution reagent (Promega, Southampton, UK) in the dark at 37°C for up to 4 hours. The reaction was stopped with 10% SDS and the absorbance was measured at 490 nm using a Hidex Sense microplate reader (LabLogic Systems Ltd, Sheffield, UK). Background absorbance was measured in wells with medium only.
Animals and Tumors
Animal experiments were performed in accordance with the local ethical review panel and the UK Home Office Animals (Scientific Procedures) Act 1986. Male C57BL/6J mice, 6–8 weeks old (Charles River, Harlow, UK), were injected with 5 x 106 TRAMP-C2 cells in 0.1 mL PBS subcutaneously into the right flank. The tumor length (L), width (W) and depth (D) were measured using callipers and the volume was calculated using the ellipsoid shape formula: (π/6) x L x W x D. When tumors were palpable and well established with an approximate volume of 100 mm3, mice were randomly divided into eight treatment cohorts. Mice in each cohort where treated intratumorally with vehicle (PBS, n = 13), 10 μg/dose IL-15 or cyto-IL-15 (n = 10, each), 100 μg/dose of each of the abs or each of the cyto-abs (n = 6, each), combination of IL-15 and abs (combo) or combination of cyto-IL-15 and cyto-abs (cyto-combo) (n = 10, each). The last cohort was treated with 10 µg/dose IL-15 intraperitoneally (IL-15 i.p., n = 6). IL-15 was given at two doses injected at days 0 and 3, while antibodies were injected as a single dose at day 0. Day 3 was chosen in order to mitigate any potential injection-related adverse effects. Mice were monitored for weight loss, hunched posture, discomfort and development of rashes. When tumors reached a maximum diameter of 15 mm (survival endpoint) they were excised and snap frozen.
Histology and Immunofluorescence
Frozen tumor sections (8 µm thick) were cut axially from two regions for each tumor, one in the centre of the tumor and one 1 mm apart. To assess immune cell infiltration, acetone-fixed sections were stained with a rabbit monoclonal anti-CD4 antibody [EPR19514] (1/200, Abcam, Cambridge, UK) and Alexa Fluor 546 goat anti-rabbit secondary antibody (3/1000, ThermoFisher), and a rat monoclonal anti-CD8 antibody [YTS169.4] (1/100, Abcam) and Alexa Fluor 488 donkey anti-rat secondary antibody (1.5/1000, ThermoFisher). Sections were also stained with a rabbit monoclonal anti-CD3 antibody [SP7] (1/100, Abcam) and Alexa Fluor 546 goat anti-rabbit secondary antibody, and Alexa Fluor 488-conjugated mouse monoclonal anti-NK1.1 antibody [PK136] (1/25, BioLegend). Non-immune-specific immunoglobulins (same classes and concentrations as the primary antibodies) were used as negative isotype controls, whereas mouse spleens were used as positive controls. Fluorescent staining was visualised under a BX51 microscope (Olympus Optical, London, UK) using a QImaging camera (Cairn Research, Faversham, UK). For each tumor (n = 5 per treatment cohort), images were acquired from five randomly selected areas for each of the two tumor sections for each antibody.
To quantify the degree of necrosis, sections were stained with haematoxylin and eosin (H&E) and images were acquired using a bright-field Hamamatsu NanoZoomer 2.0RS digital slide scanner (Hamamatsu Photonics, Hamamatsu City, Japan). Tumor necrotic areas and fluorescent areas for each stain were defined and analysed using ImageJ software35 as previously described.36 Necrosis was expressed as a percentage of the whole tumor section area, while fluorescent staining as a percentage of the total image area.
RNA Sequencing
Tumors treated with vehicle, IL-15 or cyto-IL-15 (n = 3/group) were used for RNA sequencing. Tumors were dissociated using a gentleMACS Dissociator according to manufacturer’s instructions and RNA was extracted using an RNeasy Mini Kit (Qiagen, Manchester, UK). RNA library preparations, sequencing reactions and bioinformatics analyses were conducted at Genewiz, LLC (New Jersey, USA). The method is described in detail in Supplementary Methods (Supplementary File).
Statistical Analysis
Data were analysed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA). Statistical significance of differences was determined by one- or two-way ANOVA with Sidak’s or Dunnett’s multiple comparisons post-tests, with a 5% level of significance. Results are presented as mean ±1 standard error of the mean (SEM). For the RNA sequencing, the Wald test was used to generate p-values and Log2 fold changes. Genes with adjusted p-values <0.05 and absolute log2 fold changes >1 were defined as differentially expressed genes for each comparison.
Results
Characterization of Cytotopically-Modified Proteins
The cytotopically-modified IL-15 was detected with Western blot analysis using an anti-IL-15 antibody. To confirm the presence of the PTL3146 peptide, IL-15 and antibodies were also modified using a FAM-labelled peptide, which could be visualised under UV light (Supplementary Methods, Supplementary File). Figure S2A (Supplementary File) shows an increase in the molecular mass of IL-15 from 18 to 20 kDa after the modification. Cyto-IL-15 is also detected after Silver staining (20 kDa), whereas the FAM-labelled peptide is visual under UV light (Figure S2B). The FAM-labelled peptide is also successfully bound to CTLA-4 and PD-L1 antibodies (Figure S2C).
To investigate the ability of the cytotopically-modified proteins to bind to cell membranes via the peptide and not via a receptor, cells that do not express the relevant receptors were used; red blood cells (RBCs) for IL-15 and naïve Jurkat cells for CTLA-4 and PD-L1. The cell-binding assay is described in Supplementary File (Supplementary Methods, Cell binding assay). RBCs incubated with non-modified IL-15 showed very low fluorescent intensity staining the same as the control where no protein was added. On the contrary, incubation of the cells with cyto-IL-15 led to a significant increase in fluorescent intensity (Figure 1A). To investigate whether the binding via the peptide was maintained over time, after excess or unbound protein has been removed, naïve Jurkat cells were incubated with IL-15 or cyto-IL-15 and binding was measured after 30 min or 24 h as described in Supplementary File (Supplementary Methods, Cell binding assay). Binding of cyto-IL-15 was maintained even after 24 h (increased fluorescent intensity compared with control), whereas no binding was seen with non-modified IL-15 (Figure S3, Supplementary File). Similarly, the modified antibodies cyto-CTLA-4 and cyto-PD-L1 increased fluorescent intensity staining after incubation with naïve Jurkat cells, whereas no shift in intensity was seen when cells were incubated with non-modified antibodies (Figure 1A).
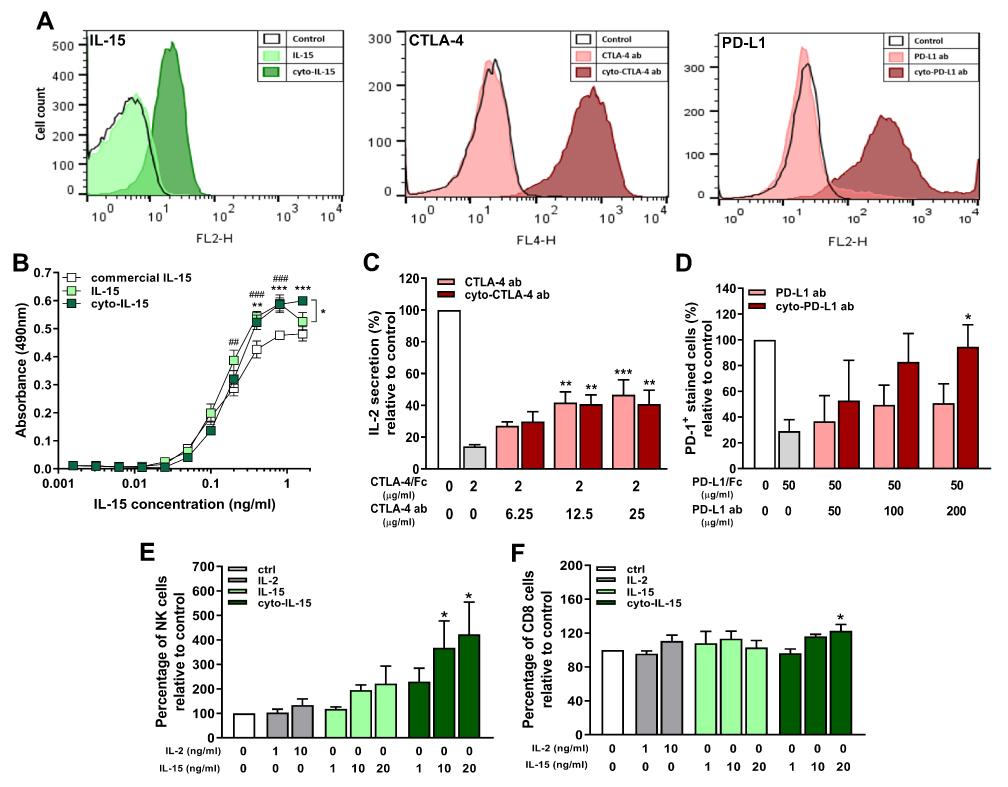 |
Figure 1 In vitro characterization of cytotopically-modified proteins. (A) Cell membrane binding of cytotopically-modified IL-15 on RBCs, and cytotopically-modified CTLA-4 or PD-L1 antibodies on naïve Jurkat cells, detected by flow cytometric analysis using fluorescent-labelled antibodies to these proteins. (B) Proliferation of CTLL-2 cells treated with different forms of IL-15 (n = 3 independent experiments performed in quadruplicates). Results are means ±1 SEM (*cyto-IL-15 versus commercial IL-15 or cyto-IL-15 versus IL-15, #IL-15 versus commercial IL-15, *p <0.05, **or ##p <0.01, ***or ###p <0.001 two-way ANOVA with Tukey multiple comparisons post-test). (C) IL-2 secretion by activated Jurkat cells inhibited with CTLA-4/Fc protein and reconstituted with varying concentrations of CTLA-4 or cyto-CTLA-4 antibodies. Results are expressed relative to IL-2 secretion by activated control cells (n = 4). (D) PD-1 binding in EL4 cells inhibited with PD-L1/Fc protein and reconstituted with varying concentrations of PD-L1 or cyto-PD-L1 antibodies. Results are expressed relative to PD-1 expression in control cells (n = 4). (E, F) NK and CD8 T cell expansion in a human non-adherent PBMC population treated with varying concentrations of IL-2, IL-15 or cyto-IL-15 (n = 3). (C–F) Results are means +1 SEM (*p <0.05, **p <0.01, ***p <0.001 one-way ANOVA with Sidak’s multiple comparisons post-test). |
The activity of IL-15 after modification was investigated measuring CTLL-2 cell proliferation. The non-modified and modified versions of IL-15 led to similar cell proliferation at lower doses; however, at higher doses (1.6 ng/mL), there was significantly more proliferation with cyto-IL-15 (p <0.001). Both IL-15 and cyto-IL-15 led to significantly higher CTLL-2 proliferation than commercial IL-15 (p <0.01, Figure 1B). The activity of IL-15 was also assessed in non-adherent PBMCs as described in Supplementary Methods (Cell expansion assay) and Table S1 (Supplementary File). The flow cytometry gating strategy is shown in Figure S4A (Supplementary File). NK cell expansion was significantly increased when cells were incubated with cyto-IL-15. More specifically, a 3.7-fold increase was seen at 10 ng/mL and a 4.2-fold increase at 20 ng/mL compared with control (p <0.05). Non-modified IL-15 at the same concentrations also increased NK cell expansion by approximately 2-fold compared with control but the result did not reach statistical significance (Figure 1E). A small increase in the expansion of CD8 cells (1.2-fold) was also seen in PBMCs incubated with 20 ng/mL cyto-IL15 compared with control (p <0.05, Figure 1F).
The activity of CTLA-4 antibody after modification was assessed by measuring IL-2 secretion in activated Jurkat cells as described in Supplementary Methods (CTLA-4 activity assay, Supplementary File). CTLA-4/Fc protein inhibited activation of Jurkat cells by binding to B7-1, and hence inhibited IL-2 secretion by ~86%. Addition of CTLA-4 or cyto-CTLA-4 antibodies, at concentration higher than 12.5 µg/mL, significantly restored IL-2 secretion to approximately 40% via CTLA-4/Fc protein blockade (p <0.01, Figure 1C). The activity of PD-L1 antibody after modification was accessed by measuring PD-1 expression in EL4 cells as described in Supplementary Methods (PD-L1 activity assay, Supplementary File). PD-L1/Fc protein blocked PD-1 leading to reduced fluorescent staining of ~30%, whereas addition of 200 µg/mL cyto-PD-L1 antibody significantly restored staining to 95% via PD-L1/Fc protein blockade (p <0.05, Figure 1D).
Immunotherapy with Cytotopically-Modified IL-15 Causes Significant Prostate Tumor Growth Delay
Mice with TRAMP-C2 prostate subcutaneous tumors were injected intratumorally with non-modified or cytotopically-modified versions of IL-15, CTLA-4 and PD-L1 antibodies. Human IL-15 was used for treatment as the human and mouse IL-15 genes have approximately 73% sequence homology.37 The mean tumor volume at treatment onset (day 0) was 106 ± 7 mm3 across all tumors. Both cyto-IL-15 and cyto-combo treatments significantly reduced tumor growth in comparison with vehicle by 52 (p <0.01) and 58% (p <0.001) respectively, at day 14 post-treatment. Combination of IL-15 and antibodies in their non-modified forms also caused a growth delay by 48% compared with vehicle at day 14 (p <0.01). However, neither intraperitoneal nor intratumoral injection of non-modified IL-15 led to significant changes in tumor growth. Similarly, treatment with antibodies or modified antibodies did not delay growth until day 14 post-treatment. At day 17, cyto-IL-15, combo and cyto-combo led to a significant growth delay in comparison with vehicle of 37 (p <0.05), 37 (p <0.05) and 46% (p <0.01) respectively. A significant delay in growth was also seen with cyto-abs at day 17 by 57% compared with vehicle (p <0.01, Figure 2A). Non-modified antibodies, cyto-IL-15 and cyto-combo were the only treatments that significantly increased median survival to 27 (p <0.05), 28 (p <0.01) and 25 days (p <0.01) respectively, compared with vehicle (17 days) (Figure 2B). Moreover, the combination of cytotopically-modified IL-15 and antibodies increased survival significantly (p <0.05) compared with the non-modified agent combination. All treatments were well tolerated and none of the mice suffered any side effects, such as rashes or weight loss. Moreover, no injection-related adverse effects were observed and no mice died as a result of any of the treatments.
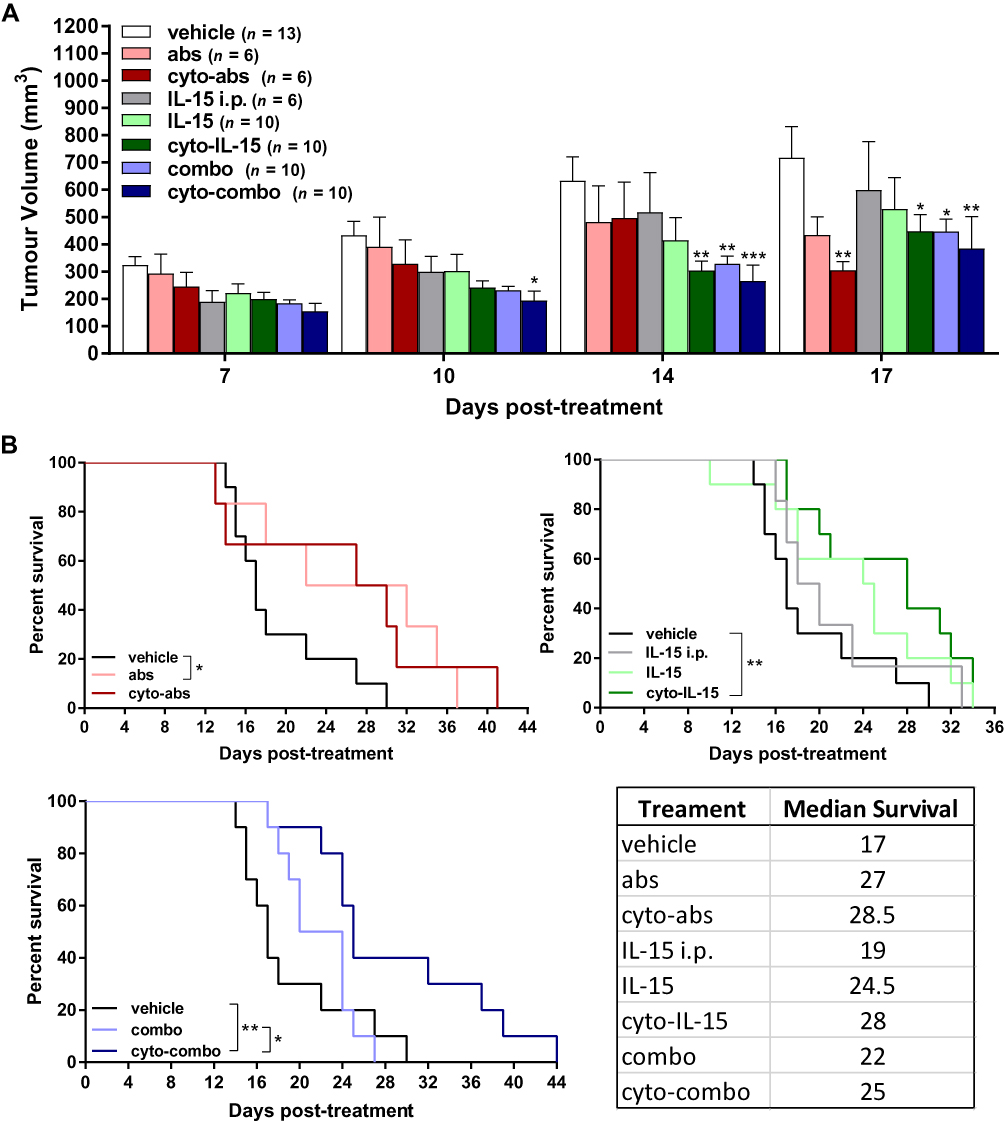 |
Figure 2 Effect of modified IL-15 and antibodies on growth of TRAMP-C2 subcutaneous prostate tumors. (A) Tumor volumes up to day 17 post-treatment. Data are means + 1 SEM for all the tumors per group and comparisons are relative to vehicle (*p <0.05, **p <0.01, ***p <0.001 two-way ANOVA with Dunnett’s multiple comparisons post-test). (B) Survival curves of mice post-treatment (*p <0.05, **p <0.01, comparisons of equality of two survival curves using Log-rank (Mantel-Cox) test). Table shows the median survival of each group. |
Cytokine Concentration in Tumors Treated with Immunotherapeutic Agents
Release of cytokines in tumors was measured using a cytokine bead array assay in tumor homogenates as described in Supplementary Methods (Cytokine bead array assay, Supplementary File). In tumors injected with cyto-combo, the release of CCL2 was significantly decreased by approximately 4-fold (p <0.05), whereas IL-1β production was increased by more than 2-fold compared with vehicle (p <0.05) (Figure 3A and B). Release of GM-CSF, IL-10 and IFN-α was significantly increased in tumors injected with combination by 4.2- (p <0.01), 5.4- (p <0.05) and 2.9-fold (p <0.05), respectively, compared with vehicle (Figure 3C–E). No significant differences were observed in the release of the rest of the cytokines measured, such as IFN-γ, TNF-α, CXCL1, CCL5, IL-6 and CXCL10, whereas IL-2 and IFN-β levels of production were not detectable (data not shown). Moreover, intratumoral treatment with antibodies or cyto-abs, or treatment with IL-15 intraperitoneally had no effect on cytokine release in the tumors (data not shown).
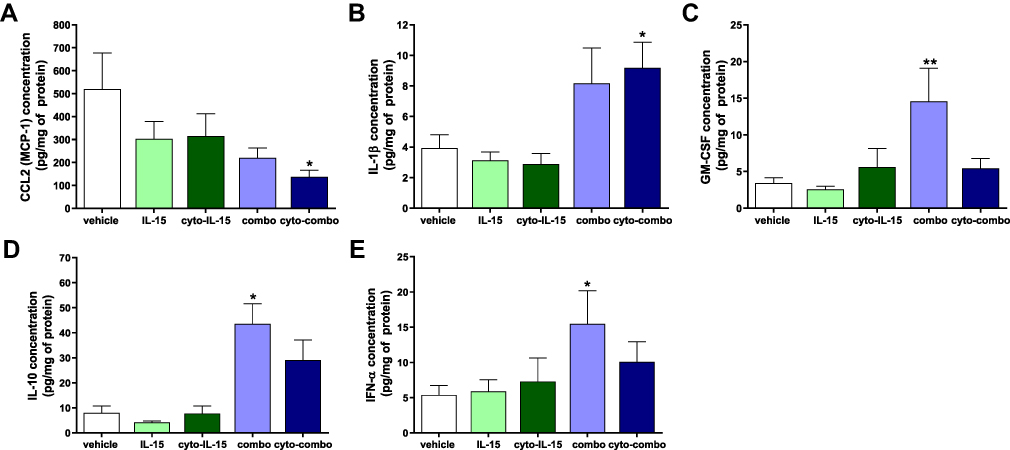 |
Figure 3 Assessment of cytokines in TRAMP-C2 prostate tumor homogenates. CCL2 (A), IL-1β (B), GM-CSF (C), IL-10 (D) and IFN-α (E) concentration in tumor homogenates derived from tumors treated with vehicle, IL-15, cyto-IL15, combo or cyto-combo. Results are means +1 SEM of duplicate measurements made from all tumors in each cohort (n = 10) corrected for protein concentration. Comparisons are relative to vehicle (*p <0.05, **p <0.01, one-way ANOVA with Dunnett’s multiple comparisons post-test). |
To detect whether any of the injected IL-15 or cyto-IL15 was retained and transferred to blood circulation, the concentration of IL-15 was measured in the mouse blood plasma collected from treated mice at the survival endpoint using an IL-15 ELISA. IL-15 levels were mostly not detectable, with concentration ranging from 0 to 7 pg/mL, and no significant differences were observed between treatment cohorts (Figure S5A, Supplementary File).
Treatment with Cyto-IL-15 and Cyto-Combo Induced Tumor Necrosis and Increased Immune Cell Infiltration
Figure 4 shows representative histological sections of excised tumors at survival endpoints after treatment with vehicle, IL-15, cyto-IL15, combo and cyto-combo stained with H&E (Figure 4A), CD4 and CD8 antibodies (Figure 4B), and CD3 and NK1.1 antibodies (Figure 4C). Most NK and CD8 T cells were observed close to or inside necrotic regions. Moreover, areas with NK cells mainly lacked CD3 cells. This is more clearly depicted in Figure S6 (Supplementary File), where two areas of the same section of a tumor treated with cyto-combo are shown, one has mostly CD3 cells (left) and the other mostly NK cells (right).
 |
Figure 4 Histological assessment of TRAMP-C2 prostate tumors. (A) Composite images and magnified images of H&E-stained sections indicating necrotic regions. (B, C) RGB images from tumor sections stained with (B) CD4 (red) and CD8 (green), and (C) CD3 (red) and NK1.1 (green) antibodies. Nuclei were stained with DAPI (blue). |
Quantification of tumor necrosis using the H&E stained sections is shown in Figure 5A. Cyto-IL15, combo and cyto-combo caused a significantly higher and increasing degree of necrosis (32 ± 6% (p <0.05), 34 ± 9% (p <0.01), and 43 ± 8% (p <0.0001), respectively) compared with vehicle (20± 8%). CD4 staining was also increased in these treatment cohorts (1.5% (p <0.0001) for cyto-IL-15, 1% (p <0.01) for combo and 1.6% (p <0.0001) for cyto-combo) compared with 0.6% in the vehicle (Figure 5B). However, CD8 and NK1.1 staining were only increased in the cohorts receiving modified IL-15 or combination of modified immunotherapies; 0.13% (p <0.001) for cyto-IL-15 and 0.11% (p <0.01) for cyto-combo compared with 0.05% in vehicle for CD8, and 0.50% (p <0.0001) for cyto-IL-15 and 0.44% (p <0.001) for cyto-combo compared with 0.10% in vehicle for NK1.1 (Figure 5C and E). Moreover, all the above markers (necrosis, CD4, CD8 and NK1.1) were significantly higher in tumors treated with cytotopically-modified agents compared with their non-modified versions (cyto-IL-15 versus IL-15 and cyto-combo versus combo). CD3 staining was slightly decreased in the cyto-IL-15 and cyto-combo groups compared with the other cohorts, but this was not statistically significant (Figure 5D).
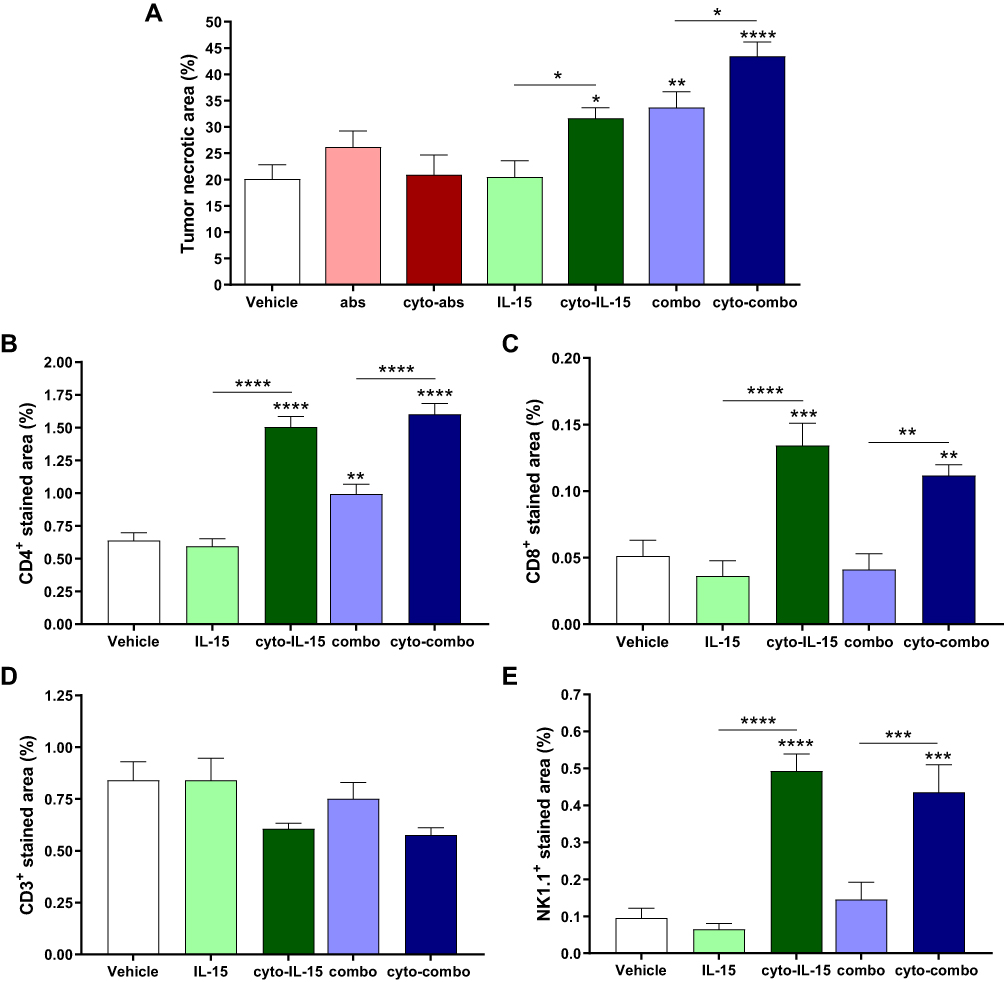 |
Figure 5 Quantification of necrotic area and immune cell infiltration in TRAMP-C2 tumors. (A) Necrotic area. Results are means +1 SEM of two sections per tumor for all tumors. (B) CD4 positive-stained area, (C) CD8 positive area, (D) CD3 positive area, and (E) NK1.1 positive-stained area. Results are means +1 SEM of 10 images per tumor for n = 5 per group. Comparisons are relative to vehicle unless otherwise indicated (*p <0.05, **p <0.01, ***p <0.001, ****p <0.0001 one-way ANOVA with Dunnett’s multiple comparisons post-test). |
To investigate whether the increase of NK and CD8 T cells observed in the tumors was due to increased production of these cells in the spleen, splenocytes were isolated from mouse spleens collected at survival endpoints from treated mice, stimulated, and flow cytometry analysis was performed (Supplementary Methods: Splenocyte ex vivo stimulation, Figure S4B, and Table S1, Supplementary File). NK cell number significantly increased by almost 2-fold (p <0.05) in the cyto-IL-15 cohort compared with vehicle (Figure S5B, Supplementary File), whereas no significant differences were measured in the numbers of CD8 T cells (data not shown).
Cyto-IL-15 Treatment Leads to Significant Changes in Gene Expression in Prostate Subcutaneous Tumors
To investigate whether cyto-IL-15 treatment had an impact on gene expression, a comparison of genes was performed between tumors treated with vehicle, IL-15 or cyto-IL-15. Genes with an adjusted p-value <0.05 and absolute log2 fold change >1 were considered as differentially expressed genes. When vehicle-treated tumors were compared with IL-15-treated tumors only eight genes were differentially expressed (3 upregulated and 5 downregulated). However, when vehicle and cyto-IL-15 cohorts were compared, a total of 1130 genes were significantly differentially expressed (866 upregulated and 264 downregulated). Similarly, 1095 genes were differentially expressed (869 upregulated and 226 downregulated) when tumors from the IL-15 and cyto-IL-15 cohorts were compared. A list of all the differentially expressed genes in all three comparison sets is provided in Table S2 (Supplementary File). Figure 6A shows heatmaps of the top 30 differentially expressed genes (8 genes in the case of vehicle versus IL-15). While IL-15 treatment caused very few, almost negligible, changes in gene expression compared with vehicle, cyto-IL-15 led to dramatic gene expression changes compared with vehicle or IL-15. More than 1000 genes, including Sema3e, Wnt4, Pdcd4, Bbip1, Yap1, Shoc2, Lamp3, Ndn, Fst, Erfe, Krt80 and Gabra3, were differentially expressed in both comparisons. The gene ontology analysis showed that the differentially expressed genes in both vehicle versus cyto- IL-15 and IL-15 versus cyto-IL-15 comparisons were involved in processes such as cell adhesion, cell proliferation, cell migration and axon guidance (Figure 6B). In the comparison of IL-15 versus cyto-IL-15, several differentially expressed genes were also involved in angiogenesis.
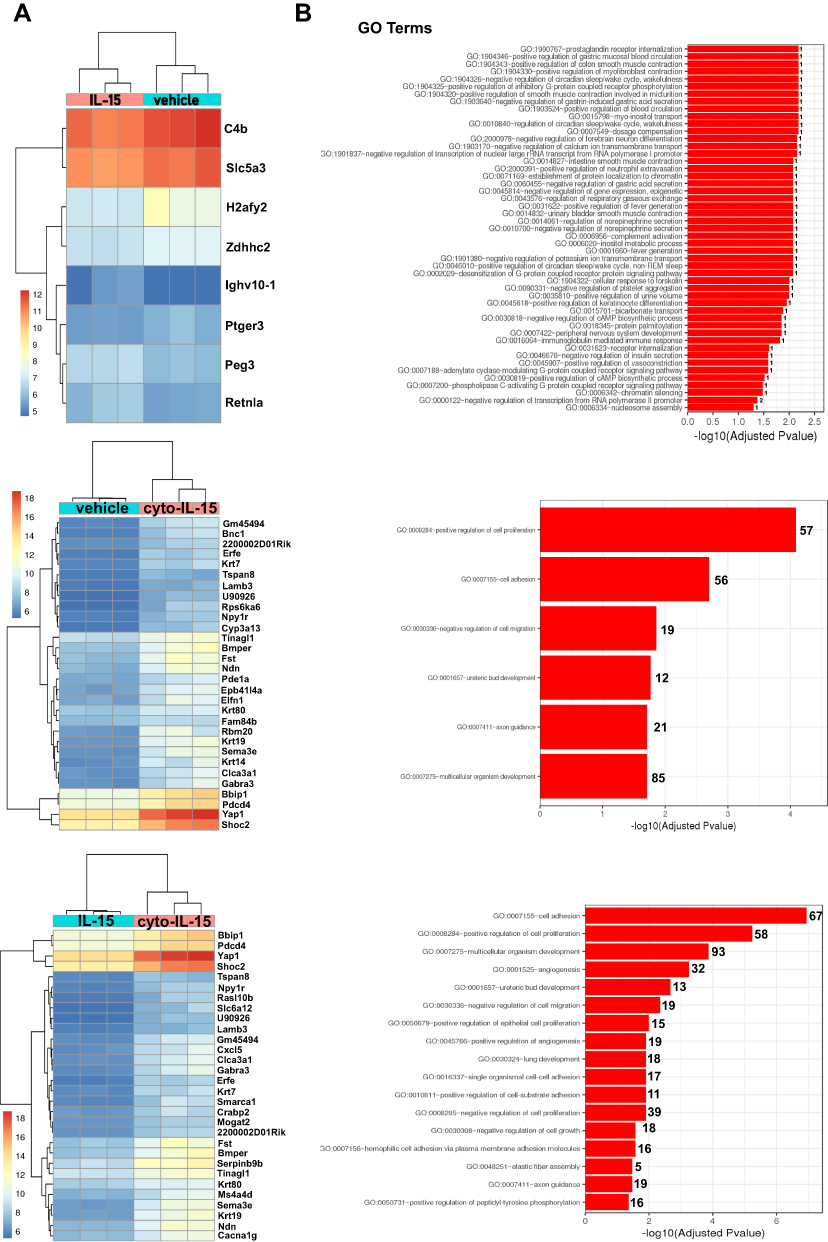 |
Figure 6 Effects of cyto-IL15 treatment on gene expression in TRAMP-C2 tumors. Differential gene expression analysis followed by gene ontology analysis were performed between tumors treated with vehicle or IL-15 (top panel), with vehicle or cyto-IL-15 (middle panel), and with IL-15 or cyto-IL-15 (bottom panel) (n = 3/cohort). (A) Bi-clustering heatmaps of the log2 transformed expression values in each sample showing the expression profiles of the top 30 differentially expressed genes (all 8 differentially expressed genes are shown in the vehicle versus IL-15 comparison). (B) Gene ontology (GO) enrichments of all significantly differentially expressed genes (adjusted p-value < 0.05) for each of the three comparison sets. The numbers next to the bars indicate the number of significantly differentially expressed genes involved in each biological process. |
The protein expression levels of one of the most upregulated genes, Pdcd4, were also investigated in tumor homogenates from mice treated with either vehicle, IL-15 or cyto-IL-15 using Western blot analysis (Supplementary Methods: SDS-PAGE electrophoresis and Western blot analysis, Supplementary File). A significantly higher Pdcd4 protein expression (p <0.05) was seen in the cyto-IL-15-treated tumors compared both with vehicle or IL-15-treated tumors (Figure S7, Supplementary File).
Discussion
In the present study, novel membrane-localising forms of IL-15, and CTLA-4 and PD-L1 antibodies were engineered to enable them to anchor to cell membranes. The efficacy of these agents was investigated in a prostate cancer murine model. Our key findings indicate that cyto-IL-15 or cyto-combo significantly prolonged mouse survival by delaying tumor growth and inducing necrosis, without any side effects. The observed necrosis was associated with enhanced recruitment and expansion of NK and T cells in the tumors. Moreover, cyto-IL-15 had an extensive impact on tumor gene expression compared with its non-modified form.
The technology of cytotopically-modified agents has been used previously to create a membrane-localizing form of the human complement receptor type 1, Mirococept, for inhibiting complement activation. Mirococept can prolong the survival of donor kidneys in rats and has been used to prevent delayed graft function in a clinical trial of renal transplantation.38,39 A membrane-localising hirudin-like peptide, Thrombalexin 1, has also been developed and used to inhibit thrombin and prevent acute antibody-mediated thrombosis in donor organs in a rat model of hyperacute rejection.33
In our study, treatment with cyto-IL-15 or a combination of cyto-IL-15 and cyto-antibodies led to both a significant growth delay of TRAMP-C2 prostate tumors in mice and an increase in survival compared with control. This effect was greater than treatment with cyto-abs alone or with non-localising forms of the same agents, as these did not lead to significant differences compared with control, either in terms of tumor growth or in terms of mouse survival. Antibodies or cyto-antibodies also have an effect on either tumor growth or survival but this effect is more delayed compared with the effect of cyto-IL-15 and cyto-combo. For tumor growth, effects are observed mainly from day 14 (day 10 for cyto-combo); however, for cyto-abs, changes are only visible on day 17. This delayed action of the checkpoint inhibitor antibodies can be explained by the different mechanism of killing cancer cells compared to IL-15. IL-15 leads to expansion and activation of NK cells and the generation of CD8 cytotoxic T lymphocytes.40 Activated NK cells release secretory lysosomes containing cytotoxic proteins, such as granzymes and perforin, that kill tumor cells, whereas CTLs kill target cells by releasing granzymes A and B or by induction of Fas ligand-mediated apoptosis. NK and CTLs also exert antitumor effects by releasing IFN-γ and TNF-α. However, activated T cells upregulate inhibitory checkpoints such as CTLA-4 and PD-1, which in turn prevent CD28 co-stimulation by competing for B7 binding and interfere with T cell receptor signalling by binding to PD-L1/PD-L2, respectively, leading eventually to abrogation of the activated T cell response. Moreover, continuous exposure to IFN-γ upregulates PD-L1 on cancer cells, which in turn suppresses the proliferation and functions of NK cells and CTLs, and induces T cell apoptosis.41–43 Blocking PD-L1 and CTLA-4 using checkpoint inhibitors, such as the abs or cyto-abs used in our study, can relieve the exhaustion of CD8 T cells and renew their priming, and thereby further eliminate antigen-expressing tumor cells. Hence, it appears that the antibodies reach their full potential later on during treatment than IL-15, but since most of the mice in the vehicle group reach the maximum tumor size around day 17 it is difficult to make comparisons after that point.
Overall, our findings are in agreement with a previous study in TRAMP-C2 subcutaneous tumors showing that murine IL-15 treatment decreased tumor growth and prolonged mice survival and that these effects were augmented when IL-15 was co-administrated together with anti-CTLA-4 and anti-PD-L1 antibodies.30 However, to achieve this level of efficacy, the mice received a higher total dose of IL-15 (75 µg) and antibodies (600 µg each) compared to the total dose in our study (20 µg for IL-15 and 100 µg for each antibody). Another study in TRAMP-C2 tumors showed that treatment with murine IL-15 combined with anti-CD40 antibody, which increased IL-15Rα expression on DCs, resulted in more than 70% tumor remissions.44
In a clinical trial in patients with metastatic melanoma or renal cell cancer, intravenously administered IL-15, increased NK and CD8 T cell populations in circulating blood, and cleared lung lesions in two patients; however, it led to several dose-limiting toxicities, such as fever, hypotension and thrombocytopenia, which were associated with inflammatory cytokine secretion.45 This study suggested that to minimize toxicity and increase efficacy of IL-15 treatment, the maximum serum concentration of IL-15 and excess cytokine release must be reduced, while optimal IL-15 concentration is maintained for a longer period as free IL-15 has a short half-life. One approach to extend the half-life of free IL-15 and improve its efficacy was to use an IL-15 superagonist/IL-15Rα fusion complex (ALT-803) shown to enhance the number and activity of NK and T cells.46 ALT-803 was used in patients with hematologic malignancies who relapse after allogeneic hematopoietic cell transplantation. Despite ALT-803 being well tolerated, especially when administered subcutaneously to avoid systemic toxicities, responses were observed in less than 20% of the patients.47 Subcutaneous administration of IL-15 (without IL-15Rα) was also well tolerated in patients with refractory solid tumors and increased circulating NK and CD8 T cells, although it led to no objective responses.48 Several studies, reviewed by Marabelle et al,49 have shown that high concentrations of immunostimulating drugs in the blood can be prevented by local injection leading to lower toxicities. Intratumoral delivery of these agents allows higher concentrations in the tumor microenvironment than systemic infusions, resulting in improved efficacy. However, there are practical limitations if repeated injections are required to trigger the adaptive immune response. In our study, we enhanced the efficacy of IL-15 by injecting it in the tumor microenvironment where cancer cells are in abundance while minimizing toxicities (low serum concentrations). Moreover, by tethering IL-15 to cell membranes we improved its efficacy, with only two injections of the drug, potentially by prolonging its retention in the tumor site.
The cyto-IL-15-mediated antitumor immune responses seen in our study were likely due to expansion and recruitment of NK, CD8 and CD4 T cells in the tumor microenvironment, as showed by increased staining in histological tumor sections. This is in agreement with preclinical and clinical studies, which demonstrated that after administration of recombinant (r)IL-15 or ALT-803, the main mechanism attributing to the potential of IL-15 to promote antitumor responses is the ability to prolong expansion and activation of NK cells, CD8 memory T cells and in some cases CD4 T cells.27,45,50,51 However, response to IL-15 and subsequent activation of T cells often results in increased production of pro-inflammatory cytokines, known as cytokine release syndrome. This usually involves an overproduction of IL-6, IL-10, TNF-α and IFN-γ and can lead to detrimental effects in patients, ranging from fever and heart problems to vascular leakage and death.52 Treatment with ALT-803 resulted in marked elevations of IFN-γ, TNF-α, and IL-10 in mice, but without any toxicities.51 Administration of rIL-15 in patients with malignant melanoma increased IL-6, IL-8, IFN-γ, TNF-α and IL-1β levels, which coincided with acute clinical toxicities, such as fever and blood pressure changes.45 In our study, when cytokines were measured in tumor extracts at the experimental endpoint, IL-10, GM –CSF and IFN-α were mildly elevated only in the non-modified combination cohort, whereas IL-1β was increased in the modified combination but without any toxicities (no weight loss or development of rashes). No cytokine levels were changed at the end of IL-15 monotherapy, suggesting that if there were any changes during NK and T cell activation, these changes were transient and did not cause side effects.
When gene expression was investigated, we found that cyto- IL-15 led to significant changes compared with vehicle or non-modified IL-15. Among the most upregulated genes was Sema3e (semaphorin-3e) with an approximate 10-fold change in both comparisons. Sema3e is an axon guidance molecule expressed by DCs that binds to plexin-D1 expressed by NK cells. It has been suggested that Sema3e production is involved in regulating NK cell activation and functions, or in DC homeostasis in NK-DC crosstalk.53 Another gene, and also protein expression, upregulated by cyto-IL-15 was the tumor suppressor gene programmed cell death protein 4 (Pdcd4) (~5 fold-change), which is known to deregulate DNA-damage response and induce apoptosis in tumor cells.54,55 Most of the differentially expressed genes in our study were involved in processes such as cell adhesion, proliferation, migration and angiogenesis (in the comparison of IL-15 versus cyto-IL-15). A previous study in a murine prostate cancer model has also shown that IL-15 is involved in the regulation of cell migration, proliferation, and blood vessel formation.56 Moreover, RNA sequencing of IL-15 cytokine-induced killer cells generated from PBMCs showed upregulation of members of the Wnt signalling pathway (especially Wnt4), which is known to be involved in cell-cell adhesion, cell proliferation and differentiation.57 Similarly, in our study, several members of the Wnt family (Wnt4, Wnt11 and Wnt7) were upregulated, and cell adhesion and proliferation were the most significant GO terms. Furthermore, a study has shown that IL-15 stimulates the migratory response of NK cells by chemotaxis and increases their adhesion to vascular endothelium, which is the primary step for their extravasation and recruitment to tissue.58 Hence, we can speculate that the effect of cyto-IL-15 in upregulating genes involved in cell adhesion observed in our study could also be part of the role of IL-15 in augmenting NK cell adhesion and enabling their recruitment to the tumor.
Conclusion
We have demonstrated that intratumor administration of the cell membrane-anchoring IL-15, by providing a higher-localised effective dose and a potentially prolonged duration of action, generated an effective immune response that led to significant increases in both antitumor effect and survival of tumor-bearing animals without any toxicities in a prostate cancer model. Administration of IL-15 in combination with blockade of CTLA-4 and PD-L1 led to similar effects, which were slightly improved when the localisable forms of the agents were used, but not superior to that of cyto-IL-15 alone. A pharmacokinetic study to fully assess the intratumoral half-life of the cytotopically modified IL-15 will have to be performed in the future. However, this study provides the scientific basis for clinical trials where cyto-IL-15 will be used as a monotherapy administered directly into the tumors of patients with prostate cancer or other solid tumors.
Abbreviations
AICD, activation-induced cell death; CRPC, castration-resistant prostate cancer; CTLA-4, cytotoxic T lymphocyte antigen 4; DCs, dendritic cells; DMEM, Dulbecco’s Modified Eagle’s culture medium; FBS, fetal bovine serum; GO, gene ontology; H&E, haematoxylin and eosin; IL-2, interleukin-2; IL-15, interleukin-15; NK cells, natural killer cells; PAP, prostatic acid phosphatase; PBMCs, peripheral blood mononuclear cells; PD-1, programmed cell death protein 1; Pdcd4, programmed cell death protein 4; PD-L1, programmed death ligand 1; PSA, prostate-specific antigen; RBCs, red blood cells; RT, room temperature; SEM, standard error of the mean; Sema3e, semaphorin-3e; TCEP, tris(2-carboxyethyl)phosphine; TILs, tumor infiltrating lymphocytes; TRAMP, transgenic adenocarcinoma of the mouse prostate; Tregs, regulatory T cells; γc, common gamma chain.
Data Sharing Statement
The RNA sequencing datasets of the current study are available from the corresponding author on reasonable request.
Ethics Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance the Animals (Scientific Procedures) Act 1986 and with the ethical standards of the institution at which the studies were conducted under Project Licence Number (PPL) P731DA7F1.
No ethics approval was required for the use of blood from anonymized leukodepletion cones from healthy volunteers as no DNA sequencing was performed using the blood.
Acknowledgments
The authors would like to thank Dr G. Giustarini for performing the staining and flow cytometry analysis for the splenocytes ex vivo stimulation experiment (Supplementary data), and the staff at the Biological Services Unit (KCL) for animal care and maintenance.
Disclosure
Efthymia Papaevangelou, Dorota Smolarek, Richard A Smith, Prokar Dasgupta, and Christine Galustian report a patent, GB 1,913,804.9, pending to King's College London and the Prostate Cancer Research Centre Charity. The authors report no other possible conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Cheng HH, Lin DW, Yu EY. Advanced clinical states in prostate cancer. Urol Clin North Am. 2012;39(4):561–571. doi:10.1016/j.ucl.2012.07.011
3. Bruno TC, French JD, Jordan KR, et al. Influence of human immune cells on cancer: studies at the University of Colorado. Immunol Res. 2013;55(1–3):22–33. doi:10.1007/s12026-012-8346-y
4. Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60(1):199–215. doi:10.1111/j.1365-2559.2011.04033.x
5. Westdorp H, Skold AE, Snijer BA, et al. Immunotherapy for prostate cancer: lessons from responses to tumor-associated antigens. Front Immunol. 2014;5:191. doi:10.3389/fimmu.2014.00191
6. Vitkin N, Nersesian S, Siemens DR, Koti M. The tumor immune contexture of prostate cancer. Front Immunol. 2019;10:603. doi:10.3389/fimmu.2019.00603
7. McArdle PA, Canna K, McMillan DC, McNicol AM, Campbell R, Underwood MA. The relationship between T-lymphocyte subset infiltration and survival in patients with prostate cancer. Br J Cancer. 2004;91(3):541–543. doi:10.1038/sj.bjc.6601943
8. Lanciotti M, Masieri L, Raspollini MR, et al. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. Biomed Res Int. 2014;2014:486798. doi:10.1155/2014/486798
9. Liu Y, Saeter T, Vlatkovic L, et al. Dendritic and lymphocytic cell infiltration in prostate carcinoma. Histol Histopathol. 2013;28(12):1621–1628. doi:10.14670/HH-28.1621
10. Silvestri I, Cattarino S, Agliano AM, Collalti G, Sciarra A. Beyond the immune suppression: the immunotherapy in prostate cancer. Biomed Res Int. 2015;2015:794968. doi:10.1155/2015/794968
11. Wurz GT, Kao CJ, DeGregorio MW. Novel cancer antigens for personalized immunotherapies: latest evidence and clinical potential. Ther Adv Med Oncol. 2016;8(1):4–31. doi:10.1177/1758834015615514
12. Matera L. The choice of the antigen in the dendritic cell-based vaccine therapy for prostate cancer. Cancer Treat Rev. 2010;36(2):131–141. doi:10.1016/j.ctrv.2009.11.002
13. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17(11):3520–3526. doi:10.1158/1078-0432.CCR-10-3126
14. Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13(6):1810–1815. doi:10.1158/1078-0432.CCR-06-2318
15. McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med. 2013;2(5):662–673. doi:10.1002/cam4.106
16. Arora S, Velichinskii R, Lesh RW, et al. Existing and emerging biomarkers for immune checkpoint immunotherapy in solid tumors. Adv Ther. 2019;36(10):2638–2678. doi:10.1007/s12325-019-01051-z
17. Hansen AR, Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann Oncol. 2018;29(8):1807–1813. doi:10.1093/annonc/mdy232
18. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi:10.1056/NEJMoa1500596
19. Wu YM, Cieslik M, Lonigro RJ, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173(7):1770–1782 e1714. doi:10.1016/j.cell.2018.04.034
20. Bracarda S, Altavilla A, Hamzaj A, et al. Immunologic checkpoints blockade in renal cell, prostate, and urothelial malignancies. Semin Oncol. 2015;42(3):495–505. doi:10.1053/j.seminoncol.2015.02.004
21. Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 2001;14(2):105–110.
22. Giri JG, Kumaki S, Ahdieh M, et al. Identification and cloning of a novel IL-15 binding protein that is structurally related to the alpha chain of the IL-2 receptor. EMBO J. 1995;14(15):3654–3663. doi:10.1002/j.1460-2075.1995.tb00035.x
23. Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol. 1999;17:19–49. doi:10.1146/annurev.immunol.17.1.19
24. Carson WE, Fehniger TA, Haldar S, et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest. 1997;99(5):937–943. doi:10.1172/JCI119258
25. Marks-Konczalik J, Dubois S, Losi JM, et al. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A. 2000;97(21):11445–11450. doi:10.1073/pnas.200363097
26. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. doi:10.1038/ni1263
27. Berger C, Berger M, Hackman RC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. 2009;114(12):2417–2426. doi:10.1182/blood-2008-12-189266
28. Fehniger TA, Cooper MA, Caligiuri MA. Interleukin-2 and interleukin-15: immunotherapy for cancer. Cytokine Growth Factor Rev. 2002;13(2):169–183. doi:10.1016/S1359-6101(01)00021-1
29. Sakellariou C, Elhage O, Papaevangelou E, et al. Prostate cancer cells enhance interleukin-15-mediated expansion of NK cells. BJU Int. 2020;125(1):89–102. doi:10.1111/bju.14893
30. Yu P, Steel JC, Zhang M, et al. Simultaneous inhibition of two regulatory T-cell subsets enhanced Interleukin-15 efficacy in a prostate tumor model. Proc Natl Acad Sci U S A. 2012;109(16):6187–6192. doi:10.1073/pnas.1203479109
31. Munger W, DeJoy SQ, Jeyaseelan R
32. Roychowdhury S, May KF
33. Karegli J, Melchionna T, Farrar CA, et al. Thrombalexins: cell-localized inhibition of thrombin and its effects in a model of high-risk renal transplantation. Am J Transplant. 2017;17(1):272–280. doi:10.1111/ajt.13951
34. Smith RAG, Cassels R. Preparation and properties of a reversible conjugate of immunoglobulin G with the active centre of human tissue plasminogen activator. Fibrinolysis. 1988;2(4):189–195. doi:10.1016/0268-9499(88)90012-4
35. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
36. Papaevangelou E, Almeida GS, Box C, deSouza NM, Chung YL. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J Cancer. 2018;143(4):992–1002. doi:10.1002/ijc.31392
37. Anderson DM, Johnson L, Glaccum MB, et al. Chromosomal assignment and genomic structure of Il15. Genomics. 1995;25(3):701–706. doi:10.1016/0888-7543(95)80013-C
38. Patel H, Smith RA, Sacks SH, Zhou W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol. 2006;17(4):1102–1111. doi:10.1681/ASN.2005101116
39. Kassimatis T, Qasem A, Douiri A, et al. A double-blind randomised controlled investigation into the efficacy of Mirococept (APT070) for preventing ischaemia reperfusion injury in the kidney allograft (EMPIRIKAL): study protocol for a randomised controlled trial. Trials. 2017;18(1):255. doi:10.1186/s13063-017-1972-x
40. Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012;33(1):35–41. doi:10.1016/j.tips.2011.09.004
41. Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15. doi:10.1111/j.1365-2567.2009.03123.x
42. Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234(6):8509–8521. doi:10.1002/jcp.27782
43. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–214. doi:10.1016/j.cell.2015.03.030
44. Zhang M, Ju W, Yao Z, et al. Augmented IL-15Ralpha expression by CD40 activation is critical in synergistic CD8 T cell-mediated antitumor activity of anti-CD40 antibody with IL-15 in TRAMP-C2 tumors in mice. J Immunol. 2012;188(12):6156–6164. doi:10.4049/jimmunol.1102604
45. Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82. doi:10.1200/JCO.2014.57.3329
46. Rhode PR, Egan JO, Xu W, et al. Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol Res. 2016;4(1):49–60. doi:10.1158/2326-6066.CIR-15-0093-T
47. Romee R, Cooley S, Berrien-Elliott MM, et al. First-in-human Phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131(23):2515–2527. doi:10.1182/blood-2017-12-823757
48. Miller JS, Morishima C, McNeel DG, et al. A first-in-Human Phase I Study Of Subcutaneous Outpatient Recombinant Human IL15 (rhIL15) in adults with advanced solid tumors. Clin Cancer Res. 2018;24(7):1525–1535. doi:10.1158/1078-0432.CCR-17-2451
49. Marabelle A, Kohrt H, Caux C, Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res. 2014;20(7):1747–1756. doi:10.1158/1078-0432.CCR-13-2116
50. Waldmann TA, Lugli E, Roederer M, et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood. 2011;117(18):4787–4795. doi:10.1182/blood-2010-10-311456
51. Kim PS, Kwilas AR, Xu W, et al. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget. 2016;7(13):16130–16145. doi:10.18632/oncotarget.7470
52. Shimabukuro-Vornhagen A, Godel P, Subklewe M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. doi:10.1186/s40425-018-0343-9
53. Alamri A, Soussi Gounni A, Kung SKP. View point: semaphorin-3E: an emerging modulator of natural killer cell functions? Int J Mol Sci. 2017;18:11. doi:10.3390/ijms18112337
54. Matsuhashi S, Manirujjaman M, Hamajima H, Ozaki I. Control mechanisms of the tumor suppressor PDCD4: expression and functions. Int J Mol Sci. 2019;20:9. doi:10.3390/ijms20092304
55. Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, Colburn NH. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res. 2008;68(5):1254–1260. doi:10.1158/0008-5472.CAN-07-1719
56. Rohena-Rivera K, Sanchez-Vazquez MM, Aponte-Colon DA, Forestier-Roman IS, Quintero-Aguilo ME, Martinez-Ferrer M. IL-15 regulates migration, invasion, angiogenesis and genes associated with lipid metabolism and inflammation in prostate cancer. PLoS One. 2017;12(4):e0172786. doi:10.1371/journal.pone.0172786
57. Wang W, Meng M, Zhang Y, et al. Global transcriptome-wide analysis of CIK cells identify distinct roles of IL-2 and IL-15 in acquisition of cytotoxic capacity against tumor. BMC Med Genomics. 2014;7:49. doi:10.1186/1755-8794-7-49
58. Allavena P, Giardina G, Bianchi G, Mantovani A. IL-15 is chemotactic for natural killer cells and stimulates their adhesion to vascular endothelium. J Leukoc Biol. 1997;61(6):729–735. doi:10.1002/jlb.61.6.729
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.